

Abstract
In some bacteria Lys-tRNALys is used both in translation and for the specific addition of Lys to phosphatidylglycerol in the cytoplasmic membrane. This reaction is catalyzed by the membrane protein MprF, and the lysyl-phosphatidylglycerol formed contributes to the resistance of these bacteria to various cationic antibacterial molecules. Obtaining proteins and reconstituting an in vitro system mimicking membrane conditions is a major challenge to studying the function of membrane proteins, especially when labile substrates such as Lys-tRNALys are required. Here we report methods to obtain a stable enriched membrane fraction containing MprF, and the techniques necessary to quantitatively monitor its activity in vitro and in vivo.
1. Introduction
One of the major mechanisms by which bacteria adapt to variations in the physicochemical conditions of their habitat takes place at the level of the cellular envelope that modulates the flow of ions, nutrients, proteins, and antibiotics into and out of the cell. Reduction of the net negative charge of the cell envelope is a strategy used by numerous bacteria to decrease their affinity for cationic antimicrobial molecules. This change in charge is accomplished by the transfer of positively charged groups to the negatively charged constituents of the cell envelope (1). Teichoic acid in the cell wall of Gram-positive bacteria, lipopolysaccharide in the outer membrane of Gram-negative bacteria and phospholipids in bacterial cytoplasmic membranes, together determine the net negative charge of the bacterial envelope. A decrease in the net negative charge of these components can be achieved through four different pathways: i) attachment of D-Ala to teichoic acid (2), ii) acylation of the lipopolysaccharide with positively charged molecules (i.e. modification of lipid A with 4-amino-4-deoxy-L-arabinose), iii) biosynthesis of phosphatidylethanolamine (PE) (3) and iv) lysylation of phosphatidylglycerol (PG) (4). PG lysylation activity, first discovered in Staphylococcus aureus (5), involves transfer of a Lys moiety from the 3’-end of tRNALys to the free distal hydroxyl group of the glycerol moiety of PG (6, 7). The specificity of the staphylococcal protein was investigated, showing that the Lys analog S-(2-aminoethyl)-l-cysteine can be efficiently processed when attached to E. coli tRNALys but not tRNACys (7). However, the biological significance of PG lysinylation was still not understood. The protein MprF (Multipeptide resistance factor) was recently identified as an integral membrane protein responsible for this transferase activity and was recognized as a novel virulence factor in certain pathogenic organisms. In S. aureus MprF confers resistance to diverse pore forming cationic peptides produced by the immune system such as defensins secreted by neutrophils and protegrins produced by leukocytes. A staphylococal mprF deletion strain exhibited an attenuated resistance to human neutrophiles killing and a reduced virulence in mice (4). In addition MprF confers resistance to lanthionine-containing peptides (lantibiotics) secreted by certain bacteria such as nisin produced by Lactococcus lactis (4) and also to several other positively charged antibiotics (aminoglycosides, betalactamines and glycopeptides)(8, 9). The MprF pathway plays a role in the virulence of Listeria monocytogenes and may also provide a selective advantage to non-pathogenic organisms during colonization of their natural habitats (10).
While the physiological significance of the MprF pathway has recently been explored, and qualitative in vitro TLC assays were set up using radiolabeled Lys (11), there is to date no convenient in vitro assay to quantitatively monitor the Lys-tRNALys PG transferase activity. Here we report general methods to express and obtain an enriched cellular fraction containing active MprF. A quantitative in vitro assay was established to monitor Lys-tRNALys PG transferase activity, and this method was applied to optimize detergent and substrate concentrations for future studies.
