

Abstract
Hepatocyte growth factor (HGF) is a plasminogen-like protein with an α chain linked to a trypsin-like β chain without peptidase activity. The interaction of HGF with c-met, a receptor tyrosine kinase expressed by many cells, is important in cell growth, migration, and formation of endothelial and epithelial tubes. Stimulation of c-met requires two-chain, disulfide-linked HGF. Portions of an α chain containing an N-terminal segment and four kringle domains (NK4) antagonize HGF activity. Until now, no physiological pathway for generating NK4 was known. Here we show that chymases, which are chymotryptic peptidases secreted by mast cells, hydrolyze HGF, thereby abolishing scatter factor activity while generating an NK4-like antagonist of HGF scatter factor activity. Thus, chymase interferes with HGF directly by destroying active protein and indirectly by generating an antagonist. The site of hydrolysis, Leu480, lies in the α chain on the N-terminal side of the cysteine linking the α and β chains. This site appears to be specific for HGF because chymase does not hydrolyze other plasminogen-like proteins, such as macrophage-stimulating protein and plasminogen itself. Mast cell/neutrophil cathepsin G and neutrophil elastase generate similar fragments of HGF by cleaving near the chymase site. Mast cell and neutrophil peptidases are secreted during tissue injury, infection, ischemia, and allergic inflammation, where they may oppose HGF effects on epithelial repair. Thus, HGF possesses an “inactivation segment” that serves as an Achilles' heel attacked by inflammatory proteases. This work reveals a potential physiological pathway for inactivation of HGF and generation of NK4-like antagonists.
HGF2 is a mitogen, motogen, and morphogen for epithelial and mesenchymal cells (1). It is also known as scatter factor because it disperses epithelial cells in culture (2). Genetic deletion in mice suggests that HGF is critical for embryonic development (3). Adult mesenchymal cells also produce HGF, which is thought to regulate tissue regeneration and repair after injury (4, 5), epithelial to mesenchymal transitions, and formation of vessels and other tube-like structures (6). HGF production is inducible. Circulating levels rise markedly in several types of tissue injury, as in liver damage (7), arterial thrombosis (8), or acute rejection of a transplanted lung (9). When used as a drug, HGF stimulates lung regrowth after pneumonectomy (10), diminishes lung fibrosis (11) and allergic airway remodeling (12), and opposes myocardial ischemia-reperfusion injury (13) and pulmonary hypertension-associated vascular remodeling (14).
Mature HGF is a disulfide-linked, heterodimeric protein related to plasminogen, with α and β chains originating from an inactive, single-chain precursor (see Fig. 1). Although HGF is proteolytically incompetent because of mutations in the peptidase domain, it retains a serine peptidase-like mode of activation by cleavage at Arg494 in the precursor, changing conformation to allow productive binding, dimerization, and activation of its receptor, c-met (15). Mature α and β chains of HGF each can bind to c-met, although individually they have little receptor activating ability (15). A fragment of the α chain comprised of the N-terminal hairpin and four kringle domains (NK4) antagonizes c-met-mediated HGF effects, such as scatter factor activity, and also inhibits angiogenesis by independent mechanisms (16). NK4 is composed of the first 447 residues (pyrGlu32–Val478 of HGF) of the α chain. NK4 was generated originally by fragmentation with pancreatic elastase, which hydrolyzes HGF at Val478 (17). Subsequently, recombinant NK4 or NK4-expressing vectors were used to inhibit invasion, metastasis, and angiogenesis in tumor models (reviewed in Ref. 16).
FIGURE 1. HGF activation and inactivation by peptidases.
