

SUMMARY
Androgen, acting via the androgen receptor (AR), is central to male development, differentiation and hormone-dependent diseases such as prostate cancer. AR is actively involved in the initiation of prostate cancer, the transition to androgen independence, and many mechanisms of resistance to therapy. To examine genetic variation of AR in cancer, we created mice by germ-line gene targeting in which human AR sequence replaces that of the mouse. Since shorter length of a polymorphic N-terminal glutamine (Q) tract has been linked to prostate cancer risk, we introduced alleles with 12, 21 or 48 Qs to test this association. The three “humanized” AR mouse strains (h/mAR) are normal physiologically, as well as by cellular and molecular criteria, although slight differences are detected in AR target gene expression, correlating inversely with Q tract length. However, distinct allele-dependent differences in tumorigenesis are evident when these mice are crossed to a transgenic prostate cancer model. Remarkably, Q tract variation also differentially impacts disease progression following androgen depletion. This finding emphasizes the importance of AR function in androgen-independent as well as –dependent disease. These mice provide a novel genetic paradigm in which to dissect opposing functions of AR in tumor suppression vs. oncogenesis.
Keywords: androgen receptor, prostate cancer, mouse models, glutamine tract, CAG repeat
BACKGROUND
Androgen Receptor and Prostate Cancer
Androgen, acting via its receptor (androgen receptor, AR), is a critical component in development, differentiation and disease of the prostate. Although many genetic and environmental factors impact the etiology of prostate cancer, all tumors initially depend on androgen for growth [1]. Risk due to androgen exposure is cumulative with age and is a product of both the level of the hormone and the efficacy of the receptor. Therefore genetic variation in AR structure, or epigenetic variation in AR levels, may impact initiation of disease [2]. Because of the tumor dependence on androgen, a first line of defense in metastatic disease is ablation of this steroid signaling pathway, by compounds that block androgen synthesis and antagonize AR action. This hormone therapy initially succeeds, but ultimately cancer recurs in a hormone-resistant state refractory to further treatment. Despite resistance, AR levels often remain high and the AR signaling pathway appears to be intact [3]. This suggests that while the disease may become androgen independent, it is not AR independent. A term recently coined to emphasize this critical distinction is “androgen-depletion independent” [4].
Several mechanisms underlie the development of hormone refractory prostate cancer, and they highlight the integral role of AR in disease progression as well as in initiation, and underscore gaps in our understanding of AR function. One class of mechanisms are genetic alterations in Ar, including somatic mutation and gene amplification [5, 6]. Mutant ARs are found that are promiscuous in ligand binding (e.g., T877A), hypersensitive to low androgen levels, or altered in cofactor interactions. An example of the latter (E231G) is sufficient to induce cancer when expressed as a transgene in mouse prostate, confirming that AR can function as an oncogene [7]. While these mutant ARs may offer some selective advantage against therapy, and have provided insights into AR structure and function, for the most part they occur infrequently or in a subset of tumor cells. A second class of mechanisms is more common, in which AR signaling is retained without changes in the AR gene. These involve AR activation in a ligand-independent manner by cross-talk with kinase pathways [8], or by altered levels, or modification, of coactivators or corepressors [9]. Either altered cofactor levels or increased expression of AR can augment AR activity at low hormone concentrations, such as existing after androgen ablation or deriving from intraprostatic conversion of adrenal testosterone precursors [10]. Finally, some modes of hormone resistance bypass AR altogether, relying on alternative growth and survival pathways, such as conferred by bcl-2 [11]. In these cases, Ar may be silenced by epigenetic mechanisms, notably promoter hypermethylation [12]. This multiplicity of mechanisms reflects the complex nature of AR regulation, in which a precise balance is maintained between the opposing functions of differentiation and proliferation, or tumor suppression and oncogenesis [13].
AR and the Glutamine Tract
As a member of the superfamily of ligand-activated nuclear receptor transcription factors, AR function relies on a highly conserved central DNA binding domain (DBD), a moderately conserved C-terminal ligand binding domain (LBD), and an N-terminal transactivation domain (NTD) that bears little similarity amongst receptors and is unusually large in the case of AR [14]. Because Ar is on the X chromosome, mutations in males are phenotypically evident and have allowed extensive receptor structure/function correlations to be drawn from cases of androgen insensitivity [15]. In a simplified view, the LBD initiates hormone response upon ligand binding, the DBD positions AR on target gene promoters, and the NTD recruits a diversity of factors that coordinate transcriptional activity. Interaction between amino and carboxyl (N/C) termini occurs in several receptors but is exceptionally important for AR, both for ligand binding and for optimal transactivation, via differential effects of coactivators, chaperones, ubiquitin ligases and kinases [16, 17]. Previously we demonstrated a role of the NTD in transcriptional specificity of promoter activation for both the mouse sex-limited protein (Slp) gene and for Ar itself, via selective interactions with other transcription factors [18].