2. Expression of MprF in Escherichia coli, preparation of membrane fractions, and analysis of lipids by TLC
2.1 Cloning of MprF from Bacillus subtilis and its expression in E. coli
MprF is not naturally expressed in E. coli, providing an ideal system for heterologous production of the protein. The ORF yfiW-yfiX from B. subtilis (a sequencing error led to misannotation of the gene as split into two ORFs) was amplified by PCR (primers: fwd, 5′-ctgcagtctagaagcttagacggagtcttttttgctt-3′, rev, 5′-ccatggaattcggatccatgctgattaaaaagaatgctttat-3′) and cloned under the control of the T7 promoter at the BamHI/HindIII sites of the vector pet33b (kanR, Novagen). Expression of the protein was attempted in three strains derived from E. coli BL21 DE3; Rosetta2 DE3 (Novagen), and strains C41 and C43 which both contain the plasmid pRARE2 (CamR, Novagen) encoding tRNAs for translation of rare codons. Strains C41 and C43 are widely used to express toxic or membrane proteins and generally give better results than the conventional BL21 strain (12).
Overproduction of B. subtilis MprF was performed in an autodinduction medium or in LB with induction of expression by addition of 1 mM IPTG at OD600nm = 0.6. One liter of autoinduction medium was reconstituted with independently sterilized solutions by mixing 930 ml LB without salt, 1 ml 1 M MgSO4, 1ml filtered sterilized trace metal solution (50 μM FeCl3, 20 μM CaCl2, 10 μM MnCl2 and ZnCl2, 2 μM CoCl2, CuCl2, NiCl2, MoSO4, SeCl2 and H3BO3), 20 ml carbon source solution containing 25% glycerol, 2.5% Glucose and 10% lactose (w/v) and 50 ml of a buffer containing 1 M KH2PO4, 1 M Na2HPO4 and 0.5 M (NH4)2SO4. Both media contained 25 mg/l chloramphenicol and 50 mg/l kanamycin and were inoculated at 1/100 of the final media volume with a starter culture, and then cultured at 37 °C overnight under vigorous shaking. Cells were harvested by centrifugation and were frozen at −80 °C. Expression levels of MprF were assessed by measuring the specific activity of Lys-tRNALys PG transferase in crude extracts (section 3.1 below). The highest yields of active MprF were obtained by using the strain C41 DE3 grown in autoinduction medium.
2.2 Membrane fraction preparation
Membrane fractions were prepared by differential centrifugation and all the following steps were performed at 4 °C. To prepare crude cell extracts, 1.6 g of frozen cell paste were sonicated 8 times 30 s in 5 ml of buffer A (50 mM Tris-HCl pH 8.0, 1 mM DIFP, 1 mM PMSF and 3 mM β-mercaptoethanol) with a 3 mm probe mounted on a Branson sonifier 450. Cellular debris were sedimented by centrifugation for 10 min at 10,000 × g. The debris pellet was re-extracted twice with 5 ml of buffer A and all the supernatants were pooled together. During this step, 62% of the total activity was lost (Table 1). However, all the activity of the 10,000 × g supernatant containing the cellular membranes was recovered by ultracentrifugation at 250,000 × g for 45 min. The membrane pellet was then washed 3 times with 4 ml of buffer A before dispersion in 1.5 ml of buffer A by brief sonication on the “low” power setting (Branson Sonifier 450). The MprF activity of this fraction was enriched 2.3-fold (Table 1) and remained stable at −80 °C for several months upon addition of 0.6 ml of buffer A containing 70 % glycerol. Analysis by SDS page of the membrane fraction revealed a weak band at 100 kDa and an intense band at 50 kDa indicating proteolytic degradation of the full length protein.
Table 1.