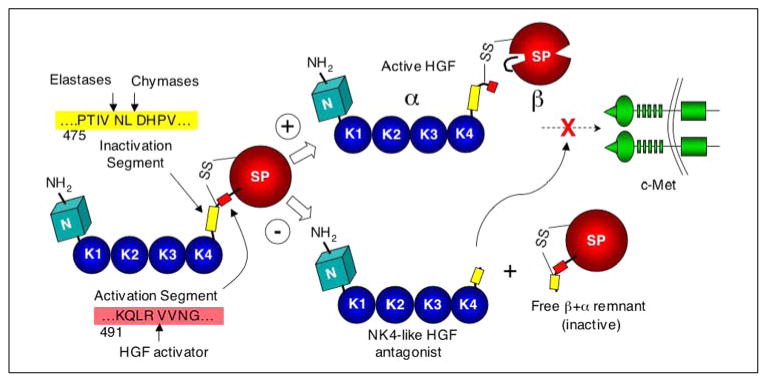
This diagram highlights the domain organization of HGF and proposed pathways of proteolytic regulation. The major recognized motifs include N-terminal (N), kringle (K1 – 4), and serine peptidase (SP) domains. Single-chain HGF is activated by hydrolysis of a site in an activation segment, as shown. This changes the receptor (c-met)-interacting site to allow productive binding. The resulting fragments, α and β, remain disulfide-linked. In contrast, hydrolysis by chymases, cathepsin G, or elastases at vulnerable sites within a 17-residue “inactivation segment” abolishes HGF activity and dissociates HGF into two unlinked fragments, one of which is an HGF antagonist. The + and − symbols identify pathways leading to stimulation and inhibition, respectively, of signaling via c-met. The mechanism by which NK4-like fragments antagonize actions of HGF is unknown. They may block the interaction between two c-met receptor-bound HGF α domains, thereby preventing receptor dimerization by a pair of ligands; alternatively, they may block direct contact between c-met and a secondary interacting site in the α domain of HGF.
HGF may influence development of mast cells, which like HGF itself are implicated in responses to injury, tissue repair, remodeling, and angiogenesis. Murine mast cell precursors express c-met during in vitro differentiation (18) and respond to HGF with β1 integrin-mediated migration. By releasing heparin, mast cells may raise circulating HGF after thrombotic events, like myocardial infarction (19). Heparin also may be a cofactor in HGF-induced angiogenesis (reviewed in Ref. 8). Thus, HGF may influence mast cell behavior and vice versa. In this regard, it is notable that secreted products of in vitro differentiated mast cells ablate HGF activity (18).
The present work focuses on major inflammatory cell peptidases, including the human mast cell chymotryptic peptidase, chymase, a mouse enzyme, mast cell protease (MCP)4, with similar tissue distribution and enzymatic properties (20–22), and human neutrophil elastase and cathepsin G. Cathepsin G possesses tryptic, chymotryptic, and metase activity (23) and is expressed by mast cells as well as by neutrophils. Human chymase and cathepsin G are expressed in a subset of mast cells inhabiting the dermis and certain other microenvironments, such as airway submucosa (24, 25). These enzymes accumulate in secretory granules and are released with other mediators by antigen-bound IgE or other stimuli, such as neuropeptides, anaphylatoxins, and bacterial surface proteins. Elastase and cathepsin G are released by activated neutrophils, where association with the cell surface or with high local concentrations in quantally released granules affords protection from circulating antipeptidases (26, 27). Thus, these enzymes are secreted in various types of inflammation. The present work reveals that chymase, cathepsin G, and neutrophil elastase, despite divergent cleavage specificities, hydrolyze HGF in a short but vulnerable inactivation segment, ablating bioactivity and generating an antagonistic fragment.
Materials and Methods
Mast Cell and Leukocyte Peptidases
Recombinant human prochymase was expressed, activated, and purified as described (21). The concentration of active chymase was determined with the substrate succinyl-l-Ala-Ala-Pro-Phe-4-nitroanilide as described (21). Chymase MCP4 was purified from mouse ears and assayed as described (21). Mast cell β-tryptase was purified from human lung (28). Human neutrophil elastase and cathepsin G were purchased from Elastin Products (Owensville, MO). The molar ratios of elastase and cathepsin G were calculated from protein content and assumed to be fully active.
Incubation of Peptidases with HGF, Macrophage-stimulating Protein (MSP), and Plasminogen
HGF and related kringle-rich, plasminogen-like proteins were incubated with peptidases. Human HGF was kindly provided by Genentech (South San Francisco, CA) or was purchased from Peprotech (Rocky Hill, NJ). These preparations are recombinant material expressed in Chinese hamster ovary and insect cells, respectively. Recombinant human MSP was from R&D Systems (Minneapolis, MN). Plasma-derived human plasminogen, porcine pancreatic elastase, and bovine chymotrypsin were from Sigma. Incubations with proteases were carried out in phosphate-buffered saline, pH 7.4, with or without heparin (10 μg/1 μg of HGF; bovine lung, Sigma, catalog number H4898, 150 units/mg), and were stopped by adding SDS-PAGE sample buffer and heating to 70 °C for 10 min.
