

Abstract
The growth and virulence of Mycobacterium tuberculosis depends on its ability to scavenge host iron, an essential and limited micronutrient in vivo. In this study we show that ferric iron accumulates both intra- and extra-cellularly in the primary lung lesions of guinea pigs aerosol-infected with the H37Rv strain of Mycobacterium tuberculosis. Iron accumulated within macrophages at the periphery of the primary granulomatous lesions while extra-cellular ferric iron was concentrated in areas of lesion necrosis. Accumulation of iron within primary lesions was preceded by an increase in expression of heavy chain (H) ferritin, lactoferrin and receptors for transferrin, primarily by macrophages and granulocytes. The increased expression of intra-cellular H ferritin and extra-cellular lactoferrin, more so than transferrin receptor, paralleled the development of necrosis within primary lesions. The deposition of extra-cellular ferric iron within necrotic foci coincided with the accumulation of calcium and phosphorus and other cations in the form of dystrophic calcification. Primary lung lesions from guinea pigs vaccinated with Mycobactrium bovis BCG prior to experimental infection, had reduced iron accumulation as well as H ferritin, lactoferrin and transferrin receptor expression. The amelioration of primary lesion necrosis and dystrophic calcification by BCG vaccination was coincident with the lack of extra-cellular ferric iron and lactoferrin accumulation. These data demonstrate that BCG vaccination ameliorates primary lesion necrosis, dystrophic mineralization and iron accumulation, in part by down-regulating the expression of macrophage H ferritin, lactoferrin and transferrin receptors, in vivo.
Keywords: Transferrin receptor, CD71, ferritin, lactoferrin, Mycobacterium tuberculosis, dystrophic mineralization, calcification, guinea pig
INTRODUCTION
Mycobacterium tuberculosis (M. tuberculosis), the causative agent of human tuberculosis, is an intracellular pathogen that is spread primarily between individuals by aerosolized respiratory secretions. The initial foci of mixed inflammation that develop in the lung following aerosol exposure are called primary lesions (1-4). A clinical and pathologic feature that characterizes tuberculosis lesions resulting from primary infection is calcifiation of granulomas of the lung and draining lymph node which often appear on chest radiographs as discrete mineralized densities (5). Dystrophic calcification replaces foci of necrosis which represents irreversible tissue damage that can persist for the life of the patient. The importance of these lesions is that in some cases, they may harbor small numbers of viable but dormant bacilli that may serve as the source of reactivation tuberculosis during times of lowered host resistance (4-7). While well organized or calcified granulomas may prevent bacilli from spreading from the initial site of infection, they may also represent a functional or mechanical barrier to both immune effector cells and penetration by sterilizing concentrations of anti-tuberculosis drugs (4, 6-9).
Like in humans, the morphogenesis of primary lesions in immunologically naïve guinea pigs infected with M. tuberculosis, progresses from small foci of mixed inflammation to lesions that develop central necrosis and dystrophic calcification (6, 8-12). Vaccination of guinea pigs with Mycobacterium bovis BCG (BCG) prior to aerosol infection results in delayed development of primary lesions that are generally smaller and rarely develop necrosis and dystrophic calcification (6, 12, 13). The morphologic differences between the primary lesions from BCG-vaccinated and non-vaccinated guinea pigs therefore, affords the unique opportunity to not only study the mechanisms of action of BCG, but to better understand the progression and pathogenesis of the primary lesions of tuberculosis.
Dystrophic calcification is a pathologic process that involves the intra- and extra-cellular precipitation of calcium phosphate in areas of tissue necrosis (14-17). The hydroxyapatite mineral complex that forms is similar to that found in bones and teeth and develops following initiation (or nucleation) and progresses by a process referred to as propagation. The initiation of intra-cellular calcification occurs with the influx of calcium within mitochondria of dead or dying cells, whereas extra-cellular phospholipid vesicles from cytoplasmic membranes or organelles of dying cells can serve as the nidus for extra-cellular calcification (18, 19). Of particular importance in the context of the current study is that extra-cellular but not intra-cellular iron has also been shown to serve as an initiator of dystrophic calcification (20).
Iron is a micronutrient required by essentially all living organisms, since it is structurally incorporated into a wide variety of proteins that are crucial for normal metabolic processes, including cell respiration, growth and DNA synthesis (21, 22). Macrophages throughout the body play an important role in iron metabolism through the ability to store and release iron at times of excess and deficiency, respectively (23, 24). Through protein binding, iron is rendered non-toxic and the in vivo availability to infectious pathogens is limited (25). Non-protein bound or free iron is extremely toxic causing cytotoxicity through the formation of reactive oxygen species and so is transported and stored by the host iron-binding proteins transferrin, ferritin, and lactoferrin respectively (26-29).
M. tuberculosis, like other pathogenic bacteria have evolved complex mechanisms to scavenge host iron even intra-cellularly from macrophages which are considered the first line of defense following infection. M. tuberculosis scavenges iron through the production and secretion of iron chelating siderophores (30, 31). Much of the understanding of the role of iron in the growth and virulence of M. tuberculosis has come primarily from in vitro studies (32-35). Less is known about the competition between host and pathogen for iron in vivo and what role iron metabolism plays in the pathogenesis of tuberculosis (36, 37). Moreover, the mechanisms and significance of dystrophic calcification in primary lesions of human tuberculosis is poorly understood and thus warrants further study through the use of appropriate animal models.
In this study using the guinea pig model of tuberculosis, we show that extra-cellular ferric iron accumulates within foci of necrosis in primary lesions coincident with the development of dystrophic calcification. The accumulation of iron within primary lesions was preceded by the expression of transferrin receptors (CD71), H ferritin and lactoferrin by macrophages and granulocytes, which was ameliorated by BCG vaccination. These in vivo data corroborate in vitro findings and supports the hypothesis that iron metabolism by macrophages is reflective of activation status and in part, determines resistance to M. tuberculosis infection.
MATERIALS AND METHODS
Experimental animals
Specific pathogen-free albino Hartley-strain guinea pigs (Charles River Breeding Laboratories, Inc., Wilmington, MA) were randomly assigned to a vaccination treatment group and were housed individually or in pairs in HEPA filtered cages with separate ventilation and held under barrier conditions in a biosaftey level III animal laboratory. Animals were provided commercial chow in stainless-steel feeders and tap water ad libitum. The guinea pigs were maintained in a temperature- and humidity-controlled environment and exposed to a 12-hour light/dark cycle. All procedures were reviewed and approved by the Colorado State University Institutional Animal Care and Use Committee.
Aerosol infection
Mycobacterium tuberculosis H37Rv strain (TMC#102; Trudeau Institute, Saranac Lake, NY) was grown in Proskauer-Beck liquid medium containing 0.05% Tween 80 to mid-log phase, aliquoted, and frozen at −80°C until used for infection (MRL Laboratories, Colorado State University). A thawed aliquot of M. tuberculosis was diluted in sterile water to 106 CFU/ml for a total working stock volume of 20 ml. Guinea pigs (approximately 9 months of age) from each treatment group were aerosolized using the Madison infection chamber (University of Wisconsin Machine Shop, Madison, WI) with a starting volume of 15 ml of working stock. The estimated retention of mycobacteria was between 25 and 30 CFU/guinea pig as judged by enumerating primary pulmonary lesions in non-vaccinated guinea pigs at five weeks post-infection (38). Non-infected controls were aerosolized with sterile water only.
