

Summary
The fatty acid biosynthesis pathway is an attractive but still largely unexploited target for development of new anti-bacterial agents. The extended use of the anti-tuberculosis drug isoniazid and the antiseptic triclosan, which are inhibitors of fatty acid biosynthesis, validates this pathway as a target for anti-bacterial development. Differences in subcellular organization of the bacterial and eukaryotic multi-enzyme fatty acid synthase systems offer the prospect of inhibitors with host vs. target specificity. Platensimycin, platencin, and phomallenic acids, newly discovered natural product inhibitors of the condensation steps in fatty acid biosynthesis, represent new classes of compounds with antibiotic potential. An almost complete catalogue of crystal structures for the enzymes of the type II fatty acid biosynthesis pathway can now be exploited in the rational design of new inhibitors, as well as the recently published crystal structures of type I FAS complexes.
Introduction
A necessary but not sufficient requirement for an effective antibacterial agent is that it target an essential reaction or pathway in the infectious organism. The most widely used antibiotics exert their effects on bacterial cell wall synthesis, protein synthesis, and DNA replication. For a number of reasons, the indispensable fatty acid synthase (FAS) pathway is now an especially attractive target for anti-bacterial agents. The rapid emergence of resistance to antibiotics that have been in use for decades increases the value of agents that act orthogonally against targets like FAS, since extant resistance mechanisms should be ineffective against them. Fatty acid biosynthesis has been validated as an antibiotic target through the demonstrated effectiveness of isoniazid and triclosan, whose primary target is an enzyme in the bacterial fatty acid biosynthesis pathway. The overall high degree of conservation in many of the component enzymes of the FASII system holds the prospect for development of broad spectrum antibiotics. The subcellular organization of components of the fatty acid biosynthesis pathway is different in mammals (type I FAS) than the type II FAS of bacteria, plants and parasites, which increases the probability that effective anti-bacterial agents will be target-specific (type I FASs are generally single chain, multidomain homodimers or two chain heterodimers carrying all proteins of the pathway, while type II FAS component proteins are dissociated). There is further unique substrate specificity in the type II FAS enzyme(s) of the important infectious agent M. tuberculosis, which in conjunction with a type I FAS is responsible for biosynthesis of mycolic acids, essential components of the cell envelope. Finally, the availability in recent years of an almost complete catalogue of three dimensional structures for representative enzymes in the fatty acid biosynthesis pathway provides a basis for rational drug design [1], and the very recent determinations of the crystal structures of two type I fungal FASs offer the first view of the higher order organization of a host type I FAS [2,3,4].
A general scheme for type II fatty acid biosynthesis is shown in Figure 1. The composition of the substrate and product pools (the R groups) varies among organisms and offers the possibility for designing high specificity into any effective inhibitor leads targeting the reactions of this pathway. Currently, the only enzyme of this pathway that is successfully inhibited by antibiotics in clinical use is FabI, the enoyl-ACP (acyl carrier protein) reductase that is inhibited by isoniazid and related compounds. The antiseptic agent triclosan also targets FabI. Two natural product inhibitors of FabH and FabB/F, thiolactomycin and cerulenin (FabB/F only) [5], have been thoroughly characterized over a number of years and have been used as templates in attempts to develop more effective inhibitors of these enzymes. This review surveys work done since 2005 in discovering and developing inhibitors of fatty acid biosynthesis [6], and in elucidating the chemistry and structural biology of FAS and its components, which has relevance to the development of inhibitors.
Figure 1. Scheme for the type II FAS reaction pathway.