Electrophoresis, Blotting, and N-terminal Sequencing
Results of incubation of HGF, MSP, and plasminogen with chymases were monitored by reducing and non-reducing gradient SDS-PAGE (Invitrogen). Coomassie Blue-stained gels were scanned and analyzed by densitometry. Chymase-generated fragments separated by non-reducing SDS-PAGE were transferred to polyvinylidene fluoride membrane. Blotted bands of interest were excised and subjected to N-terminal sequencing by Midwest Analytical (St. Louis, MO).
Purification and Sequencing of Peptidase-generated Fragments of HGF
Unreduced fragments of HGF arising from incubation with human chymase were loaded onto a 2.1 × 250 mm C4 reverse-phase HPLC column (Alltech, Deerfield, IL) in 5% acetonitrile, 95% H2O, 0.1% trifluoroacetic acid and eluted with a two-slope gradient of increasing concentrations of acetonitrile in H2O, 0.1% trifluoroacetic acid. The eluted proteins were detected by monitoring of absorbance at 280 nm, collected, and analyzed by SDS-PAGE. A purified fragment was co-incubated with HGF in the scatter factor assays described below. In separate experiments, chymase-generated fragments of HGF were reduced in 40 mm dithiothreitol, alkylated by incubation in 80 mm iodoacetamide, then separated on the C4 column. The major early eluting peak was collected and further analyzed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy (Voyager DE-STR, Applied Biosystems, Foster City, CA) at UCSF's Biomolecular Resource Center. The material was further probed by HPLC-linked tandem mass spectroscopy (QSTAR XL, Applied Biosystems). To characterize fragments of HGF generated by neutrophil elastase and cathepsin G, digested products were subjected directly to MALDI-TOF mass spectroscopy. Also, to compare the heparin-binding characteristics of HGF and products of chymase-mediated hydrolysis, partial digests were loaded onto a heparin HPLC column (TosoHaas, Montgomeryville, PA) and eluted with a linear gradient of 0.5–2 m NaCl in 10 mm bis-Tris-HCl, pH 6.1.
Scatter Factor Activity
To assess the bioactivity of chymase-hydrolyzed HGF, the scatter factor activity of cleaved protein was compared with that of native protein in Madin-Darby canine kidney (MDCK) cell assays (17). The antagonistic potential of an NK4-like fragment liberated by chymase was assessed in the same assay by incubating the purified fragment with mature, active HGF. Briefly, cells were plated on 6-well culture dishes at 17,000 cells/well 48 h before incubation with HGF. For inhibition studies, cells were preincubated with the NK4-like fragment for 30 min at 37 °C before addition of HGF. After incubation for 18 h, cells were fixed for 30 min in 4% paraformaldehyde and then washed in 70% ethanol and air-dried for light microscopy.
Results
Hydrolysis of HGF by Mast Cell Chymases
As determined by reducing SDS-PAGE prior to incubation with peptidases, the HGF in this study is mature, two-chain HGF mixed with smaller amounts of single-chain pro-HGF. As shown in Fig. 2, human chymase and mouse MCP4 incubated at molar ratios of enzyme:HGF of 1:70 selectively hydrolyze HGF in a time- and heparin-dependant fashion, generating major bands of 30 and 45 kDa. N-terminal sequencing of an unreduced 30-kDa protein reveals two sequences of similar molarity: Val-Val-Asn-Gly-Ile, corresponding to HGF residues 495–499, which is the N terminus of the activated serine peptidase domain, and Asp-His-Pro-Val-Ile, corresponding to residues 481–485 in the proposed inactivation segment (see Fig. 3). These findings indicate that the 30-kDa fragment is a heterodimer comprised of a remnant of the α domain disulfide-linked via Cys487 to intact, activated β (serine peptidase) domain. These data also suggest that the major site of hydrolysis is Leu480. Therefore, the prominent 45-kDa band is an NK4-like protein apparently ending with Leu480, i.e. at the site cleaved by chymase. Based on disappearance of single-chain HGF in digests run on reducing gels (not shown), chymase cleaves pro-HGF as well as the two-chain active form. Co-incubation with heparin modestly accelerates the loss of the HGF parent band. MCP4 activity in the presence of heparin is more selective than in its absence, because faint bands in addition to NK4-like and β+α remnant bands are apparent. However, heparin does not overtly influence selectivity of human chymase.
FIGURE 2. Selective hydrolysis of HGF by mast cell and neutrophil peptidases.