Polymorphisms of Ar that influence its function could modify susceptibility to androgen-dependent diseases such as prostate cancer. This has fueled interest in two polyamino acid tracts in the NTD comprised of varying numbers of glutamine (Q) or glycine (G) residues [19]. Glutamine rich regions have functional importance in several transcription factors, such as Sp1 and glucocorticoid receptor [20]. The AR glutamine tract, encoded by a CAG repeat, varies in the normal human population between 9 and 37 residues, while the G tract (GGN repeats) varies from 8 to18 residues [21]. The Q tract has received most scrutiny because expansion of the CAG repeat beyond 40 codons is associated with the late onset neurodegenerative disease, spinal and bulbar muscular atrophy (SBMA, Kennedy disease) [22]. Expanded Q tracts result in decreases in both mRNA and protein levels [23], and there is an inverse relationship between Q tract length and AR transactivation in transfection [24, 25]. The greater transcriptional efficacy of shorter Q tract length is likely to encompass several activities, including increased N/C interaction and differential association with p160 coactivators or SWI/SNF chromatin remodeling components [25, 26]. One AR coactivator, the Ras-related protein RAN/ARA24, may interact directly with the Q tract, and both this interaction and AR activation diminish with increasing Q tract length [27].
Expansion of the Q tract in man results in SBMA, but whether variation within the normal range influences androgen-dependent disease is an unresolved question [21]. Significant effort has focused on detecting an effect in prostate cancer, particularly because shorter tracts occur in the higher risk African-American population [2]. While several studies link shorter Q tract length with increased risk, earlier age of onset, or greater aggressivity of disease (e.g., [2, 28]), other studies have found no association with prostate cancer (e.g. [29, 30]). These conflicting results may be due to genetic heterogeneity within the human population, small patient sample sizes, or complicating effects of gene-gene or gene-environment interactions. An experimental model of oncogenesis and progression is necessary to test the role of Q tract variation in mechanistic detail, both in cancer and in other androgen-dependent syndromes.
Modeling Prostate Cancer in Mice
Mouse models of human cancer are proving highly informative, especially in providing biological samples not readily available from patients. Mice do not normally get prostate cancer but transgenic paradigms recapitulate essential features of the disease course. One of the earliest and most informative models, developed by Dr. Norman Greenberg, is TRAMP (Transgenic Adenocarcinoma of the Mouse Prostate), in which oncogenic SV40 T-antigen is targeted to prostate epithelium by the probasin enhancer and promoter [31]. TRAMP males develop intraepithelial neoplasia and well-differentiated cancer histologically similar to human prostate cancer [32]. These tumors metastasize to distant sites, display similar changes in growth factor axes and progress to androgen independence [33]. Tumors in mice as young as 10 weeks can be detected by magnetic resonance imaging (MRI), are palpable somewhat later, and can be delayed by castration, although not ultimately prevented [34]. These observations support the value of these mice for studying early events in prostate cancer, for evaluating strategies for prevention, and for targeting new treatments to distinct cancer stages. While T-antigen may be a more potent initiator than in human prostate cancer, subsequent oncogenic steps similarly abrogate Rb and p53 function. More recent models that inactivate the tumor suppressor PTEN may bear greater similarity in some respects to the human disease, recapitulating biological and molecular changes during tumor progression in a gene-dose dependent manner [35].
Regardless of what the “first hit” is, these models all show varying degrees of heterogeneity, as in man, at the level of initiation of disease, despite the genetic homogeneity of mice, demonstrating the stochastic nature of subsequent tumorigenic events. These events are subject to strain background, indicating genetic effects at all stages of cancer, and also may be epigenetic, reflecting alterations in cell functions, perhaps exacerbated by inflammation, nutrition or hormonal parameters [36]. With our experience in both androgen action and mouse genetics [18], we initiated studies on AR function in prostate cancer by first “humanizing” mice to carry hAr rather than mAr alleles [37]. These studies, as described below, confirm that Q tract differences affect initiation and progression of prostate cancer. Perhaps more remarkably, these Q tract differences in mice also impact progression following androgen depletion, confirming the importance of AR in androgen-independent disease and suggesting that AR function may predispose differential response to treatment.
