

Abstract
Plasmacytoid predendritic cells (pDCs) are the main producers of type I interferon (IFN) in response to Toll-like receptor (TLR) stimulation. Phosphatidylinositol-3 kinase (PI3K) has been shown to be activated by TLR triggering in multiple cell types; however, its role in pDC function is not known. We show that PI3K is activated by TLR stimulation in primary human pDCs and demonstrate, using specific inhibitors, that PI3K is required for type I IFN production by pDCs, both at the transcriptional and protein levels. Importantly, PI3K was not involved in other proinflammatory responses of pDCs, including tumor necrosis factor α and interleukin 6 production and DC differentiation. pDCs preferentially expressed the PI3K δ subunit, which was specifically involved in the control of type I IFN production. Although uptake and endosomal trafficking of TLR ligands were not affected in the presence of PI3K inhibitors, there was a dramatic defect in the nuclear translocation of IFN regulatory factor (IRF) 7, whereas nuclear factor κB activation was preserved. Thus, PI3K selectively controls type I IFN production by regulating IRF-7 nuclear translocation in human pDCs and could serve as a novel target to inhibit pathogenic type I IFN in autoimmune diseases.
Plasmacytoid pre-DCs (pDCs) are the main type I IFN producers in humans and mice (1). They play a key role in innate antiviral immune responses but can also evolve into potent APCs and be important players in adaptive response (1, 2). Activation of pDCs through Toll-like receptor (TLR) 7 and 9 can trigger both types of response, including large quantities of type I IFN production and/or DC differentiation (1). Synthetic CpG-containing oligonucleotides of the types A and B (CpG-A and -B) selectively induce type I IFN production and DC differentiation, respectively (3), whereas some microbial stimuli such as influenza virus (Flu), HSV, or CpG-C can simultaneously induce both responses (1). Two factors seem to be key for the induction of large quantities of type I IFN in pDCs: (a) the ability of the TLR ligand to bind its receptor in the early endosomal compartments (4, 5) and (b) the phosphorylation and nuclear translocation of the transcription factor IFN regulatory factor (IRF) 7 (6). This last step was shown to depend on IL-1 receptor–associated kinase 1 (7) and IκB kinase (IKK) α (8) in mouse pDCs. However, the molecular switch regulating type I IFN production versus DC differentiation in pDCs is not fully elucidated and could have important clinical implications, considering the link between a dysregulated TLR-induced IFN response and autoimmune diseases (9, 10).
The phosphatidylinositol-3 kinase (PI3K) pathway is involved in a variety of biological processes, including cell survival and proliferation, B and T cell receptor signaling, and activation of G protein–coupled receptors, such as chemokine receptors (11). PI3K contains regulatory subunits (p85 α and β) and catalytic subunits (p110 α, β, γ, and δ). PI3K γ and δ are preferentially expressed in cells of hemopoietic origin, whereas expression of PI3K α and β is ubiquitous. Accordingly, knockout mice for p110 α and β show embryonic lethality, whereas knockout mice for p110 γ and δ are viable and fertile and show altered phenotypes exclusively when their immune system is under acute stress (12). The PI3K pathway has been shown to be activated by various TLR ligands and can function as a positive or negative regulator of TLR responses depending on the cell type and the TLR ligand used (13). Inhibition of PI3K in mouse myeloid DCs and macrophages increased IL-12 production in response to TLR stimulation (13), a result compatible with the in vivo observation of a skewed Th1 response in PI3K p85α−/− mice (14) and susceptibility to microbial-induced sepsis in mice through an increased production of innate cytokines (15). In mouse CD4+ T cells, MyD88 was recently shown to activate PI3K and to enable CpG-mediated proliferation but not survival (16). In mouse macrophages, however, CpG oligodeoxynucleotide (ODN) promoted survival through TLR9 and the PI3K pathway (17). The function of PI3K in pDCs has not been evaluated. Cell type specificity of PI3K, as well as discrepancies in the role of PI3K between cell lines and primary cells (11), strengthened the need to study this pathway using human primary cells. In this report, we show that PI3K activation is an important early step in the signaling pathway leading to IRF-7 nuclear translocation and type I IFN production after TLR7 and 9 activation of human pDCs that differentially regulate the IRF-7 and NF-κB signaling pathways.
RESULTS AND DISCUSSION
TLR ligands induce PI3K-dependent Akt phosphorylation in primary human pDCs
To assess the activity of PI3K in primary human pDCs, we measured phosphorylated Akt (p-Akt), a downstream target of PI3K (11). p-Akt was not detected at significant levels in freshly sorted pDCs and was not induced by serum-containing medium (Fig. 1), as opposed to other cell-culture systems in which serum could induce PI3K activation (18). However, p-Akt was up-regulated at both 20 and 90 min of culture in the presence of CpG-C or Flu (Fig. 1, A and B). This increase was PI3K dependent because it could be blocked by the specific PI3K inhibitor LY294002 (LY) at both time points and for both TLR ligands (Fig. 1, A and B). TLR9 signaling could lead to PI3K activation in different cell types, such as CD4+ T cells (16), mouse macrophages (17), or splenic DCs (19). After TLR9 triggering, Akt phosphorylation was observed 30 min after CpG stimulation (16, 19), which was comparable to our data on human pDCs. This rapid response, together with the ability of MyD88 to associate to the p85 subunit of PI3K (16), supports a direct TLR-induced activation of PI3K rather than indirect activation through a TLR-induced autocrine loop.
Figure 1.