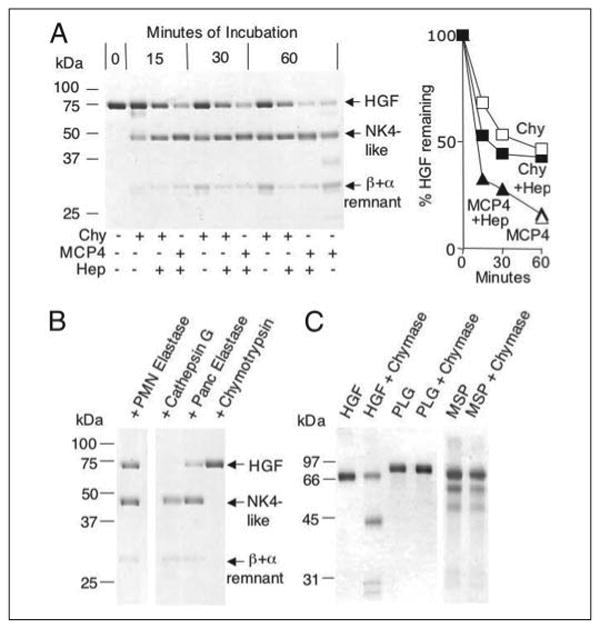
A, shows non-reducing SDS-PAGE of products generated from human HGF by the chymotryptic peptidases human chymase (Chy) and mouse MCP4, with time-dependent generation of an NK4-like major fragment and a β chain attached to an α remnant. Incubation intervals are indicated. Some incubation mixtures contain heparin (Hep). Incubation of HGF alone for 60 min does not yield visible fragmentation (not shown). The graph shows results of densitometry. B, shows results of incubation with neutrophil (PMN) elastase, cathepsin G, pancreatic elastase, and chymotrypsin, revealing that PMN elastase and cathepsin G generate an NK4-like fragment similar to classical NK4 generated by pancreatic elastase. Chymotrypsin has comparatively little activity. C, shows results of incubating chymase with HGF-related proteins, including MSP and plasminogen (PLG).
FIGURE 3. Identification of cleavage sites and comparison of inactivation segments.
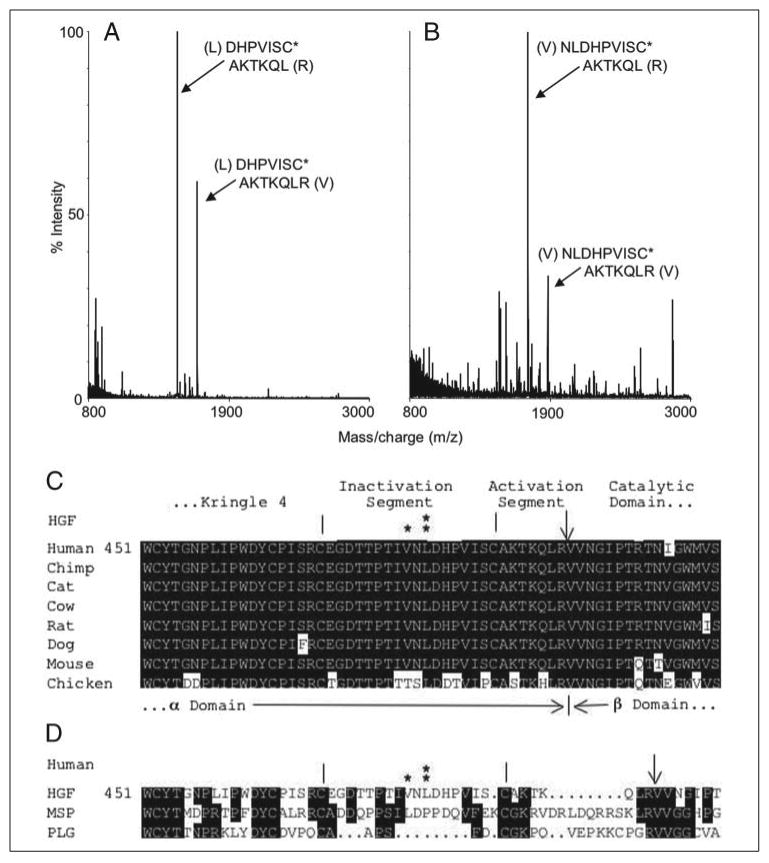
A, MALDI-TOF mass spectrum of the major HPLC-purified peptides generated from chymase-cleaved HGF. Peaks representing mono-protonated peptides DHPVISC*AKTKQL (m/z = 1496.80) and DHPVISC*AKTKQLR (m/z = 1652.91) are noted. C*, carbamidomethyl cysteine. The identity of these peptides was validated by tandem mass spectrometry. B, mass spectrum of neutrophil elastase-generated fragments of HGF, including peaks consistent with NLDHPVISC*AKTKQL (m/z 1723.94) and NLDHPVISC*AKTKQLR (m/z 1880.06). With results of SDS-PAGE, these findings suggest that the major site of HGF hydrolysis by chymase and neutrophil elastase is Leu480 and Val478, respectively. C, sequence alignment of seven mammalian HGFs and chicken HGF between residues 451 and 510, which includes the proposed protease-cleaved inactivation and activation segments bracketed by the end of the fourth kringle and beginning of the catalytic domain. Major sites of hydrolysis by elastase and chymase are shown by * and **, respectively. An arrow points to the site of hydrolysis of single-chain HGF by its activator to generate the active, two-chain form, in which the α chain is disulfide-linked to β chain via Cys487. Hydrolysis at any site between the two cysteines (vertical bars), which flank the 17-residue inactivation segment, unlinks α from β and generates a free, NK4-like fragment of α chain. Cleavage within the kringle domain itself does not generate a free fragment because of triple looping via Cys–Cys linkages. Note absolute conservation of the chymase cleavage site. D, compares sequence of human HGF, MSP, and plasminogen (PLG) in the same regions. Note that chymase and elastase hydrolysis sites in the HGF inactivation segment are not conserved in otherwise-homologous MSP and plasminogen.
