

Summary
Huntington’s disease is a genetic neurological disorder that is triggered by the dissociation of the huntingtin protein (htt) from its obligate interaction partner Huntingtin-interacting protein 1 (HIP1). The release of htt permits HIP-protein interactor (HIPPI) to bind to its recognition site on HIP1 to form a HIPPI/HIP1 complex that recruits Procaspase-8 to begin the process of apoptosis. The interaction module between HIPPI and HIP1 was predicted to resemble a death-effector domain (DED). Our 2.8 Å crystal structure of the HIP1 371-481 sub-fragment that includes F432 and K474 important for HIPPI binding is not a DED, but is a partially opened coiled-coil. The HIP1 371-481 model reveals a basic surface we hypothesize is suitable for binding HIPPI. There is an opened region next to the putative HIPPI site that is highly negatively charged. The acidic residues in this region are highly conserved in HIP1 and a related protein, HIP1R from different organisms, but are not conserved in the yeast homolog of HIP1, sla2p. We have modeled ∼85% of the coiled-coil domain by joining our new HIP1 371-481 structure to the HIP1 482-586 model (PDB code: 2NO2). Finally, the middle of this coiled-coil domain may be intrinsically flexible and suggests a new interaction model where HIPPI binds to a “U” shaped HIP1 molecule.
Keywords: HIP1, HIPPI, Apoptosis, Huntington’s disease, Clathrin
Introduction
Huntington’s disease is a late-onset neurodegenerative genetic disorder caused by the expansion of a polyglutamine tail at the N-terminus of the huntingtin protein (htt).1 When this polyglutamine tail is greater than 36 residues, the affinity of mutant htt for its obligate binding partner, Huntingtin-interacting protein 1 (HIP1), is greatly diminished, causing them to dissociate from each other. 2 The lost htt/HIP1 interaction causes the death of medium spiny neurons through apoptosis. 3 HIP1 belongs to the HIP1/R protein family, including the yeast homologue, Sla2p. This protein family interacts with clathrin and adaptor proteins via its central coiled-coil domain 4,5,6 and links clathrin coated pits (CCP) to membrane phospholipids by its N-terminal ANTH domain. 7,8,9 It also links CCP to actin cytoskeleton by its C-terminal THATCH domain.10,11 HIP1/R family protein also promotes clathrin assembly by interacting with clathrin light chain 4,6,12 HIP1 is highly expressed in neurons and is an important component of clathrin coated vesicles. 13HIP1 knockout mice show neurological deficits and cultured neurons derived from HIP1 knockout mice have a defect in clathrin-mediated internalization. 14 On the other hand, HIP1 over-expression induces cellular toxicity in cultured neuronal cells. 15 Importantly, HIP1 has been identified as a marker for several human cancers, including colon, prostrate and brain cancer.16,17,18,19 The wide-ranging roles of HIP1 were demonstrated when this protein was found to traffic androgen receptor into the nucleus to regulate gene transcription. 20
HIP1 has been demonstrated to be a pro-apoptotic protein. Sequence analysis identified a pseudo death effector domain (pDED) in HIP1. 15 The over-expression of the HIP1 pDED alone was sufficient to induce apoptosis, which was partially reversed when wild type but not mutant Huntingtin was coexpressed. 15 A yeast two-hybrid screen pulled out a HIP1 interacting protein named HIPPI (HIP1 protein interactor) that also has pDED domain. 3 Based on these findings, a model was proposed where the pDED of HIP1 interacted with the pDED of HIPPI to recruit Procaspase-8 for activation to turn on the activity of Caspase-3 to cause apoptotic death. 3,15
Fas or the tumour necrosis factor receptor mediates apoptotic death. Caspase-8 is brought to the adaptor protein Fas-associated death domain (FADD) by the interaction of DEDs present in both Caspase-8 and FADD. 21 This oligomerization event ultimately leads to the formation of the death-inducing signal complex. 22 The NMR structure of the FADD DED is a six-helix bundle with a conserved phenylalanine positioned in the middle of one of the two hydrophobic patches important for caspase-8 binding. 23 The HIP1 and HIPPI pDEDs share 39.2% similarity and 26.6% identity with each other while conventional DEDs share 41.7% averaged similarity and 29.5% averaged identity.3 The HIP1 pDED has the conserved phenylalanine that is shared with other known DEDs but this phenylalanine is not present in the pDED of HIPPI. Interestingly, there is one charged lysine residue that is shared exclusively between HIP1 and HIPPI, but not in other DEDs. 3 This particular lysine has been shown by yeast two-hybrid to be important for the HIP1-HIPPI interaction. 3 To find out if the proposed pDED in HIP1 is truly a DED (or has DED-like structure), we crystallized a sub-fragment of HIP1 that included the presumed pseudo death-effector domain.