TLR triggering activates the PI3K pathway in human pDCs. Purified pDCs were cultured with (A) 1 μM CpG-C or (B) Flu (multiplicity of infection [MOI] = 2) for 20 or 90 min with or without the PI3K inhibitor LY at 1 μM. Cells were stained with anti–p-AKT, as specified in Materials and methods. Representative histograms of at least three separate experiments are shown.
Selective involvement of PI3K for type I IFN production by TLR-activated pDCs
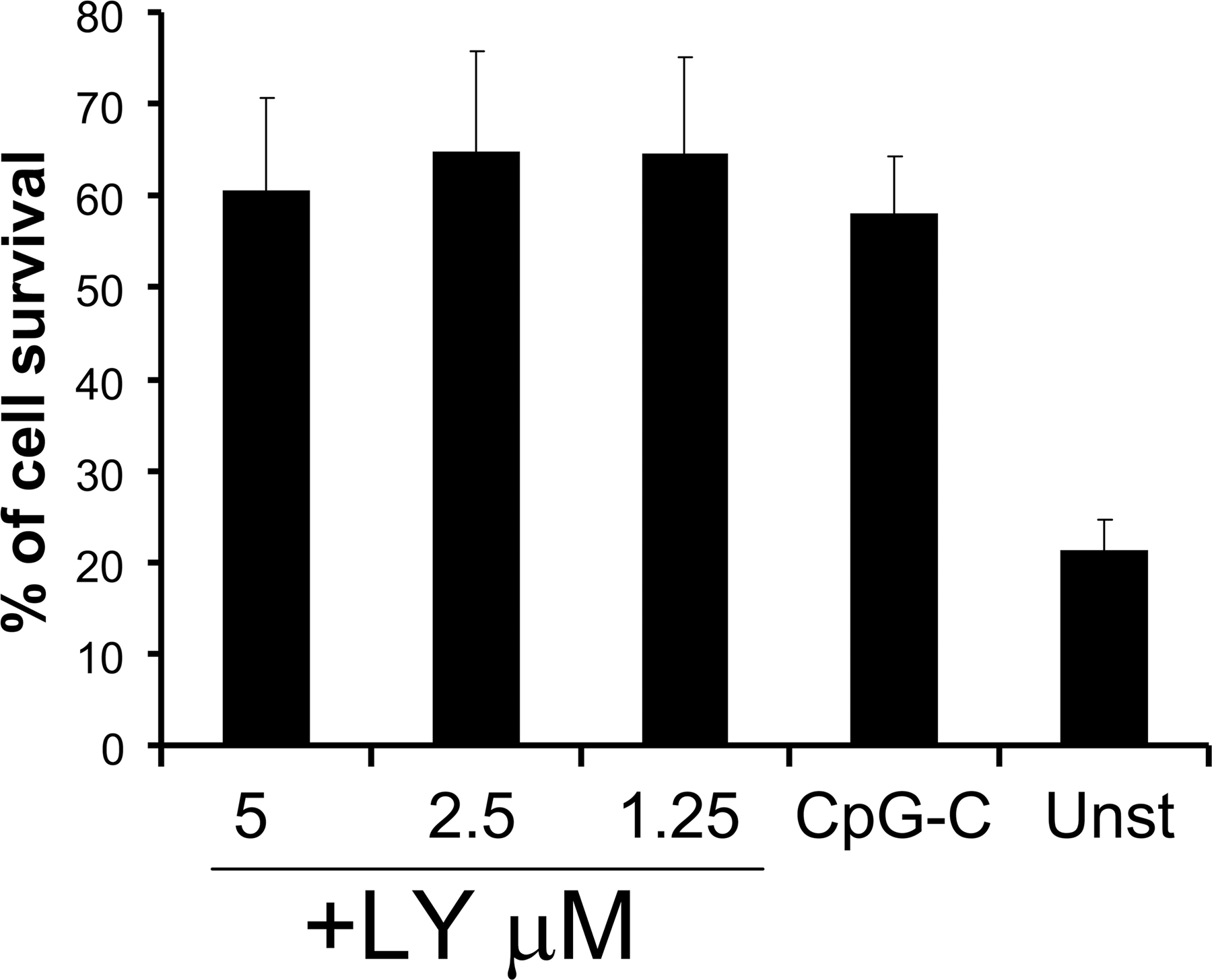
The selective inhibition of PI3K in TLR2, 4, and 9-stimulated mouse DCs and macrophages enhanced IL-12 production, suggesting that PI3K may negatively regulate TLR-induced inflammatory responses in APCs (13). To address its role in human pDCs, purified cells were stimulated with TLR9 (CpG-C, HSV) or TLR7 (Flu) ligands with or without the pharmacological inhibitors of PI3K, LY, and wortmannin. These TLR ligands induced high levels (20–30 ng/ml) of IFN-α production by freshly sorted pDCs (Fig. 2 A). This response was strongly inhibited by LY in a dose-dependent manner, with a maximal effect at 1.25 μM LY for both TLR7 and 9 ligands (Fig. 2 A). A 50% inhibition of IFN-α was still observed with LY concentrations as low as 0.08 μM for TLR9 (Fig. 2 A). Similarly, strong inhibition of IFN-α was observed in CpG-A–stimulated pDCs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070763/DC1). Importantly, no negative effect on pDC viability was observed at any of the concentrations used (Fig. S2). Similar results were obtained with wortmannin (not depicted), another inhibitor targeting the PI3K pathway.
Figure 2.

