

Abstract
Toll-like receptors (TLRs) are pattern recognition receptors of innate immunity. TLRs initiate inflammatory pathways that may exacerbate chronic inflammatory diseases like atherosclerosis. En face laser scanning confocal microscopy (LSCM) of isolated aortic segments revealed the distribution of intimal TLR2 expression and the atheroprotective outcomes resulting from a TLR2 deficiency. TLR2 expression was restricted to endothelial cells in regions of disturbed blood flow, such as the lesser curvature region, in atherosclerosis-prone, low-density lipoprotein receptor–deficient (LDLr −/−) mice. Diet-induced hyperlipidemia in LDLr −/− mice increased this regional endothelial TLR2 expression. Bone marrow (BM) reconstitution of LDLr −/− and LDLr −/− TLR2 −/− mice created chimeric mice with green fluorescent protein (GFP) expression in BM-derived cells (BMGFP+). Lesser curvature BMGFP+ leukocyte accumulation, lipid accumulation, foam cell generation and endothelial cell injury were all increased by hyperlipidemia, whereas hyperlipidemic double mutant BMGFP+ LDLr −/− TLR2 −/− mice had reduced BMGFP+ leukocyte accumulation, lipid accumulation, foam cells, and endothelial cell injury. This is the first report of in vivo site-specific expression of endothelial cell TLR2. Expression of this receptor on endothelial cells contributed to early atherosclerotic processes in lesion-prone areas of the mouse aorta.
Epidemiological and experimental studies have established a causal relationship between hyperlipidemia, inflammation, and atherosclerotic disease (1, 2). Toll-like receptors (TLRs) are pattern-recognition receptors of innate immunity that initiate inflammatory pathways critical in host responses to pathogens (3). Elicitation of such inflammatory pathways requires TLR activation via exogenous microbial-derived ligands, with each TLR binding a distinct subset of exogenous agonists (4). TLR2 detects a range of exogenous microbial components, such as Gram-positive–derived lipoteichoic acid and bacterial lipoproteins. Accumulating evidence also suggests the existence of host-derived endogenous TLR2 ligands, many of which are found within atherosclerotic lesions. Candidate endogenous TLR2 ligands include the following: the extracellular matrix components, including biglycan and hyaluronan; components of modified low-density lipoprotein (LDL); the nuclear chromatin protein high-mobility group box 1; and serum amyloid A.
Atherosclerosis is a chronic inflammatory process with myriad cellular and humoral effectors (5). Lesions are characterized by accumulation of lipids and macrophage-derived foam cells within the intima layer of the vessel wall. The disease process can be categorized into initiating and progressive stages that, in some instances, may lead to vulnerable plaque rupture. Different atherogenic stimuli and cell effectors participate at different stages of lesion development. Lesion initiation occurs by stimuli that arise from both the vascular wall (local) and from the systemic circulation. However, the site-specificity of lesions emphasizes the importance of local factors in their initiation, specifically hemodynamic shear stress (6, 7). In sites of lesion predilection, disturbed blood flow coupled with hyperlipidemia results in endothelial cell activation, which is a prerequisite to fatty streak formation (1).
We reported that total deletion of TLR2 in hyperlipidemic LDL receptor–deficient (LDLr −/−) mice is atheroprotective, whereas BM cell–specific deletion of TLR2 has no effect on lesion severity in the absence of a defined exogenous TLR2 agonist (8). Our in vitro studies established that human coronary artery endothelial cells exposed to laminar flow express lower levels of TLR2 compared with endothelial cells exposed to static or disturbed flow (7). A flow-dependent effect on endothelial TLR4 activation is not observed. These data implicate a role for endothelial TLR2 up-regulation at sites of disturbed blood flow, like the lesion-prone regions of the aorta. We hypothesize that endothelial TLR2 activation contributes to endothelial cell activation, thereby intensifying the early steps of atherogenesis, such as lipid accumulation and leukocyte accumulation.
The lesser curvature of the murine aortic arch is a well-documented site of disturbed blood flow and lesion predilection (9). In contrast, the ventral and dorsal margins of the lesser curvature region extending to the greater curvature experience laminar flow and are lesion resistant (10). En face laser scanning confocal microscopy (LSCM) was used to create images that spanned the aortic lesser curvature. Composite images were created by mapping areas of the intimal surface and stitching together the contiguous image fields. The resultant composite images covered a relatively large aortic surface area and allowed for visualization of the cellular events occurring within both laminar and disturbed blood flow regions of the aortic arch.
Herein, we demonstrate enhanced aortic endothelial TLR2 expression that was confined to regions that experience disturbed blood flow. In atherosclerosis-susceptible LDLr −/− mice, consumption of a high-fat diet (HFD) increased endothelial TLR2 expression within a very restricted region of the lesser curvature of the aortic arch. BM reconstitution of LDLr −/− and double-mutant LDLr −/− TLR2 −/− recipient mice with BM constitutively expressing GFP (BMGFP+) generated mouse chimeras (BMGFP+ LDLr −/− and BMGFP+ LDLr −/− TLR2 −/−), that allowed for quantitation of intimal BMGFP+ leukocyte accumulation. During lesion initiation, BMGFP+ LDLr −/− mice fed a HFD had BMGFP+ leukocyte accumulation that was confined to the lesser curvature. BMGFP+ LDLr −/− TLR2 −/− recipient mice also demonstrated increased intimal BMGFP+ leukocyte accumulation with continued consumption of the HFD that was also confined to the lesser curvature region. However, relative to BMGFP+ LDLr −/− recipient mice, BMGFP+ LDLr −/− TLR2 −/− had significantly reduced BMGFP+ leukocyte accumulation. TLR2 deficiency was also associated with fewer endothelial cell changes, reduced lipid accumulation, and less foam cells within the lesser curvature region. Analysis of the time course of these changes indicates that endothelial TLR2 expression both preceded and contributed to atherosclerotic lesion development in hyperlipidemic LDLr −/− mice.