Q TRACT LENGTH EFFECTS IN HUMANIZED ANDROGEN RECEPTOR MICE
The significance of the NTD, and specifically the Q tract, in prostate cancer is supported indirectly by comparison of ARs between species. Rodents rarely develop prostate cancer naturally, whereas dogs share with man susceptibility to age- and hormone-dependent prostatic disease. Mouse and human ARs are identical in the DBD and LBD, but differ 15% in protein sequence of the NTD, likely underlying the weaker activity of mAR in vitro [38]. Further, the mouse Q tract is displaced about 100 amino acids and is disrupted by histidines, and there is only a short G tract. Dogs, in contrast, have polymorphic AR Q tracts in both the human and rodent positions. While dogs are a useful physiological model, they lack the advantages of the mouse with respect to facile molecular genetics, small size and short lifespan. Therefore to address mechanisms by which Q tract length may affect prostate cancer risk, and whether other sequence differences in human vs. mouse AR play a role in disease susceptibility, we chose to replace the mouse AR with that of man. These humanized mice also provide enhanced relevance for preclinical testing of novel treatments targeting the human AR.
In order to exchange the human for the mouse AR, and to introduce Q tract length variations, we swapped the entire human and murine NTDs by homologous recombination in embryonic stem (ES) cells. The targeting vector was created from mouse chromosomal DNA containing the AR first exon, which encompasses nearly all of the NTD, plus several kb of upstream flanking information and a contiguous portion of intron 1 downstream, into which a neomycin gene was inserted as a selectable marker. Human AR cDNA sequence encoding amino acids 35 to 466 (including both Q and G tracts) was substituted for the equivalent region of mouse DNA (Fig. 1). Three versions of this vector were created, with Q tracts of 12, 21 or 48 CAG codons. We chose extremes of the normal range in the population, where the median tract length is 21Qs, to optimize the ability to obtain informative differences. Although 48Q in man may cause SBMA, transgenic ARs with as many as 65Qs produce no phenotype in mice, where gene defects often must be more severe to model a human syndrome [39]. These three vectors were introduced into mouse ES cells, and correctly recombined clones were identified. Following a second transfection with a vector expressing cre-recombinase to remove the selectable marker, ES clones were introduced into mouse blastocysts, and chimeric progeny used to establish three lines of humanized AR mice with Ar alleles containing short (12Q), median (21Q) and long (48Q) glutamine tracts. Transcription, processing and translation of this locus leads to an AR with only eight amino acids of mouse rather than human sequence. The humanized mice and their engineered gene are called h/mAR to reflect the chimeric origin.
Figure 1. Structure of the humanized AR gene.

The vector for gene targeting in mouse embryonic stem cells encompassed all of mAR exon 1 with surrounding 5′ flank and downstream intron 1 sequences. Into the construct, human AR cDNA (hatched region) encoding amino acids 35 to 466 (dependent on length of polyamino acid tracts) was substituted for the corresponding mouse sequence. The human AR sequence differs about 15% in this N-terminal region, including an extensive glycine tract (G) and a variable glutamine tract (12, 21 or 48 Qs were inserted) that is also farther 5′-ward than the mouse Q tract. The DBD and LBD are nearly identical between man and mouse in protein sequence. Approximate intron locations are marked by bars below the receptor map. The recombination essentially reconstructs the human AR under control of mouse regulatory sequences.
The humanized mice were compared to wild type for physiological effects due to human AR or the Q tract variations [37]. Both sexes of mice appear normal in behavior, growth and body weight. All three strains are fertile, with similar frequency of litters and numbers of pups, and no differences in lifespan. The androgen axis is within the normal range of variation in serum testosterone, LH and FSH levels, and the testis histology and subcellular localization of AR in all three genotypes is normal. As a direct indication of androgen action, seminal vesicle (SV) weight was determined with age. At 6 months, the 48Q-h/mAR mice lag slightly in SV weight; at two years, SV weight in the 12Q-h/mAR mice is significantly greater. This result confirms biologically that AR activity correlates inversely with Q tract length, and suggests these effects sum over time to detectable phenotypes.
AR with 48Qs can lead to SBMA in man, but there is no evidence of neuromuscular deficiency increasing with age in 48Q-h/mAR mice, as assessed by tests of grip strength. In contrast, mice with 113Qs created with a derivative of our targeting vector show a profound Kennedy disease phenotype [40]. Although fertility is normal in these mice, analysis of testes at the molecular level reveals some variations. For example, Hsd17b3, a marker of mature Leydig cell function, has somewhat lower mRNA levels in h/mAR mice with 48Qs than in those with 12 or 21Qs, but levels are still within a normal range and are much higher than in androgen-insensitive tfm mice. More interestingly, although there is no significant difference in AR mRNA or soluble protein levels between genotypes, there is evidence that the 48Q-h/mAR protein has a tendency to aggregate, as substantial amounts are found in the pellet fraction of whole cell lysates. Nevertheless, the 48Q-h/mAR still has sufficient potency for male health and fertility, suggesting any altered activity may be compensated by feedback mechanisms within the organism.