Here we report the crystal structure (2.8 Å) of HIP1 sub-fragment 371-481 that has determinants important for the binding of HIPPI. The HIP1 371-481 crystal structure addresses a longstanding idea that the HIPPI binding region of HIP1 may resemble the death-effector domain. 3,15 Our HIP1 model reveals that there is a basic region that can serve as an interaction surface for HIPPI. HIP1 371-481 is a coiled-coil dimer that is continuation of the HIP1 482-586 structure recently solved in our laboratory. 24 Interestingly, the two coils in the HIP1 371-481 dimer are not tightly intertwined between E431 and H482. This opened coiled-coil structure is consistent with our previous observation that the N-terminal region of HIP1 482-586 is splayed opened. 24 This supports our working hypothesis that helices near the center of the coiled-coil domain are incapable of twisting tightly together because of electrostatic repulsions. We reassembled our new HIP1 371-481 model with the HIP1 482-586 structure 24 to reconstruct about 85% of the HIP1 coiled-coil domain. The analysis of the assembled HIP1 shows the putative HIPPI interaction surface is faced opposite the clathrin light chain-binding region. The information from the HIP1 371-481 sub-fragment that includes a basic surface suitable for HIPPI has generated new insights into how the HIP1/HIPPI heterocomplex is formed.
Structure
The HIP1 371-481 sub-fragment produced tetragonal crystals (P43) that diffracted to a maximum resolution of ∼2.7 Å at the Advanced Light Source (beamline MBC 4.2.2). These crystals were highly mosaic (∼2 degrees) due to the disordered packing of protomers in the unit cell. A search for different conditions yielded very thin two-dimensional needles that diffracted poorly. We tested about 100 small molecule and detergent additives to improve the original P43 crystals, but diffraction quality did not get better. The high mosaicity was not caused by rapidly cooling the crystals in liquid nitrogen for data collection because crystals mounted in capillaries were also very mosaic. In the end, we decided to grow selenomethionine P43 crystals and performed MAD experiments on the best ones. The experimental phases obtained were sufficient to generate an interpretable electron density maps to 2.8 Å, but model refinement was limited, giving high r-values at 2.8 Å (r=26.5% and rfree=32.4%). Throughout this paper we call our model HIP1 371-481 (human numbering, accession number NP 005329) even though the first two amino acids at positions 369 and 370 are left over from the cleaved GST tag and some C-terminal residues were not visible. The MAD data collection and refinement statistics are given in Table 1.
Table 1.
Data collection and refinement statistics
| A. Crystal | |
|---|---|
| Space group | P43 |
| Cell dimensions (Å) | 72.9 × 72.9 × 106.5 |
| B. Data collection statistics | Absorption peak | Inflection point | Remote |
|---|---|---|---|
| Wavelength (Å) | 0.97892 | 0.97907 | 0.96407 |
| No. observed reflections | 53,721 | 54,774 | 54,846 |
| No. unique reflections | 15,348 | 15,256 | 15,272 |
| Resolution limits (Å) | 43.07-2.70 (2.80-2.70)a | 46.35-2.70 (2.80-2.70) | 43.00-2.70 (2.80-2.70) |
| Mosaicity (degrees) | 1.97 | 1.94 | 1.94 |
| Completeness (%) | 99.6 (99.8) | 99.7 (99.7) | 99.6 (99.9) |
| Redundancy | 3.5 (3.4) | 3.6 (3.5) | 3.6 (3.4) |
| Rmerge (%) | 9.9 (52.3) | 8.5 (45.0) | 8.2 (44.0) |
| I/σ (I) | 6.2 (1.6) | 7.5 (1.9) | 7.7 (2.0) |
| C. Refinement statistics | |
|---|---|
| R factor (%) | 26.5 |
| Free R factor (%) | 32.4 |
| rmsd in bond lengths (Å) | 0.009 |
| rmsd in bond angles (degrees) | 1.19 |
| Ramachandran plot most favored (%) | 92.3 |
| Additional allowed (%) | 7.1 |
| Generously allowed (%) | 0.0 |
| Disallowed (%) | 0.6 |
Values in parentheses are for the data in the outermost shell.
The asymmetric unit contains two HIP1 371-481 dimers (labeled Dimer 1 and Dimer 2) that are oriented into an antiparallel “A” configuration (Figure 1). A single HIP1 dimer has two parallel right-handed helices (Helix 1 and Helix 2) whose N-terminal regions are twisted together into a left-handed coiled-coil (see Figure 1). Each HIP1 dimer copy has an uneven C-terminal end because residues 441-482 of Helix 2 are not visible in the crystal. The end of Helix 2 was not proteolytically cleaved off because there is only a single MALDI-TOF peak that gives the expected molecular weight of the monomer (data not shown). The opened region in the HIP1 371-481 sub-fragment and in the HIP1 482-586 model 24 may be an intrinsic property of this portion of the HIP1 coiled-coil domain. We do not favor the possibility that the opened region was caused by the crystallization solutions because the conditions used to grow crystals of the two HIP1 sub-fragments were very different.
Figure 1.
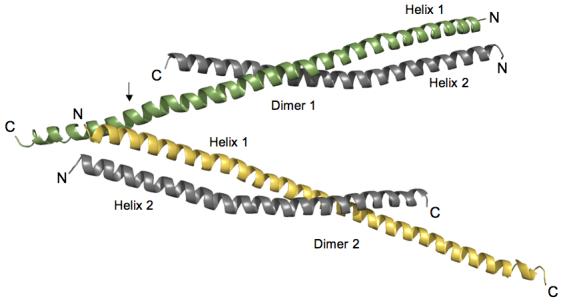
Orientation of HIP1 371-481 dimers in the crystal stabilizes the C-terminal region. Two dimers of HIP1 371-481 are arranged in an antiparallel “A” configuration. Helix 1 (green in Dimer 1) in each dimer shown is longer than Helix 2 (grey in Dimer 1) because of a crystal packing contact indicated by the arrow. At this point R380 and Y381 in Helix 1 of Dimer 2 (yellow) forms a pocket for D446 protruding from Helix 1 of Dimer 1 (green). The indicated lattice contact point faces away from the dimerization interfaces in the two dimers shown in the figure. The N- and C-termini of each helix are labeled N and C. The helices were rendered using PyMol [http://www.pymol.org].