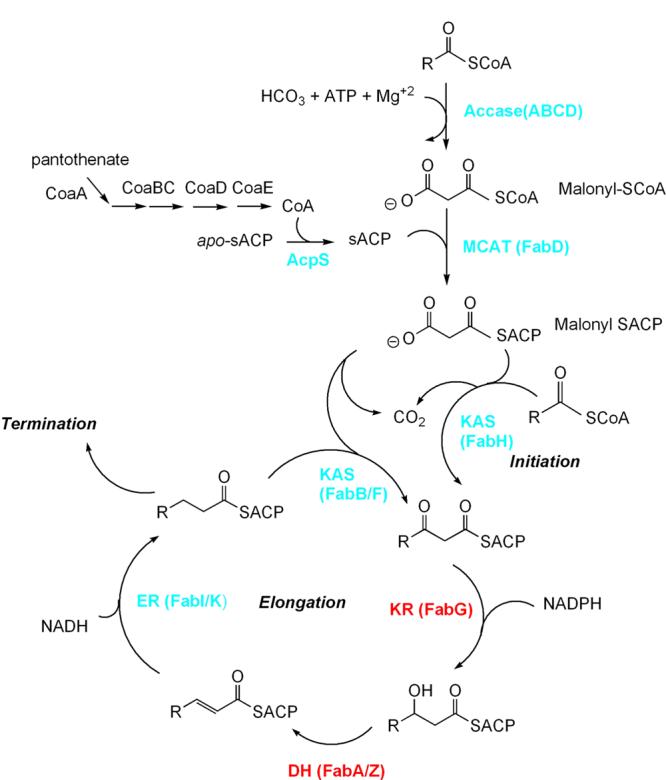
Enzyme names are in red and cyan, the latter indicating enzymes for which inhibitors exist.
New Natural Product Inhibitors of Fatty Acid Biosynthesis
A promising and significant recent development in finding inhibitors of fatty acid biosynthesis is the discovery of platensimycin [7,8]. The discovery of platensimycin was facilitated by the development of a targeted, differential sensitivity assay exploiting antisense RNA [9]. Depletion of fabF/H transcript levels through expression of an inducible, plasmid-borne antisense fabF transcript, sensitized Staphylococcus aureus to inhibitors that target these proteins. Use of this strain in a screen of 250,000 natural product extracts ultimately led to the discovery of platensimycin (Figure 2A). This natural product from Streptomyces platensis represents a new chemical class of antibiotic with promising inhibitor activity toward Gram-positive bacteria (MIC of ∼1μg/ml toward S. aureus, Enterococcus faecalis and S. pneumoniae). The sensitivity of S. aureus to platensimycin is inversely correlated with fabF expression levels, confirming FabF as the target. In cell-free, single enzyme assays, platensimycin inhibits FabF with an IC50 of 48 nm and 160 nM for S. aureus and E. coli respectively, but has only weak inhibitor activity toward FabH (IC50 = 67 μM).
Figure 2.

Structures of natural product (A) and chemically synthesized (B) type II FAS inhibitors (see text for more details).
In vitro, platensimycin binds to FabF relatively weakly, which led to the discovery that it preferentially targets the acyl-thioester intermediate of the FabF pathway (paralleling the cases of isoniazid [10] and triclosan [11], which act through binding with a FabI-NAD reaction intermediates). In a crystal structure of a Cys-163-Gln FabF mutant, which simulates this intermediate, platensimycin was observed to bind at the active site of FabF with the carboxylic acid group lying in the malonate binding site coplanar with the amide sidechain of Gln163 (Fig 3). Platensimycin has little activity toward wild type E. coli, and probably other Gram negative bacteria, due to efflux, but it is as active against antibiotic resistant strains of S. aureus and Enterococcus faecalis as against the wild types. It also showed high efficacy in a mouse model of disseminated S. aureus infection.
Figure 3. Platensimycin in the active site of FabF.