P13K inhibition selectively inhibits TLR7- and 9-mediated IFN-α response in human pDCs. (A) Purified pDCs were cultured with 1 μM CpG-C, HSV (MOI = 5), or Flu (MOI = 1) either alone or in combination with various concentration of the PI3K inhibitor LY (in micromolars) for 16 h. IFN-α production was evaluated by ELISA. Means of one experiment with 3 independent donors (representative of >15 donors) are shown. (B) Purified pDCs were cultured with 1 μM CpG-C alone or in the presence of 2 μM of LY inhibitor for 2 and 5 h. The expression levels of IFN-α, -ω, and -β were measured by real-time PCR. The mean of three independent donors is shown. (C) Purified pDCs were cultured with CpG-C or Flu, either alone or in the presence of LY inhibitor. The supernatants were collected after 5 h, after which the cells were washed twice and restimulated with CpG-C or Flu for another 12 h. IFN-α production was evaluated by ELISA. The mean of three independent donors is shown. Data were analyzed using a two-tailed Student's t test. *, P < 0.05.
Specificity of signaling inhibitors can be an issue, especially in cultures exceeding several hours. To exclude nonspecific effects caused by the potential toxicity of using PI3K inhibitors that could affect important functions of pDCs, we performed two types of experiments. First, we cultured pDCs for shorter periods of 2 and 5 h, and analyzed the ability of PI3K inhibitors to inhibit the IFN-α response at the transcriptional level. After 2 h, we detected significant IFN-α, -β, and IFN-ω messenger RNA in the presence of CpG-C, which was almost completely inhibited by LY (Fig. 2 B). The same magnitude of inhibition was observed at 5 h of culture (Fig. 2 B). Second, we attempted to reverse the inhibition of IFN-α production by washing out the inhibitor. After 5 h of culture, CpG-induced IFN-α production was inhibited in the presence of LY (Fig. 2 C). Washing out the inhibitor after the first 5 h enabled pDCs to recover their ability to produce large amounts of IFN-α during the subsequent 12 h (Fig. 2 C).
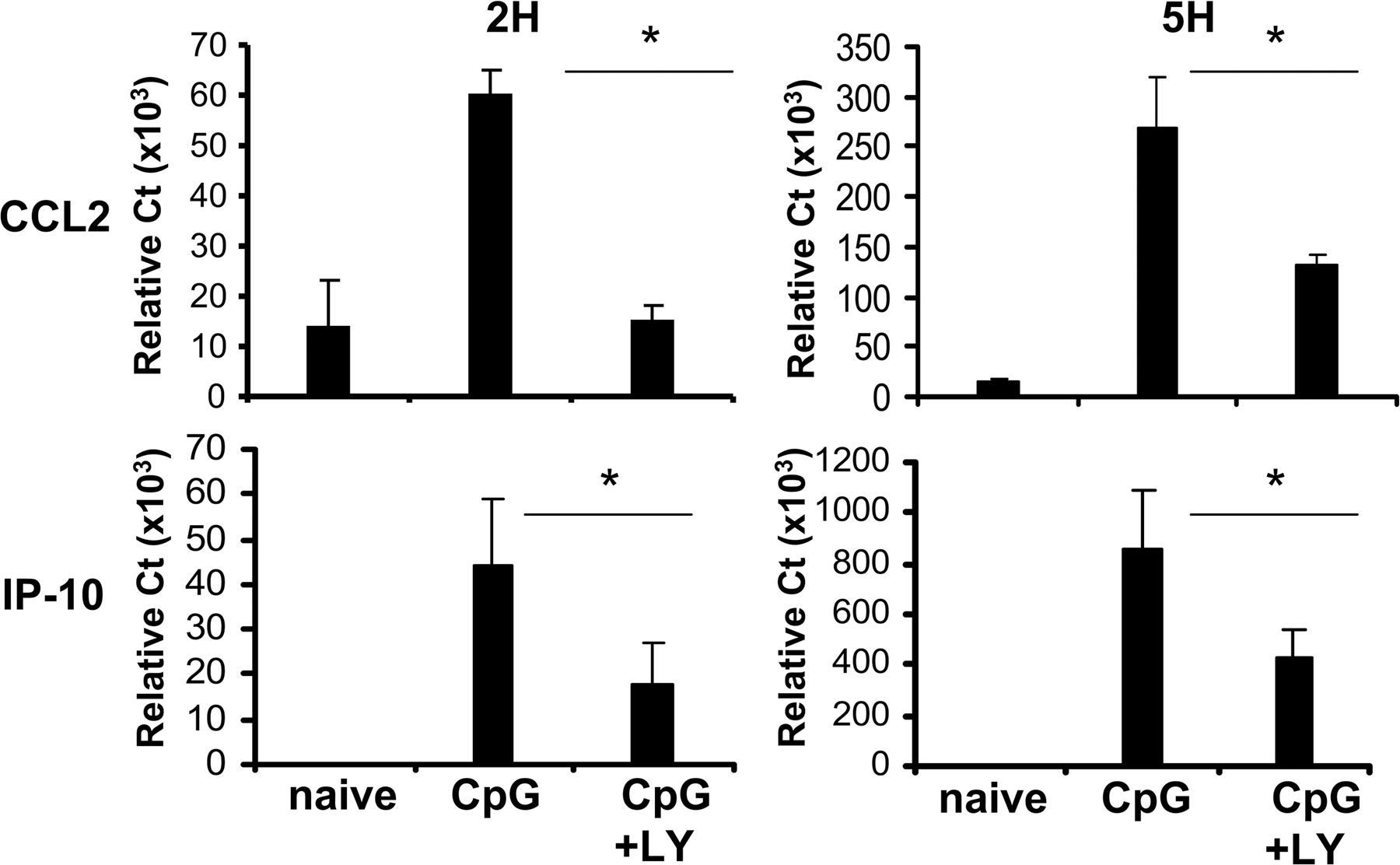
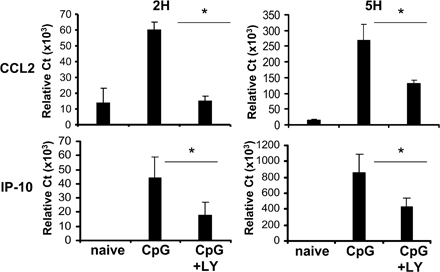
Autocrine IFN-α signaling was shown to account for a portion of the induction of chemokines, such as CC chemokine ligand (CCL) 2 and IFN-γ–inducible protein 10 (IP-10), in response to TLR9 activation (20). Consistent with a strong inhibition of IFN-α production, PI3K inhibition induced a 70% reduction in the expression of CCL2 and IP-10 in CpG-activated pDCs (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070763/DC1).