Hydrolysis of HGF by Other Peptidases
As shown in Fig. 2, neutrophil elastase and cathepsin G hydrolyze HGF with a pattern similar to that of chymase and MCP4, including generation of an NK4-like fragment. Elastase was used in a molar ratio of enzyme:HGF of 1:38. Cathepsin G, which is weaker than chymase toward most substrates, was used in a ratio of 1:5. Chymotrypsin, used in a molar ratio of 1:12, exhibits little activity, consistent with the lack of preferred aromatic residues in the proposed inactivation segment (see Fig. 3). Mast cell tryptase does not cleave HGF (not shown), even after prolonged incubations with molar ratios of enzyme:substrate as high as 1:2.
Characterization of Peptidase-generated Fragments by Mass Spectroscopy
To confirm that Leu480 is a site of cleavage by chymase (as suggested by results of N-terminal sequencing of the blotted β+α remnant fragment) and to identify other potential fragments of HGF generated by inflammatory peptidases, we analyzed peptidase-generated products by mass spectroscopy. In the case of the chymase digest, MALDI-TOF mass spectroscopy of the purified principal peptide peak confirms Leu480 to be the major site of hydrolysis. Two major fragments co-eluted on the C4 column. One matches the mass of the Asp481-Arg494 segment (see Fig. 3) expected from cleavage at Leu480 of two-chain HGF. The other matches Asp481-Leu493, which is a desArg version of the other fragment. This is consistent with chymase-mediated hydrolysis at Leu480 and Leu493 or with prior removal of Arg494 from a portion of two-chain HGF. As analyzed by the MALDI-TOF approach, elastase generates major peaks matching masses of Asn479-Arg494 and Asn479-Leu493, consistent with the main site of cleavage being Val478, which is also the major site cleaved by pancreatic elastase (17, 29) and is two residues away from the site attacked by chymase. The finding of the desArg form of these peptides in the digests of elastase as well as chymase is consistent with a pre-existing population of two-chain HGF lacking Arg494 generated by a carboxypeptidase during the production of the recombinant enzyme. Cathepsin G, which is less specific than chymase or elastase, generates a variety of small fragments, consistent with hydrolysis at multiple sites within the inactivation segment.
Failure of Chymase to Hydrolyze HGF Homologues
As shown in Fig. 2, chymase does not fragment human plasminogen and MSP, which are the other major members of the plasminogen subfamily of kringle-containing proteins with serine peptidase domains. Thus, human chymase sensitivity is specific to HGF among its close relatives. However, MCP4 can cleave human but not mouse plasminogen (not shown), consistent with somewhat differing substrate preferences between human chymase and MCP4.