Prostates of h/mAR mice appear normal in all lobes and AR epithelial localization is similar for all alleles [37]. Low levels of hyperplasia and prostatic intraepithelial neoplasia (PIN) increase with age, but not in a manner that appears to differ between these AR genotypes. To examine transcriptional regulation, prostate expression of several AR target genes was quantified by real-time RT-PCR in 6 months old mice (Fig. 2). AR mRNA levels show some slight inverse correlation with Q tract length, but the trend is not statistically significant. Nkx3.1, a prostate-specific homeobox gene directly regulated by AR [41], shows significantly lower levels in 48Q-h/mAR mice, suggesting in accord with in vitro data that a longer Q tract decreases AR activation. Androgen-regulated probasin [42] expresses at highest levels in mice with the 12Q-h/mAR allele. Clusterin mRNA is repressed by AR [43] and is significantly higher in 48Q-h/mAR mice. Overall, the h/mAR alleles do not produce large differences in target gene expression, but trends are consistent with greater transactivational strength of the 12Q allele (e.g., for probasin and Nkx3.1) and weaker activity of the 48Q allele (both in reduced activation of Nkx3.1 and reduced repression of clusterin). This supports the hypothesis that Q tract length impacts prostate cancer by differential transcription of critical androgen-dependent genes. In TRAMP the probasin-driven T-antigen transgene is an obvious oncogenic target. AR-driven events are also causal in man although as yet unidentified; candidates may emerge from the recent discovery within prostate tumors of chromosomal translocations to AR-responsive promoters [44].
Figure 2. Prostate gene expression is sensitive to AR Q-tract length.

Prostate mRNAs from 3–8 mice at 6 months of age per genotype were quantified by real-time RT-PCR. Results are relative to levels in wild type mAR mice. Nkx3.1 and probasin are up-regulated by AR, clusterin is repressed. Modified from data in Albertelli et al. [37].
Q TRACT LENGTH EFFECTS IN h/mAR PROSTATE CANCER
Since prostate cancer does not occur spontaneously in mice even with h/mAR alleles, we crossed these lines with TRAMP mice to investigate the effect of Q tract length in oncogenesis. Tumor development was followed by abdominal palpation of the progeny, and in some cases by MRI, until mice were moribund to allow comparison of the full course of disease and collection of survival data. In TRAMP mice, the T-antigen oncogene begins to express at puberty in prostatic epithelium due to androgen induction of the probasin promoter. Despite this relatively synchronous oncogene activation, additional stochastic events are required for tumorigenesis, accounting for heterogeneity in time of disease onset [32]. Most TRAMP studies have been done in a C57BL/6 x FVB cross. For our studies, we used the C57BL/6 background, for its purported greater androgen sensitivity. In this relatively homogeneous genetic background, cancer onset is clearly influenced by AR’s Q tract length. We initially compared disease status in mice at 29 weeks of age (Fig. 3, left panel). At this time point, about half of the 21Q-h/mAR mice (and their mAR littermates, not shown) have a palpable tumor or have already died from prostate cancer. In contrast, 85% of the 12Q-h/mAR mice evidence disease, whereas less than a third of the 48Q-h/mAR mice do. In the 12Q group, few mice have died by 29 weeks, even though most have a palpable tumor, suggesting progression of disease as well as initiation differs from the median 21Q allele. The lower death rate at 29 weeks is also found in the 48Q-h/mAR mice. From this comparison we infer that mAR and 21Q-h/mAR lead to similar prostate cancer onset and progression, whereas the short Q tract has an earlier initiating but more slowly progressing disease course and the long Q tract is protective, having both later onset and slower disease course. This hints that variation in Q tract length affects functional capacities of AR that drive androgen-dependent prostate cancer.
Figure 3. Q tract length in h/mARxTRAMP mice affects prostate tumorigenesis.

Mice were abdominally palpated weekly to track tumor development. The status of each genotype at 29 weeks of age is shown for intact mice (A) and mice castrated at 12 weeks of age (B). Mice that died prior to 29 weeks of age are represented by the black portion of the bar, those with a palpable tumor are represented by hatching, and those with no palpable tumor are in white. Modified in part from data in Albertelli et al. [37] and unpublished studies.