Protein Expression and Purification: The plasmid encoding the N-terminal GST tagged HIP1 370-644 was kindly provided by the McPherson group. GST-HIP1 371-481 was made by altering the HIP1 cDNA to introduce a stop codon (TAG) at residue 482. Site-directed mutagenesis was performed according to the QuikChange mutagenesis protocol (Stratagene). The sequence was confirmed by DNA sequencing (IMBI, Indiana University) and then transformed into Rosetta 2 (DE3) pLysS cells (Novagen). The recombinant protein GST-HIP1 371-481 construct was expressed and purified as described previously 24 with some modifications. Briefly, GST-HIP1 371-481 was over-expressed at 37°C in M9 minimum medium. After reaching an O.D. 600 of 0.5 units, selenomethionine (50 μg/ml final concentration) and IPTG (100μg/ml final concentration) were added to the bacterial culture. Cells were then incubated for 16 h at 30°C before being harvested and frozen for use. Cell pellets were thawed on ice and then resuspended gently in 45 ml of PBS (10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.3), 140 mM NaCl, 2.7 mM KCl), supplemented with 0.25 ml of 1 M DTT, 0.25 ml of protease inhibitor cocktail (Sigma), and 2 ml of PMSF (17.4 mg/ml in 2-propanol) and incubated on ice for ∼15min. After sonication, 2.5 ml of 20% (v/v) Triton X-100 was added and the lysate was incubated at room temperature by rotating for ∼30 min. The crude bacterial lysate was cleared by centrifugation (19 k, 4°C) for 15 min and then the supernatant was mixed with ∼5 ml of glutathione Sepharose 4B (Amersham) resin suspended in PBS. This slurry was rocked gently at room temperature for 2 h before being transferred into a small column. The packed column was washed with 50 ml of PBS supplemented with 0.25 ml of 1M DTT until no more background protein was detected by staining with Coomassie brilliant blue. The GST-HIP1 371-481 was eluted from the column with 50 ml of 3 mg/ml L-glutathione (sigma) in (PBS) at pH 8.0 and dialyzed against the same buffer. The GST tag was removed by rocking the protein with sequencing grade thrombin (Novagen) at 37°C for 3 h. The digested sample was then dialyzed against 10 mM HEPES (pH 8.5), 20 mM Imidazole, 2 mM tris(2-carboxyethyl)-phosphine, 1% (v/v) glycerol. The protein was passed through an anion-exchange column (POROS 20 HQ) equilibrated in buffer A (10 mM HEPES, 20 mM imidazole, 2 mM tris(2-carboxyethyl)-phosphine, 1% (v/v) glycerol pH 8.5). The target protein was eluted with a linear gradient of buffer B (10 mM HEPES (pH 8.5), 20mM imidazole, 2 mM tris(2-carboxyethyl)-phosphine, 1% (v/v) glycerol, 250 mM NaCl). We confirmed the single selenomethionine substitution by electrospray mass spectroscopy.
Crystallization and data collection: The protein was crystallized by the hanging drop method in reservoir buffer (15% PEG3350, 0.1 M succinate pH 7.0, 0.2 M potassium sodium tartrate, and 0.01 M NiCl2). The crystal grew in the tetragonal space group P43 (a=72.9, b=72.9, c=106.5, α=β=γ=90°), with two dimers in the asymmetric unit. The crystals were highly mosaic (∼2.9 degrees) along the c-axis. Exhaustive screening for alternative crystallization conditions or small molecule additives did not improve crystal quality. The crystals were looped out and quickly dipped in buffer containing ethylene glycol and then plunged in liquid nitrogen. The HIP 371-481 structure was solved by multi-wavelength anomalous dispersion (MAD). A 3-wavelength data set was obtained from a single crystal on beam line 4.2.2 at the Advanced Light Source, Lawrence Berkeley National Laboratory. The data were collected at 100 K in 1.0 degree oscillations using a NOIR-1 CCD detector. Wavelengths λ1 (1.0215Å) and λ2 (1.0213 Å) were at the peak and inflection, respectively, of the K-edge of selenium and λ3 (1.0373 Å) was a high energy remote. An inverse beam experiment was done at each wavelength, with E1=phi0 and E2=phi180 degrees, for a total oscillation of 90 degrees for each phi. The kappa angle was optimized to prevent overlaps in the long axis. Data were integrated and scaled using D*TREK.
Phasing and refinement: The MAD data was phased using a Bayesian approach 31 taking the high-energy remote (λ3) as the ‘native’ wavelength and the other two as ‘derivative’ wavelengths. The single selenium site in each monomer was found by SOLVE (http://www.solve.lanl.gov) and the experimental map was improved using RESOLVE 32. Model building was performed using O 33,34 the model was refined using CNS 34. Alternating rounds of positional, grouped B factor and simulated annealing were performed in reference to 2Fo-Fc and Fo- Fc maps and a bulk-solvent correction was applied near the end of refinement. The HIP1 371-481 structure refined against all the data from 30-2.8 Å with an R-factor of 26.5% and an Rfree of 32.4% at 2.8Å resolution.