RESULTS
Aortic endothelial cell TLR2 expression was confined to areas of disturbed blood flow
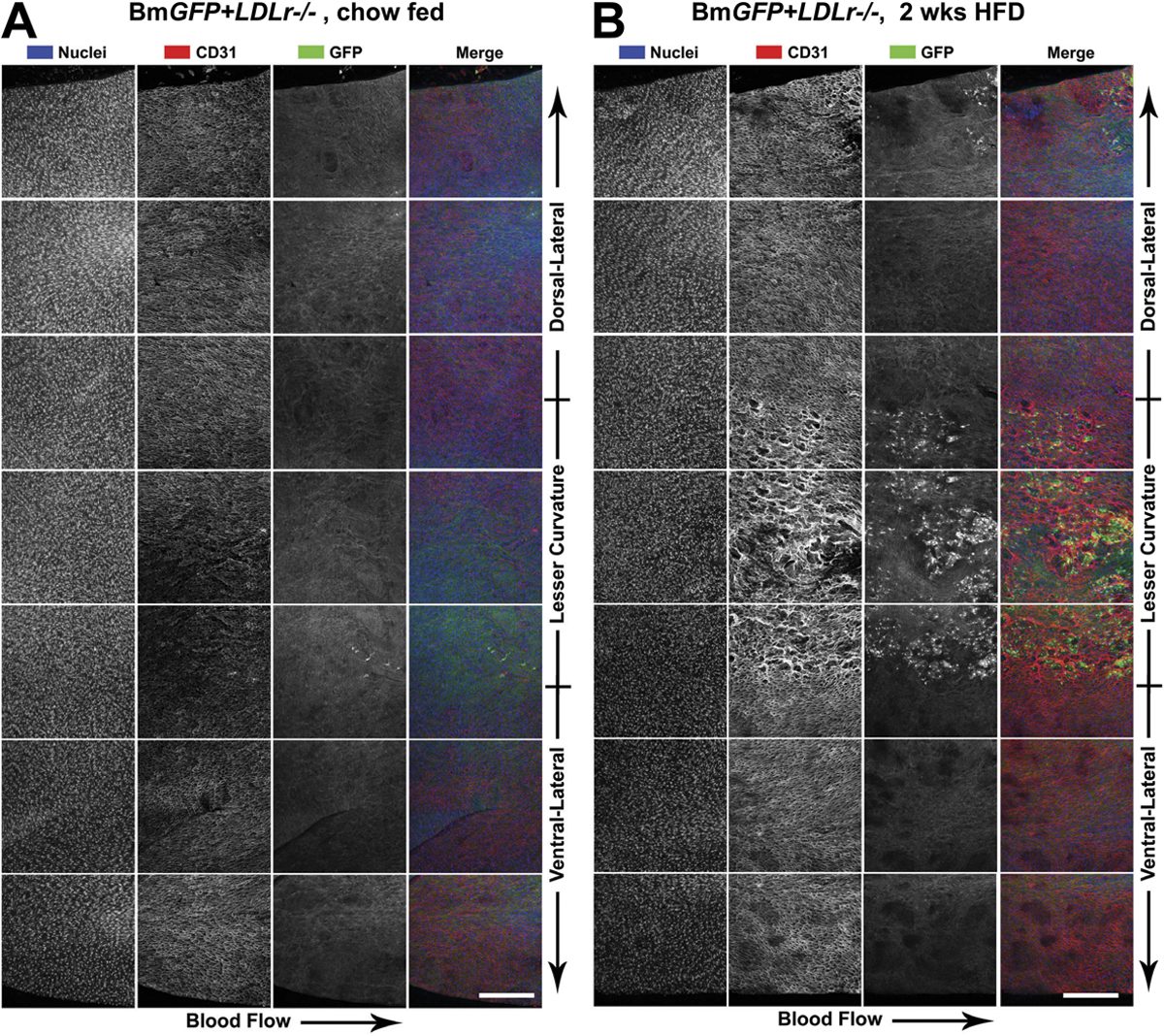
Endothelial cell TLR2 expression was first evaluated by traditional sectioning methods in aortic rings excised from chow- and HFD-fed atherosclerotic-susceptible female LDLr −/− mice (Fig. 1, A and B). In these aortic cross sections, TLR2 expression (green) was confined to the endothelial layer, which was stained bright red with anti-CD31 (anti–platelet-endothelial cell adhesion molecule-1). Perturbations of the endothelial layer after 4 wk of diet-induced hyperlipidemia were observed concomitant with an increase in endothelial TLR2 expression (Fig. 1 B). Because these cross sections did not allow for an observation of TLR2 expression or endothelial cell–specific changes over a large surface area of the aorta, an en face approach was used to allow for more extensive examination of early lesional events on the luminal surface of the aorta.
Figure 1.

Aortic TLR2 expression was confined to endothelial cells within regions of disturbed blood flow. Aortic segments were cut from the ascending aorta of C57BL/6 and LDLr −/−mice. Endothelial TLR2 expression was visualized in the cross sections with LSCM. Cross sections of the aortic arch from chow-fed (A) or 4-wk HFD-fed (B) LDLr −/− mice demonstrated regional TLR2 expression (green) that was confined to the endothelium that was also stained with anti-CD31 (bright red). Insets show higher magnification of the lesser curvature of the ascending aorta. Bars, 500 μm.
Hyperlipidemia increased endothelial TLR2 expression within areas of disturbed blood flow
The distribution of endothelial TLR2 expression during early lesion development was next assessed in hyperlipidemic atherosclerotic-susceptible female LDLr −/− mice by en face LSCM. The lesser curvature region and beyond were mapped by creating contiguous images vertically spanning the lesser curvature, which were later stitched together as a large composite “circumferential strip” image (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071096/DC1). Each circumferential strip image allowed for an analysis of large areas of the luminal surface that spanned the entire circumferential surface, from the greater to the lesser curvature.
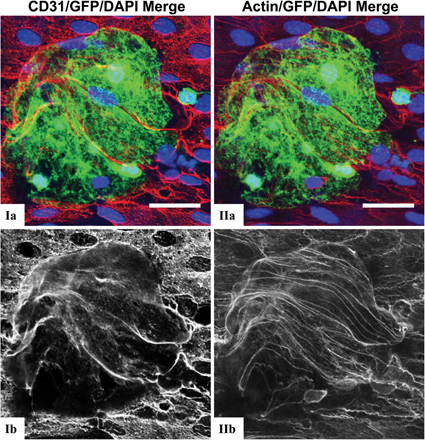
Aortae were harvested after 0, 1, 2, and 4 wk of consuming the HFD, at a time when lesions could not be visualized by traditional en face staining of pinned aortas. Circumferential strip images centering on the lesser curvature of the proximal aortic arch revealed that TLR2 expression (green) was confined to the lesser curvature and was progressively increased by 18-fold with 4 wk of hyperlipidemia (from 4,955 ± 360 to 89,587 ± 2,130 μm2; Fig. 2 A). High-resolution LSCM of this region after 4 wk (Fig. 2 B) revealed a marked alteration of the endothelium (red), as made evident by the large vacuole-like endothelial cell boundaries that were often positive for TLR2 expression (yellow). These endothelial changes were not observed in regions known to experience laminar flow and were related to the extent of endothelial TLR2 expression in disturbed flow regions as seen by comparing the separate grayscale images of TLR2 and CD31 staining (Fig. 2 A). The distribution of TLR2 staining was not dependent on tissue permeabilization, demonstrating that the lack of subendothelial TLR2 staining was not an artifact of the staining procedure (Fig. 3, A and B). Cytoskeleton actin staining via rhodamine phalloidin demonstrated a pattern of endothelial cell changes that was similar to CD31 staining (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20071096/DC1), and confirmed that these morphological changes were not specific to CD31 expression. These images show that endothelial cells overlying clusters of leukocytes are significantly distended or stretched and appear to have lost CD31 staining at some cell junctions. As expected, aortic tissue segments from LDLr −/− TLR2 −/− mice fed the atherogenic diet for 1 or 4 wk were negative for TLR2 staining (Fig. 2 A). Additionally, the changes in endothelial cell morphology after 4 wk of hyperlipidemia were mitigated by TLR2 deficiency (Fig. 2 A) and, most likely, were the result of the marked reduction in foam cells in these aortae. Thus, endothelial cell TLR2 expression in LDLr −/− mice was confined to areas of disturbed blood flow. Endothelial TLR2 expression was increased with continued exposure to hyperlipidemia, and TLR2 deficiency led to less prominent hyperlipidemic-induced changes in endothelial cell morphology.
Figure 2.