In addition to large amounts of type I IFNs, TLR activation of pDCs can induce the production of proinflammatory cytokines such as IL-6 and TNF-α. In contrast to the strong inhibition of type I IFN, TNF-α and IL-6 production by pDCs in response to both TLR9 or 7 ligands was not significantly affected by the addition of LY, even at high (5 μM) concentrations of the inhibitor (Fig. 3, A and B; and Fig. S1). This was confirmed at the transcriptional level (Fig. 3 C). Similarly, the pDCs differentiation into mature DCs, as assessed by surface expression of the co-stimulatory molecules CD80 and CD86, was not significantly affected by PI3K inhibitors (Fig. 3 D; and Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20070763/DC1). These data demonstrate that PI3K is selectively involved in the IFN-α pathway but not in the signaling events required for TNF-α or maturation induction. Moreover, they show that important functional pathways are conserved in pDCs despite PI3K inhibition, which, together with the conserved viability of pDCs, demonstrate that the observed effect on IFN-α was not caused by the overall toxicity of the inhibitor. Importantly, the PI3K inhibitor wortmannin was previously used as a means to inhibit autophagy in Flu-activated mouse pDCs without any effect in type I IFN production, suggesting major species-specific differences in the regulation of type I IFN production in human compared with mouse pDCs (21). In contrast to our data, inhibition of IFN-α in human pDCs using specific inhibitors of TLRs (22), or after cross-linking of surface ILT7 (23) or BDCA2 (24), induced a parallel decrease in TNF-α and IL-6 production, suggesting a different molecular mechanism.
Figure 3.

P13K inhibition does not affect inflammatory cytokines or maturation of pDCs in response to TLR7/9 triggering. Purified pDCs were cultured with (A) 1 μM CpG-C immunostimulatory sequence (ISS) or HSV (MOI = 5) or (B) Flu (MOI = 1) either alone or in combination with various concentrations of the PI3K inhibitor LY (in micromolars) for 16 h. IL-6 and TNF-α production were evaluated by ELISA. Means of one experiment with 3 independent donors (representative of >15 donors) are shown. (C) Purified pDCs were cultured with 1 μM CpG-C alone or in the presence of 2 μM of LY inhibitor for 2 and 5 h. The expression levels of IL-6 and TNF-α were measured by real-time PCR. The mean of three independent donors is shown. (D) Cells were stimulated as indicated and characterized for CD80 and CD86 expression by flow cytometry analysis. Data shown are representative of at least 10 donors (see Fig. S4).
To further define the function of the different subunits of PI3K in human pDCs, we addressed (a) their expression profile and (b) their respective contribution to regulate type I IFN in pDCs. First, we show that freshly purified and activated pDCs preferentially expressed the p85α regulatory subunit and the p110δ catalytic subunit (Fig. 4 A). Second, the PI3K δ–specific inhibitor IC87114 (25) inhibited IFN-α production in a dose-dependent manner (Fig. 4 B), whereas when we cultured pDCs in the presence of the PI3K γ–specific inhibitor AS604850, we did not observe any effect on IFN-α production unless used at high concentration (>20 μM), where its specificity for the γ subunit is lost (26). These results demonstrate that the PI3K δ subunit is the essential subunit involved in the production of IFN-α by pDCs.
Figure 4.

PI3K δ is essential for IFN-α production by pDCs in response to TLR stimulation. (A) Expression of class I A p110 (α, β, and δ) and class I B (γ) PI3K subunits in human pDCs. Purified pDCs were cultured for 6 h as indicated, and RNA was extracted and analyzed by quantitative PCR. Expression levels are expressed after normalization to β-actin. Data are shown as the mean with standard deviation from three independent donors. (B) Purified pDCs were cultured with 1 μM CpG-C, either alone or in combination with various concentrations of the PI3K inhibitors p110 γ AS 604850, p110 δ IC 87114, or LY for 16 h. IFN-α production was measured by ELISA. The mean of three independent donors is shown.
PI3K inhibition does not affect the uptake and endosomal location of CpG ODN
CpG ODN requires both uptake and localization into appropriate endosomal compartments to signal through TLR9. It is possible that PI3K is required for one or both of these steps, rather than for signal transduction downstream of TLR9. To test the role of PI3K in uptake, we used fluorescent CpG ODN and demonstrated that inhibiting PI3K with LY or wortmannin did not have any effect on CpG uptake, as measured by flow cytometry (Fig. S5 A, available at http://www.jem.org/cgi/content/full/jem.20070763/DC1). PI3K was described to be important for phagocytosis and endocytosis in various cellular models (27), partly by contributing to phagosome formation and maturation (27, 28). In addition, it was previously shown that blocking PI3K resulted in a complete blockade of CpG ODN uptake in mouse myeloid DCs and TLR9-transfected HEK 293 cells (29). Differences in the cell type used could account for this discrepancy.