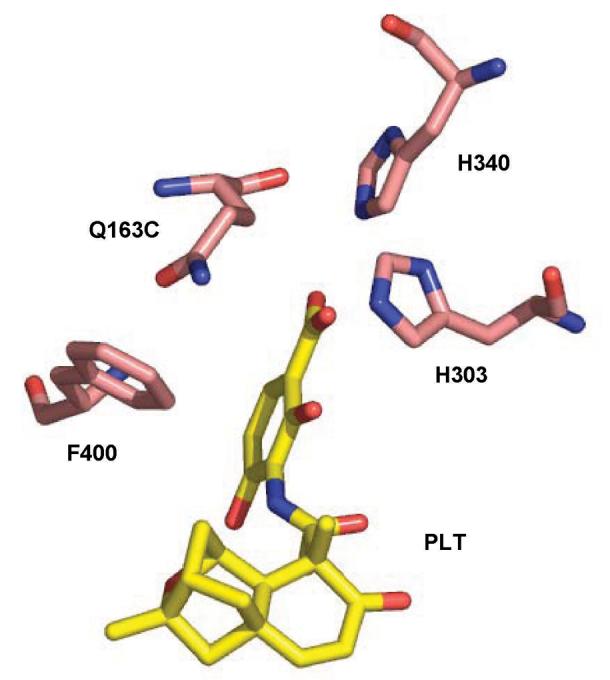
The carboxylate of platensimycin lies in the malonate binding site (H303, H340) coplanar with the sidechain of Q163, which substitutes for C163 and simulates an acyl-linked sidechain.
Continued natural product screening has led to the discovery of a platensimycin analog, platencin [12] (Figure 2A), which differs in the N–acyl substituent on the 3-amino-2,4-dihydroxybenzoic acid. Single enzyme assays revealed that it inhibits both FabH and FabF comparably (1.95 μg/ml and 3.91 μg/ml respectively). As such it is a better FabH inhibitor than platensimycin but a poorer FabF inhibitor. The in vitro activity of platencin is comparable with platensimycin with a range of Gram-positive bacteria, and better with vancomycin-resistant Enterococcus faecium and efflux-negative Escherichia coli.
Natural product screening with the same antisense assay has also led to the discovery of phomallenic acids (Figure 2A), a different class of FabH/F inhibitor [9,13]. These acetylenic acids were confirmed to inhibit FASII, and the most active of the series (phomallenic acid C) has MICs in the range of 0.77 – 3.9 μg/ml against S. aureus and H. influenzae. From analysis of the reaction intermediate profile in a FASII assay, it appears that phomallenic acids target both FabH and FabF in S. aureus.
Using a high throughput FASII elongation assay system with long chain acyl-CoA substrates, Merck researchers also screened natural product libraries and found a moderately active bischloroanthrabenzoxocinone inhibitor (Figure 2A) [14]. This compound (BABX) was effective against cell-free S. aureus and E. coli FASII assay systems (IC50 = 11.4 and 35.3 μg/ml) and had promising MIC against S. aureus and permeable E. coli (0.2 – 0.4 μg/ml). Using the mechanistically characterized inhibitors cerulenin and triclosan for reference, BABX was inferred to inhibit FabB/F in the elongation cycle.
Screening natural product libraries for inhibitors of other enzymes in the type II FAS has also been reported. Screens for FabI (the enoyl ACP reducutase) inhibitors lead to recurrent isolation of unsaturated long chain fatty acids [15]. An examination of their effects on this enzyme and on the viability of several bacteria has revealed they inhibit the S. aureus and E. coli FabI by binding to free enzyme and to its NADH complex. However, they are relatively poor inhibitors and show high MICs against the bacteria. A noteworthy characteristic of this inhibition is that it is specific for unsaturated fatty acids, saturated fatty acids showing no inhibitory activity. Corytuberine (Figure 2A) was discovered by screening a natural product library for inhibitors of Helicobacter pylori malonyl-CoA acyl-transferase (MCAT or FabD), the enzyme that catalyzes the transfer of the malonyl group from malonyl-CoA to holo-ACP [16]. This inhibitor has modest activity against this MCAT in an enzyme assay (IC50= 33 μM), and was found to be an uncompetitive inhibitor (the antibacterial activity of corytuberine was not reported).