Hyperlipidemia increased endothelial TLR2 expression within the lesser curvature. LDLr −/− or LDLr −/− TLR2 −/− mice were fed the HFD for up to 4 wk, and their aortae were harvested for a qualitative analysis of endothelial TLR2 expression. The tissues were examined as illustrated in Fig. S1. (A) Increasing weeks of HFD consumption (1–4 wk) resulted in a progressive increase of endothelial TLR2 expression (green), from 4,955 ± 360 (T0) to 89,587 ± 2130 μm2 (T4), and it altered endothelial cell morphology (red) in these circumferential strips. Nuclei are blue in all images. Both events were confined to the lesser curvature regions within the aortic arch, and they are shown in grayscale images below the merged color images. Comparable images from LDLr −/− TLR2 −/− mice fed the HFD for 1 or 4 wk demonstrated the specificity of the TLR2 staining and also illustrated reduced endothelial cell changes. (B) Higher resolution en face LSMC of endothelial TLR2 expression from the 4-wk time point of the areas marked I and II within the insets in A. Note the enlarged vacuole-like endothelial cell boundaries and profuse endothelial TLR2 expression (green; demarcated by the white arrowheads). Bars: (A) 150 μm; (B) 50 μm. Fig. S1 is available at http://www.jem.org/cgi/content/full/jem.20071096/DC1.
Figure 3.

Aortic TLR2 staining in the presence or absence of tissue permeabilization. (A) 20× lesser curvature circumferential strips taken from BMGFP+ LDLr−/− mice fed the HFD for 4 wk revealed no changes in TLR2 staining distribution in the presence or absence of tissue permeabilization. (B) Higher resolution 100× confocal images of lesser curvature regions from Fig. 3 A. Colocalization of GFP (green) and TLR2 staining (red), if it occurred, would have appeared orange to yellow on the higher power images. Bars: (A) 150 μm; (B) 25 μm.
TLR2 deficiency decreased intimal leukocyte accumulation
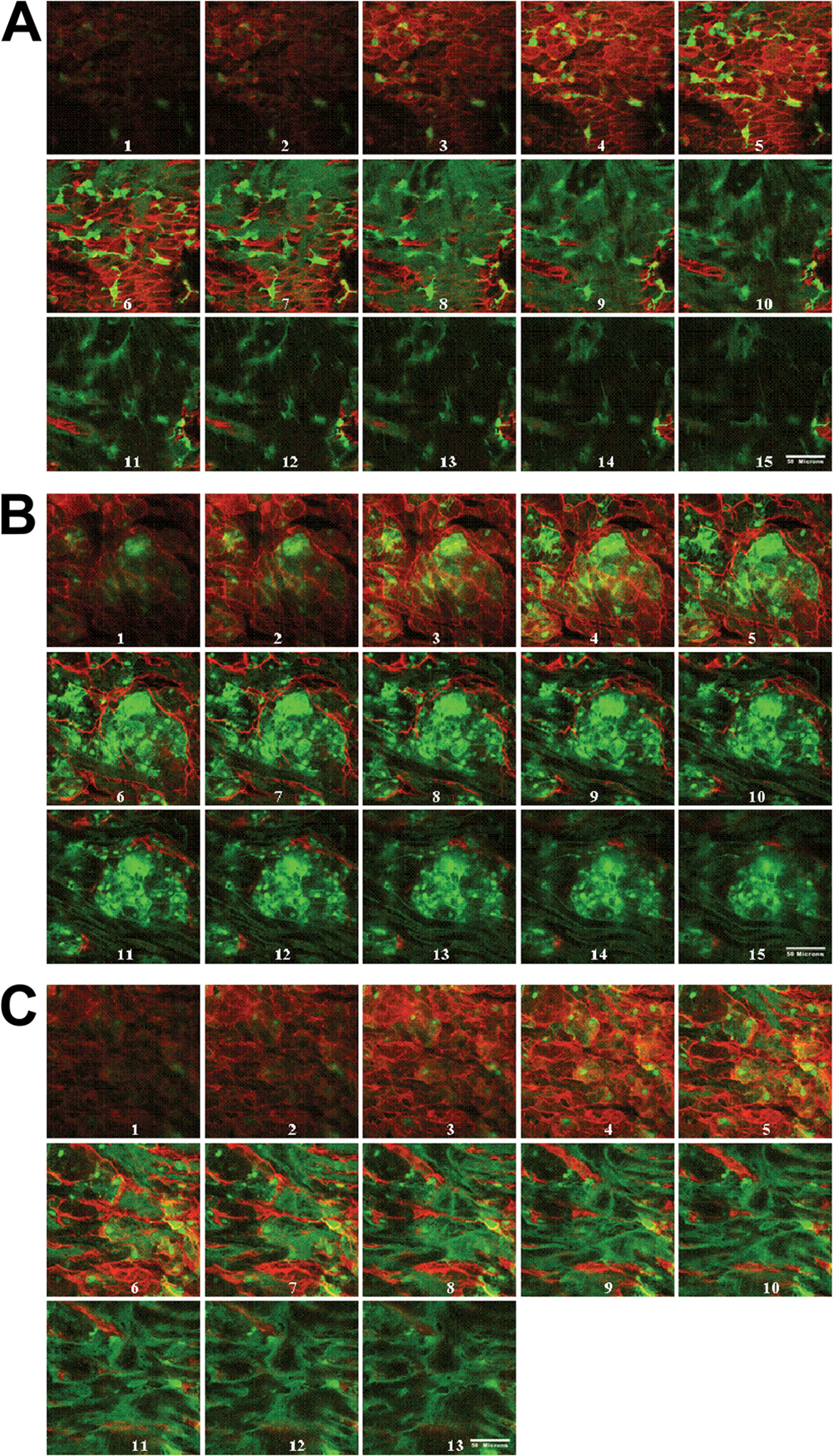
Because BM-derived leukocyte accumulation into the intima is a critical event in atherosclerotic lesion development, we tracked leukocyte cell entry during early lesion development. BM transplantation (BMT) was performed in lethally irradiated female C57BL/6, LDLr −/−, or LDLr−/−TLR2−/− recipient mice that were reconstituted with donor BM extracted from mice constitutively expressing GFP. Normolipidemic BMGFP+ C57BL/6 mice demonstrated a BMGFP+ cell infiltrate in the lesser curvature region (Fig. 4 A). These BMGFP+ cells displayed long cellular processes and were CD68+, identifying them as macrophages or dendritic cells derived from circulating monocytes (Fig. 4 B). The dendritic cell marker CD11c could also be visualized in these sections associated with the BM GFP+ cells (unpublished data). The GFP fluorescence provided an enhanced signal-to-noise ratio that was much higher than could be achieved with antibody staining and avoided the possibility of staining artifacts as well. BMGFP+ leukocyte accumulation was also observed in chow-fed BMGFP+ LDLr −/− mice, and upon exposure to hyperlipidemia, the BMGFP+ leukocytes appeared as enlarged foam cell–like macrophages that no longer displayed the long cellular processes characteristic of macrophages or dendritic cells found in the normolipidemic chimeric BMGFP+ C57BL/6 aortas (Fig. 4 A). These enlarged foam cells were not observed in the normolipidemic wild-type BMGFP+ C57BL/6 chimeras, and this is consistent with the idea that vascular tissue–resident macrophages differentiate into foam cells upon exposure to hyperlipidemia. Direct demonstration that the BMGFP+ cells were beneath the endothelial cells (and not just adherent to the luminal surface) was obtained by examination of cross-sectional images of z-series stacks (Fig. S3, A–C, available at http://www.jem.org/cgi/content/full/jem.20071096/DC1). Computer-generated 3D renditions of these z-series stacks confirmed that the endothelial cell nuclei were above the BMGFP+ cells and the elongated smooth muscle cell nuclei were beneath the BMGFP+ cells (Fig. S4). In BMGFP+ LDLr −/− mice, the number of BMGFP+ leukocytes increased upon exposure to hyperlipidemia (Fig. S5, A and B). Importantly, in normolipidemic BMGFP+ C57BL/6 and hyperlipidemic BMGFP+ LDLr −/− aortas, intimal BMGFP+ leukocytes were confined within the lesser curvature region and not found in the dorsal or ventral areas of the aorta.