We and others have shown recently that the nature of the pDC response to TLR9 strongly depends on the intracellular compartment where the receptor/ligand interaction occurs (4, 5). In human pDCs, the production of IFN-α is associated with the trafficking of CpG in the early endosomal compartment, whereas maturation in APCs required accumulation of the CpG within the late endosomal compartment (5). We therefore investigated whether PI3K inhibition would interfere with the localization of the CpG in the early endosome compartment, a situation that is predicted to hamper IFN-α response. Transferrin receptor and Lamp-1 were used as markers of early and late endosomes, respectively. As previously described (5), in the absence of PI3K inhibition, fluorescent CpG-C colocalized with transferrin receptor– as well as Lamp-containing endosomal compartments (Fig. S5 B). This pattern of distribution was not affected by PI3K inhibitors, indicating that PI3K does not interfere with the intracellular trafficking of CpG in primary pDCs (Fig. S5, B and C). Importantly, this shows that although we cannot exclude that blocking PI3K may have some effect on endosomal trafficking, it did not prevent the localization of the CpG in the early endosome that is essential for triggering IFN-α at time points where inhibition of IFN-α was almost complete by gene expression analysis (Fig. 2 B). Furthermore, the concentration of LY was similar to the one used to demonstrate inhibition of IFN-α at a similar time of stimulation (Fig. 2 B). These data show that PI3K does not interfere with the uptake and distribution of the TLR ligands and suggest that it could be an important player in the signaling pathway downstream of TLR7 or 9 activation.
PI3K is required for IRF-7 nuclear translocation but not NF-κB phosphorylation in TLR-activated pDCs
In mouse pDCs, IFN-α production depends on the activation and translocation of IRF-7 to the nucleus (6). Moreover the strong up-regulation of IRF-7 messenger was suggested to be key for the high magnitude of IFN-α response upon TLR7/9 ligation in human pDCs (30). We thus investigated whether PI3K alters this pathway by looking at both transcriptional up-regulation of IRF-7 and its ability to migrate to the nucleus upon activation. First, we observed that freshly sorted pDCs constitutively expressed IRF-7 messenger RNA, and that its level was increased 2 and 5 h after CpG stimulation (Fig. 5 A). This transcriptional up-regulation of IRF-7 was not affected in the presence of PI3K inhibitor (Fig. 5 A). We then studied the nuclear translocation of IRF-7. Using confocal microscopy, we found that IRF-7 protein was expressed in the cytoplasm of unstimulated pDCs and did not colocalize with the DAPI nuclear staining (Fig. 5 B). MHC class II surface staining was used to visualize the pDCs. After stimulation with CpG, the majority of IRF-7 translocated to the nucleus, as assessed by the colocalization of the IRF-7 (red) and DAPI (blue) stainings, as well as the reduction of detectable IRF-7 staining in the cytoplasmic compartment (Fig. 5 B). This process was dramatically decreased in the presence of a PI3K inhibitor, with the majority of the IRF-7 staining remaining in the cytoplasm (Fig. 5 B). The total number of cells showing nuclear staining of IRF-7 returned to baseline levels in the presence of LY (Fig. 5 C). Similar results were obtained with both IFN-inducing classes of CpG, A and C, as well as with HSV (not depicted).
Figure 5.

PI3K is critical for the nuclear translocation of IRF-7 in pDCs but does not block IRF-7 up-regulation. (A) 105 purified pDCs were cultured with or without 1 μM CpG-C ISS alone or in the presence of 5 μM of LY inhibitor. IRF-7 expression was evaluated 2 and 5 h after stimulation by real-time PCR. The mean of three independent donors is shown. (B) 2 × 105 purified pDCs were left untreated or stimulated with CpG alone or in the presence of LY inhibitor for 3 h. Cells were visualized using the membrane staining of class II molecule (FITC), whereas the nucleus was identified using DAPI. IRF-7 nuclear translocation was visualized by immunofluorescence with IRF-7 antibody (Alexa Fluor 555, red). Representative cells of at least four independent donors are shown. Bar, 5 μm. (C) Between 50 and 70 cells from at least four different donors were analyzed for IRF-7 translocation in the nuclei. Cells were considered positive when at least 20% of the IRF-7 fluorescence was localized in the nucleus. (D, left) Purified pDCs were cultured with 1 μM CpG-C ISS with or without 1 or 5 μM of the PI3K inhibitor LY, or 0.5 μM of NF-κB inhibitor for 90 min (representative histograms are shown). (right) Expression intensity values as mean fluorescent intensity (MFI). The mean of three experiments is shown. *, P < 0.05. (E) Purified pDCs were cultured with 1 μM CpG-C ISS with or without 5 μM of the PI3K inhibitor LY for 4 h. Nuclear extracts were analyzed for the binding activity of NF-κB p50 and p65 family members. Data are shown as the fold of increase to unstimulated (mean ± SEM) of three separate experiments.