BCG vaccination
Guinea pigs were vaccinated with 1 × 104 Mycobacterium bovis (BCG, strain Pasteur) or mock vaccinated with saline, intra-dermally 4 weeks prior to aerosol exposure to M. tuberculosis.
Euthanasia and sample collection
At intervals of 5, 10, 15, 20, 30 and 60 days post-infection, guinea pigs were euthanized humanely by an overdose (1 ml per 0.75 kg body weight) of sodium pentobarbital (Sleepaway; Fort Dodge Laboratories Inc.) by intraperitoneal injection. Following euthanasia, the left pulmonary lobes were infused in situ with 5 ml of 4% paraformaldehyde for 48 hours and stored in 70% ethanol. At the time of processing, all tissues were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E), Pearl's Prussian blue for ferric iron, Turnball's for ferrous iron or Von Kossa's for calcium.
Immunohistochemistry
Paraffin sections, 5−6 μm, of lung and tracheobronchial lymph nodes were deparaffinized, rehydrated and subjected to antigen retrieval by incubation in Target Retrieval solution, pH 9.0 (DAKO, Carpentaria, CA) for 25 min at 90° C, followed by a 20 min cooling period at room temperature. The sections were then treated with 0.3% hydrogen peroxide in water for 15 min to quench endogenous peroxidase activity. Following a rinse in Tris buffered saline with 1% Tween-20 (TTBS), the slides were subjected to two blocking steps: (i) 15 min incubation with 0.15 mM glycine in PBS, and 30 min incubation with 1% normal horse serum with a rinse in TTBS in between. The slides were then incubated with rabbit polyclonal antibody to H ferritin (Novus Biologicals, Inc., Littleton, CO) at 1:500 dilution in blocking buffer or with rabbit polyclonal antibody to lactoferrin at 1:300 dilution (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 2 hours at room temperature, or with goat polyclonal antibody specific for CD71 (Santa Cruz Biotechnology) at a 1:20 dilution overnight at 4° C, followed by several rinses in TBSS. The cross-reactivity of primary antibodies against guinea pig H ferritin, lactoferrin and CD71 was confirmed by western blot analysis of extracts of isolated guinea pig alveolar macrophages and the human monomyelocytic cell line THP-1 cells (data not shown). This was followed by 30 min incubation with biotinylated goat-anti rabbit-IgG (Vector Laboratories) or alkaline-phosphatase conjugated donkey anti-goat-IgG (Santa Cruz Biotechnology), and visualization of bound antibody by the ABC-method (Vectastain; Vector Laboratories) and diaminobenzidine for H ferritin and transferrin, and with BCIP/NBT-substrate (Dako; Carpentaria, CA) for CD71. The sections were counterstained with Meyer's hematoxylene (Scytek Laboratories; Logan, Utah), mounted with coverslips, and examined on an Olympus BX41 light microscope. Photomicrographs were acquired with an Olympus Q-Color 3 camera and associated computer software. The number of positive cells were scored as follows: 1 = scattered, rare positive cells, 2 = small numbers, often clustered, of positive cells, 3 = moderate numbers of positive cells, 4 = large numbers of positive cells.
Tissue Analyses
Grossly visible primary lung lesions for chemical analysis were dissected from paraformaldehyde fixed lungs of BCG- or sham-vaccinated guinea pigs. Random sections of paraformaldehyde fixed lung from non-infected animals were used as negative controls. Tissues were dried overnight in a drying oven at ≈ 85°C, weighed, and ashed overnight in a muffle furnace at ≈600°C. The ashed samples were allowed to cool, and then dissolved in nitric acid. The solutions were sonicated to complete dissolution. The resulting acid solution was diluted with deionized water for copper, iron and zinc analyses, and diluted with 5% lanthanum oxide for calcium and magnesium analyses. Calcium, copper, iron, magnesium and zinc concentrations were determined via flame atomic absorption spectrophotometry. The concentration of phosphorus was determined by reacting the acid solution with ammonium molybdate and measuring the absorbancies using UV/VIS spectrophotometry. All tissue concentrations were reported as parts per million (PPM) on a dry weight basis.
Statistics
Results are expressed as mean ± standard deviation (SD). A statistical software package (Graphpad® Prizim 4.02, San Diego, CA) was used for data analysis and graphics. Differences between groups were compared by two-way analysis of variance. Bonferroni post-test was used to assess differences within and between treatment groups. The values of P ≤ 0.05 (*), P ≤ 0.001 (**) were considered significant.
RESULTS
The morphogenesis of the primary lung lesions of naïve guinea pigs experimentally infected with M. tuberculosis by low-dose aerosol notably differs from primary lesions in BCG vaccinated guinea pigs (6, 12, 13). The stains Pearl's Prussian blue for ferric iron, Turnball's stain for ferrous iron (data not shown) and Von Kossa's stain for calcium were used to compare the accumulation of iron and calcium within the primary lung lesions of sham-vaccinated and BCG-vaccinated guinea pigs prior to low dose aerosol challenge. At 30 days post-infection in sham-vaccinated guinea pigs, neither ferric iron nor calcium was evident within early primary lesions with acute inflammation and central necrosis (data not shown). However, the accumulation of intra- and extra-cellular ferric iron and calcium occurred between 30 and 60 days post-infection (FIG. 1). Primary lesions of sham-vaccinated guinea pigs at 60 days post-infection had punctuate extra-cellular deposits of ferric iron (arrows, FIG. 1 A, inset) and calcium (arrows, FIG. 1B) mixed with necrotic debris. Dead or degenerate cells within necrotic lesions had diffuse cytoplasmic calcification, often with intact cytoplasmic margins (arrowheads, FIG. 1B). As lesions progressed, ferric iron accumulated intra-cellularly in macrophages within the cell rich periphery of primary lesions (white arrow FIG. 1C) or was incorporated into central foci of dystrophic calcification and concentrated at the periphery of the calcified lesion at the interface with the acellular but non-mineralized zone of the lesion (black arrow, FIG. 1C). More organized lesions had diffuse calcification of necrotic debris that was well delineated from the cellular and acellular zones of the lesion (arrow FIG. 1D). In contrast, the primary lesions in BCG vaccinated animals (FIG. 3) had neither intra-cellular nor extra-cellular iron accumulation at 60 days post infection (FIG. 1 E and F). Only intra-cellular ferric iron was evident at 60 days post-infection with no evidence of extra-cellular iron accumulation (FIG. 1 F). The lack of extra-cellular iron accumulation paralleled the lack of central necrosis and mineralization (FIG. 1 E and F).
FIG. 1.