Figure 4.

Characterization of BMGFP+ cell infiltrate in healthy and atherosclerotic aortic tissue. C57BL/6 and LDLr −/− mice underwent BM transplantation, receiving BM from GFP+ donor mice. (A) Aortic tissue segments from chimeric mice that had consumed the HFD diet for 5 wk revealed BMGFP+ cell infiltrate (green) below the endothelial layer (red) within the lesser curvature. In the normolipidemic BMGFP+ C57BL/6 recipient mice, intimal BMGFP+ cells had long cellular processes, as expected for tissue macrophages or dendritic cells. In hyperlipidemic BMGFP+ LDLr −/− recipient mice, intimal BMGFP+ cells were swollen with a foam cell–like appearance. DAPI-stained nuclei are not shown. (B) The intimal BMGFP+ cells (green) were positive for CD68 (red), which is a marker for monocyte-derived macrophages or dendritic cells. In this image, the endothelial cells are blue. A comparison of the CD68 and the GFP images indicated that the GFP was fairly uniformly distributed within the intracellular compartment of the BMGFP+ cells. Bars: (A) 50 μm; (B) 25 μm.
Other groups have shown that BM-derived endothelial progenitor cells can repopulate the endothelial layer in disturbed flow regions (12). To determine if BMGFP+ cell endothelial progenitor cells could be identified, the sections were also stained for stem cell antigen or Sca-1. Although some Sca-1+ cells were observed, very few of them were also GFP+ (unpublished data).
The time course of intimal BMGFP+ leukocyte accumulation was evaluated in BMGFP+ LDLr −/− chimeric mice fed the HFD. Analogous to the composite circumferential strip images described previously, the luminal surface was mapped by creating contiguous images horizontally spanning the lesser curvature (Fig. S1). The resultant “lesser curvature strip” images allowed for a quantitation of BMGFP+ leukocyte accumulation in the entire lesser curvature region within each aortic segment (Fig. 5 A). Four mice fed the HFD were killed at 2 wk intervals for up to 10 wk. The rate of lesser curvature BMGFP+ leukocyte infiltrate, quantified as fractional GFP+ area or GFP+ cell count (Fig. 5 B), increased during the first 8 wk of consuming the HFD. By 10 wk, en face LSCM could not detect changes in BMGFP+ leukocyte accumulation. Because lesion development and leukocyte accumulation would be expected to continue, a likely explanation for the apparent plateau of leukocyte accumulation is a lack of sensitivity of en face LSCM to detect increased BMGFP+ leukocyte accumulation that occurred in the z axis direction (in and out of the plane of the endothelium) with the imaging parameters that were used. As described in the Materials and methods, the z axis resolution of the 20× lesser curvature image scans was ∼4 μm. BMGFP+ infiltrate that occurred beyond 4 μm would not be detected in the image scans. Such volume changes in a lesion would be expected to occur during advanced lesion development. However, these phenomena were difficult to quantitatively assess with en face LSCM, unless extensive z-series scans were done during the 20× mapping routine. The extensive imaging that such scans would require precluded us from performing this analysis.
Figure 5.

BMGFP+ leukocyte infiltrate progressively increased within the lesser curvature during hyperlipidemia. LDLr −/− mice underwent BM transplantation, receiving BM from GFP+ donor mice. The chimeras were used to evaluate the rate of BMGFP+ leukocyte accumulation and endothelial cell changes during HFD consumption. (A) Representative lesser curvature composite images of aortic segments collected at 2 wk intervals demonstrated a progressive increase in BMGFP+ leukocyte accumulation (green) that was accompanied by morphological changes in the endothelium (red). Nuclei are blue. Both events were confined to the lesser curvature region. The green and red color channels are also shown as single grayscale images. Bar, 500 μm. (B) BMGFP+ leukocyte fractional areas expressed as the percentage of total area (gray boxes) and numbers of nuclei per mm2 (gray diamonds) was quantified from the lesser curvature composite images. Four mice were examined per group per time point.
Like cellular TLR2 expression, BMGFP+ leukocyte accumulation was confined to the lesser curvature region and was accompanied by a progressive alteration of endothelial cell morphology, which was seen by comparing the separate grayscale images of GFP fluorescence and CD31 staining (Fig. 5 A). The time course of hyperlipidemic-induced BMGFP+ leukocyte accumulation appeared to differ from that of hyperlipidemic-induced endothelial cell TLR2 expression. Increased endothelial cell TLR2 expression was evident before 4 wk of HFD consumption (Fig. 2 A), and appeared to precede major BMGFP+ leukocyte accumulation that was evaluated after 4 wk of hyperlipidemia in the study shown in Fig. 5.
Rates of BMGFP+ leukocyte accumulation were also evaluated in a second study in which female BMGFP+ LDLr −/− TLR2 −/− and BMGFP+ LDLr −/− chimeric mice were fed the HFD. Groups of 3 chimeric mice were killed at 2-wk time points for up to 8 wk. As compared with the BMGFP+ LDLr −/− mice, the BMGFP+ LDLr −/− TLR2 −/− mice had qualitatively less BMGFP+ leukocyte infiltrate after 4 wk (Fig. 6 A). BMGFP+ leukocyte accumulation, quantified by fractional GFP+ leukocyte area, GFP+ leukocyte counts, GFP+ leukocyte cluster area, and GFP+ leukocyte nuclei per cluster, were all significantly decreased in TLR2−/− chimeras (Fig. 6 B). Importantly, the BMGFP+ leukocytes in these BMGFP+ LDLr −/− TLR2 −/− chimeras were obtained from TLR2 expressing GFP+ donors, yet intimal BMGFP+ leukocyte accumulation was still reduced.
Figure 6.

Endothelial cell TLR2 deficiency attenuated HFD-induced BMGFP+ leukocyte accumulation. LDLr −/− or LDLr −/− TLR2 −/− mice underwent BM transplantation, receiving BM from GFP+ donor mice. The chimeric mice were fed the atherogenic diet for up to 8 wk. At 2-wk intervals, 3–4 mice per group were killed and their aortae were analyzed for BMGFP+ cell accumulation and changes in endothelial cell morphology within the lesser curvature of the proximal aortic arch. (A) BMGFP+ LDLr −/− TLR2 −/− recipient mice had a qualitative decrease in lesser curvature BMGFP+ leukocyte accumulation (green and grayscale images) after 4 or 8 wk of HFD consumption. Nuclei are blue in the merged images. Bars, 250 μm. (B) After 8 wk of hyperlipidemia, the BMGFP+ LDLr −/− TLR2 −/− recipient mice had significantly reduced intimal BMGFP+ leukocyte accumulation within the lesser curvature region, as quantitated as GFP+ area, GFP+ cell count (GFP+ nuclei), and GFP+ cell density (GFP+ nuclei/cluster; *P < 0.05). Refer to the Materials and methods for a more complete description of these terms.
These same mice were also bled, and total plasma cholesterol, TNF-α, and monocyte chemoattractant protein-1 (MCP-1) were measured. In both BMGFP+ LDLr −/− and BMGFP+ LDLr −/− TLR2 −/− mice, consumption of the HFD increased total plasma cholesterol by approximately threefold, and these changes were comparable in both groups (Table I). MCP-1 measured in plasma samples taken during the HFD feeding period also were similar in both groups (BMGFP+ LDLr −/−, 57.7 ± 12.1 pg/ml vs. BMGFP+ LDLr −/− TLR2 −/−, 50.4 ± 7.1 pg/ml). However, a significant decrease in serum TNF-α was found in the TLR2−/− mice (BMGFP+ LDLr −/−, 12.9 ± 2.5 pg/ml vs. BMGFP+ LDLr −/− TLR2 −/−, 6.9 ± 1.5 pg/ml; P < 0.05). TNF-α production is regulated by TLR activation (13). Additionally, TLR2 deficiency was associated with elevated body weight gain, as seen by a twofold greater gain in body weight during the HFD feeding period (BMGFP+ LDLr−/−, 2.3 ± 0.3 g vs. BMGFP+ LDLr−/−TLR2−/−, 5.3 ± 0.7 g weight gain; P < 0.05).