FASII inhibitors from structure-based design and chemical library screens
The availability of crystal structures for almost all enzymes in the FASII pathway has made it possible to apply the methods of rational drug design to the development of inhibitors. The initiating FabH was the target of efforts based on crystal structure information incorporated into a comprehensive knowledge database for the enzyme in conjunction with chemical library screening and refinement [17]. A series of benzoylaminobenzoic acids (Figure 2B i) were found to have sub-μM range IC50s in a cell-free assay of E. faecalis FabH, and these were further improved by modifications based on the FabH crystal structure. These compounds bind at the active site of FabH. Cross-genus activity of the best of these inhibitors toward FabH from four clinically important pathogens varied widely (IC50 = 0.004 - >100μM). Attempts to use amino acid sequence alignments to improve activity toward poor targets were not useful and crystal structures of complexes of the inhibitors with poorly inhibited FabH were necessary to investigate the basis for the differences in inhibitor activity. Disappointingly, there is almost an inverse relationship in this series of compounds between inhibitor activity toward FabH in vitro and IC50 toward six microbial targets, apparently due to penetration barriers or efflux.
A search of the 3D National Cancer Institute database for compounds with structural similarity to thiolactomycin, a known natural product inhibitor of FabF that binds at the malonyl-ACP site, identified cyclic sulfones as possible FabH leads [18]. A synthetic structure-activity screen led to reversible inhibitors with low μM IC50s against E. coli FabH (Figure 2B ii), but weak activity against the M. tuberculosis and P. falciparum enzymes. Crystal structures and homology models of these enzymes were compared and potential reasons for these differing activities were considered.
Mechanism-based inhibitors directed at the transacylation step of FabH have been developed, primarily as tools for understanding the enzyme catalyzed pathway. Disulfide linked alkyl-coenzyme A (CoA) compounds (Figure 2B iii) were shown to inhibit E. coli and Mycobacterium tuberculosis FabH through a disulfide exchange of the inhibitor with the active site cysteine side chain of these enzymes [19]. The specificity of the alkyl chain length in the inhibition reaction matched the known differential specificity of these two enzymes toward acyl-CoA substrates. A surprising conclusion from solution and crystal structure studies of the E. coli FabH with methyl-CoA disulfide is that the homodimeric FabH has differential half-sites reactivity toward this inhibitor despite virtually complete identity of the two subunits in the unreacted enzyme. The resolution of this conundrum is proposed to lie in the partially disordered state observed in an unliganded form of FabH [20]. Binding of the first inhibitor, and presumably substrate, ligand induces order in the disordered regions of the apo-enzyme, and this imposes a kinetic differentiation on the binding of ligands to the two subunits. This property of structural disorder in FabH is likely to be critical to understanding the mechanism and properties of this, and possibly other enzymes, in the FASII pathway.
The enoyl-acyl-ACP reductase, FabI, continues to attract attention as a target in the FASII pathway, to some extent due to its proven susceptibility to isoniazid and triclosan[21]. However, enthusiasm for development of inhibitors of FabI is somewhat tempered by the existence of a structurally and mechanistically unrelated enzyme, FabK, that can catalyze the same reaction as FabI in some important pathogens [22]. A recently developed inhibitor of FabI API-1252 (Fig. 2B iv), showed MIC50s of 4 – 15 ng/ml against S. aureus and S. epidermidis drug resistant strains[23], none of which carries the FabK gene
Ethionamide and prothionamide, which are structurally similar to isoniazid, have been used for some time in the treatment of M. tuberculosis, M. leprae and M.avium infections and are assuming increasing importance with the growing incidence of isoniazid resistance. Their mechanism of action has been assumed to be the same as isoniazid, which is a pro-drug that requires enzymatic activation by a catalase-peroxidase (KatG) to react with NAD+ and form the adduct which is the proximal inhibitor of FabI. It has now been shown that ethionamide and prothionamide act through formation of similar adducts, but are activated by a flavin monooxygenase EthA, rather than KatG [24].