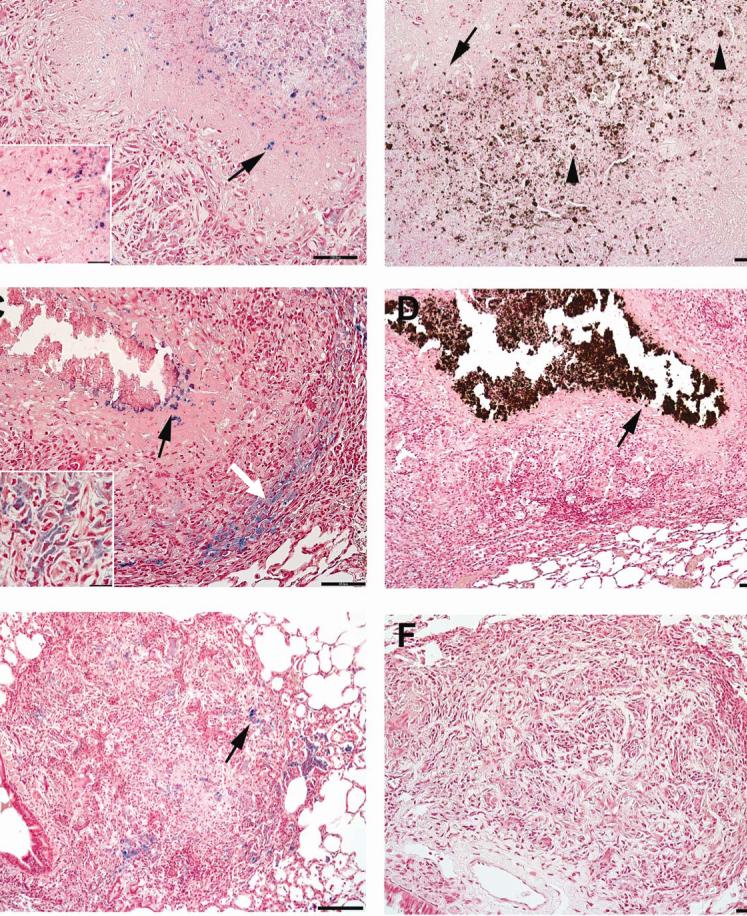
The pattern of ferric iron accumulation and calcification within the primary lung lesions in guinea pigs sham vaccinated with saline prior to infection by low-dose aerosol with the H37Rv strain of M. tuberculosis differs depending on progression of primary lesions. (A) In less advanced lesions 60 days post-infection, extra-cellular (inset) aggregates of ferric iron (arrows) are scattered amongst the necrotic cellular debris within the center of primary lesions. (B) In the same lesion, the pattern of extra-cellular calcium deposition (arrows) is similar to that of ferric iron, being concentrated within the areas of central necrosis. (C) In more advanced lesions 60 days post-infection, extra-cellular ferric iron is at the periphery (black arrow) of the area of calcified necrotic debris, while the intra-cellular (inset) ferric iron is within macrophages at the periphery granuloma (white arrow). (D) In the more advanced lesions 60 days post-infection, the intra- and extra-cellular calcification coalesce to form a well delineated center of calcified necrotic debris (arrow). Primary lung lesions in guinea pigs BCG-vaccinated prior to infection by low-dose aerosol with the H37Rv strain of M. tuberculosis fail to progress to necrosis at 30 days (E) and , calcification and extra-cellular ferric iron accumulation by day 60 (F) with Von Kossa's stain for calcium. Pearl's Prussian blue for ferric iron (A, C, E), Von Kossa's for calcium (B, D, F). A, B, D, Bars = 130 μm, 100× magnification. C, E, F Bars = 55.0 μm, 200× magnification.
FIG. 3.
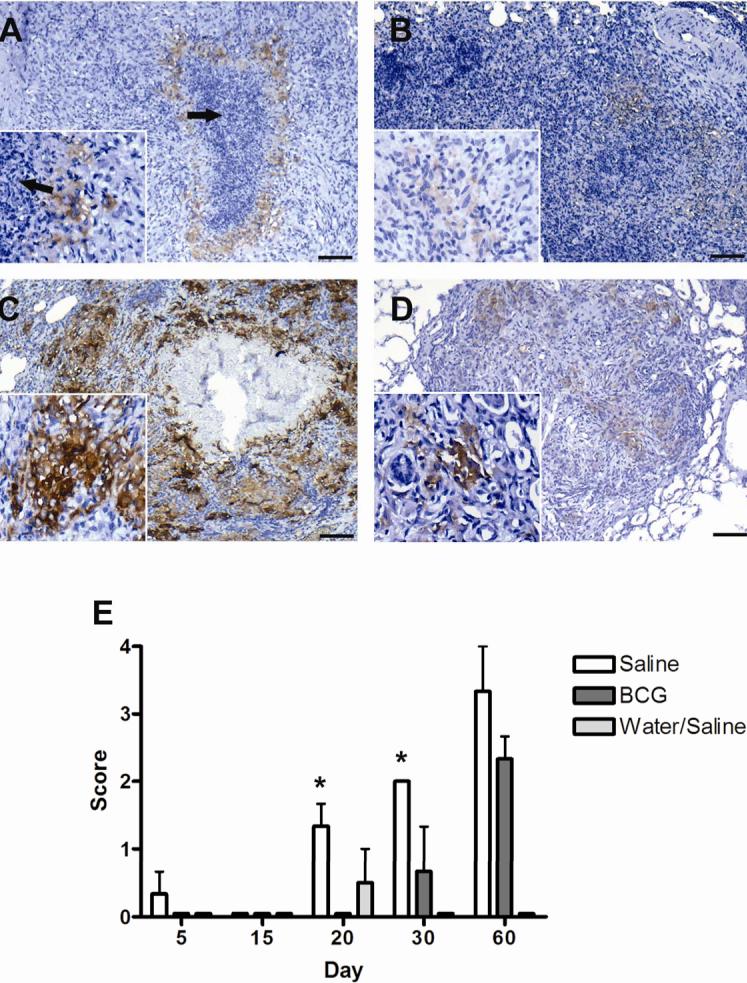
Expression of ferritin heavy chain, (H ferritin) as demonstrated by immunohistochemistry, by macrophages within primary lung lesions of guinea pigs challenged with a low-dose aerosol of the H37Rv strain of M. tuberculosis, without (A, C) or with prior BCG-vaccination (B,D). Lesions from sham-vaccinated animals had few cells in surrounding foci of lesion necrosis (arrows) that showed weak immunoreactivity at day 30 (inset) (A) that intensified by day 60 (C). Immunoreactivity was less in lesions from BCG-vaccinated animals at both day 30 (B) and day 60 (D) but differences were not statically different at day 60 (E). Expression of ferritin heavy chain was not significantly increased compared to non-infected controls, until 20 days post-infection where it was first seen in animals sham-vaccinated with saline. Expression was significantly increased over BCG-vaccinated animals at days 20 and 30 but was not different at day 60 post-infection. Expression of ferritin was not seen in BCG-vaccinated animals until 30 days post-infection. Ferritin showed no significant expression in non-infected animals (water aerosolized). Data represents the mean ± standard deviation from 1 representative experiment. n = 3. Asterisks denote values significantly different between BCG- and sham-vaccinated controls. * P≤ 0.05, ** P≤ 0.01. Bars = 340.0 μm, 40× magnification, insets 200× magnification.