Purification of an NK4-like Protein Generated by Chymase
Fig. 4 shows chromatographic separation and purification of the NK4-like fragment from the β+α remnant fragment. The size disparity of the two peaks is because of a large difference in the extinction coefficient between the two major fragments, which are more nearly equal on a molar basis. The peak 1 protein was used in scatter factor assays, as noted below. Not shown are the results of heparin affinity chromatography, which reveals that chymase-generated NK4-like protein binds much more strongly than the free β chain. This suggests that the heparin affinity of HGF derives principally from the NK4 portion of the α chain. The shared affinity of HGF and chymase for heparin may promote the formation of a ternary complex and accelerate chymase-mediated HGF inactivation and generation of NK4-like protein, as seen in Fig. 2.
FIGURE 4. Purification of a chymase-generated NK4-like fragment of HGF.

This figure shows an HPLC chromatogram of unreduced fragments generated by incubation of HGF with human chymase. As shown in the inset, aliquots of major peaks 1 and 2 were subjected to non-reducing SDS-PAGE. Peak 1 contains a ∼45-kDa protein representing an NK4-like fragment generated by chymase. This purified protein was used to inhibit HGF scatter factor activity as in Fig. 5. Peak 2 contains the ∼30-kDa HGF β domain plus disulfide-linked α fragment remaining after removal of the NK4-like fragment from the native protein.
Effect of Cleavage by Chymase and Incubation with NK4-like Fragment on Cell Scattering Activity of HGF
As shown in Fig. 5, chymase destroys the ability of HGF to alter cell motility and morphology. Furthermore, incubation with the chymase-generated NK4-like fragment fully antagonizes HGF effects on MDCK cells. This antagonism occurs at ratios of NK4-like protein:HGF as low as 250:1. NK4 is shorter than chymase-generated NK4-like protein by two residues. The major proposed sites of hydrolysis involved in activation and inactivation of HGF and generation of antagonistic fragments are summarized in Fig. 1.
FIGURE 5. Anti-scatter factor activity of the NK4-like fragment.
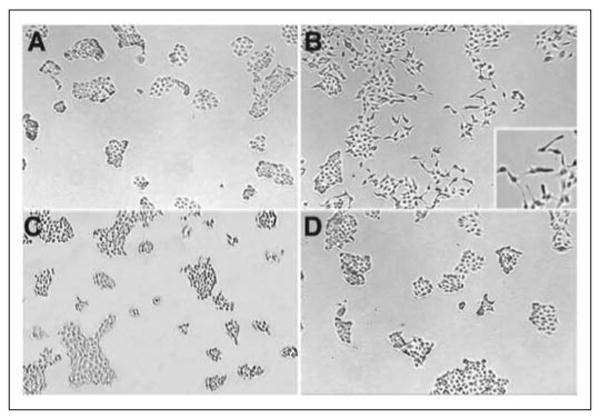
These micrographs show results of incubating MDCK cells with medium alone (A), intact HGF (B), human chymase-cleaved HGF (C), and intact HGF plus the purified NK4-like fragment generated by chymase (D). Note that HGF loosens cell clumps, elongates and scatters individual cells from sheet-like aggregates, and induces the formation of tube-like structures (inset). The NK4-like fragment opposes these effects.
Discussion
This work reveals that mast cell chymase hydrolyzes HGF, ablating scatter factor activity and simultaneously generating an antagonistic fragment. The Leu480 hydrolysis site is selective in that related members of the plasminogen subfamily have little homology in this region and resist cleavage. The site of vulnerability is not in a classical kringle domain but in a region connecting K4 to the “activation segment,” which, as shown in Fig. 1, contains the Arg494 site of hydrolytic activation. We propose that Leu480 is part of a 17-residue “inactivation segment” comprised of residues 470 – 486, which are not connected to other parts of HGF by disulfide bridges. Conservation of Leu480 and surrounding residues in mammalian HGFs hints that changes in this segment are poorly tolerated, possibly because susceptibility to hydrolytic inactivation and release of an antagonistic fragment is important for regulating HGF function.