We looked at the packing of protomers in the unit cell to see if lattice contacts stabilized the C-terminus of Helix 1. The arrow in Figure 1 points to where the C-terminus of Helix 1 of Dimer 1 crosses Helix 1 of Dimer 2 (R380 and Y381 in Helix 1 (Dimer 2) forms a pocket for D446 from Helix 1 (Dimer 1)). These crystal contacts do not interfere with the dimerization interface (see Figure 1), which means they are not responsible for opened coiled-coil. Rather, the opened HIP1 371-481 structure may be due to weak affinity between the C-terminal segments of Helix 1 and Helix 2. This is supported by our analysis of surface potential that indicates the opened region (E445, D446, E448, E456, E458, and E465, human numbering) is highly negatively charged (see Figure 2). Five out of six acidic residues are conserved in HIP1 and HIP1R from different organisms. For example, D446, E448, E456 and E458 (human numbering) are present in HIP1 from mouse and human and D446, E456 and E458 are conserved in HIP1R from human, rabbit, mouse and rat (E448 in mouse and rat is alanine). Interestingly, none of the acidic residues in the opened region are conserved in the yeast homolog of HIP1, sla2p. This suggests sla2p is structurally different than the HIP family proteins, at least in this region. We predict a significant amount of electrostatic repulsive tension would be localized in this highly acidic patch (see Figure 2(b)). The unfavorable juxtaposition of like charges in a coiled-coil motif is an important feature for kinesin activity. Electrostatic repulsive forces in the neck domain provide the flexibility needed for this molecular motor to “walk” 25.
Figure 2.
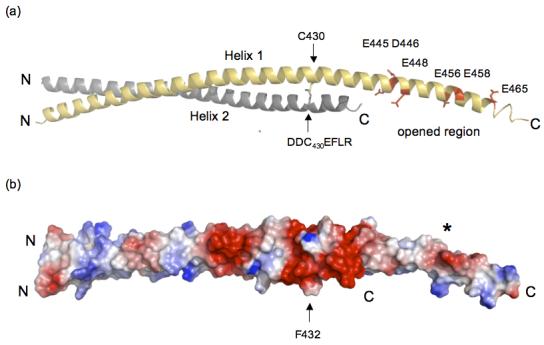
(a) The opened region is highly negatively charged and is close to an interhelical disulfide bridge. Dimer 2 is shown and the colors of Helices 1 and 2 are the same as in Figure 1. There is a concentration of negatively charged residues in the open region (E445, D446, E448, E456, E458 and E465) indicated in red in Helix 1 (yellow). The C430 disulfide bridge between Helix 1 (grey) and Helix 2 (indicated by arrows) is directly upstream from the acidic opened region and is in the DDC430EF432LR stretch. The C-terminal end of Helix 2 is disordered and is not visible (see text). The ribbon model was created using PyMol [http://www.pymol.org]. (b) Surface potential analysis of HIP1 371-481 shows the opened region has a high negative potential (red surface marked by the asterisk). The position of the dimer is the same as in (a) and the arrow is F432 that is one residue away from the C430 disulfide bridge. The C430 disulfide bridge is in a region that forms an acidic ring around the HIP1 371-481 dimer (see red colored region around the arrow pointing to F432). The N- and C-termini in (a) and (b) are labeled N and C. The surface potentials (blue, positive; red, negative) were calculated using PyMol.
We observed a disulfide bridge next to the opened region, in the DDC430EF432LR stretch (see Figure 2(a)). This disulfide bridge is completely buried in the dimer interface, which explains why the reducing agent in our purification and crystallization buffers did not break this bond. The C430 disulfide bridge is positioned in a cluster of negative charges that form a ring around the HIP1 dimer (see Figure 2(b)). Moreover, the C430 disulfide bond is at position a in the heptad pattern. A disulfide bond at this position is disruptive compared to when the bond is placed at position d26. Thus, the C430 disulfide bond may serve to increase the plasticity of the surrounding structure by bringing together repulsive surfaces and by introducing local distortions in this region of HIP1. Cysteine-430 is conserved in mouse and human, but is not in HIP1R (N instead) or the yeast homolog of HIP1, sla2p (V instead). There is only one other cysteine in HIP1 (only in mouse and human) at the other end of the coiled-coil domain at position-613 (human numbering). Although it would be intriguing to speculate that the C430 disulfide bond is functionally significant in vivo, we cannot rule out the possibility that this bond is an artifact of expression and purification.
Possible HIPPI binding surface
Two amino acids in HIP1 has been implicated in HIP1-induced cell toxicity 3,15. When F432 is replaced with glycine, cell toxicity is dramatically reduced, but a conservative F to Y substitution does not alter HIP1-induced apoptosis 15. Past work suggested that K474 that is in our HIP1 371-481 sub-fragment was important for recruiting HIPPI 3. Gervais et al. observed that mutating K409 in the pDED of HIPPI to leucine (K474/L409 combination) decreased the HIP1/HIPPI interaction, but they found that wild type binding was restored when K474 in HIP1 was changed to leucine (L474/L409) 3. These workers saw an intermediate phenotype when K409 was mutated to D409 (K474/D409), but surprisingly, full activity was seen when the lysine in HIP1 and HIPPI were both replaced with aspartic acid (D474/D409) 3.