Going further upstream from the fatty acid synthesis condensation and elongation reactions, inhibitors have been found for AcpS and for M. tuberculosis AccD5, one of 6 known acyl-CoA carboxylases in M. tuberculosis. AcpS catalyzes the transfer of the 4'-phosphopantetheinyl group from CoA to the inactive apoACP (generating the active holoACP) and the latter catalyzes formation of methylmalonyl CoA (via carboxylation of propionyl-CoA), the substrate for multimethyl branched chain fatty acids biosynthesis, critical for M. tuberculosis envelope formation. A high throughput screen for inhibitors of B. subtilis AcpS yielded anthranilic acid inhibitors, with moderate activity (12-32 μM) (Figure 2B v) [25]. These compounds had moderate biological activity 32 μg/ml against some bacteria (MRSA S. aureus among others), but appeared to kill Bacillus subtilis though a process other than inhibition of AcpS (such as disruption of the cell-membrane). An iterative process involving synthetic modification, enzymatic assays and X-ray complex structure determination led to inhibitors with improved activity against AcpS (1 μM) and slightly reduced antibacterial activity (32 μg/ml). Treatment of B. subtilis with these led to an accumulation of the apo-ACP, consistent but not proving AcpS as the target.
Lead inhibitors of AccD5 were sought through in silico screens of the NCI diversity set and the ChemDB database at the University of California, Irvine [26]. A compound (NCI 65828) with a competitive Ki = 13.1 μM was identified in the NCI set (Fig. 2b) and used as the basis for screening 3000 chemical homologs, the most effective of which had an IC50 of 25 μM.
Conclusions
Platensimycin and platencin represent a new class of potential antibiotics and their discovery is a triumph for natural product drug discovery and for new methods that expand screening sensitivity and specificity [27]. The fact that they inhibit a new target, fatty acid biosynthesis, increases their significance in the race to stem multidrug resistance against antibiotics that have been in use for many years.
Several technical aspects of the well documented discovery and characterization of platensimycin deserve mention. The use of an assay measuring inhibition of cell growth led to identification of compounds with potent antibacterial activity not restricted by poor cell-penetration or efflux. Furthermore, the development of screening protocols that sensitize test organisms by use of small interfering RNA provided a methodology with increased sensitivity in the screen and also rapid determination of the likely target of any new ‘hits’. The discovery that platensimycin binds tightly to the acyl-enzyme intermediate of the FabF pathway but not to the unliganded enzyme, re-emphasizes the need to understand intermediates on a targeted pathway in detail [28] (isoniazid and triclosan both inhibit FabI by forming ternary complexes with the enzyme-NAD+ complex).
The success of platensimycin/platensin as a basis for generating structurally related new clinical drug faces remains to be seen [27, 29]. Efficacy in a murine model of a disseminated S. aureus infection was only accomplished by continuous high dose parenteral infusion. Other issues and uncertainties include the frequency of resistance, toxicity, stability and regulatory approval. The cost of development is likely to be a major factor in exploiting these new discoveries, given the low probability of success and the limited investment return for antibiotics [29]
The type II FAS inhibitors which have come from structure-based drug design and screening chemical libraries have arguably been less exciting than those that came from natural product screening. These latter methods have typically generated compounds with relatively poor inhibition indexes against the target enzyme (μM versus nm). Many of the more potent compounds show poor efficacy against bacteria, suggesting that cell penetration and/or efflux are constraints on their further development. We would argue that natural product libraries continue to provide a more chemically diverse pool of potentially unique drug leads than synthetic libraries, and that selective pressure ensures that compounds from the former penetrate cells and are potent enzyme inhibitors. Additional limitations with structure-based discovery may arise as a consequence of modeling the unliganded enzyme and not a ligand complex or intermediate (as noted above). The apparent disorder→ order transition that occurs in E. coli FabH on binding inhibitor or substrate suggests significant structural changes may occur during the processes catalyzed by the type II FAS enzymes. There is also very little known about the interactions among the enzymes of the type II FAS (in contrast to the type I where structures of the complex are now available). Preliminary evidence for higher order organization of the components of the type II FAS has been provided and may be essential for activity [30]. Such organization seems likely by analogy to the FASI complex structure, but it may be transient and dependent on cellular location. The role of both protein conformational changes and protein –protein interactions in the type II FAS offers both a challenge and an opportunity for developing new inhibitors.