Table I.
Plasma total cholesterol in BMGFP+ LDLr −/− and LDLr −/− TLR2 −/− chimeric mice
| Weeks of HFD consumptiona
|
|||||||
|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 8 | 10 | HFD-ave.b | |
| LDLr−/− | 241 ± 18 | 768 ± 75 | 1,064 ± 54 | 982 ± 91 | 1,064 ± 29 | 983 ± 97 | 972 ± 38 |
| n = 3 | n = 4 | n = 4 | n = 4 | n = 4 | n = 4 | n = 20 | |
| LDLr−/−TLR2−/− | 230 ± 8 | 784 ± 122 | 829 ± 45 | 936 ± 25 | 891 ± 69 | 1,013 ± 126 | 887 ± 37 |
| n = 3 | n = 3 | n = 4 | n = 3 | n = 4 | n = 3 | n = 17 | |
All values are reported as milligrams per deciliter ± the SEM. n, number of mice used for each time point
HFD-ave. was calculated from the cholesterol measurements taken during the HFD feeding period (2–10-wk time points).
Liver and splenic tissue samples were taken from hyperlipidemic BMGFP+ LDLr −/− and BMGFP+ LDLr −/− TLR2 −/− chimeric mice and analyzed for GFP+ protein at 5, 7, or 9 wk after BMT. ELISA analysis of GFP+ protein showed no differences in BM cell tissue reconstitution rates in BMGFP+ LDLr −/− versus BMGFP+ LDLr −/− TLR2 −/− recipient mice, indicating that a TLR2 deficiency in these recipient mice had no impact on BM-derived cell accumulation in the spleen or liver (unpublished data). Thus, the differences seen in intimal BMGFP+ leukocyte accumulation were most likely caused by intrinsic differences in the vascular beds of BMGFP+ LDLr −/− and BMGFP+ LDLr −/− TLR2 −/− mice.
TLR2 deficiency decreased early lipid accumulation in lesions
Aortic segments from female BMGFP+ LDLr −/− and BMGFP+ LDLr −/− TLR2 −/− chimeric mice were analyzed for lipid accumulation by en face LSCM. Nile red, which is a lipophilic dye that fluoresces red in neutral and/or polar lipids (11), identified lipid accumulation in the intima of the lesser curvature. Within 2 wk of consuming the HFD, BMGFP+ LDLr −/− TLR2 −/− chimeric mice had reduced lipid (red) accumulation within the lesser curvature compared with BMGFP+ LDLr −/− chimeras (Fig. 7 A). Additionally, BMGFP+ LDLr −/− chimeras had numerous cells costained with Nile red and GFP (orange and yellow), indicating that GFP protein and lipid are colocalized in foam cells. These events were far fewer in BMGFP+ LDLr −/− TLR2 −/− chimeras (Fig. 7 A, high magnification images), indicating fewer lipid-laden foam cells. Interestingly, lipid accumulation was observed that was not associated with BMGFP+ leukocytes, suggesting that intimal lipid retention was spatially and/or temporally distinct from increased leukocyte entry and foam cell generation. Quantitation of BMGFP+ foam cells (yellow and orange) or Nile red+ lipid (red, plus orange, and yellow) revealed significantly decreased lipid accumulation in BMGFP+ LDLr −/− TLR2 −/− chimeras (Fig. 7, B and C). Comparing the time course of BMGFP+ leukocyte accumulation (Fig. 6 B) and Nile red+ lipid accumulation (Fig. 7 C) demonstrated that TLR2 deficiency reduced intimal lipid accumulation 6 wk before significant decreases in leukocyte accumulation were detected. Such observations hint at the temporal events of lesion development.
Figure 7.

Endothelial cell TLR2 deficiency decreased early intimal lipid and foam cell accumulation. Aortic segments from BMGFP+ LDLr −/− chimeric mice fed the HFD were examined for the colocalization (yellow and orange) of Nile red lipid accumulation (red) and leukocyte accumulation (green) in BMGFP+ foam cells during early lesion development. (A) After 2 or 4 wk of consuming a HFD, compared with BMGFP+ LDLr −/− mice (Ia and IIa), lipid-laden BMGFP+ foam cells were reduced in BMGFP+ LDLr −/− TLR2 −/− recipient mice (Ib and IIb). Inset areas bounded by white boxes (120 × 120 μm) are shown at higher magnification to the right of each image. Bars, 25 μm. (B) BMGFP+ LDLr −/− TLR2 −/− chimeric mice had significantly reduced BMGFP+ foam cell (yellow and orange) number and (C) Nile red+ lipid accumulation (red, yellow and orange), as quantified from within the lesser curvature region. Three to four mice were analyzed for both groups and at each time point. *, P < 0.05.
TLR2 expression in early atherosclerotic lesions was predominantly endothelial cell derived
To clearly define the cells expressing TLR2, aortic segments were analyzed with LSCM using z-series optical sectioning. Although cultured GFP+ TLR2+/+ BM-derived macrophages stained positive with the TLR2-specific antibody (unpublished data), GFP+ BM-derived cells in en face aortic segments from BMGFP+ LDLr −/− chimeric mice were not positive for TLR2. Areas positive for TLR2 (red) were distinct from regions of BMGFP+ leukocyte accumulation (green; Fig. 3 B and Fig. 8, A and B). Although TLR2+ endothelial cells and BMGFP+ leukocytes were found in the same restricted region of the lesser curvature, TLR2 expression did not colocalize with BMGFP+ leukocytes. Analysis of z-series confocal image stacks in these early lesions further demonstrated colocalization of CD31+ and TLR2+ staining (purple) and confirmed that TLR2 expression was largely confined to the endothelial cells, whereas the BMGFP+ leukocytes occupied a distinct region within the intima, but below the endothelium (Fig. 8 C). The absence of TLR2 expression in BM-derived leukocytes was consistent with our previous report that leukocyte TLR2 expression did not participate in lesion formation in vivo in response to endogenous TLR2 agonists (8).
Figure 8.

Luminal TLR2 expression was confined to endothelial cells. Aortic segments from BMGFP+ LDLr −/− chimeric mice fed the HFD for 4 wk were examined to identify regions of TLR2 expression (red) and BMGFP+ leukocyte (green) accumulation. Endothelial cells (CD31) are blue in all images. (A) A circumferential composite image centered on the lesser curvature region demonstrated that areas of endothelial TLR2 staining (blue + red = purple) did not colocalize with BMGFP+ leukocytes. Inset areas bounded by white boxes (120 × 120 μm) are shown at higher magnification to the right of each image (B) A better appreciation of the distribution of TLR2, CD31, and GFP fluorescence can be seen with each fluorescence channel shown separately in grayscale. (C) A cross-sectional view of aortic TLR2, CD31, and BMGFP+ leukocyte accumulation further confirmed that TLR2 expression was confined to endothelial cells. Z-series image stacks spanning a depth of 9.5 μm were collected from the lesser curvature region. White lines i–iii denote the x-y locations where z-series imaging were performed. TLR2 expression (red) and BMGFP+ leukocyte (green) events did not colocalize; however, TLR2 expression was colocalized with CD31+ endothelial cells (blue+ red = purple). Bars, 25 μm.