Intact h/mAR-TRAMP mice thus corroborate some Q tract effects predicted by epidemiological studies. Given differences in genetic complexity as well as prostatic disease between mouse and man, it may be that the h/mAR alleles, rather than precisely modeling Q tract effects as in man, more effectively represent androgen axes that differ subtly in overall strength. In the homogeneous genetic background of the mouse, it is possible to demonstrate physiologically that the 48Q-h/mAR mice are at the low-end of the normal range for some androgen-dependent traits, and similarly, that the 12Q-h/mAR mice are at the high-end. Unlike the intriguing physiological variations of the h/mAR mice that rarely reach statistical significance, dramatic differences are exerted dependent upon the Q tract length in early prostate tumor growth in the context of the TRAMP oncogene. Apparently when the normal homeostatic balance is perturbed by the stress of cancer, subtle variations are amplified and distinct patterns of disease progression are produced. These subtle allelic differences in AR efficacy are highlighted against the genetic homogeneity of inbred mice relative to man.
Demonstrating that the AR Q tract length affects androgen-dependent tumorigenesis led us to question whether Q tract length might also affect androgen-independent disease, since AR is apparently still active even following androgen depletion. To address this, h/mAR-TRAMP males were castrated at 12 weeks of age, when PIN is prevalent but disease is not yet overt. Mice were followed for tumor development by abdominal palpation, and as an initial assessment, their status compared at 29 weeks of age as for the intact cohort (Fig. 3, right panel). Remarkably, genotype-dependent differences occur, in directions opposite to those found in intact mice. This is most notable for the 12Q-h/mAR allele. Not only do tumors develop much later in 12Q mice, but the time from detection by palpation until death (length of disease), which is very brief in castrated TRAMP mice generally, is significantly prolonged. This pronounced Q tract effect on hormone-independent prostate cancer, as well as hormone-dependent disease, was unexpected and is currently being pursued in our lab. It will be important to elucidate the mechanism(s) by which Q tract length affect AR function in the absence of ligand. This observation also may have implications for Q tract effects that may impact differential response to clinical treatment.
Studying Mechanisms of Androgen Resistance in Humanized AR Mice
The h/mAR mice present the ability to assess in a preclinical situation how different therapies influence the course of disease. One aspect to track is the type of AR mutations that occur during tumor progression. In a previous study, the location of mutations found in mouse AR in the TRAMP model were dependent on the hormonal status of the tumor [45]. That is, in intact mice mutations more frequently occurred in the LBD, while in castrated mice mutations were found more commonly in the NTD. These mutations often lead to greater AR activity under particular conditions (gain-of-function), and potentially highlight sites of interaction with critical cofactors that might themselves serve as novel therapeutic targets to complement androgen ablation. To identify directly relevant sites in human rather than mouse AR, especially in the divergent NTDs, we are comparing mutations in tumors of h/mAR-TRAMP mice following different treatment regimens.
Initially, we compared tumorigenesis in mice castrated at 12 weeks of age to mice treated from 12 weeks with the antiandrogen bicalutamide (compounded into chow for a daily dose of 25 mg/kg). Since mice, unlike man, lack adrenal androgen synthesis, castration provides more complete androgen ablation and lowers serum testosterone to essentially undetectable levels. Although treatment of intact mice with bicalutamide at the early time point of 12 weeks provides no ultimate survival benefit as all TRAMP mice succumb to cancer, comparison of mice at 29 weeks of age provides insight into differences in disease progression (Fig. 4). For the 21Q h/mAR allele, fewer mice have a palpable tumor by 29 weeks of age following castration than after bicalutamide or no treatment. However, the differences are small, perhaps largely due to small numbers in the pilot study. More intriguing is that among those with tumors, most of those in the castrated group are already dead, whereas most in the bicalutamide-treated group are alive. This may reflect the very rapid course of disease, once tumorigenesis begins, in the absence of androgen, compared to slower disease progression in the presence of antagonist. Previous studies in TRAMP mice have suggested that castration synchronizes the growth of pre-existing androgen-independent lesions and may delay primary tumor growth, but fails to eliminate metastatic disease [46]. The striking variation in disease course with level of hormone or presence of antagonist suggests that AR differentially affects disease dependent on both quantitative and qualitative differences of the ligand-occupied LBD. Mechanisms underlying these differences may be discerned by characterizing AR levels, subcellular localization and alternative signaling pathways in these tumors, as well as by identifying AR mutations arising under different selective conditions. The Q tract variant mice provide an added experimental dimension in which AR efficacy can be intrinsically modulated to test response against extrinsic agents. Ultimately this may provide better understanding of androgen resistance in prostate cancer and allow design of more effective treatments.
Figure 4. h/mAR mice respond differentially to treatment.