Acknowledgement
The authors' work in this area is supported by a grant from the NIH (AI52230).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
H. Tonie Wright, Dept. of Biochemistry and Institute of Structural Biology and Drug Discovery Virginia Commonwealth University 800 E. Leigh St. Suite 212 Richmond, VA USA 23219-1540 xrdproc@vcu.edu.
Kevin A. Reynolds, Department of Chemistry Portland State University Portland, OR USA 97207 reynoldsk@pdx.edu
References
- 1.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–83. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- •• 2.Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Structure of fungal fatty acid synthase and implications for iterative substrate shuttling. Science. 2007;316:254–260. doi: 10.1126/science.1138248. The 3.1Å resolution crystal structure of the α6β6 FASI from Thermomyces lanuginosus showing the higher order organization of this complex. Interactions of the different functional domains, the dimeric compartmentalization of the reaction cavity, and the probable mode of ACP migration during the reaction cycle are persuasively inferred from the structure. [DOI] [PubMed] [Google Scholar]
- •• 3.Leibundgut M, Jenni S, Frick C, Ban N. Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase. Science. 2007;316:288–290. doi: 10.1126/science.1138249. Structure of S. cerevisiae FASI, virtually identical to that of T. lanuginosus, with ACP oriented at the active site of the ketoacyl-synthase condensing enzyme domain. [DOI] [PubMed] [Google Scholar]
- •• 4.Lomakin I, Xiong Y, Steitz TA. The crystal structure of yeast fatty acid synthase, a cellular machine with eight active sites working together. Cell. 2007;129:319–332. doi: 10.1016/j.cell.2007.03.013. The 4Å resolution structure of yeast FASI shows ACP within the central chamber of the assembly and the phosphopantetheinyl transferase outside, implying formation of holo-ACP prior to FASI complex assembly. [DOI] [PubMed] [Google Scholar]
- 5.Price AC, Choi KH, Heath RJ, Li Z, White SW, Rock CO. Inhibition of beta-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J Biol Chem. 2001;276:6551–6559. doi: 10.1074/jbc.M007101200. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, White SW, Rock CO. Inhibiting Bacterial Fatty Acid Synthesis. J Biol Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- •• 7.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358–361. doi: 10.1038/nature04784. Identification from natural product screen of platensimycin as an inhibitor of FabF/B in gram positive pathogenic bacterial targets, including antibiotic resistant ones (MIC ∼1μg/ml). Platensimycin binds at active site of E. coli FabF in the malonyl binding site. [DOI] [PubMed] [Google Scholar]
- • 8.Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K, Wang J. Isolation, Structure, and Absolute Stereochemistry of Platensimycin, A Broad Spectrum Antibiotic Discovered Using an Antisense Differential Sensitivity Strategy. J Am Chem Soc. 2006;128:11916–11920. doi: 10.1021/ja062232p. Chemical and structural characterization of platensimycin. [DOI] [PubMed] [Google Scholar]