We used the published mutagenesis data to search for the HIPPI interaction surface in our HIP1 371-481 crystal structure. Specifically, we mapped the positions of F432 and K474 in our HIP1 371-481 model (Figure 3(a)) to see if we could identify any unique surface feature between these two amino acid landmarks. Note that the position of K474 is approximate because the electron density for K474 was fragmented. We discovered that the molecular surface (∼58 Å long) between F432 and K474 is highly positively charged (see asterisk, Figure 3(b)). This suggests any HIPPI surface that contacts the positive potential surface in Figure 3(b) will have opposite charge. Furthermore, this suggests that K409 in HIPPI does not interact with the basic region shown in Figure 3. We wonder if the putative HIPPI binding surface extends beyond the region marked by F432 and K474. We observed that a narrow groove formed by V402, L416 and R423 goes 48 Å beyond F432 towards the N-terminus. This narrow groove is positioned between Helix 1 and Helix2, and is not part of the dimerization interface.
Figure 3.
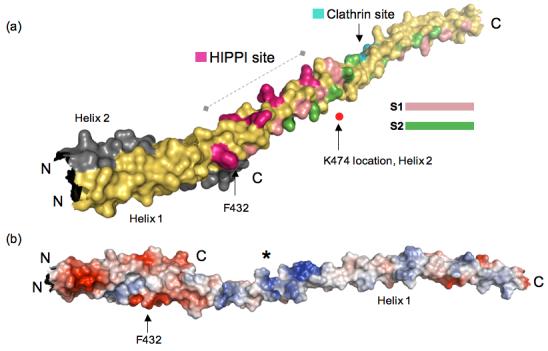
(a) Reassembled HIP1 coiled-coil domain using HIP1 371-481 (PDB code: 2QA7) and HIP1 482-586 24 (PDB code: 2NO2) models. Helix 1 of 2QA7 is in yellow and Helix 2 is in grey (ends just after F432, C-terminus labeled C). The N-terminus of 2QA7 has been cut slightly shorter for this figure. The C-terminal half of the assembled model is 2NO2, but only Helix 1 of this dimeric structure is shown in this figure for the sake of clarity. The surface bracketed by F432 and K474 indicated by the dashed line is a surface suitable for HIPPI binding. The position of K474 as it would be in Helix 2 (grey) is marked by the red dot. The residues forming the putative HIPPI binding surface between F432 and K474 are indicated in purple. The S1 (pink) and S2 (green) hydrophobic paths we described previously in 2NO2 24 are mapped out in the assembled HIP1 model to show that the HIPPI site is not in the dimerization interface, but is solvent exposed. The DLLRKN clathrin light chain-binding region (Clathrin site) is indicated in teal, and is barely visible because it is on the opposite face of Helix 1. The surface model was generated using PyMol [http://www.pymol.org]. (b) Surface potential analysis of the assembled model reveals the putative HIPPI site in (a) is highly basic (indicated by the asterisk) and is exposed to the solvent. This model is rotated relative to the model in (a) to show the basic region and is not on the same scale as the model in (a). The N- and C-termini are labeled N and C. The surface potentials (blue, positive; red, negative) were calculated using PyMol.
To visualize the topology of ∼85% of the central coiled-coil domain of the HIP1 molecule, we reassembled the HIP1 371-481 and HIP1 482-586 crystal structures (Figure 3(a)). We point out that this “divide-and-conquer” approach was the only way to resolve the coiled-coil domain because longer HIP1 constructs would not give quality crystals (unpublished data), presumably because of excessive heterogeneity due to flexibility. Our reassembled HIP1 coiled-coil domain in Figure 3 shows the spatial relationship between the molecular surface involved clathrin light chain binding 24 and the basic stretch defined by F432 and K474. To join the two HIP1 model, we built a short helical connector (residues 470 to 481) because fragmented electron density prevented us for building sequence in beyond S469 of Helix 1. The first half of the assembled HIP1 model in Figure 3(a) consists of Helix 1 (yellow) and Helix 2 (grey) from the HIP1 371-481 crystal structure (PDB code: 2QA7). For the sake of clarity, the other half of the assembled HIP1 model shows only Helix 1 of the HIP1 482-586 model (PDB code: 2NO2). Note that in Figure 3(a) the approximate position of K474 that would be in Helix 2 (grey) is indicated as a red dot because this same lysine in Helix 1 (yellow) is not visible the way the model is shown.
The assembled HIP1 model reveals the surface suitable for HIPPI binding is the continuation of the S3 path we previously described to be involved in the binding of clathrin light chain 24. From this structural information, we conclude that the basic surface (putative HIPPI binding site) in Figure 3 is fully exposed to solvent and is not a part of the coiled-coil dimer interface. The basic region between F432 and K474 is ∼80 Å (straight line distance) from the DLLRKN region. The reassembled HIP1 model shows there is sufficient room for both clathrin and HIPPI to bind at the same time to HIP1 because our proposed clathrin and HIPPI interaction surfaces are facing on opposite sides of the helix (another set of surfaces are also present in the second helix of the HIP1 dimer).