Enrichment of the Lys-tRNALys PG transferase activity
| Protein (mg) | Activity (pmol/min) | Specific Activity (pmol/min/mg) | Enrichment | Yield (%) | |
|---|---|---|---|---|---|
| Cell crude extract | 132 | 189 | 1.43±0.17 | 1 | 100 |
| Supernatant 10,000 × g | 100 | 72 | 0.72±0.03 | 0.5 | 38 |
| Membrane fraction | 22 | 71 | 3.26±0.38 | 2.3 | 38 |
2.3 Preparation of total lipids
Biosynthesis of lysylated-PG (Lys-PG) in wild-type B. subtilis or in E. coli strains producing MprF can be monitored by analyzing their lipid composition by TLC. For this purpose, total lipids were extracted from 15 − 30 OD of harvested cells using the Bligh and Dyer extraction method (13) modified as follows. The cell pellet was resuspended in 0.2 ml of 120 mM sodium acetate pH 4.5 by vortexing. Low pH stabilizes the ester linkage of Lys to PG and enhances the extraction yield of polar lipids. A sonication step can be added to increase the yield of extraction when lipids are prepared from B. subtilis. To 0.2 ml of the resuspended cells, 0.75 ml chloroform:methanol (1:2, v:v) were added and the lipids were extracted by vortexing the mix for 10 min at room temperature. 0.25 ml of chloroform and 0.25 ml of 120 mM sodium acetate pH 4.5 were successively added and mixed by vortexing. The organic and aqueous phases were separated by centrifugation and the organic (lower) phase containing the lipids was collected and dried under vacuum. The dried lipids were finally resuspended in 50 μl of chloroform:methanol (2:1, v:v).
Phospholipids are the main lipids of bacterial membranes and their formation and relative abundance can be determined by in vivo radioactive labelling, and their relative composition analyzed by TLC. The protocol above can be scaled up for preparation of lipids from litres of media or scaled down for in vivo [32P] labelling of phospholipids (Fig. 1). In the latter case, 2 ml of media supplemented with 2 × 106 cpm of [32P]PPi (PerkinElmer) were inoculated with 20 μl of an overnight pre-culture, all solvent volumes necessary for the lipid extraction were decreased two-fold and the sonication step omitted.
Figure 1.
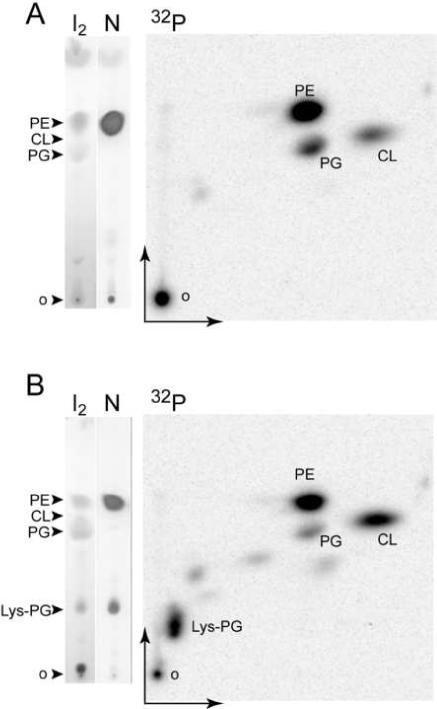
TLC analysis of lipids from E. coli. Lipids were extracted from E. coli C43 transformed with pet33b (A) and from E. coli C43 expressing B. subtilis Mprf (B). TLC plates were developed in a one dimensional (vertical) or a two dimensional solvent system (see text section 2.4). Total lipids were revealed with iodine (I2), amino lipids with ninhydrine (N) and [32P]-radiolabelled phospholipids by phosphorimaging (32P). PE: phosphoethanolamine, PG: phosphatidylglycerol, CL: cardiolipin, KPG: lysyl-phosphatidylglycerol, o: TLC origin.
2.4 TLC analysis of lipids
5 − 10 μl of the unlabeled lipid preparation or a suitable amount of radiolabeled [32P]phospholipids were spotted on a 10 cm long 250 μ HLF silicagel TLC plate (Analtech). The TLC was developed in one or two dimensions using successively the solvents chloroform:methanol:water (14:6:1, v:v:v) in the first dimension and chloroform:methanol:acetic acid (13:5:2, v:v:v) in the second dimension (Fig. 1). Each dimension ran for 20 min; a longer migration time resulted in compression of the migrating lipids at the solvent front. Unsaturated fatty acid chains (57% of the total E. coli fatty acid chains) were visualized by exposure of the dried TLC to iodine vapors. Detection of amino groups was performed with ninhydrin, and of phosphate groups by using molybdenum blue (Sigma) (14). Radiolabeled phospholipids were revealed by phosphorimaging (Fig. 1).