As shown in Figs. 2 and 3, PI3K appears to be essential for IFN-α response but not for other inflammatory cytokines and DC differentiation, two responses that were shown in the mouse to be mostly NF-κB dependent (31). We show that in parallel to IRF-7 activation, CpG-C also induced phosphorylation of NF-κB, as assessed by flow cytometry (Fig. 5 D). Interestingly, although the NF-κB phosphorylation was inhibited using a specific NF-κB inhibitor, we did not observe any significant effect of the PI3K inhibitor LY (Fig. 5 D). To confirm that the NF-κB pathway was not affected after PI3K inhibition, we analyzed pDC nuclear extracts for the binding activity of NF-κB p50 and p65 subunits. No difference was detected in the absence or presence of LY (Fig. 5 E). These data suggest an absence of cross talk between the PI3K and NF-κB pathways in human pDCs . This may not be the case in other cell types, as PI3K could favor NF-κB activity in cell lines (32).
Our results provide a molecular link between PI3K activity and the regulation of type I IFN production by pDCs, and identify PI3K as an essential component of the pathway leading to IFN production in pDCs. IRF-7 was shown to be essential for IFN-α production by pDCs in mice (6) and to form a complex with MyD88 and TNF receptor–associated factor 6 for the induction of type I IFN (33). However, the factors regulating its phosphorylation and subsequent translocation to the nucleus are not completely elucidated, and no data are available in human pDCs. Recently, two independent studies showed that intracellular osteopontin (34), as well as IKK-α (8), were required for IRF-7 nuclear translocation and type I IFN production in mouse pDCs. Our data show that PI3K is a key component of the signal transduction pathway that controls IRF-7 nuclear translocation and subsequent type I IFN production by human pDCs. In addition, a previous report has suggested that PI3K could act as a negative regulator during the initial phase of innate response to microbial pathogens (13). On the contrary, our results suggest that PI3K is essential for pDCs to respond properly to viruses by favoring early production of type I IFN. Which is the specific target of PI3K, and whether the PI3K pathway regulates the function and/or recruitment of TNF receptor–associated factor 6, osteopontin, or IKK-α, will be important questions to be addressed in future studies.
The dissection of the molecular mechanisms controlling the innate functions of pDCs could uncover new ways to manipulate these cells in pathological conditions, such as autoimmune or infectious diseases. There is increasing evidence for a role of IFN-α in the development of autoimmunity, and pDCs, through their production of high levels of type I IFN, were implicated in the pathophysiology of various autoimmune diseases, such as systemic lupus erythematosus, psoriasis, or Sjögren's disease (9, 10). PI3K inhibitors, in particular those specifically targeting the δ subunit as nonspecific PI3K inhibitors could generate toxicity, offer a unique way to selectively block type I IFN production while preserving NF-κB–dependent responses, which could have important proinflammatory or regulatory effects through modulation of T cell responses.
MATERIALS AND METHODS
Reagents.
Oligonucleotides CpG-C C274 and CpG-A D19 were prepared as previously described (3). UV-inactivated HSV-1 was a gift from R. Pyles (University of Texas Medical Branch, Galveston, TX). Heat-inactivated influenza virus (H1N1, strain A/PR/8/34) was obtained from the American Type Culture Collection. PI3K inhibitors (LY and wortmannin), and the NF-κB inhibitor (IKK-2 IV) were purchased from Calbiochem. PI3K γ inhibitor (AS 604850) was purchased from Echelon. PI3K δ inhibitor (IC 87114) was synthesized as previously described (patent application US 20050261317 A1).
Human IFN-α, IL-6, and TNF-α ELISA sets were purchased from PBL Biomedical Laboratories; anti-CD123, anti-CD80, anti-CD86, anti-CD71, and anti-CD107a were purchased from BD Biosciences; and anti–BDCA-2 was purchased from Miltenyi Biotec.
Isolation and stimulation of purified human cell subsets.
Buffy coats were obtained from the Stanford Blood Center and cells were used under internal Institutional Review Board–approved protocols, or they were obtained from adult healthy donors at the Saint-Antoine Crozatier Blood Bank, where all donors signed informed consent to allow the use of their blood for research purposes. This study was approved by the Institut Curie Internal Review Board and by the French National Blood Agency. pDCs were isolated either by using positive selection using BDCA-4–conjugated beads or by using negative depletion (Miltenyi Biotec), as previously described (35). pDCs were 94–99% BDCA2+ CD123+, as determined by flow cytometry.
Detection of p-AKT and NF-κB by flow cytometry.
Negatively purified pDCs were stimulated with CpG-C, and cells were immediately fixed with 4% paraformaldehyde for 15 min at 37°C. Cells were then washed, permeabilized with PermBuffer III (BD Biosciences) for 30 min on ice, and stained with either Alexa Fluor 647 anti–human NF-κB p65 (pS529) or with Alexa Fluor 647 anti–human AKT (pS473; BD Biosciences) for 30 min and analyzed by flow cytometry.
NF-κB transcription factor binding activity.
Negatively purified pDCs were stimulated and nuclear extracts were prepared. NF-κB activities were measured using TransAM NF-κB kits (Active Motif) according to the manufacturer's instructions.
Real-time quantitative PCR (TaqMan) analysis.
PCR reactions were performed as described previously (3). In brief, threshold cycle (CT) values for each gene were normalized to the housekeeping gene ubiquitin or β-actin using the formula Eq. 1.8 (HSKGENE) (100,000), where HSK is the mean CT of triplicate housekeeping gene runs, GENE is the mean CT of duplicate runs of the gene of interest, and 100,000 is arbitrarily chosen as a factor to bring all values above 0.
Confocal microscopy.