- •• 9.Young K, Jayasuriya H, Ondeyka JG, Herath K, Zhang C, Kodali S, Galgoci A, Painter R, Brown-Driver V, Yamamoto R, Silver LL, Zheng Y, Ventura JI, Sigmund J, Ha S, Basilio A, Vicente F, Tormo JR, Pelaez F, Youngman P, Cully D, Barrett JF, Schmatz D, Singh SB, Wang J. Discovery of FabH/FabF inhibitors from natural products. Antimicrob Agents Chemother. 2006;50:519–526. doi: 10.1128/AAC.50.2.519-526.2006. Development and validation of assay incorporating target-specific antisense RNA sensitization of bacteria. A new class of FabF inhibitors, phomallenic acids, were discovered and the screening assay was used in the discovery of platensimycin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Jr., Sacchettini JC. Modification of the NADH of the Isoniazid Target (InhA) from Mycobacterium tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 11.Qiu X, Janson RI, Smyth MG, Payne DJ, Abdel-Meguid SS. Molecular basis for triclosan activity involves a flipping loop in the active site. Prot Sci. 1999;8:2529–2532. doi: 10.1110/ps.8.11.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 12.Wang J, Kodali S, Lee SH, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, Gonzalez I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF, Singh SB. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Nat. Acad. Sci USA. 2007;104:7612–7616. doi: 10.1073/pnas.0700746104. 2007. Discovery of a close analogue of platensimycin by the same methods and having higher inhibitor activity toward FabH than platensimycin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 13.Ondeyka JG, Zink DL, Young K, Painter R, Kodali S, Galgoci A, Collado J, Tormo JR, Basilio A, Vicente F, Wang J, Singh SB. Discovery of bacterial fatty acid synthase inhibitors from a Phoma species as antimicrobial agents using a new antisense-based strategy. J Nat Prod. 2006;69:377–380. doi: 10.1021/np050416w. Chemical characterization and biological assay of phomallenic acids. [DOI] [PubMed] [Google Scholar]
- 14.Kodali S, Galgoci A, Young K, Painter R, Silver LL, Herath KB, Singh SB, Cully D, Barrett JF, Schmatz D, Wang J. Determination of Selectivity and Efficacy of Fatty Acid Synthesis Inhibitors. J Biol Chem. 2005;280:1669–1677. doi: 10.1074/jbc.M406848200. [DOI] [PubMed] [Google Scholar]
- 15.Zheng CJ, Yoo JS, Lee TG, Cho HY, Kim YH, Kim WG. Fatty acid synthesis is a target for antibacterial activity of unsaturated fatty acids. FEBS Lett. 2005;579:5157–5162. doi: 10.1016/j.febslet.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Han C, Hu L, Chen K, Shen X, Jiang H. Characterization and inhibitor discovery of one novel malonyl-CoA:Acyl carrier protein transacylase (MCAT) from Helicobacter pylori. FEBS Lett. 2006;580:697–702. doi: 10.1016/j.febslet.2005.12.085. [DOI] [PubMed] [Google Scholar]
- •17.Nie Z, Perretta C, Lu J, Su Y, Margosiak S, Gajiwala KS, Cortez J, Nikulin V, Yager KM, Appelt K, Chu S. Structure-based design, synthesis, and study of potent inhibitors of beta-ketoacyl-acyl carrier protein synthase III as potential antimicrobial agents. J Med Chem. 2005;48:1596–1609. doi: 10.1021/jm049141s. Structure based screening and improvement of lead inhibitors of FabH. [DOI] [PubMed] [Google Scholar]
- 18.Alhamadsheh MM, Waters NC, Huddler DP, Kreishman-Deitrick M, Florova G, Reynolds KA. Synthesis and biological evaluation of thiazolidine-2-one 1,1-dioxide as inhibitors of Escherichia coli beta-ketoacyl-ACP-synthase III (FabH) Bioorg Med Chem Lett. 2007;17:879–883. doi: 10.1016/j.bmcl.2006.11.067. [DOI] [PubMed] [Google Scholar]