3. n vitro assays of Lys-tRNALys phosphatidylglycerol transferase activity
In our in vitro assay, Lys-tRNALys is steadily provided to MprF by an excess of E. coli lysyl-tRNA synthetase (LysRS) in the presence of tRNALys, free [14C]Lys, ATP and PG according to the following scheme:
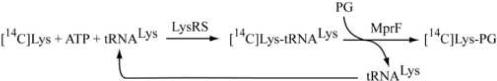
PG is not soluble in water and forms liposomes upon sonication or vortexing. Since MprF was isolated in a membrane fraction and the amount of embedded PG was not sufficient to promote the saturation of MprF, PG liposomes were added to the reaction media as substrates for the reaction. However, without addition of exogenous PG and in the presence of larger amounts of membrane extract, a very weak MprF activity (0.02 pmol/min/μg) can be measured that results from the lysinylation of PG present in the membrane extract. The length of pre-incubation necessary to allow fusion of cellular membranes with PG liposomes, as well as the concentration of PG, were both critical in establishing a reproducible in vitro assay. The use of detergent (Triton X100) was also helpful for the efficient reconstitution of numerous membrane activities and was investigated in more detail (section 3.4 below).
3.1 Steady-state assays for Lys-tRNALys PG transferase activity
PG liposomes were prepared by resuspending dried egg yolk PG (Sigma P0514) in buffer B containing 100 mM Hepes-NaOH pH 7.2, 10 mM MgCl2, 30 mM KCl and then vortexing for 15 min at room temperature. Total tRNA from E. coli was used as substrate and was prepared from an E. coli strain overexpressing tRNALys (15). 20% of the total tRNA from this strain could be aminoacylated with [14C]Lys as determined from aminoacylation plateau levels. LysRS and stock solutions of membrane fractions were diluted with buffer B. Reconstitution of the reaction medium was performed by the sequential addition of 1) the liposome solution, 2) water, 3) detergent (Triton X-100) and 4) the membrane fraction. The mix was pre-incubated at 37 °C (section 3.2) and the Lys-tRNALys PG transferase reaction was started by addition of an aminoacylation mix containing ATP, Lys-tRNALys, LysRS, buffer and salt (see concentrations below). The aminoacylation mix was pre-incubated for 5 min at 37 °C to pre-form Lys-tRNALys. Initiation of the reaction by addition of the membrane fraction or short pre-incubation times of the membrane fraction with PG resulted in biphasic kinetics. Once reconstituted, the Lys-tRNALys PG transferase reaction media contained 100 mM Hepes-NaOH pH 7.2, 10 mM MgCl2, 30 mM KCl, 8 mM ATP, 14 μM [14C]Lys (230 cpm/pmol), 2 mg/ml PG liposomes, 1.76 mg/ml Triton X-100, 5 μM tRNALys, 0.2 μM affinity tag purified E. coli LysRS (16) and 0.4 mg/ml of the membrane protein fraction with or without B. subtilis MprF (section 2.3). It is worth noting that the amount of LysRS used (0.2 μM, kcat = 108 min−1) ensures that the rate of Lys-tRNALys synthesis (21 μM min−1) is not the limiting step of the overall reaction scheme since the rate of formation