Coiled-coil plasticity and implication for HIP1/HIPPI binding
The flexibility suggested by the new HIP1 371-481 structure reported here and by our HIP1 482-586 model 24 has new implications for how HIPPI interacts with HIP1. Gervais et al. transiently transfected a variety of HIP1 and HIPPI deletion constructs in 293 cells and assessed the HIP1-HIPPI interaction by co-immunoprecipitation 3. They could not reduce the binding of HIPI to HIPPI when the pDED of HIP1 was deleted, but the binding between HIP1 and HIPPI decreased slightly when the HIPPI pDED was taken out 3. Apparently, the HIPPI myosin-like domain is important because the interaction is completely lost when a segment of HIPPI containing both the myosin-like domain (MLD) and pDED was removed. In addition, these workers showed that the HIP1 C-terminal talin-like domain was crucial for the tight binding of HIPPI 3. We believe that the mutagenesis data by Gervais et al. suggest there are determinants in the central coiled-coil and in the talin-like domains of HIP1 that are required for HIPPI to productively interact with HIP1. Therefore, an important question to address is how HIPPI can interact with determinants that are widely spaced apart. One possible way this can happen is if the structure of HIPPI is sufficiently extended to touch both regions in HIP1 to form a stable HIP1/HIPPI complex. An appealing structural alternative is that the distant HIPPI binding determinants are brought close together by folding back the HIP1 molecule. The ability of HIP1R to form a folded back structure 12 suggests that HIP1 may adopt a similar “U” shaped topology. We hypothesize that the association of huntingtin protein may control the binding of HIPPI to HIP1 by “locking” the HIP1 molecular into a rigid, extended conformation. According to this model the binding of htt to HIP1 buries some of the determinants for recruiting HIPPI and prevents the HIP1 molecule from folding back on itself to bring the critical talin-like domain at the C-terminus of HIP1 close to determinants near the center of the HIP1 coiled-coil domain. We are currently evaluating our proposed HIP1/HIPPI binding scenario by mutating residues in the basic region shown in Figure 3. We are also neutralizing acidic residues in the open region that might impact the overall flexibility of the coiled-coil domain of HIP1 to see if these point mutations can impact the ability of HIPPI to bind HIP1.
Hydrophobic residues occupy the a- and d-positions of stable coiled-coils, while less stable coiled-coils have hydrophilic amino acids at these positions 27,28,29. We therefore looked to see if the a- and d-positions in the opened region of the assembled HIP1 model (2QA7 and 2NO2 combined) were occupied by non-hydrophobic amino acids (see Figure 4). For this analysis, the assembled HIP1 model (residues 371-587) was divided into three regions, designated A, B, and C (see Figure 4). In our 2QA7 and 2NO2 24 crystal structures, regions A and C have coiled-coil structures, but the strands in region B are separated. The a- and d-positions in region A are occupied by stabilizing residues (I, L, M and V), but there is a destabilizing serine (S398) in the middle and two destabilizing residues (R423 (a), and C430 (a)) near the end of this region. Our crystal structure shows R423 in one helix is rotated away from R423 in the second helix to avoid an unfavorable electrostatic clash. We also found two coiled-coil stabilizing clusters 29 (LILL and LVLL) on either side of S398 (Figure 4). These two clusters may offset any negative impact S398 has on coiled-coil stability in region A. We note that the other coiled-coil segment (region C) of the assembled HIP1 does not have any of the stabilizing clusters described by Kwok and Hodges 29. Apparently, region C can tolerate unfavorable residues in a- and d-positions (E563(a), R577(a), and R587(d), Figure 4) because this part of HIP1 is coiled-coil 24. Finally, we looked at the a-d profile in region B to see if its hydrophobic core was disrupted. Region B contained seven destabilizing residues at a- and d-positions along the entire region (Figure 4). We identified a stretch of residues near the end of region B that resembled a LLIL stabilizing cluster, except glycine occupies the last position (L513(d)L(a)I(d)G524(a), see Figure 4). We predict that G524 could contribute to destabilizing this region of HIP1 because it is located in an a-position of the heptad repeat 28. We conclude that the hydrophobic core of region B is disrupted by hydrophilic amino acids, which can cause the coiled-coil domain of HIP1 to be more flexible.
Figure 4.
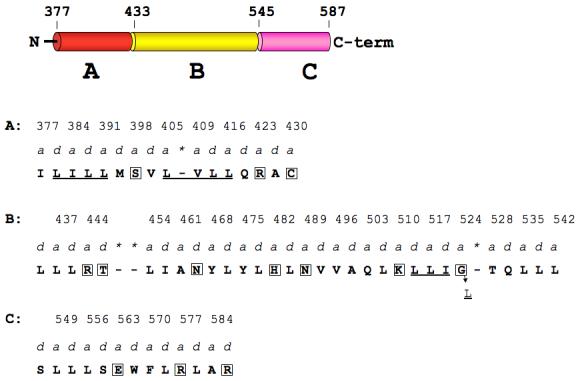
Hydrophobic core of the opened region of assembled HIP1 is disrupted by hydrophilic amino acids in a- and d-positions of the heptad repeat. The domain map of the assembled HIP1 model (371-587) is divided into coiled-coil regions A (red) and B (yellow) and an opened segment, designated region C (pink). N and C-term are the N- and C-terminal ends. Amino acids that occupy a- and d-positions (predicted by COILS 35) in regions A, B, and C are in bold capital letters and the a-position residue numbers are indicated above the a labels. Asterisks indicate skips in the heptad repeat flagged by COILS. Boxed amino acids in A, B, and C are disruptive to the hydrophobic core (see text). Region A: there are two coiled-coil stabilizing clusters (underlined, see text) separated by S398. Region B: the underlined LLI sequence may represent an altered coiled-coil cluster with glycine at position 524 instead of leucine. Changing G524 to L (indicated by the arrow) would create a stabilizing coiled-coil cluster (LLIL) seen in myosin 29.