Dystrophic calcification consists predominately of calcium phosphate, but includes a complex mixture of elements. To determine what effect BCG vaccination had on elements of mineralized lesions, we analyzed isolated primary lung lesions from M. tuberculosis infected guinea pigs for calcium, phosphorus, iron, magnesium, copper and zinc in sham-vaccinated and BCG vaccinate animals. The difference in the progressive accumulation dystrophic calcification seen histologically in lesions from M. tuberculosis infected guinea pigs was reflected by the concentration of mineral deposits in isolated primary lung lesions (FIG. 2). At day 30 of infection, the concentration of calcium and phosphorus in primary lesions from sham- and BCG-vaccinated animals was not different from controls, but concentrations increased significantly in sham-vaccinated animals by 60 days (FIG. 2 A, B). Iron concentrations in primary lesions from BCG -vaccinated animals were increased above sham-vaccinated and normal controls at 30 days of infection, but decreased to control levels by day 60 (FIG. 4 C). Similar to total calcium, phosphorus and iron, magnesium concentrations in isolated primary lesions from sham vaccinated animals increased significantly above those of non-infected controls and those from BCG-vaccinated animals (data not shown). Copper and zinc concentrations were also measured in primary lesions but did not vary significantly over time between sham-vaccinated, BCG-vaccinated or non-infected controls (data not shown).
FIG. 2.
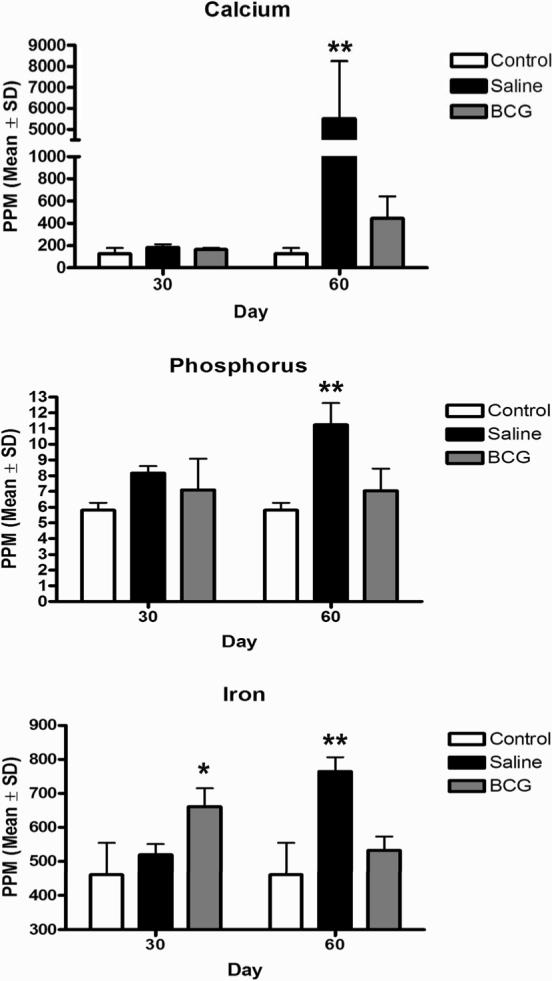
Trace mineral analysis of isolated of primary lung lesions of guinea pigs challenged with a low-dose aerosol of the H37Rv strain of M. tuberculosis or sterile water, without or with prior BCG-vaccination. The marked increase in calcium (A), phosphorus (B) and total iron (C) content of primary lesions at day 60 post-infection was ameliorated by BCG vaccination which reflects the lack of dystrophic calcification and ferric iron deposition seen microscopically. Data represents the mean ± standard deviation parts per million (PPM) from 2 separate experiments. Control n = 10, BCG- and sham-vaccinated n = 8. Asterisks denote values significantly different between BCG- and sham-vaccinated controls. * P≤ 0.05, ** P≤ 0.01.
FIG. 4.
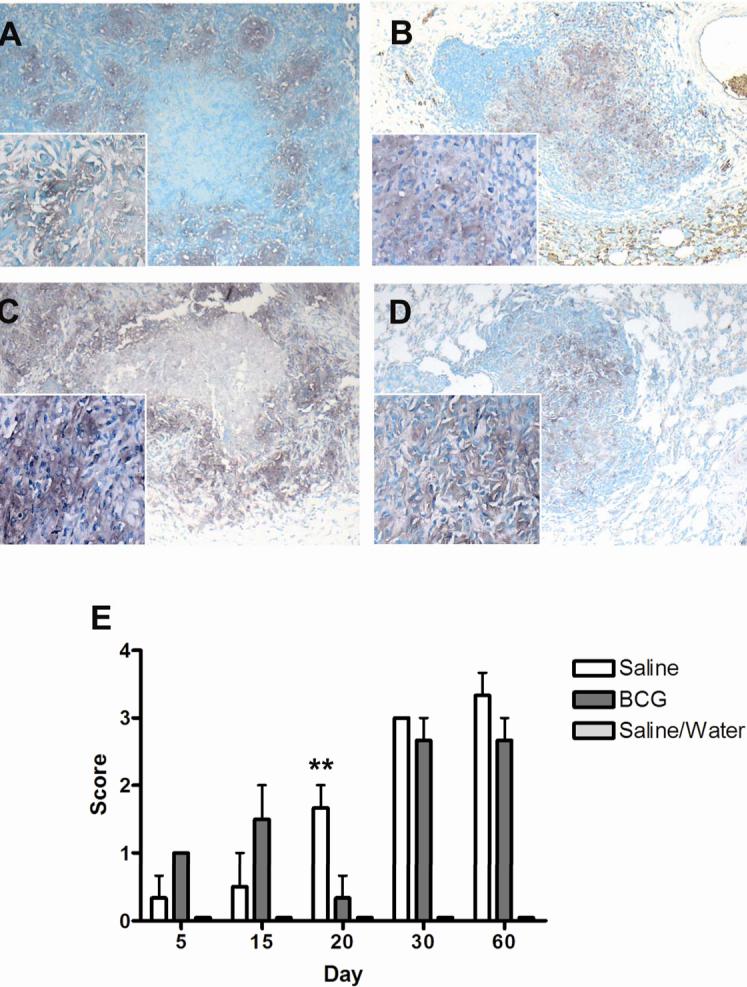
Expression of transferrin receptor (CD71) as demonstrated by immunohistochemistry, by macrophages within primary lung lesions of guinea pigs challenged with a low-dose aerosol of the H37Rv strain of M. tuberculosis, without (A, C) or with prior BCG-vaccination (B,D). Transferrin receptor expression in primary lesions in both BCG- and sham-vaccinated animals increased significantly from day 5 to 60 post-infection. Expression was highest in BCG-vaccinated animals at days 30 and 60 but based on scores, differences were not statistically different from sham-vaccinated animals (E). Data represents the mean ± standard deviation from 1 representative experiment. n = 3. Asterisks denote values significantly different between BCG- and sham-vaccinated controls. * P≤ 0.05, ** P≤ 0.01. Bars = 340 μm, 40× magnification, insets 200× magnification.