of Lys-PG in all subsequent experiments never exceeded 2 μM min−1. Two techniques were employed to monitor the kinetics of Lys-PG synthesis, a TLC procedure to determine the relative amount of Lys-PG compared to the total amount of Lys, and an extraction procedure that in combination with standard scintillation methods determines simultaneously the amounts of Lys-tRNALys and Lys-PG (Fig. 2). Aliquots were periodically removed from the reaction media between 2 − 32 min after initiation. For TLC analysis, aliquots of 1.5 μl were added to 5.6 μl of chloroform:methanol (1:2, v:v) and the resulting mixtures spotted on silicagel plates and developed as described in section 2.4. [14C]Lys and derivatives were visualized by phosphorimaging. Free [14C]Lys and [14C]Lys-tRNALys remained at the TLC origin whereas [14C]Lys-PG migrated with an Rf = 0.28. The concentration of [14C]Lys-PG in the reaction was derived from quantification of the ratio of [14C]Lys-PG:(free [14C]Lys+ [14C]Lys-tRNALys) multiplied by the initial [14C]Lys concentration (14 μM) (Fig. 2A). The simultaneous measurement of [14C]Lys-PG formation and the level of aminoacylation of tRNALys was performed by the quantification of [14C]Lys-tRNALys and [14C]Lys-PG after their simultaneous extraction from each time course as follows. 8 μl of the reaction media were added to 500 μl chloroform:methanol:120 mM potassium acetate pH 4.5 (1:2:0.8, v:v:v). In this solvent, [14C]Lys-PG was stable for several hours at room temperature (data not shown). At the end of the time course, 130 μl of chloroform and 130 μl of 120 mM potassium acetate pH 4.5 were successively added and mixed by vortexing for 20 s. Phases were then separated by centrifugation at room temperature and the lower phases containing [14C]Lys-PG transferred to a scintillation vial, dried in an oven at 50 °C and the radioactivity then determined by scintillation counting. The level of [14C]Lys-tRNALys in the aqueous phase was determined by precipitation of the material by adding 25 μl 3 M potassium acetate pH 4.5 and 1 ml ethanol followed by incubation at −80°C. The pellet obtained after centrifugation was air dried for 30 min at room temperature, resuspended in 50 μl 60 mM potassium acetate pH 4.5 and spotted on a 3 MM Whatman filter disc. Free [14C]Lys was removed from the filter disc by washing two times with 5% TCA and radioactivity determined by scintillation counting.
Figure 2.
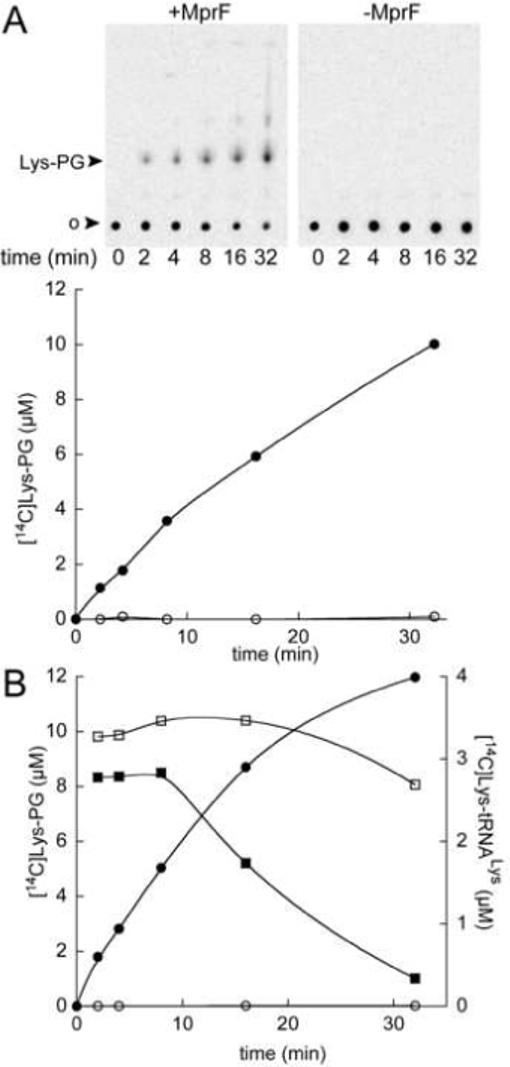
In vitro kinetic assays for [14C]Lys-PG synthesis. (A) TLC assay. Lower panel: [14C]Lys-PG concentration derived from the phosphorimaging quantitation of the TLC. (B) Extraction and scintillation counting assay (see text section 3.1). [14C]Lys-PG concentrations are represented by circles and Lys-tRNALys concentrations by squares. Both methods (A, B) were applied simultaneously in the presence of a membrane fraction from the E. coli C43 strain expressing B. subtilis MprF (+MprF, filled symbols) or without expression of MprF (-MprF, open symbols).