Evaluation of intracellular localization of CpG was performed as previously described (5). IRF-7 detection was performed as follows: purified pDCs (2 × 105/200 μl in 96-well round-bottom plates) were stimulated with 1 μM CpG-A or CpG-C either alone or in the presence of 5 μM of the PI3K inhibitor LY for 3 h. Cells were first stained with anti–human MHC class II–FITC and subsequently fixed with 2% paraformaldehyde and then permeabilized with 100% ice-cold methanol for 10 min at −20°C. Samples were labeled with rabbit polyclonal anti–human IRF-7 (Santa Cruz Biotechnology, Inc.). Anti–rabbit IgG Alexa Fluor 555 (Invitrogen) was used as secondary antibody. Cells were seeded on glass slides by cytospin and mounted using Prolong antifade with DAPI (Invitrogen).
Images were acquired using a confocal microscope (LSM 510 META; Carl Zeiss, Inc.) and a 63×/1.4 NA objective, with the pinhole set for a section thickness of 0.8 μm. Images were acquired sequentially using separate laser excitation to avoid any cross talk between the fluorophore signals.
Online supplemental material.
Fig. S1 shows the effect of PI3K inhibition on pDCs stimulated with CpG-A and CpG-B. Fig. S2 shows the effect of LY on pDC survival. Fig. S3 shows the relative expression of the IFN-inducible genes CCL2 and IP-10 in CpG-activated pDCs in the presence of LY. Fig. S4 shows the cumulative data on the effect of PI3K inhibition on pDC maturation. Fig. S5 shows the endosomal localization of CpG in pDCs in the presence of the PI3K inhibitor. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070763/DC1.
Supplemental Material
Acknowledgments
We would like to thank Philippe Benaroch, Tyler Jacks, and our colleagues at Dynavax Technologies for their critical reading of the manuscript. We thank Holly L. Aaron for invaluable assistance with confocal analysis.
This work was supported by the Alliance for Lupus Research (F.J. Barrat) and an Institut National de la Santé et de la Recherche Médicale Avenir grant (to V. Soumelis).
C. Guiducci, T. Matray, R.L. Coffman and F.J. Barrat are full-time employees of Dynavax Technologies. The authors have no other conflicting financial interests.
F.J. Barrat and V. Soumelis contributed equally to this work.
References
- 1.Liu, Y.J. 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23:275–306. [DOI] [PubMed] [Google Scholar]
- 2.Colonna, M., G. Trinchieri, and Y.J. Liu. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226. [DOI] [PubMed] [Google Scholar]
- 3.Duramad, O., K.L. Fearon, J.H. Chan, H. Kanzler, J.D. Marshall, R.L. Coffman, and F.J. Barrat. 2003. IL-10 regulates plasmacytoid dendritic cell response to CpG-containing immunostimulatory sequences. Blood. 102:4487–4492. [DOI] [PubMed] [Google Scholar]
- 4.Honda, K., Y. Ohba, H. Yanai, H. Negishi, T. Mizutani, A. Takaoka, C. Taya, and T. Taniguchi. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 434:1035–1040. [DOI] [PubMed] [Google Scholar]
- 5.Guiducci, C., G. Ott, J.H. Chan, E. Damon, C. Calacsan, T. Matray, K.D. Lee, R.L. Coffman, and F.J. Barrat. 2006. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J. Exp. Med. 203:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda, K., H. Yanai, H. Negishi, M. Asagiri, M. Sato, T. Mizutani, N. Shimada, Y. Ohba, A. Takaoka, N. Yoshida, and T. Taniguchi. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 434:772–777. [DOI] [PubMed] [Google Scholar]
- 7.Uematsu, S., S. Sato, M. Yamamoto, T. Hirotani, H. Kato, F. Takeshita, M. Matsuda, C. Coban, K.J. Ishii, T. Kawai, et al. 2005. Interleukin-1 receptor–associated kinase-1 plays an essential role for Toll-like receptor (TLR)7– and TLR9-mediated interferon-α induction. J. Exp. Med. 201:915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshino, K., T. Sugiyama, M. Matsumoto, T. Tanaka, M. Saito, H. Hemmi, O. Ohara, S. Akira, and T. Kaisho. 2006. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature. 440:949–953. [DOI] [PubMed] [Google Scholar]
- 9.Kanzler, H., F.J. Barrat, E.M. Hessel, and R.L. Coffman. 2007. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 13:552–559. [DOI] [PubMed] [Google Scholar]
- 10.Colonna, M. 2006. Toll-like receptors and IFN-alpha: partners in autoimmunity. J. Clin. Invest. 116:2319–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deane, J.A., and D.A. Fruman. 2004. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu. Rev. Immunol. 22:563–598. [DOI] [PubMed] [Google Scholar]
- 12.Rommel, C., M. Camps, and H. Ji. 2007. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. 7:191–201. [DOI] [PubMed] [Google Scholar]
- 13.Fukao, T., and S. Koyasu. 2003. PI3K and negative regulation of TLR signaling. Trends Immunol. 24:358–363. [DOI] [PubMed] [Google Scholar]
- 14.Fukao, T., M. Tanabe, Y. Terauchi, T. Ota, S. Matsuda, T. Asano, T. Kadowaki, T. Takeuchi, and S. Koyasu. 2002. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3:875–881. [DOI] [PubMed] [Google Scholar]