The proposed inactivation segment is one of few regions in which proteolysis will divide HGF into free, inactive fragments. This is because most of HGF, including kringle and peptidase domains, is fortified by overlapping Cys–Cys links. Hydrolysis at most sites will produce nicking without separation of fragments. The peptides between kringles are theoretically vulnerable but appear to be protease-insensitive likely because they are short and inaccessible to bulky peptidases. The observation that elastases cleave human HGF at a site just two residues away from the site cleaved by chymase, along with the finding that cathepsin G cleaves in the same vicinity, invites speculation that these sites lie in an interdomain region in which neutral and hydrophobic residues are exposed. Crystal-derived structures of HGF do not address this point because the inactivation peptide is not included in existing structures. Identification of Leu at the site of hydrolysis itself argues for an unusual degree of exposure, because Leu is not favored at the scissile bond in short peptides cleaved by chymotryptic peptidases generally and chymase particularly. A profiling of the substrate preferences of human chymase with a combinatorial peptide library found the cleavage of substrates with P1 Leu at the site of hydrolysis to be far less likely than when the P1 residue is Phe or Tyr (30). Profiling with selected 4-nitroanilide and angiotensin-like peptides similarly suggests preferences for P1 aromatic residues (31, 32). Notwithstanding target preferences suggested by screening short peptides, at least two other proteins, procollagenase (33) and procollagen 1α (34), are cleaved by human chymase at P1 Leu. These proteins and HGF evidently present Leu-containing segments in a more cleaveable conformation than in short peptides with little secondary structure. It bears noting that combinatorial screening suggests that the residues occupying the P4 – P2 positions (Ile-Val-Asn) of HGF adjacent to the site hydrolyzed by chymase are favorable when present in short peptides (30). This suggests that P1 side chain accessibility, combined with P4 – P2 side chains interacting favorably with the extended substrate-binding site, contribute to the ability of chymase to hydrolyze at Leu480. The inability of mast cell tryptase to hydrolyze HGF is consistent with the lack of basic residues preferred by tryptic enzymes in the inactivation segment.
The present data suggest an explanation for the observation that a product of in vitro differentiated mouse mast cells degrades the HGF α chain (18). Our data indicate that a particular mouse chymase, MCP4, hydrolyzes HGF in a pattern similar to that generated by the complex mixture of biomolecules released by degranulating mast cells. Thus, MCP4 may be the principal HGF-degrading enzyme of mouse mast cells. In comparison to the large fragment of α chain produced by human chymase, MCP4 also yields one or more smaller bands, which appear to derive from the larger band. This finding suggests hydrolysis by MCP4 at sites less susceptible to human enzyme. Furthermore, MCP4 (but not human chymase) generates angiostatin-like fragments from human plasminogen (not shown). This is not physiologically significant because of the species mismatch and because mouse plasminogen is not cleaved. Although human chymase and mouse MCP4 are similar in physical and enzymatic properties, the present data indicate that they can also differ in hydrolyzing protein targets. Imperfect matching of mouse and human substrate preferences is not surprising given previously established differences, as in hydrolysis of angiotensin (21, 35). There are no human phylogenetic equivalents to MCP4 and other β-chymases, which may be rodent-specific. The mouse chymase with closest overall structural and phylogenetic similarity to human chymase is MCP5 α-chymase (36). However, an amino acid change in mouse and rat MCP-5 alters the specificity from chymotryptic to elastolytic with little ability to cleave peptide substrates with P1 Phe, Tyr, or Leu (37, 38). Overall, the present work supports the proposal that MCP4 β-chymase is the closest functional equivalent of human α-chymase (22).
Other investigations have established that a human mast cell line (HMC-1) and in vitro differentiated mouse mast cells express HGF receptor c-met and respond to HGF (18, 39). In HMC-1 cells, c-met expression is inducible and exposure to HGF suppresses release of tumor necrosis factor-α (39). In bone marrow-derived mouse mast cells, HGF promotes developmental stage- and integrin-dependant migration (18). Thus, secreted mast cell chymase and cathepsin G may decrease cytokine release and migration responses to HGF in the vicinity of a degranulating mast cell. Perhaps more significant are the implications of mast cell and neutrophil peptidase-mediated destruction of HGF for the behavior of other cells. Given what is known of the roles of HGF in pathobiology, inactivation by inflammatory peptidases could limit proliferation, migration, or differentiation of c-met-bearing cells in acute response to infection, injury, or allergen exposure. More specifically, selective hydrolysis may oppose HGF effects on epithelial repair in pneumonia, allergic airway inflammation (12), and ischemia-reperfusion injury (13). By interrupting control of epithelial to mesenchymal transformation by HGF, proteolytic inactivation may promote lung and airway fibrosis (11). Mast cell secretion of chymase and cathepsin G may modulate the rise of plasma HGF in clot-related disorders such as myocardial infarction. This rise has been linked to clot-stimulated degranulation of mast cells, which release heparin, to which HGF binds, facilitating its role in reparative angiogenesis (8, 19). Like HGF, chymase and cathepsin G bind to heparin. Indeed, they associate tightly with heparin in secretory granules and remain bound upon secretion. The mutual attraction of HGF and peptidase to heparin may encourage formation of a ternary complex and selective degradation.