A good correlation was found between the two techniques for monitoring MprF activity (Fig. 2). TLC analysis revealed a spot migrating above [14C]Lys-PG that may correspond to the [14C]Lys-monoacyl-PG. The slight differences in the level of [14C]Lys-PG formation between the two techniques may result from the existence of other lysyl- derivatives that are extractable by the solvent method but that may not migrate during TLC analysis and stay instead at the origin. The level of Lys-tRNALys is steady for the first 10 min of the reaction allowing measurement during this time of the initial velocity of Lys-PG formation.
3.2 Optimization of pre-incubation of PG with membrane fractions
Short pre-incubations of PG liposomes with membrane extract prior to initiation of the reaction resulted in biphasic kinetics for Lys-PG formation, indicating that sufficient time had not been allowed for the fusion of PG liposomes with cellular extract membranes. Specific activity of membrane extracts was determined with the assay described above (section 3.1) after increasing the pre-incubation time of PG liposomes with membrane fractions (Fig. 3). 10 min pre-incubation before initiation of the reaction (addition of the aminoacylation mix) was necessary to obtain maximum specific activity. Longer incubation times did not significantly improve the specific activity of the extracts.
Figure 3.
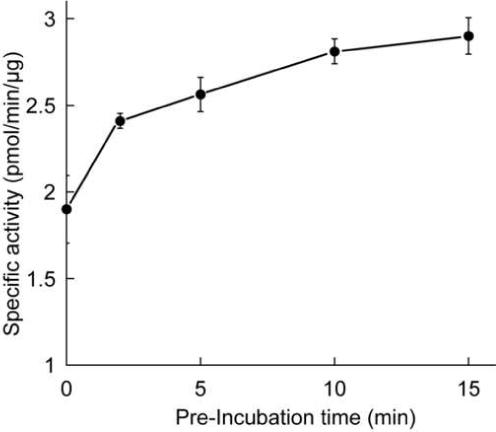
Specific activity of Lys-PG synthesis from membrane extracts as a function of reaction media pre-incubation time (see text section 3.2). n=2.
3.3 Optimal concentration of PG
Before investigating the kinetic parameters of MprF for Lys-tRNALys, it was necessary to determine whether or not MprF in the membrane preparation was saturated with PG. Saturation of MprF by PG was determined under the conditions described above (section 3.1) in the presence of 0.25 mg/ml membrane protein fraction, a saturating amount of Lys-tRNALys (10 μM) and a range of concentrations of PG (0.25−6 mg/ml). Figure 4 shows that 2 mg/ml of PG was sufficient to saturate MprF under these conditions.
Figure 4.
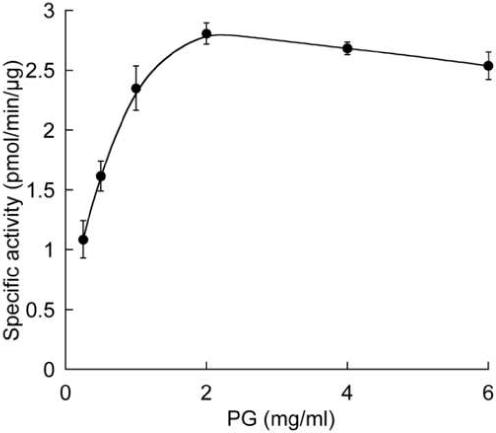
Specific activity of Lys-PG synthesis from membrane extracts as a function of PG concentration. 0.25 mg/ml membrane extract were saturated by increasing amounts of PG (see text section 3.3). n=2.