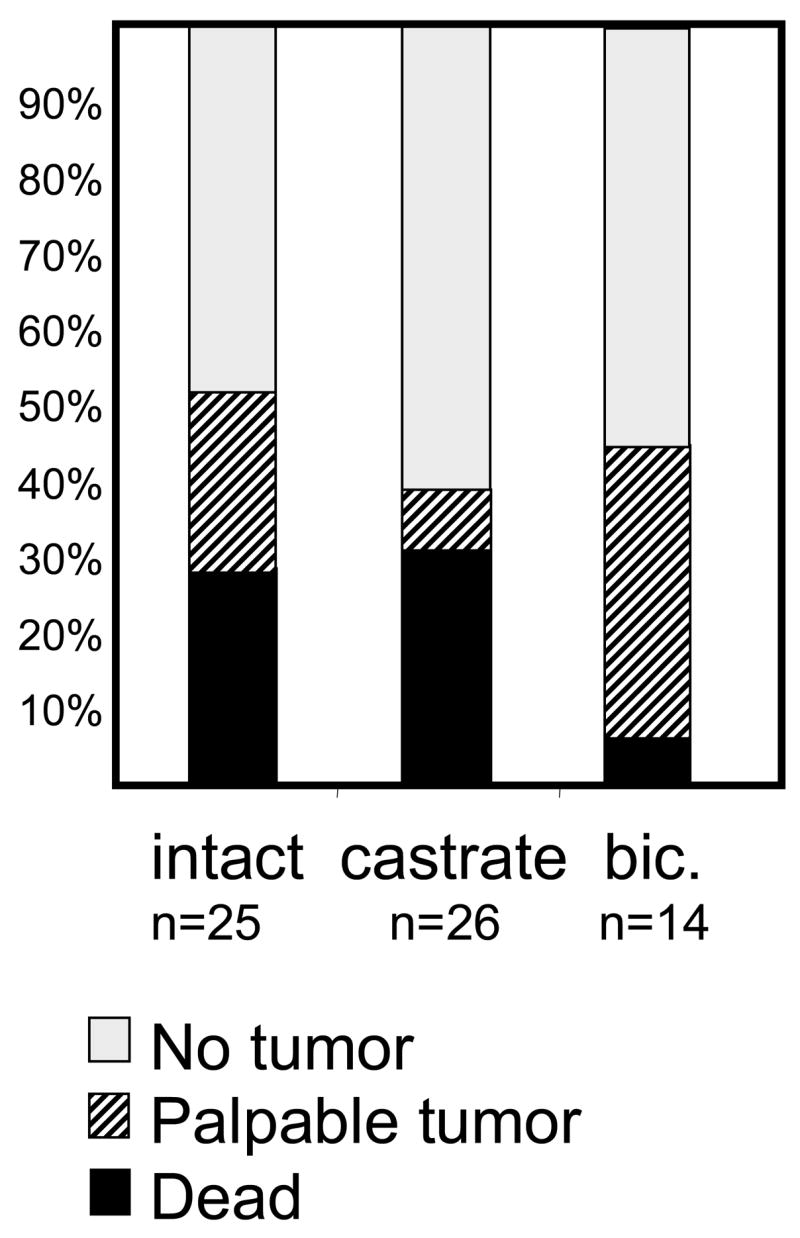
Mice were abdominally palpated weekly to track tumor development. The status of each treatment group at 29 weeks of age is shown for intact mice, mice castrated at 12 weeks of age, or mice fed the AR antagonist bicalutamide in chow from the age of 12 weeks. Mice that died prior to 29 weeks are represented by the black portion of the bar, those with a palpable tumor are represented by hatching, and those with no palpable tumor are in white.
CONCLUSIONS
We have created mice bearing human rather than mouse AR genes, with an allelic series varying in Q tract length. This provides an in vivo model to test the role of the Q tract in androgen-influenced traits and in the etiology of disease. These mice demonstrate that slight differences in AR function within the normal range of phenotypic variation can profoundly affect prostate cancer biology. Q tract length associations in human epidemiological studies may reflect small differences in AR target gene expression that are cumulative over time. These differences may be more readily apparent in the homogeneous genetic background of the mouse than in man. Furthermore, differences are also apparent in tumor progression in the absence of androgen, suggesting that Q tract length impacts androgen-independent as well as –dependent disease. These mice provide a novel genetic paradigm to dissect how AR function is involved in all stages of prostate cancer progression and how this function may be critical in response to treatment.