DISCUSSION
The cellular changes that occurred within the intimal layer during early atherosclerotic lesion development were characterized by en face LSCM. This analysis provided a direct comparison of the cellular events that occurred within regions of disturbed versus laminar blood flow, and allowed for detection of early atherogenic events. Because the aortic segments analyzed included anatomical landmarks with consistent disturbed blood flow areas, image data could be collected in a reproducible manner. Because the imaged tissue was intact, LSCM visualized cellular morphologies that were not appreciated with traditional microscopy methods that involve tissue sectioning.
In disease-free hyperlipidemic LDLr −/− mice, endothelial TLR2 expression was up-regulated in aortic regions of disturbed blood flow, specifically in the lesser curvature of the aorta. This was consistent with our previous report of flow-dependent regulation of TLR2 expression in cultured human coronary artery endothelial cells where disturbed flow increased and laminar flow decreased both endothelial cell TLR2 expression and TLR2 activation (7). These data also support the concept that areas of lesion predilection are primed for disease, even in the absence of atherogenesis (12, 13, 14, 15). In the absence of factors that contribute to atherosclerotic disease progression, up-regulation of endothelial TLR2 in disturbed flow regions may mediate homeostatic processes that maintain the integrity of the vessel wall (15).
With the early progression of disease in LDLr −/− mice, endothelial TLR2 expression dramatically increased, and this increase was confined to endothelial cells exposed to disturbed blood flow. TLR2 expression was not detected in intimal leukocytes in BMGFP+ LDLr −/− chimeric mice. We speculate that TLR2 expression was minimal in early lesional intimal leukocytes. This is consistent with our previous report that a TLR2 deficiency in BM-derived cells had no impact at 10 wk on lesion size in hyperlipidemic LDLr −/− chimeric mice exposed to only unknown endogenous TLR2 agonists (8). In advanced human atherosclerotic plaques obtained from patients undergoing endarterectomy, Edfeldt et al. detected TLR2 (and TLR1 and 4) in both the endothelium and areas infiltrated with inflammatory cells (16). The presence of leukocyte TLR expression in such advanced lesions suggests that TLR expression by exogenous agonists can participate in advanced disease. Thus, more work is needed to identify the time course of lesional cell TLR2 expression and the cellular effect of endogenous vs. exogenous agonists, as well as determine the relevance of our findings to human lesion development.
Concomitant in time and location with the changes in endothelial TLR2 expression, endothelial cell morphology was progressively disrupted by hyperlipidemia. These changes were not observed beyond the boundaries of the lesser curvature, such as the regions exposed to laminar blood flow (10). The apparent luminal protrusion of endothelial cells was a likely result of subendothelial accumulation of lipid and leukocytes. These changes occurred within 4 wk of the diet-induced hyperlipidemia and are among the earliest events of atherogenesis. One can speculate that such morphological changes in endothelial cells could further exacerbate the hemodynamics of disturbed blood flow and initiate a positive-feedback cycle that accelerates the early events of atherogenesis. Lipid accumulation in early atherosclerotic lesions within regions distinct from leukocyte accumulation has been described in human aortic (17) and coronary (18) specimens and in rabbit aortae (19). These data support the idea that extracellular intimal lipid accumulation could at times be spatially and temporally distinct from leukocyte accumulation during early atherogenesis.
Within 4 wk of HFD consumption, a deficiency of vascular wall TLR2 resulted in decreased intimal lipid accumulation and reduced the hyperlipidemic-induced morphological changes of the endothelial cells. These events could have delayed the initiation of lesion development, and add new insight to our earlier study of reduced atherosclerosis measured by traditional methods after 10 or 14 wk of HFD consumption (8). Supporting this was the slower time course of decreased intimal leukocyte accumulation, as compared with intimal lipid accumulation, that we observed in mice deficient in vascular wall TLR2. Because lesion development occurs in distinct phases, with initiation more a consequence of endothelial cell activation and progression facilitated by leukocyte entry and foam cell formation (20), endothelial cell TLR2 can be considered a key contributor to lesion initiation.
The atheroprotective effects of endothelial cell TLR2 deficiency were consistent with our previous studies that found no atherogenic role of BM cell-derived TLR2 in the absence of an injected TLR2 exogenous agonist (8). Collectively, these results suggested that endogenous TLR2 agonists stimulate atherogenic processes via activation of endothelial cell TLR2. Potential endogenous TLR2 agonists include: fibronectin extra domain A (EDA), hyaluronan fragments, biglycan, high-mobility box chromosomal protein 1, components of oxidized lipoproteins, and serum amyloid A. It is noteworthy that these TLR2 agonists are found in atherosclerotic lesions (20), which makes them all likely candidates.
A similar association between TLR2 and disease was reported in models of heart and kidney ischemia/reperfusion (I/R) injury (21, 22, 23, 24). Leemans et al. found that TLR2 deficiency resulted in reduced renal dysfunction and tubular damage after I/R. After generating BM transplant chimeric mice, the protection provided by TLR deficiency was only observed when TLR2 was absent in non–BM-derived cells, and this is identical to our findings on atherogenesis (8). Moreover, granulocyte and macrophage numbers were reduced in kidneys from TLR2−/− mice relative to controls after I/R injury. Recent reports have also established a protective role of TLR2 deficiency in mice during I/R injury to maintain coronary endothelial function (22) or left ventricular function (23). Such reports support the idea that the generation of endogenous TLR2 agonists during tissue injury exacerbates the disease processes. Reduced macrophage recruitment in atherosclerotic lesions was also observed in apoE−/− mice with a deficiency in the common intracellular TLR signaling adaptor protein, MyD88 (25). Collectively, these data suggest that endogenous ligand signaling via TLR2 impacts region-specific disease outcomes, and that TLR2 expression on non–BM-derived cells mediates the effects of endogenous ligand TLR2 activation.
To determine whether TLR2 deficiency altered systemic factors that impacted lesion development, plasma cholesterol, TNF-α, and MCP-1 were measured. There were no changes in plasma cholesterol or MCP-1 with TLR2 deficiency; however, plasma TNF-α was reduced in TLR2−/− mice. Because macrophages are a primary source of TNF-α, and TNF-α production is a consequence of cellular TLR2 stimulation, the reduction of this cytokine could have been a consequence of decreased macrophage tissue accumulation in the TLR2−/− mice (15). These data also cannot exclude the possibility that TLR2 deficiency alters systemic factors that contribute to disease initiation.
In summary, these data implicate endothelial cell TLR2 expression in the modulation of key atherogenic events during early lesion development. Future studies are needed to identify the key endogenous TLR2 ligands involved in stimulating endothelial cells both before and during disease development and define the mechanisms contributing to endothelial cell TLR2 up-regulation in regions of disturbed blood flow.