Limiting intracellular iron availability is one mechanism by which activated macrophages control the growth of M. tuberculosis (39). We therefore used immunohistochemistry to determine what effect BCG vaccination had on the macrophage expression of transferrin receptor (CD71), H ferritin and lactoferrin in vivo. Macrophages within lesions were identified for the presence of lysozyme by immunohistochemistry (data not shown). The significant increase in ferritin expression in sham-vaccinated animals paralleled the peak of necrosis seen histologically. Macrophage H ferritin expression increased significantly from 30 to 60 days of infection within primary lung lesions from sham-vaccinated guinea pigs infected with M. tuberculosis (FIG. 3 A, C). Ferritin accumulated intra-cellularly in macrophages that encircled the necrotic centers of the lesion in sham-vaccinated animals (FIG. 3 A, C). The variable expression of H ferritin seen in BCG-vaccinated animals reflects occasional primary lesions that show non-progressive necrosis. The expression of H ferritin in lesions from BCG-vaccinated animals was delayed such that it was first seen at 30 days of infection (FIG. 3B, E). By 60 days (FIG. 3D, E) the total number of cells expressing H ferritin in the BCG- and sham-vaccinated animals was increased but differences between the groups were not statistically different (FIG. 3E).
Macrophages encircling necrotic centers in sham-vaccinated guinea pigs expressed transferrin receptor in a pattern similar to that of H ferritin (FIG. 4A, C). In the early stages of infection, the total expression of macrophage transferrin receptor was significantly increased in BCG-vaccinated animals compared to sham-vaccinated controls (FIG 4E). In BCG-vaccinated animals where necrosis was rare, transferrin receptor expression was more diffusely distributed within the primary lesions (FIG. 4B, D). The expression of transferrin receptor was seen in primary lesions as early as 5 days post-infection (FIG. 4 E). The increase in the number of total cells expressing transferrin receptor was progressive in both BCG- and sham-vaccinated animals. While BCG-vaccinated animals had the greatest expression early following aerosol challenge, differences compared to sham-vaccinated controls were not statistically different. Despite the amelioration of transferrin receptor expression within individual primary lesions in BCG-vaccinated animals, the mean scores between BCG- and sham-vaccinated animals were not statistically different at days 30 and 60 of infection.
Lactoferrin expression varied significantly between sham-vaccinated and BCG vaccinated guinea pigs. In non-vaccinated animals the expression of lactoferrin was most prominent extra-cellularly in the necrotic centers while the surrounding macrophages expressed no or only low levels of this iron-binding protein (FIG. 5A). In contrast, in BCG-vaccinated animals macrophages expressed relatively high levels of lactoferrin with a distribution similar to that of the other two iron-binding proteins (FIG. 5B). Similar to transferrin receptor expression, the highest intra-cellular lactoferrin expression was first evident in BCG vaccinated animals which was seen as early as 5 days of infection. Expression of lactoferrin in sham-vaccinated animals was not significant until day 20 of infection and then was extra-cellular within foci of early necrosis that was progressive to day 60.
FIG. 5.
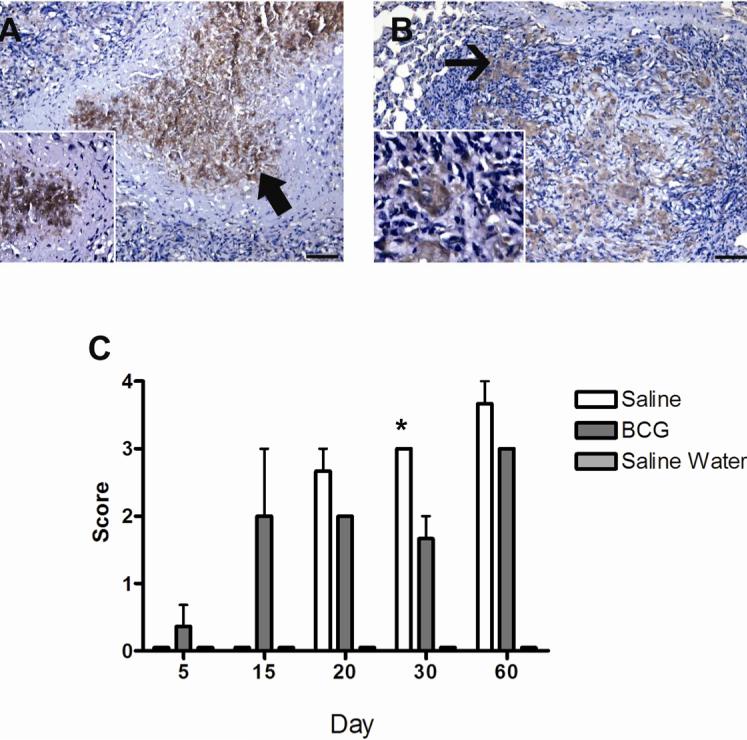
Expression of lactoferrin, as demonstrated by immunohistochemistry, by cells within the primary lesions of guinea pigs at day 60 following challenge with a low-dose aerosol of the H37Rv strain of M. tuberculosis, without (A) or with prior BCG-vaccination (B). In the non-vaccinated animals labeling was particularly prominent in the necrotic centers (arrow) and appeared to be both intracellular, in degenerating cells, and extra-cellular in the necrotic debris (inset) (A). The surrounding macrophages expressed no or only low levels of lactoferrin. In contrast, in BCG-vaccinated animals the macrophages (arrow) in primary granulomas expressed moderate to high levels of lactoferrin (B). Lactoferrin expression increased progressively in non-vaccinated animals from 20 to 60 days but was only significantly different from BCG vaccinated animals at day 30 (C). Data represents the mean ± standard deviation from 1 representative experiment. n = 3. Asterisks denote values significantly different between BCG- and sham-vaccinated controls. * P≤ 0.05, ** P≤ 0.01. Bar = 130 μm, 100× magnification, inset 200× magnification.
DISCUSSION
In this study we examined the temporal relationship between the accumulation of extra-cellular iron and calcification in the primary lesions of guinea pigs infected with M. tuberculosis. We exploited the morphologic differences between the primary lesions in BCG-vaccinated and non-vaccinated guinea pigs to better understand the mechanisms of action of BCG in vivo and the pathogenesis of dystrophic calcification of primary lesions. The visualization of calcified lesions in the lungs and lymph nodes of humans by radiography has been recognized for decades as an important criterion in the clinical diagnosis of tuberculosis (5, 6, 21). More importantly, primary lesions characterized by calcification, are resistant to treatment with first line anti-tuberculosis drugs and may therefore contribute to the development of multi-drug resistant strains of M. tuberculosis (6, 7, 40).