Conclusions
In summary, we have discovered that HIP1 371-481 is a not folded into death-effector domain, but is a coiled-coil. The C-terminus of HIP1 371-481 is splayed open, which is a continued structural feature that we first observed in the N-terminus of HIP1 482-586 24. Together, our HIP1 crystal structures raise the possibility that the center of the coiled-coil domain is highly flexible. The proposed flexibility of HIP1 is supported by the presence of destabilizing residues at a- and d-positions in the opened region of the assembled HIP1 model. We propose that a stable interaction between HIP1 and HIPPI may require HIP1 to fold back on itself to bring the F432-containing region in proximity to the talin-like domains to form a productive two-point HIPPI binding site. This may explain why htt binding must first fall off HIP1 before HIPPI can productively interact with HIP1 to trigger apoptosis. 3 Finally, the reassembled HIP1 model is useful for understanding how huntingtin (htt) and clathrin can bind to HIP1 at the same time. 13,30
Acknowledgements
We thank Michael Hayden and Peter McPherson for the pGST-HIP1h construct; Sanjay Mishra for generating constructs and protein purification; Jay Nix at beamline 4.2.2 of the Molecular Biology Consortium at the Advanced Light Source for assistance in collecting MAD data. This work was supported by NIH grant RO1 GM064387 to J.A.Y.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Protein Data Bank accession code
The coordinates have been deposited in the RCSB Protein Data Bank with accession code 2QA7. The coordinates will be released on publication.
References
- 1.Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 2.Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Walter S, Tait D, Colicelli J, Lehrach H. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet. 1997;6:487–95. doi: 10.1093/hmg/6.3.487. [DOI] [PubMed] [Google Scholar]
- 3.Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst C-A, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW. Recruitment and activation of caspase-8 by the huntingtin-interacting protein HIP-1 and a novel partner HIPPI. Nature Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- 4.Chen C-Y, Brodsky FM. Huntington-interacting protein 1 (Hip1) and Hip1-related protein (Hip1R) bind the conserved sequence of clathrin light chains and thereby influence clathrin assembly in vitro and actin distribution in vivo. J. Biol. Chem. 2005;280:6109–6117. doi: 10.1074/jbc.M408454200. [DOI] [PubMed] [Google Scholar]
- 5.Mishra SK, Agostinelli NR, Brett TJ, Mizukami I, Ross TS, Traub LM. Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins. J. Biol. Chem. 2001;276:46230–46236. doi: 10.1074/jbc.M108177200. [DOI] [PubMed] [Google Scholar]
- 6.Legendre-Guillemin V, Metzler M, Lemaire J-F, Philie J, Gan L, Hayden MR, McPherson PS. Huntingtin interacting protein 1 (Hip1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain. J. Biol. Chem. 2005;280:6101–6108. doi: 10.1074/jbc.M408430200. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Kaksonen M, Madden DT, Schekman R, Drubin DG. Interaction of Sla2p’s ANTH domain with PtdIns(4,5)P2 is important for actin-dependent endocytic internalization. Mol Biol Cell. 2005;16:717–30. doi: 10.1091/mbc.E04-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senetar MA, Foster SJ, McCann RO. Intrasteric inhibition mediates the interaction of the I/LWEQ module proteins Talin1, Talin2, Hip1, and Hip12 with actin. Biochemistry. 2004;43:15418–28. doi: 10.1021/bi0487239. [DOI] [PubMed] [Google Scholar]
- 9.Hyun TS, Rao DS, Saint-Dic D, Michael LE, Kumar PD, Bradley SV, Mizukami IF, Oravecz-Wilson KI, Ross TS. HIP1 and HIP1r stabilize receptor tyrosine kinases and bind 3-phosphoinositides via epsin N-terminal homology domains. J Biol Chem. 2004;279:14294–306. doi: 10.1074/jbc.M312645200. [DOI] [PubMed] [Google Scholar]
- 10.McCann RO, Craig SW. The I/LWEQ module: a conserved sequence that signifies F-actin binding in functionally diverse proteins from yeast to mammals. Proc Natl Acad Sci U S A. 1997;94:5679–84. doi: 10.1073/pnas.94.11.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brett TJ, Legendre-Guillemin V, McPherson PS, Fremont DH. Structural definition of the F-actin-binding THATCH domain from HIP1R. Nat Struct Mol Biol. 2006;13:121–30. doi: 10.1038/nsmb1043. [DOI] [PubMed] [Google Scholar]
- 12.Engqvist-Goldstein AEY, Warren RA, Kessels MM, Keen JH, Heuser J, Drubin DG. The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J. Cell Biol. 2001;154:1209–1223. doi: 10.1083/jcb.200106089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet. 2001;10:1807–17. doi: 10.1093/hmg/10.17.1807. [DOI] [PubMed] [Google Scholar]