MATERIALS AND METHODS
Animals
All animal use was approved by the Institutional Animal Care and Use Committee. LDLr −/− mice backcrossed onto a C57BL/6 background were purchased from The Jackson Laboratory (Ldlrtm1Her) and bred in-house. TLR2−/− mice were provided by Tularik, Inc. TLR2−/− mice were backcrossed into a C57BL/6 background and, after at least seven generations, double-mutant mice (LDLr −/− TLR2 −/−) were generated by crossing C57BL/6-TLR2−/− mice with C57BL/6-LDLr−/− mice. TLR2−/− and LDLr −/− genotyping was performed as previously described (8). The mice were weaned at 4 wk and given ad libitum access to a standard mouse chow diet (7019; Harlan Teklad). BMT was performed as previously described (8). In brief, BM recipient animals at 8–10 wk of age received 1,000 rad γ irradiation. Reconstitution of ablated hematopoietic stem cells was achieved by tail vein injection of donor BM harvested from GFP+ mice that constitutively expressed GFP (C57BL/6-Tg(ACTB-EGFP)1Osb/J; The Jackson Laboratory). Recipient and donor mice were sex and age matched. Mice were allowed to recover from the BMT for 3 wk, after which they were fed the HFD ad libitum, which contained 1.25% cholesterol and 15.8% fat (with no added cholate; 94059; Harlan Teklad). Mice were fasted before harvest and weighed, and venous blood was drawn from the retroorbital sinus. All mice were housed four per cage in autoclaved, filter-top cages with autoclaved water and kept on a 12-h light/dark cycle. Animal care and use for all procedures was done in accordance with our institutional guidelines.
Plasma measurements/ELISAs
Plasma was isolated and total cholesterol levels were measured by a colorimetric enzymatic method (Thermo Electron Corp.). Plasma TNF-α was measured using an ELISA prepared with monoclonal anti–mouse TNF-α (BD Biosciences). Plasma MCP-1 was measured by ELISA (R&D Systems). Liver and spleen GFP was extracted with T-PER tissue protein extraction reagent (Thermo Fisher Scientific), and GFP was measured using an ELISA prepared with monoclonal anti-GFP (BD Biosciences).
En face LSCM
Antibodies and reagents.
Monoclonal anti–mouse primary antibodies targeting TLR2 (16–9021 and 14–9024; eBioscience), CD68 (MCA1957GA; Serotec), and CD31 (553370; BD Biosciences) were used at 2.5–5.0 μg/ml. Secondary antibodies were used at 4.0–5.0 μg/ml, and included the following: goat anti–mouse and –rat IgG conjugated to Alexa Fluor 488, 568, or 647 (Invitrogen). Goat serum used to eliminate nonspecific antibody binding was generated in-house. An Alexa Fluor 555 monoclonal antibody labeling kit was used to directly conjugate CD31 to Alexa Fluor 555 (A20187; Invitrogen). The directly conjugated CD31-Alexa Fluor 555 was used in multiple antibody labeling protocols when other rat isotype antibodies were used, such as CD68 and TLR2. Nile red and rhodamine phalloidin were purchased from Sigma-Aldrich, DAPI was purchased from Invitrogen, and the anti-fade mounting medium ImmunO was purchased from MP Biomedicals.
Surgery.
En face LSCM of aortic tissue was used previously to map aortic areas of disease predilection (10). We modified the procedure to measure lesional BMGFP+ cell infiltrate and Nile red–positive lipid staining from large composite images of the lesser curvature region of the ascending aortic arch. This entailed segmentation of the aortic arch, using the innominate branch (the first bifurcation of the arch) as a landmark. After sequential perfusions of PBS (10 ml), 5% paraformaldehyde (10 ml for 10 min), and PBS (to flush aortae of paraformaldehyde), arch segments were prepared for cleaning and dissection by removal of the heart and aorta from the animal. With the heart and aorta excised and placed in PBS, loose aortic adventitial connective tissue and fat were removed with the aid of a dissecting microscope. Microdissection of the proximal aortic arch produced “segment 1,” containing the innominate bifurcation (Fig. S1).
Segment staining.
Segments were stained whole, as aortic rings, in 0.1-ml droplets of staining buffer. During each staining and washing step, segments were carefully handled by fine forceps, taking care to only manipulate the segment via the innominate artery branch. Segments were initially incubated in 10% normal goat serum for 60 min at room temperature to limit nonspecific antibody staining. Immunofluorescence antibody staining was performed in a stepwise manner, with each staining step accomplished in the dark at room temperature on a rotating mixer for 45 min. To allow for sufficient penetration of staining antibodies, tissue was first permeabilized with 0.2% Triton X-100 and 0.1 M glycine in PBS for 7 min at room temperature. Between staining steps, segments were washed in PBS or 0.1% Tween-20 in PBS (for CD68 or TLR2 staining). After completion of staining, segments were opened by cutting through the greater curvature and base of the innominate artery branch (Fig. S1). The opened segments were placed on a glass slide, adventitial side down, coverslipped with the anti-fade mounting media, and pressed overnight (26).
Image acquisition and analysis.
Sample visualization was performed with a confocal microscope (2100 Radiance; Bio-Rad Laboratories) using 20×, 60×, and 100× objectives. The 60× and 100× objectives were used to collect high-resolution images with an x–y resolution of ∼0.15 μm and a z-distance resolution of ∼0.80 μm. To characterize the lesional cells within the intimal layer, z-series optical sectioning was performed with the 60× or 100× objectives, creating image stacks that spanned the thickness of the intimal layer. Z-series image stacks were collected at 0.5 μm z-distance increments. In the z-series images, the forest green autofluorescence of the internal elastic lamina was easily distinguished from the bright green GFP.
The 20× objective was used to create relatively large composite images of the lesser curvature region, which were generated by stitching contiguous images that spanned the lesser curvature either vertically (circumferential strip) or horizontally (lesser curvature strip; Fig. S1). Each contiguous image had dimensions of 584 × 584 μm with pixel size 0.570 μm and a z axis resolution of ∼4 μm. All scans were performed such that contiguous images were collected with overlap that allowed for creation of the composite stitched images (Photostitch; LEAD Technologies). The circumferential composite images typically had dimensions of 2,000 μm (vertical) × 584 μm (horizontal). The lesser curvature composite images captured the entire lesser curvature zone within the arch segment with dimensions of 1,200 μm (vertical) × 1,000 μm (horizontal). In generating these 20× contiguous image maps of the lesser curvature, z-series image stacks were not collected because of the extensive additional image mapping that would be required. The 4-μm z-axis resolution proved to be sufficient to capture all intimal GFP+ cellular events during early lesion development (≤ 8 wk of HFD-feeding). However, with advanced lesions (>8 wk of HFD-feeding), the ability to detect increased GFP+ cellular infiltrate became compromised because of the thickness of the GFP+ infiltrate exceeding 4 μm.
Image quantitation of BMGFP+ leukocyte events and Nile red–stained lipid accumulation was performed by ImageJ (National Institutes of Health) and Image-Pro Plus 6.0 (Media Cybernetics). In brief, GFP+ and Nile red+ fluorescence were measured in Image-Pro Plus after segmenting background fluorescence apart from GFP+ or Nile red+ fluorescence. Discreet regions of interest (ROIs) representing the areas of positive (nonbackground) fluorescence were created. Remaining background fluorescence was further eliminated during the “Count and Measure” feature in Image-Pro Plus by using filters such as “Area,” “Aspect,” “Fractal Dimension,” and “Heterogeneity.” The outlines of GFP+ ROIs, defined as GFP+ clusters, were saved to perform a “Population Density” analysis. Colocalization of DAPI-stained and GFP+ nuclei (for leukocyte number) or Nile red+ and GFP+ ROIs (for foam cell number) were performed in ImageJ using a colocalization plug-in (RG2B Colocalization; C.P. Mauer). The resultant nuclear colocalization image was imported into Image-Pro Plus for a population density analysis of leukocyte accumulation (GFP+ nuclei/cluster), which counted GFP+ nuclei within each GFP+ cluster (GFP+ ROI). GFP+ area fraction and GFP+ nuclei per mm2 were calculated by normalizing these values to the total segment area in the lesser curvature composite strips.