The accumulation of iron within the primary lesions of M. tuberculosis infected guinea pigs was not surprising given that the macrophage is central to the pathogenesis of tuberculosis and plays an important role in normal host iron metabolism. What was unexpected was the accumulation of extra-cellular ferric iron within the foci of necrosis that coincided with the development of dystrophic calcification. In addition, ferric iron was incorporated into the mineralized matrix with a restricted peripheral distribution, a region that may be available for scavenging by persistent, viable bacilli in the non-mineralized, acellular margins of the primary lesions (7). Given that extra-cellular, but not intra-cellular, iron can serve as an initiator of dystrophic calcification, our data support the hypothesis that iron is not only important in M. tuberculosis virulence but may play a central role in the pathogenesis of primary lesion necrosis. Iron may not only initiate calcification but the initial tissue necrosis may be mediated in part by free iron which is extremely damaging to cells by catalyzing the generation of reactive oxygen intermediates such as highly toxic hydroxyl radicals (41). By-products of oxygen free radical damage are detectable in the serum of patients with active tuberculosis, however, the source of these free radicals and the role they have in the pathogenesis has yet to be determined (26, 42).
Applying special stains to histologic sections allowed us to localize accumulations and distribution of both iron and calcium within lesions and to visualize the effect BCG vaccination, prior to aerosol challenge, had on lesion morphogenesis. The analysis of isolated primary lesions for iron and other elements allowed us to quantify the chemical composition of representative lesions and to confirm the observations made histologically. The marked increase in the calcium concentration between days 30 and 60 in the sham, saline-vaccinated animals infected with M. tuberculosis is consistent with the presence of dystrophic calcification in lesions as seen with routine H&E and the Von Kossa's stain for mineralization. There were significant differences in the range of calcium concentrations in randomly selected lesions of non-vaccinated animals which reflects the heterogeneity in the amount of calcification in lesions found within and between individuals. This is routinely recognized histologically and therefore was not unexpected. Also in keeping with the histology results, compared to non-vaccinated animals, BCG vaccination significantly reduced measurable calcium concentrations in isolated lesions, to levels that were only modestly elevated compared to lung parenchyma of non-infected controls.
Dystrophic calcification is a complex of ions that is composed primarily of calcium and phosphorus as calcium phosphate (14-17). As expected, the increase in phosphorus in isolated primary lesions from M. tuberculosis infected guinea pigs sham-vaccinated with saline paralleled the increase in calcium. BCG vaccination prior to infection prevented phosphorus accumulation in isolated lesions. Additional ions are incorporated into the mineralized matrix depending on the extra-cellular concentration and the presence or absence of endogenous inhibitors of calcification (14, 43, 44).
The cation of particular interest in this study was iron. Iron is a cofactor for numerous heme and non-heme proteins that are involved in critical metabolic functions that are common to all living organisms (21, 22). M. tuberculosis like other pathogenic bacteria has evolved complex mechanisms to acquire host iron which is limited in vivo even under normal physiologic conditions (30, 31, 45, 46). It was the observations from in vitro studies, that M. tuberculosis can scavenge host iron that led us to hypothesize that iron accumulated in the primary lung lesions in the guinea pig model of tuberculosis. The accumulation of ferric iron but not ferrous iron coincided with the appearance of extra-cellular calcium visualized in specially stained tissue sections and total iron concentrations in isolated primary lesions. The deposition of extra-cellular ferric iron had a pattern similar to extra-cellular calcification prior to organization into fully calcified foci. These observations suggest that like cellular phospholipids, iron may serve as an initiator of dystrophic calcification in the guinea pig model of tuberculosis. The relationship between extra-cellular iron and soft-tissue calcification has been shown in previous studies and can be induced experimentally by the injection of various iron containing compounds (20).
The host contributes significantly to the accumulation of iron in primary lesions. The regulation of iron metabolism by macrophages is closely linked to the expression and internalization of transferrin bound iron to surface expressed transferrin receptors (32, 47). Once internalized, iron dissociates from transferrin within the acidified phagolysosome and transferrin is recycled to the cell surface (35). M. tuberculosis has been shown to acquire host iron within the phagosome by the secretion and binding of iron chelating siderophores that have a higher binding capacity for iron than transferrin or lactoferrin (34, 48, 49). Classical activation of macrophages with IFN-gamma prevents the accumulation of iron within the phagosome and down regulates the expression of surface bound transferrin (50). The increased expression of macrophage transferrin receptor expression in vivo in our study is consistent with the phenomenon of alternative macrophage activation induced by exposure of macrophages to IL-4 in vitro (50). In addition, it has been shown that BCG vaccination of guinea pigs results in a notable decrease in the expression of host ferritin levels (51). Collectively, these data are consistent with the hypothesis that the protective effect of BCG vaccination is in part due to classical macrophage activation.
In our study, ferritin and lactoferrin expression was closely tied to the presence of lesion necrosis. Ferritin is a ubiquitous protein that is expressed in essentially all cells of the body. Ferritin has a complex structure made up of two different subunits designated heavy and light chain (52, 53). Because of its complex structure, ferritin has a high capacity to store free iron by oxidizing ferrous to ferric iron and thus functions as an anti-oxidant by reducing the availability of intracellular free ferrous iron (53). Ferrous iron is highly reactive with oxygen and can participate in the generation of free radicals through the Fenton reaction. Only the heavy chain of ferritin has ferroxidase activity and therefore the anti-oxidant activity of ferritin is attributed only to the heavy chain subunit. The close association between H ferritin expression and the presence of necrosis suggests that H ferritin expression along with lactoferrin may be useful as a biomarker with prognostic or predictive value in determining the clinical outcome of tuberculosis (54).
M. tuberculosis can also acquire iron from lactoferrin and has been recently shown to utilize lactoferrin over transferrin in human macrophages in vitro (35, 49). Since lactoferrin is a component of secretory granules of both macrophages and granulocytes, it is released into the extra-cellular space following inflammatory cell degranulation at sites of inflammation (55). We have recently shown by flow cytometry and immunohistochemistry that granulocyte necrosis and degranulation are associated with the development of primary lesion necrosis, a pattern that matches that of extra-cellular lactoferrin in the current study (56). By being more resistant to oxidative modification, lactoferrin may maintain its iron binding and sequestering capacity at sites of inflammation, when released with reactive oxygen species and myeloperoxidase from stimulated and degranulating neutrophils (57). While lactoferrin can serve as a source of intra-cellular iron, it is unknown whether M. tuberculosis can acquire either free or protein bound iron from extra-cellular sources at sites of inflammation in vivo (35, 49).