- •19.Alhamadsheh MM, Musayev F, Komissarov AA, Sachdeva S, Wright HT, Scarsdale N, Florova G, Reynolds K. Alkyl-CoA disulfides as inhibitors and mechanistic probes for FabH. Chemistry and Biology. 2007 doi: 10.1016/j.chembiol.2007.03.013. in press. Detection of half sites reactivity of E. coli FabH that is linked to order-disorder transition in parts of the structure. [DOI] [PubMed] [Google Scholar]
- 20.Qiu X, Janson CA, Smith WW, Head M, Lonsdale J, Konstantinidis AK. Refined structures of beta-ketoacyl-acyl carrier protein synthase III. J Mol Biol. 2001;307:341–356. doi: 10.1006/jmbi.2000.4457. [DOI] [PubMed] [Google Scholar]
- 21.Moir DT. Identification of inhibitors of bacterial enoyl-acyl carrier protein reductase. Curr Drug Targets Infect Disord. 2005;5:297–305. doi: 10.2174/1568005054880154. [DOI] [PubMed] [Google Scholar]
- 22.Marrakchi H, Dewolf WE, Jr, Quinn C, West J, Polizzi BJ, So CY, Holmes DJ, Reed SL, Heath RJ, Payne DJ, Rock CO, Wallis NG. Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK) Biochem J. 2003;370:1055–1062. doi: 10.1042/BJ20021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlowsky JA, Laing NM, Baudry T, Kaplan N, Vaughan D, Hoban DJ, Zhanel GG. In Vitro Activity of API-1252, a Novel FabI Inhibitor, Against Clinical Isolates of Staphylocccus aureus and Staphylococcus epidermidis. Antimicrob Agents Chemother. 2007 doi: 10.1128/AAC.01254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang F, Langley R, Gulten G, Dover LG, Besra GS, Jacobs WR, Jr, Sacchetini JC. Mechanism of thioamide drug action against tuberculosis and leprosy. J Exp Med. 2007;204:73–78. doi: 10.1084/jem.20062100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joseph-McCarthy D, Parris K, Huang A, Failli A, Quagliato D, Dushin EG, Novikova E, Severina E, Tuckman M, Petersen PJ, Dean C, Fritz CC, Meshulam T, DeCenzo M, Dick L, McFadyen IJ, Somers WS, Lovering F, Gilbert AM. Use of structure-based drug design approaches to obtain novel anthranilic acid acyl carrier protein synthase inhibitors. J Med Chem. 2005;48:7960–7969. doi: 10.1021/jm050523n. [DOI] [PubMed] [Google Scholar]
- 26.Lin T-W, Melgar MM, Kurth D, Swamidass SJ, Purdon J, Tseng T, Gago G, Baldi P, Gramajo H, Tsai S-C. Structure-based inhibitor design of AccD5, an essential acyl-CoA carboxylase carboxyltransferase domain of Mycobacterium tuberculosis. Proc Nat Acad Sci USA. 2006;103:3072–3077. doi: 10.1073/pnas.0510580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habich D, von Nussbaum F. Platensimycin, a new antibiotic and “superbug challenger” from nature. ChemMedChem. 2006;1:951–954. doi: 10.1002/cmdc.200600145. [DOI] [PubMed] [Google Scholar]
- 28.Wright HT. Cofactors in fatty acid biosynthesis - active site organizers and drug targets. Structure. 2004;12:358–359. doi: 10.1016/j.str.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Pearson H. Antibiotic faces uncertain future. Nature. 2006;441:260–261. doi: 10.1038/441260a. [DOI] [PubMed] [Google Scholar]
- •30.Veyron-Churlet R, Guerrini O, Mourey L, Daffe M, Zerbib D. Protein-protein interactions within the fatty acid synthase-II system of Mycobacterium tuberculosis are essential for mycobacterial viability. Mol Microbiol. 2004;54:1161–1172. doi: 10.1111/j.1365-2958.2004.04334.x. Evidence for in vivo associations of the discrete FASII enzymes of Mycobacterium tuberculosis. [DOI] [PubMed] [Google Scholar]