3.4 Use of detergent
The use of detergents greatly facilitates in vitro reconstitution of various membrane activities (for reviews e.g. (17, 18)). Mixed lipid liposomes containing detergents constitute a more permeable bilayer and we expected that such structures would effect the in vitro reconstitution of MprF activity. Specific activity of membrane fractions was measured under the conditions described above (section 3.1) with 0.5 mg/ml of extract in the presence of 5 μM [14C]Lys-tRNALys, 2 mg/ml PG and a range of concentrations of Triton X100 (0−5.3 mg/ml). The mix of PG, detergent and membrane extract was pre-incubated for 10 min at 37 °C before initiation of the reaction by addition of the aminoacylation mix. In this first approach, the detergent was not removed from the reaction media before the initiation of the reaction, but diluted 1.4-fold upon addition of the aminoacylation mix. In order to standardize the amount of detergent added in the assay and to report the specific activity versus the ratio of Triton X-100:Lipids (w/w), the concentration of lipids in the membrane fraction was determined (19). The lipid concentration in the membrane fraction was 3.33 mg/ml (with 10.8 mg/ml protein) bringing the total amount of lipid in the assay to 2.1 mg/ml. Figure 4 shows the specific activity of MprF in the membrane fraction as a function of the ratio of Triton X100:Lipids (w/w) in the assay. Optimal conditions for the reaction were found without Triton X-100, and a suboptimal peak of activity was reached with a ratio of Triton X100:lipid of 0.8.
4. Future Applications
The results above show that Lys-tRNALys PG transferase activity can be quantitatively monitored under steady-state conditions, allowing future investigation of substrate specificity and structure-function studies of MprF proteins. The specificity of MprF for the aminoacyl- and tRNA moieties of aminoacyl-tRNA can be studied for any substrate that can be efficiently assembled either by an aminoacyl-tRNA synthetase or a flexizyme (20). Numerous Lys analogues are substrates of LysRSs (21) and can be readily used to form an array of new aa-tRNALys variants with which to investigate recognition of the aminoacyl- moiety by MprF. Moreover, transplanting an orthologous identity set into tRNALys could also provide tRNALys aminoacylated with non-cognate amino acids (22). tRNA recognition by MprF can now be investigated by determining the tRNA identity set for this protein using tRNALys variants that are substrates of LysRS. The techniques presented above can also be used to investigate the specificity of MprF for other lipids using commercially available compounds (cardiolipin, lipoteichoic acid) or lipid mixtures radiolabeled in vivo. Overall, the versatility of the techniques described here will now allow for the first time a robust and systematic investigation of the biochemical activities of the MprF superfamily, an emerging group of bacterial virulence factors.
Figure 5.
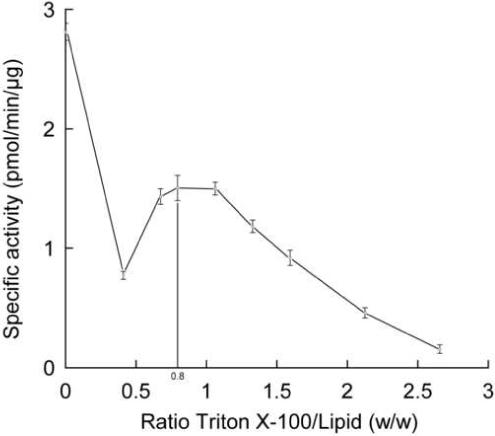
Specific activity of Lys-PG synthesis from membrane extracts as a function of the ratio of Triton X-100:Lipid (w/w). (see text section 3.4). No Triton X-100 was added to the reaction media for the × axis value = 0. n=2.