Since iron toxicity is detrimental to both host macrophages and the tubercle bacillus, yet each has a strict nutritional requirement for iron, it stands to reason that the successful accumulation and sequestration of iron would be an important determinant in the outcome of M. tuberculosis infection. We have demonstrated that the lack of necrosis and dystrophic calcification in lung lesions of guinea pigs vaccinated with BCG parallels the amelioration of iron accumulation, transferrin receptor, lactoferrin and H ferritin expression by macrophages in vivo. Iron metabolism, as our data suggests, can be influenced by immunopotentiation and may be an important consideration in the development of the next generation of tuberculosis vaccines and treatments.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grant AI054697.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ho RS, Fok JS, Harding GE, Smith DW. Host-parasite relationships in experimental airborne tuberculosis. VII. Fate of Mycobacterium tuberculosis in primary lung lesions and in primary lesion-free lung tissue infected as a result of bacillemia. J. Infect. Dis. 1978;138:237–241. doi: 10.1093/infdis/138.2.237. [DOI] [PubMed] [Google Scholar]
- 2.Dubovsky H. A historical basis for modern concepts of the pathogenesis of tuberculosis. S. Afr. Med. J. 1975;49:1105–1110. [PubMed] [Google Scholar]
- 3.Smith DW, McMurray DN, Wiegeshaus EH, Grover AA, Harding GE. Host-parasite relationships in experimental airborne tuberculosis. IV. Early events in the course of infection in vaccinated and nonvaccinated guinea pigs. Am. Rev. Respir. Dis. 1970;102:937–949. doi: 10.1164/arrd.1970.102.6.937. [DOI] [PubMed] [Google Scholar]
- 4.Canetti G. The Tubercle Bacillus in the Pulmonary Lesion of Man; Histobacteriology and its bearing on the therapy of pulmonary tuberculosis. 2 ed. Springer Publishing Company, Inc; New York: 1955. [Google Scholar]
- 5.Stead WW, Kerby GR, Schlueter DP, Jordahl CW. The clinical spectrum of primary tuberculosis in adults. confusion with reinfection in the pathogenesis of chronic tuberculosis. Ann. Intern. Med. 1968;68:731–745. doi: 10.7326/0003-4819-68-4-731. [DOI] [PubMed] [Google Scholar]
- 6.Smith DW, Balasubramanian V, Wiegeshaus E. A guinea pig model of experimental airborne tuberculosis for evaluation of the response to chemotherapy: the effect on bacilli in the initial phase of treatment. Tubercle. 1991;72:223–231. doi: 10.1016/0041-3879(91)90013-i. [DOI] [PubMed] [Google Scholar]
- 7.Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ. Location of persisting mycobacteria in the guinea pig model of tuberculosis revealed by R207910. Antimicrob. Agents. Chemother. 2007;51(9):3338–45. doi: 10.1128/AAC.00276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J. Pathol. 2006;208:261–269. doi: 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- 9.Ulrichs T, Kosmiadi GA, Trusov V, Jorg S, Pradl L, Titukhina M, Mishenko V, Gushina N, Kaufmann SH. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defense in the lung. J. Pathol. 2004;204:217–228. doi: 10.1002/path.1628. [DOI] [PubMed] [Google Scholar]
- 10.Turner OC, Basaraba RJ, Frank AA, Orme IM, Boros DL. Granulomatous Infections and Inflammations: Cellular and Molecular Mechanisms. ASM Press; Washington D.C.: 2003. Granuloma Formation in Mouse and Guinea Pig Models of Experimental Tuberculosis; pp. 65–84. [Google Scholar]
- 11.Turner OC, Basaraba RJ, Orme IM. Immunopathogenesis of pulmonary granulomas in the guinea pig after infection with Mycobacterium tuberculosis. Infect. Immun. 2003;71:864–871. doi: 10.1128/IAI.71.2.864-871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basaraba RJ, Dailey DD, McFarland CT, Shanley CA, Smith EE, McMurray DN, Orme IM. Lymphadenitis as a major element of disease in the guinea pig model of tuberculosis. Tuberculosis. (Edinb.) 2006;86:386–94. doi: 10.1016/j.tube.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Basaraba RJ, Izzo AA, Brandt L, Orme IM. Decreased survival of guinea pigs infected with Mycobacterium tuberculosis after multiple BCG vaccinations. Vaccine. 2006;24:280–286. doi: 10.1016/j.vaccine.2005.07.103. [DOI] [PubMed] [Google Scholar]
- 14.Giachelli CM. Vascular calcification: in vitro evidence for the role of inorganic phosphate. J. Am. Soc. Nephrol. 2003;14:S300–S304. doi: 10.1097/01.asn.0000081663.52165.66. [DOI] [PubMed] [Google Scholar]
- 15.Giachelli CM, Speer MY, Li X, Rajachar RM, Yang H. Regulation of vascular calcification: roles of phosphate and osteopontin. Circ. Res. 2005;96:717–722. doi: 10.1161/01.RES.0000161997.24797.c0. [DOI] [PubMed] [Google Scholar]
- 16.Wu-Wong JR, Noonan W, Ma J, Dixon D, Nakane M, Bolin AL, Koch KA, Postl S, Morgan SJ, Reinhart GA. Role of phosphorus and vitamin D analogs in the pathogenesis of vascular calcification. J. Pharmacol. Exp. Ther. 2006;318:90–98. doi: 10.1124/jpet.106.101261. [DOI] [PubMed] [Google Scholar]
- 17.Farber JL. Biology of disease: membrane injury and calcium homeostasis in the pathogenesis of coagulative necrosis. Lab. Invest. 1982;47:114–123. [PubMed] [Google Scholar]
- 18.Giachelli CM. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004;15:2959–2964. doi: 10.1097/01.ASN.0000145894.57533.C4. [DOI] [PubMed] [Google Scholar]
- 19.Kim KM. Cells, rather than extracellular matrix, nucleate apatite in glutaraldehyde-treated vascular tissue. J. Biomed. Mater. Res. 2002;59:639–645. doi: 10.1002/jbm.10038. [DOI] [PubMed] [Google Scholar]
- 20.Gabbiani G, Tuchweber B. The role of iron in the mechanism of experimental calcification. J. Histochem. Cytochem. 1963;11:799–803. [Google Scholar]
- 21.Jordan A, Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 22.Lill R, Dutkiewicz R, Elsasser HP, Hausmann A, Netz DJ, Pierik AJ, Stehling O, Urzica E, Muhlenhoff U. Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim. Biophys. Acta. 2006;1763:652–667. doi: 10.1016/j.bbamcr.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol. 2003;38:61–88. doi: 10.1080/713609210. [DOI] [PubMed] [Google Scholar]
- 24.Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 2007 doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]
- 25.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu.Rev.Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 26.Wesselius LJ, Williams WL, Bailey K, Vamos S, O'Brien-Ladner AR, Wiegmann T. Iron uptake promotes hyperoxic injury to alveolar macrophages. Am. J. Respir. Crit. Care Med. 1999;159:100–106. doi: 10.1164/ajrccm.159.1.9801033. [DOI] [PubMed] [Google Scholar]
- 27.Wessling-Resnick M. Iron transport. Annu. Rev. Nutr. 2000;20:129–151. doi: 10.1146/annurev.nutr.20.1.129. [DOI] [PubMed] [Google Scholar]