Acknowledgments
We thank Norm Greenberg, members of the Robins lab and the University of Michigan Prostate Cancer SPORE for helpful discussions throughout the years. This work was supported by grants (to D.M.R.) from the Department of Defense (DAMD17-02-1-0099) and the National Cancer Institute (SPORE in Prostate Cancer, P50-CA69568). M.A.A. was supported by a NIH training program (T32-RR07008) and O.A.M. by a postdoctoral fellowship from the Department of Defense (PC040380).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hayward SW, Cunha GR, Dahiya R. Normal development and carcinogenesis of the prostate. Ann NY Acad Sci. 1996;784:50–62. doi: 10.1111/j.1749-6632.1996.tb16227.x. [DOI] [PubMed] [Google Scholar]
- 2.Irvine RA, Yu MC, Ross RK, Coetzee GA. The CAG and GGC microsatellites of the androgen receptor gene are in linkage disequilibrium in men with prostate cancer. Cancer Res. 1995;55:1937–1940. [PubMed] [Google Scholar]
- 3.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CI. Molecular determinants of resistance to antiandrogen therapy. Nature Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 4.Roy-Burman P, Tindall DJ, Robins DM, Greenberg NM, Hendrix MJC, Moghla S, Getzenberg RH, Isaacs JT, Pienta KJ. Androgens and prostate cancer: are the descriptors valid? Cancer Biol Ther. 2005;4:4–5. doi: 10.4161/cbt.4.1.1563. [DOI] [PubMed] [Google Scholar]
- 5.Buchanan G, Greenberg NM, Scher HI, Harris JM, Marshall VR, Tilley WD. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res. 2001;7:1273–1281. [PubMed] [Google Scholar]
- 6.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature Genetics. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 7.Han G, Buchanan G, Ittmann M, Harris JM, Yu X, Demayo FJ, Tilley W, Greenberg NM. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci USA. 2005;102(4):1151–6. doi: 10.1073/pnas.0408925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nature Med. 1999;5:280–285. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 9.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61(11):4315–9. [PubMed] [Google Scholar]
- 10.Mohler JL, Gregory CW, Ford OH, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 11.Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995;55:4438–4445. [PubMed] [Google Scholar]
- 12.Kinoshita H, Shi Y, Sandefur C, Meisner LF, Chang C, Choon A, Reznikoff CR, Bova GS, Friedl A, Jarrard DR. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60:3623–3630. [PubMed] [Google Scholar]
- 13.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 14.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans R. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McPhaul MJ. Androgen receptor mutations and androgen insensitivity. Mol Cell Endocrinol. 2002;198:61–67. doi: 10.1016/s0303-7207(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 16.He B, Wilson EM. The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol Gen Metab. 2002;75:293–298. doi: 10.1016/S1096-7192(02)00009-4. [DOI] [PubMed] [Google Scholar]
- 17.Shen HC, Coetzee GA. The androgen receptor: Unlocking the secrets of its unique transactivation domain. Vitamins and Hormones. 2005;71:301–319. doi: 10.1016/S0083-6729(05)71010-4. [DOI] [PubMed] [Google Scholar]
- 18.Robins DM. Multiple mechanisms of male-specific gene expression: lessons from the mouse sex-limited protein (Slp) gene. Prog Nucleic Acid Res Mol Biol. 2004;78:1–36. doi: 10.1016/S0079-6603(04)78001-0. [DOI] [PubMed] [Google Scholar]
- 19.Choong CS, Wilson EM. Trinucleotide repeats in the human androgen receptor: a molecular basis for disease. J Mol Endocrinol. 1998;21:235–257. doi: 10.1677/jme.0.0210235. [DOI] [PubMed] [Google Scholar]
- 20.Gerber HP, Seipel K, Georgiev O, Hofferer M, Hug M, Rusconi S. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Sci. 1994;263:808–811. doi: 10.1126/science.8303297. [DOI] [PubMed] [Google Scholar]
- 21.Zitzmann M, Nieschlag E. The CAG repeat polymorphism within the androgen receptor gene and maleness. Internatl J Androl. 2003;26:76–83. doi: 10.1046/j.1365-2605.2003.00393.x. [DOI] [PubMed] [Google Scholar]
- 22.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 23.Choong CS, Kemppainen JA, Zhou ZX, Wilson EM. Reduced androgen receptor gene expression with first exon CAG repeat expansion. Mol Endocrinol. 1996;10:1527–1535. doi: 10.1210/mend.10.12.8961263. [DOI] [PubMed] [Google Scholar]
- 24.Kazemi-Esfarjani P, Trifiro MA, Pinsky L. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: possible relevance for the (CAG)n-expanded neuronopathies. Hum Mol Genet. 1995;4:523–527. doi: 10.1093/hmg/4.4.523. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Udayakumar TS, Vasaitis TS, Brodie AM, Fondell JD. Mechanistic relationship between androgen receptor polyglutamine tract truncation and androgen-dependent transcriptional hyperactivity in prostate cancer cells. J Biol Chem. 2004;279:17319–17328. doi: 10.1074/jbc.M400970200. [DOI] [PubMed] [Google Scholar]