Acknowledgments
We thank C. Hausmann and J. Ling for critical reading of the manuscript. This work was supported by Grant GM 65183 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peschel A. Trends Microbiol. 2002;10:179–86. doi: 10.1016/s0966-842x(02)02333-8. [DOI] [PubMed] [Google Scholar]
- 2.Neuhaus FC, Baddiley J. Microbiol Mol Biol Rev. 2003;67:686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao M, Helmann JD. J Bacteriol. 2004;186:1136–46. doi: 10.1128/JB.186.4.1136-1146.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA. J Exp Med. 2001;193:1067–76. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lennarz WJ, Nesbitt JA, 3rd, Reiss J. Proc Natl Acad Sci U S A. 1966;55:934–41. doi: 10.1073/pnas.55.4.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lennarz WJ, Bonsen PP, van Deenen LL. Biochemistry. 1967;6:2307–12. doi: 10.1021/bi00860a005. [DOI] [PubMed] [Google Scholar]
- 7.Nesbitt JA, 3rd, Lennarz WJ. J Biol Chem. 1968;243:3088–95. [PubMed] [Google Scholar]
- 8.Nishi H, Komatsuzawa H, Fujiwara T, McCallum N, Sugai M. Antimicrob Agents Chemother. 2004;48:4800–7. doi: 10.1128/AAC.48.12.4800-4807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komatsuzawa H, Ohta K, Fujiwara T, Choi GH, Labischinski H, Sugai M. FEMS Microbiol Lett. 2001;203:49–54. doi: 10.1111/j.1574-6968.2001.tb10819.x. [DOI] [PubMed] [Google Scholar]
- 10.Staubitz P, Peschel A. Microbiology. 2002;148:3331–2. doi: 10.1099/00221287-148-11-3331. [DOI] [PubMed] [Google Scholar]
- 11.Staubitz P, Neumann H, Schneider T, Wiedemann I, Peschel A. FEMS Microbiol Lett. 2004;231:67–71. doi: 10.1016/S0378-1097(03)00921-2. [DOI] [PubMed] [Google Scholar]
- 12.Miroux B, Walker JE. J Mol Biol. 1996;260:289–98. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 13.Bligh EG, Dyer WJ. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 14.Dittmer JC, Lester RL. J Lipid Res. 1964;15:126–7. [PubMed] [Google Scholar]
- 15.Commans S, Lazard M, Delort F, Blanquet S, Plateau P. J. Mol. Biol. 1998;278:801–13. doi: 10.1006/jmbi.1998.1711. [DOI] [PubMed] [Google Scholar]
- 16.Ataide SF, Ibba M. Biochemistry. 2004;43:11836–41. doi: 10.1021/bi0490542. [DOI] [PubMed] [Google Scholar]
- 17.Seddon AM, Curnow P, Booth PJ. Biochim Biophys Acta. 2004;1666:105–17. doi: 10.1016/j.bbamem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 18.Rigaud JL, Pitard B, Levy D. Biochim Biophys Acta. 1995;1231:223–46. doi: 10.1016/0005-2728(95)00091-v. [DOI] [PubMed] [Google Scholar]
- 19.Stern I, Shapiro B. J Clin Pathol. 1953;6:158–60. doi: 10.1136/jcp.6.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramaswamy K, Saito H, Murakami H, Shiba K, Suga H. J Am Chem Soc. 2004;126:11454–5. doi: 10.1021/ja046843y. [DOI] [PubMed] [Google Scholar]
- 21.Levengood JD, Ataide SF, Roy H, Ibba M. J Biol Chem. 2004 doi: 10.1074/jbc.M313665200. [DOI] [PubMed] [Google Scholar]
- 22.Anderson JC, Wu N, Santoro SW, Lakshman V, King DS, Schultz PG. Proc Natl Acad Sci U S A. 2004;101:7566–71. doi: 10.1073/pnas.0401517101. [DOI] [PMC free article] [PubMed] [Google Scholar]