- 28.Castranova V. Generation of oxygen radicals and mechanisms of injury prevention. Environ. Health Perspect. 1994;102(Suppl 10):65–68. doi: 10.1289/ehp.94102s1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghio AJ, Turi JL, Yang F, Garrick LM, Garrick MD. Iron homeostasis in the lung. Biol. Res. 2006;39:67–77. doi: 10.4067/s0716-97602006000100008. [DOI] [PubMed] [Google Scholar]
- 30.Macham LP, Ratledge C, Nocton JC. Extracellular iron acquisition by mycobacteria: role of the exochelins and evidence against the participation of mycobactin. Infect. Immun. 1975;12:1242–1251. doi: 10.1128/iai.12.6.1242-1251.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratledge C. Iron, mycobacteria and tuberculosis. Tuberculosis.(Edinb.) 2004;84:110–130. doi: 10.1016/j.tube.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE. Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools. Impact of interferon-gamma and hemochromatosis. J. Biol. Chem. 2002;277:49727–49734. doi: 10.1074/jbc.M209768200. [DOI] [PubMed] [Google Scholar]
- 33.Wagner D, Maser J, Lai B, Cai Z, Barry CE, III, Honer zu BK, Russell DG, Bermudez LE. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis- containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J. Immunol. 2005;174:1491–1500. doi: 10.4049/jimmunol.174.3.1491. [DOI] [PubMed] [Google Scholar]
- 34.Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat. Chem. Biol. 2005;1:149–153. doi: 10.1038/nchembio717. [DOI] [PubMed] [Google Scholar]
- 35.Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE. The nature of extracellular iron influences iron acquisition by Mycobacterium tuberculosis residing within human macrophages. Infect. Immun. 2004;72:2022–2028. doi: 10.1128/IAI.72.4.2022-2028.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gangaidzo IT, Moyo VM, Mvundura E, Aggrey G, Murphree NL, Khumalo H, Saungweme T, Kasvosve I, Gomo ZA, Rouault T, Boelaert JR, Gordeuk VR. Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 2001;184:936–939. doi: 10.1086/323203. [DOI] [PubMed] [Google Scholar]
- 37.Schaible UE, Collins HL, Priem F, Kaufmann SH. Correction of the iron overload defect in beta-2-microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J. Exp. Med. 2002;196:1507–1513. doi: 10.1084/jem.20020897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiegeshaus EH, McMurray DN, Grover AA, Harding GE, Smith DW. Host-parasite relationships in experimental airborne tuberculosis. 3. Relevance of microbial enumeration to acquired resistance in guinea pigs. Am. Rev. Respir. Dis. 1970;102:422–429. doi: 10.1164/arrd.1970.102.3.422. [DOI] [PubMed] [Google Scholar]
- 39.Bullen JJ, Rogers HJ, Spalding PB, Ward CG. Natural resistance, iron and infection: a challenge for clinical medicine. J. Med. Microbiol. 2006;55:251–258. doi: 10.1099/jmm.0.46386-0. [DOI] [PubMed] [Google Scholar]
- 40.Dhillon J, Mitchison DA. Influence of BCG-induced immunity on the bactericidal activity of isoniazid and rifampicin in experimental tuberculosis of the mouse and guinea-pig. Br. J. Exp. Pathol. 1989;70:103–110. [PMC free article] [PubMed] [Google Scholar]
- 41.Mladenka P, Simunek T, Hubl M, Hrdina R. The role of reactive oxygen and nitrogen species in cellular iron metabolism. Free Radic. Res. 2006;40:263–272. doi: 10.1080/10715760500511484. [DOI] [PubMed] [Google Scholar]
- 42.Jack CI, Jackson MJ, Hind CR. Circulating markers of free radical activity in patients with pulmonary tuberculosis. Tuber. Lung Dis. 1994;75:132–137. doi: 10.1016/0962-8479(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 43.Makowski GS, Ramsby ML. Amorphous calcium phosphate-mediated binding of matrix metalloproteinase-9 to fibrin is inhibited by pyrophosphate and bisphosphonate. Inflammation. 1999;23:333–360. doi: 10.1023/a:1020209616428. [DOI] [PubMed] [Google Scholar]
- 44.Gupta LC, Singla SK, Tandon C, Jethi RK. Mg2+: a potent inhibitor of collagen-induced in vitro mineralization. Magnes. Res. 2004;17:67–71. [PubMed] [Google Scholar]
- 45.Krithika R, Marathe U, Saxena P, Ansari MZ, Mohanty D, Gokhale RS. A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2069–2074. doi: 10.1073/pnas.0507924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou CJ, Wisedchaisri G, Monfeli RR, Oram DM, Holmes RK, Hol WG, Beeson C. Functional studies of the Mycobacterium tuberculosis iron-dependent regulator. J. Biol. Chem. 2004;279:53554–53561. doi: 10.1074/jbc.M407385200. [DOI] [PubMed] [Google Scholar]
- 47.Batista A, Millan J, Mittelbrunn M, Sanchez-Madrid F, Alonso MA. Recruitment of transferrin receptor to immunological synapse in response to TCR engagement. J. Immunol. 2004;172:6709–6714. doi: 10.4049/jimmunol.172.11.6709. [DOI] [PubMed] [Google Scholar]
- 48.Lounis N, Truffot-Pernot C, Grosset J, Gordeuk VR, Boelaert JR. Iron and Mycobacterium tuberculosis infection. J. Clin. Virol. 2001;20:123–126. doi: 10.1016/s1386-6532(00)00136-0. [DOI] [PubMed] [Google Scholar]
- 49.Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J. Leukoc. Biol. 2007;81:195–204. doi: 10.1189/jlb.0606405. [DOI] [PubMed] [Google Scholar]
- 50.Kahnert A, Seiler P, Stein M, Bandermann S, Hahnke K, Mollenkopf H, Kaufmann SH. Alternative activation deprives macrophages of a coordinated defense program to Mycobacterium tuberculosis. Eur. J. Immunol. 2006;36:631–647. doi: 10.1002/eji.200535496. [DOI] [PubMed] [Google Scholar]
- 51.Tree JA, Elmore MJ, Javed S, Williams A, Marsh PD. Development of a guinea pig immune response-related microarray and its use to define the host response following Mycobacterium bovis BCG vaccination. Infect. .Immun. 2006;74:1436–1441. doi: 10.1128/IAI.74.2.1436-1441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Theil EC, Sayers DE, Brown MA. Similarity of the structure of ferritin and iron dextran (imferon) determined by extended X-ray absorption fine structure analysis. J. Biol. Chem. 1979;254:8132–8134. [PubMed] [Google Scholar]
- 53.Torti SV, Torti FM. Iron and ferritin in inflammation and cancer. Adv. Inorg. Biochem. 1994;10:119–137. [PubMed] [Google Scholar]
- 54.Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, Mollenkopf HJ, Ziegler A, Kaufmann SH. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J. Mol. Med. 2007 doi: 10.1007/s00109-007-0157-6. [DOI] [PubMed] [Google Scholar]
- 55.LaForce FM, Boose DS. Release of lactoferrin by polymorphonuclear leukocytes after aerosol challenge with Escherichia coli. Infect. Immun. 1987;55:2293–2295. doi: 10.1128/iai.55.9.2293-2295.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ordway D, Palanisamy G, Henao-Tamayo M, Smith EE, Shanley C, Orme IM, Basaraba RJ. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J. Immunol. 2007;179:2532–41. doi: 10.4049/jimmunol.179.4.2532. [DOI] [PubMed] [Google Scholar]
- 57.Winterbourn CC, Molloy AL. Susceptibilities of lactoferrin and transferrin to myeloperoxidase-dependent loss of iron-binding capacity. Biochem. J. 1988;250:613–616. doi: 10.1042/bj2500613. [DOI] [PMC free article] [PubMed] [Google Scholar]