- 15.Williams, D.L., C. Li, T. Ha, T. Ozment-Skelton, J.H. Kalbfleisch, J. Preiszner, L. Brooks, K. Breuel, and J.B. Schweitzer. 2004. Modulation of the phosphoinositide 3-kinase pathway alters innate resistance to polymicrobial sepsis. J. Immunol. 172:449–456. [DOI] [PubMed] [Google Scholar]
- 16.Gelman, A.E., D.F. LaRosa, J. Zhang, P.T. Walsh, Y. Choi, J.O. Sunyer, and L.A. Turka. 2006. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 25:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sester, D.P., K. Brion, A. Trieu, H.S. Goodridge, T.L. Roberts, J. Dunn, D.A. Hume, K.J. Stacey, and M.J. Sweet. 2006. CpG DNA activates survival in murine macrophages through TLR9 and the phosphatidylinositol 3-kinase-Akt pathway. J. Immunol. 177:4473–4480. [DOI] [PubMed] [Google Scholar]
- 18.Tsuruta, F., N. Masuyama, and Y. Gotoh. 2002. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J. Biol. Chem. 277:14040–14047. [DOI] [PubMed] [Google Scholar]
- 19.Park, Y., S.W. Lee, and Y.C. Sung. 2002. Cutting Edge: CpG DNA inhibits dendritic cell apoptosis by up-regulating cellular inhibitor of apoptosis proteins through the phosphatidylinositide-3′-OH kinase pathway. J. Immunol. 168:5–8. [DOI] [PubMed] [Google Scholar]
- 20.Megjugorac, N.J., H.A. Young, S.B. Amrute, S.L. Olshalsky, and P. Fitzgerald-Bocarsly. 2004. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J. Leukoc. Biol. 75:504–514. [DOI] [PubMed] [Google Scholar]
- 21.Lee, H.K., J.M. Lund, B. Ramanathan, N. Mizushima, and A. Iwasaki. 2007. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 315:1398–1401. [DOI] [PubMed] [Google Scholar]
- 22.Barrat, F.J., T. Meeker, J. Gregorio, J.H. Chan, S. Uematsu, S. Akira, B. Chang, O. Duramad, and R.L. Coffman. 2005. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 202:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao, W., D.B. Rosen, T. Ito, L. Bover, M. Bao, G. Watanabe, Z. Yao, L. Zhang, L.L. Lanier, and Y.J. Liu. 2006. Plasmacytoid dendritic cell–specific receptor ILT7–FcεRIγ inhibits Toll-like receptor–induced interferon production. J. Exp. Med. 203:1399–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dzionek, A., Y. Sohma, J. Nagafune, M. Cella, M. Colonna, F. Facchetti, G. Gunther, I. Johnston, A. Lanzavecchia, T. Nagasaka, et al. 2001. BDCA-2, a novel plasmacytoid dendritic cell–specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon α/β induction. J. Exp. Med. 194:1823–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadhu, C., B. Masinovsky, K. Dick, C.G. Sowell, and D.E. Staunton. 2003. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J. Immunol. 170:2647–2654. [DOI] [PubMed] [Google Scholar]
- 26.Camps, M., T. Ruckle, H. Ji, V. Ardissone, F. Rintelen, J. Shaw, C. Ferrandi, C. Chabert, C. Gillieron, B. Francon, et al. 2005. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 11:936–943. [DOI] [PubMed] [Google Scholar]
- 27.Gillooly, D.J., A. Simonsen, and H. Stenmark. 2001. Phosphoinositides and phagocytosis. J. Cell Biol. 155:15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vieira, O.V., R.J. Botelho, L. Rameh, S.M. Brachmann, T. Matsuo, H.W. Davidson, A. Schreiber, J.M. Backer, L.C. Cantley, and S. Grinstein. 2001. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishii, K.J., F. Takeshita, I. Gursel, M. Gursel, J. Conover, A. Nussenzweig, and D.M. Klinman. 2002. Potential role of phosphatidylinositol 3 kinase, rather than DNA-dependent protein kinase, in CpG DNA-induced immune activation. J. Exp. Med. 196:269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai, J., N.J. Megjugorac, S.B. Amrute, and P. Fitzgerald-Bocarsly. 2004. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J. Immunol. 173:1535–1548. [DOI] [PubMed] [Google Scholar]
- 31.O'Keeffe, M., R.J. Grumont, H. Hochrein, M. Fuchsberger, R. Gugasyan, D. Vremec, K. Shortman, and S. Gerondakis. 2005. Distinct roles for the NF-kappaB1 and c-Rel transcription factors in the differentiation and survival of plasmacytoid and conventional dendritic cells activated by TLR-9 signals. Blood. 106:3457–3464. [DOI] [PubMed] [Google Scholar]
- 32.Yang, C.H., A. Murti, S.R. Pfeffer, J.G. Kim, D.B. Donner, and L.M. Pfeffer. 2001. Interferon alpha/beta promotes cell survival by activating nuclear factor kappa B through phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 276:13756–13761. [DOI] [PubMed] [Google Scholar]
- 33.Kawai, T., S. Sato, K.J. Ishii, C. Coban, H. Hemmi, M. Yamamoto, K. Terai, M. Matsuda, J. Inoue, S. Uematsu, et al. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5:1061–1068. [DOI] [PubMed] [Google Scholar]
- 34.Shinohara, M.L., L. Lu, J. Bu, M.B. Werneck, K.S. Kobayashi, L.H. Glimcher, and H. Cantor. 2006. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat. Immunol. 7:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duramad, O., K.L. Fearon, B. Chang, J.H. Chan, J. Gregorio, R.L. Coffman, and F.J. Barrat. 2005. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J. Immunol. 174:5193–5200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}