- 26.Buchanan G, Yang M, Cheong A, Harris JM, Irvine RA, Lambert PF, Moore NL, Raynor M, Neufing PJ, Coetzee GA, Tilley WD. Structural and functional consequences of glutamine tract variation in the androgen receptor. Hum Mol Genet. 2004;13:1677–1692. doi: 10.1093/hmg/ddh181. [DOI] [PubMed] [Google Scholar]
- 27.Hsiao PW, Lin DL, Nakao R, Chang C. The linkage of Kennedy’s neuron disease to ARA24, the first identified androgen receptor polyglutamine region-associated coactivator. J Biol Chem. 1999;274:20229–20234. doi: 10.1074/jbc.274.29.20229. [DOI] [PubMed] [Google Scholar]
- 28.Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens CH, Kantoff PW. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci USA. 1997;94:3320–3323. doi: 10.1073/pnas.94.7.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lange EM, Chen H, Brierley K, Livermore H, Wojno KJ, Langefeld CD, Lange K, Cooney KA. The polymorphic exon 1 androgen receptor CAG repeat in men with a potential inherited predisposition to prostate cancer, Cancer Epidemiol. Biomarkers & Prevention. 2000;9:439–442. [PubMed] [Google Scholar]
- 30.Mir K, Edwards J, Paterson PJ, Hehir M, Underwood MA, Bartlett JM. The CAG trinucleotide repeat length in the androgen receptor does not predict the early onset of prostate cancer. Brit J Urol Internatl. 2002;90:573–578. doi: 10.1046/j.1464-410x.2002.02981.x. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci USA. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ, Maddison LA, Foster BA, Greenberg NM. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate. 2003;55:219–317. doi: 10.1002/pros.10215. [DOI] [PubMed] [Google Scholar]
- 33.Foster BA, Kaplan PJ, Greenberg NM. Peptide growth factors and prostate cancer: new models, new opportunities. Cancer Metastasis Rev. 1999;17:317–324. doi: 10.1023/a:1006162410436. [DOI] [PubMed] [Google Scholar]
- 34.Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997;57:4687–4691. [PubMed] [Google Scholar]
- 35.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biology. 2003;1:385–396. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nature Reviews Genetics. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 37.Albertelli MA, Scheller A, Brogley M, Robins DM. Replacing the mouse androgen receptor with human alleles demonstrates glutamine tract length dependent effects on physiology and tumorigenesis in mice. Mol Endocrinol. 2006;20 doi: 10.1210/me.2006-0021. [DOI] [PubMed] [Google Scholar]
- 38.Chamberlain NL, Driver ED, Miesfeld RL. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994;22:3181–3186. doi: 10.1093/nar/22.15.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bingham PM, Scott MO, Wang S, McPhaul MJ, Wilson EM, Garbern JY, Merry DE, Fischbeck KH. Stability of an expanded trinucleotide repeat in the androgen receptor gene in transgenic mice. Nature Genetics. 1995;9:191–196. doi: 10.1038/ng0295-191. [DOI] [PubMed] [Google Scholar]
- 40.Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman A. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. 2006 doi: 10.1172/JCI28773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen MM, Abate-Shen C. Roles of the Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Dev Dynamics. 2003;228:767–778. doi: 10.1002/dvdy.10397. [DOI] [PubMed] [Google Scholar]
- 42.Johnson MA, Hernandez I, Wei Y, Greenberg N. Isolation and characterization of mouse probasin: an androgen-regulated protein specifically expressed in the differentiated prostate. Prostate. 2000;43:255–262. doi: 10.1002/1097-0045(20000601)43:4<255::aid-pros4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 43.July LV, Akbari M, Zellweger T, Jones EC, Goldenberg SL, Gleave ME. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. The Prostate. 2002;50:179–188. doi: 10.1002/pros.10047. [DOI] [PubMed] [Google Scholar]
- 44.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 45.Han G, Foster BA, Mistry S, Greenberg NM. Hormone status selects for spontaneous somatic androgen receptor variants in autochthonous prostate cancer that demonstrate specific ligand and cofactor dependent transcriptional activities. J Biol Chem. 2001;276:11204–11213. doi: 10.1074/jbc.M008207200. [DOI] [PubMed] [Google Scholar]
- 46.Johnson MA, Iversen P, Schwier P, Corn AL, Sandusky G, Graff J, Neubauer BL. Castration triggers growth of previously static androgen-independent lesions in the Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model. The Prostate. 2005;62:322–338. doi: 10.1002/pros.20148. [DOI] [PubMed] [Google Scholar]