- 14.Metzler M, Li B, Gan L, Georgiou J, Gutekunst CA, Wang Y, Torre E, Devon RS, Oh R, Legendre-Guillemin V, Rich M, Alvarez C, Gertsenstein M, McPherson PS, Nagy A, Wang YT, Roder JC, Raymond LA, Hayden MR. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. Embo J. 2003;22:3254–66. doi: 10.1093/emboj/cdg334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hackam AS, Yassa AS, Singaraja R, Metzler M, Gutekunst C-A, Gan L, Warby S, Wellington CL, Vaillancourt J, Chen N, Gervais FG, Raymond L, Nicholson DW, Hayden MR. Huntingtin interacting protein 1 induces apoptosis via a novel capase-dependent death effector domain. J. Biol. Chem. 2000;275:41299–41308. doi: 10.1074/jbc.M008408200. [DOI] [PubMed] [Google Scholar]
- 16.Bradley SV, Holland EC, Liu GY, Thomas D, Hyun TS, Ross TS. Huntingtin interacting protein 1 is a novel brain tumor marker that associates with epidermal growth factor receptor. Cancer Res. 2007;67:3609–15. doi: 10.1158/0008-5472.CAN-06-4803. [DOI] [PubMed] [Google Scholar]
- 17.Bradley SV, Oravecz-Wilson KI, Bougeard G, Mizukami I, Li L, Munaco AJ, Sreekumar A, Corradetti MN, Chinnaiyan AM, Sanda MG, Ross TS. Serum antibodies to huntingtin interacting protein-1: a new blood test for prostate cancer. Cancer Res. 2005;65:4126–33. doi: 10.1158/0008-5472.CAN-04-4658. [DOI] [PubMed] [Google Scholar]
- 18.Rao DS, Hyun TS, Kumar PD, Mizukami IF, Rubin MA, Lucas PC, Sanda MG, Ross TS. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J Clin Invest. 2002;110:351–60. doi: 10.1172/JCI15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan EY. HIP1 as a marker of aggressive prostate cancer. Clin Genet. 2002;62:372–5. doi: 10.1034/j.1399-0004.2002.620503_4.x. [DOI] [PubMed] [Google Scholar]
- 20.Mills IG, Gaughan L, Robson C, Ross T, McCracken S, Kelly J, Neal DE. Huntingtin interacting protein 1 modulates the transcriptional activity of nuclear hormone receptors. J Cell Biol. 2005;170:191–200. doi: 10.1083/jcb.200503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 22.Barnhart BC, Lee JC, Alappat EC, Peter ME. The death effector domain protein family. Oncogene. 2003;22:8634–44. doi: 10.1038/sj.onc.1207103. [DOI] [PubMed] [Google Scholar]
- 23.Eberstadt M, Huang B, Chen Z, Meadows RP, Ng SC, Zheng L, Lenardo MJ, Fesik SW. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature. 1998;392:941–5. doi: 10.1038/31972. [DOI] [PubMed] [Google Scholar]
- 24.Ybe JA, Mishra S, Helms S, Nix J. Crystal structure at 2.8 A of the DLLRKN-containing coiled-coil domain of huntingtin-interacting protein 1 (HIP1) reveals a surface suitable for clathrin light chain binding. J Mol Biol. 2007;367:8–15. doi: 10.1016/j.jmb.2006.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tripet B, Vale RD, Hodges RS. Demonstration of coiled-coil interaction within the kinesin neck region using synthetic peptides. J. Biol. Chem. 1997;272:8946–8956. doi: 10.1074/jbc.272.14.8946. [DOI] [PubMed] [Google Scholar]
- 26.Zhou NE, Kay CM, Hodges RS. Disulfide bond contribution to protein stability: Positional effects of substitution in the hydrophobic core of the two-stranded α-helical coiled-coil. Biochemistry. 1993;32:3178–3187. doi: 10.1021/bi00063a033. [DOI] [PubMed] [Google Scholar]
- 27.Wagschal K, Tripet B, Lavigne P, Mant C, Hodges RS. The role of position a in determining the stability and oligomerization state of alpha-helical coiled-coils: 20 amino acid stability coefficients in the hydrophobic core of proteins. Protein Science. 1999;8:2312–2329. doi: 10.1110/ps.8.11.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tripet B, Wagschal K, Lavigne P, Mant CT, Hodges RS. Effects of side-chain characteristics on stability and oligomerization state of a de Novo-designed model coiled-coil: 20 amino acid substitutions in position “d”. J. Mol. Biol. 2000;300:377–402. doi: 10.1006/jmbi.2000.3866. [DOI] [PubMed] [Google Scholar]
- 29.Kwok SC, Hodges RS. Stabilizing and destabilizing clusters in the hydrophobic core of long two-stranded α-helical coiled-coils. J. Biol. Chem. 2004;279:21576–21588. doi: 10.1074/jbc.M401074200. [DOI] [PubMed] [Google Scholar]
- 30.Strehlow AN, Li JZ, Myers RM. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum Mol Genet. 2007;16:391–409. doi: 10.1093/hmg/ddl467. [DOI] [PubMed] [Google Scholar]
- 31.Terwilliger TC, Berendzen J. Bayesian correlated MAD phasing. Acta Crystallogr D Biol Crystallogr. 1997;53:571–9. doi: 10.1107/S0907444997005398. [DOI] [PubMed] [Google Scholar]
- 32.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–72. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones TA, Zhou JY, Cowan SW, Kjeldgaard Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–53. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 35.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]