Statistical analysis
Plots are expressed as the mean ± the SEM. Two-factor analysis of variance was used to determine if differences existed between the groups at the different time points for LSCM data, weight data, cholesterol data, and ELISA data. For each dataset, a normality test (P > 0.05) and an equal variance test (P > 0.05) was performed to identify normally distributed data. If either test failed, the nonparametric analysis of variance on ranks test was performed. The analysis of factor level effects was done by the Holm-Sidak test of pairwise multiple comparisons. All statistical analyses were done with the use of the SigmaStat 3.00 (SPSS, Inc.). A value of P < 0.05 was considered significant.
Online supplemental material
Fig. S1 illustrates the preparation and LSCM image acquisition procedures for the aortic segments. Fig. S2 is a high-resolution LSCM image of the morphological changes observed in the endothelial cells in an early lesion. Fig. S3 is a collection of z-series image stacks demonstrating the extent of leukocyte penetration within the intima in disease-free and atherosclerotic mice during early lesion development. Fig. S4 is a 3D computer rendered analysis of leukocyte accumulation within the intimal layer. Fig. S5 compares early intimal leukocyte accumulation in diseased and disease-free mice. These images allow for an appreciation of the regional specificity of leukocyte accumulation within areas of disturbed blood flow as well as the magnitude of this increase with an increased disease. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20071096/DC1.
Supplemental Material
Acknowledgments
The authors wish to thank Audrey S. Black, David J. Bonnet, Joshua Bulgrien, and Karen McKeon for their excellent technical assistance, and M. Rachel Richards for her editorial comments.
This study was supported by National Institutes of Health grants HL35297 (L.K. Curtiss), HL84556 (L.K. Curtiss), and GM066119 (P.S. Tobias). A.E. Mullick was supported by a fellowship from the Western States Affiliate of the American Heart Association.
The authors have no conflicting financial interests.
Abbreviations used: BMT, BM transplantation; HFD, high-fat diet; I/R, ischemia/reperfusion; LDLr, low-density lipoprotein receptor; LSCM, laser scanning confocal microscopy; MCP, monocyte chemoattractant protein; ROI, region of interest; TLR, Toll-like receptor.
References
- 1.Steinberg, D. 2002. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat. Med. 8:1211–1217. [DOI] [PubMed] [Google Scholar]
- 2.Ross, R. 1999. Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. [DOI] [PubMed] [Google Scholar]
- 4.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. [DOI] [PubMed] [Google Scholar]
- 5.Hansson, G.K., A.-K.L. Robertson, and C. Soderberg-Naucler. 2006. Inflammation and atherosclerosis. Annu. Rev. Pathol. 1:297–329. [DOI] [PubMed] [Google Scholar]
- 6.VanderLaan, P.A., C.A. Reardon, and G.S. Getz. 2004. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 24:12–22. [DOI] [PubMed] [Google Scholar]
- 7.Dunzendorfer, S., H.K. Lee, and P.S. Tobias. 2004. Flow-dependent regulation of endothelial Toll-like receptor 2 expression through inhibition of SP1 activity. Circ. Res. 95:684–691. [DOI] [PubMed] [Google Scholar]
- 8.Mullick, A.E., P.S. Tobias, and L.K. Curtiss. 2005. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest. 115:3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tzima, E., M. Irani-Tehrani, W.B. Kiosses, E. Dejana, D.A. Schultz, B. Engelhardt, G. Cao, H. DeLisser, and M.A. Schwartz. 2005. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 437:426–431. [DOI] [PubMed] [Google Scholar]
- 10.Iiyama, K., L. Hajra, M. Iiyama, H. Li, M. DiChiara, B.D. Medoff, and M.I. Cybulsky. 1999. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 85:199–207. [DOI] [PubMed] [Google Scholar]
- 11.Greenspan, P., E.P. Mayer, and S.D. Fowler. 1985. Nile red: a selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 100:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tso, C., G. Martinic, W.H. Fan, C. Rogers, K.A. Rye, and P.J. Barter. 2006. High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler. Thromb. Vasc. Biol. 26:1144–1149. [DOI] [PubMed] [Google Scholar]
- 13.Nijhuis, M.M., G. Pasterkamp, N.I. Sluis, D.P. de Kleijn, J.D. Laman, and L.H. Ulfman. 2007. Peptidoglycan increases firm adhesion of monocytes under flow conditions and primes monocyte chemotaxis. J. Vasc. Res. 44:214–222. [DOI] [PubMed] [Google Scholar]
- 14.Hajra, L., A.I. Evans, M. Chen, S.J. Hyduk, T. Collins, and M.I. Cybulsky. 2000. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA. 97:9052–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jongstra-Bilen, J., M. Haidari, S.N. Zhu, M. Chen, D. Guha, and M.I. Cybulsky. 2006. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 203:2073–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edfeldt, K., J. Swedenborg, G.K. Hansson, and Z.Q. Yan. 2002. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 105:1158–1161. [PubMed] [Google Scholar]
- 17.Bobryshev, Y.V., and R.S. Lord. 1999. Accumulation of co-localised unesterified cholesterol and neutral lipids within vacuolised elastin fibres in athero-prone areas of the human aorta. Atherosclerosis. 142:121–131. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima, Y., H. Fujii, S. Sumiyoshi, T.N. Wight, and K. Sueishi. 2007. Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage accumulation. Arterioscler. Thromb. Vasc. Biol. 27:1159–1165. [DOI] [PubMed] [Google Scholar]
- 19.Simionescu, N., E. Vasile, F. Lupu, G. Popescu, and M. Simionescu. 1986. Prelesional events in atherogenesis. Accumulation of extracellular cholesterol-rich liposomes in the arterial intima and cardiac valves of the hyperlipidemic rabbit. Am. J. Pathol. 123:109–125. [PMC free article] [PubMed] [Google Scholar]
- 20.Mullick, A.E., P.S. Tobias, and L.K. Curtiss. 2006. Toll-like receptors and atherosclerosis: key contributors in disease and health? Immunol. Res. 34:193–209. [DOI] [PubMed] [Google Scholar]
- 21.Leemans, J.C., G. Stokman, N. Claessen, K.M. Rouschop, G.J. Teske, C.J. Kirschning, S. Akira, T. van der Poll, J.J. Weening, and S. Florquin. 2005. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 115:2894–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Favre, J., P. Musette, V. Douin-Echinard, K. Laude, J.P. Henry, J.F. Arnal, C. Thuillez, and V. Richard. 2007. Toll-Like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 27:1064–1071. [DOI] [PubMed] [Google Scholar]
- 23.Sakata, Y., J.W. Dong, J.G. Vallejo, C.H. Huang, J.S. Baker, K.J. Tracey, O. Tacheuchi, S. Akira, and D.L. Mann. 2007. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 292:H503–H509. [DOI] [PubMed] [Google Scholar]
- 24.Bjorkbacka, H., V.V. Kunjathoor, K.J. Moore, S. Koehn, C.M. Ordija, M.A. Lee, T. Means, K. Halmen, A.D. Luster, D.T. Golenbock, and M.W. Freeman. 2004. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 10:416–421. [DOI] [PubMed] [Google Scholar]
- 25.Shigeoka, A.A., T.D. Holscher, A.J. King, F.W. Hall, W.B. Kiosses, P.S. Tobias, N. Mackman, and D.B. McKay. 2007. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol. 178:6252–6258. [DOI] [PubMed] [Google Scholar]
- 26.Kiosses, W.B., N.H. McKee, and V.I. Kalnins. 1997. Evidence for the migration of rat aortic endothelial cells toward the heart. Arterioscler. Thromb. Vasc. Biol. 17:2891–2896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}