The significance of in situ generation of NK4-like protein may lie in coupling of production of an antagonist with destruction of intrinsic activity. This combination may be more powerful than either effect by itself. There may be additional effects specific to the NK4-like fragment, for NK4 antagonizes HGF through competitive inhibition of binding to c-met and also has actions apparently unrelated to c-met, including inhibition of angiogenesis. The large α chain fragment generated by chymase is similar but not identical to NK4 in that NK4 lacks the C-terminal Asn-Leu dipeptide of the chymase-generated fragment. In its capacity to inhibit HGF scatter factor activity, the NK4-like fragment appears to be at least as potent as NK4, in that inhibition is achieved at ratios of antagonist to HGF much lower than reported for NK4 (17). Cells transcribing the HGF gene sometimes generate splice variants, resulting in truncated proteins containing N-terminal domain plus kringle 1 (NK1) or N-terminal domain plus kringle 1 and 2 (NK2) (40). These shortened proteins also can act as agonists of HGF. NK1 appears to require heparin to act as an agonist. When used as a drug, NK4 inhibits tumor growth and metastasis (16). Whether NK4-like protein produced endogenously by inflammatory peptidases has similar activity remains to be determined.
These findings suggest potential physiological pathways for generating NK4-like HGF antagonists in allergic and neutrophilic inflammation. The necessary ratios of enzyme to substrate are achievable in the vicinity of migrating neutrophils and degranulating mast cells, where local concentrations of enzyme are transiently high. These ratios also can be reached on a more sustained basis in settings such as purulent bronchitis in cystic fibrosis, where disintegrating neutrophils release active peptidases and overwhelm local antipeptidase defenses.
In conclusion, this work reveals that serine peptidases released during allergic and neutrophilic inflammation, acting on HGF, couple inactivation with generation of an HGF antagonist. This is achieved by hydrolyzing an inactivation segment that is conserved in mammals and vulnerable to cleavage by peptidases of varying specificity. Regulation and dysregulation of HGF activity by inflammatory peptidases may be important in diverse types of inflammation.
Acknowledgments
We thank Dr. Steve Hall of UCSF's Biomolecular Resource Center for assistance in evaluating mass spectroscopy data.
Footnotes
This work was supported by Grant HL024136 from the National Institutes of Health and by the Northern California Institute for Research and Education.
The abbreviations used are: HGF, hepatocyte growth factor; NK4, N-terminal and kringle 1 – 4 domain protein; MSP, macrophage-stimulating protein; MCP, mast cell protease; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; HPLC, high performance liquid chromatography; MDCK, Madin-Darby canine kidney.
References
- 1.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 2.Weidner KM, Arakaki N, Hartmann G, Vandekerckhove J, Weingart S, Rieder H, Fonatsch C, Tsubouchi H, Hishida T, Daikuhara Y, et al. Proc Natl Acad Sci U S A. 1991;88:7001–7005. doi: 10.1073/pnas.88.16.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- 4.Yanagita K, Matsumoto K, Sekiguchi K, Ishibashi H, Niho Y, Nakamura T. J Biol Chem. 1993;268:21212–21217. [PubMed] [Google Scholar]
- 5.Verghese GM, McCormick-Shannon K, Mason RJ, Matthay MA. Am J Respir Crit Care Med. 1998;158:386–394. doi: 10.1164/ajrccm.158.2.9711111. [DOI] [PubMed] [Google Scholar]
- 6.Sonnenberg E, Meyer D, Weidner KM, Birchmeier C. J Cell Biol. 1993;123:223–235. doi: 10.1083/jcb.123.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gohda E, Tsubouchi H, Nakayama H, Hirono S, Sakiyama O, Takahashi K, Miyazaki H, Hashimoto S, Daikuhara Y. J Clin Investig. 1988;81:414–419. doi: 10.1172/JCI113334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumori A. Cardiovasc Drugs Ther. 2004;18:321–326. doi: 10.1023/B:CARD.0000041252.33870.74. [DOI] [PubMed] [Google Scholar]
- 9.Aharinejad S, Taghavi S, Klepetko W, Abraham D. Lancet. 2004;363:1503–1508. doi: 10.1016/S0140-6736(04)16148-5. [DOI] [PubMed] [Google Scholar]
- 10.Sakamaki Y, Matsumoto K, Mizuno S, Miyoshi S, Matsuda H, Nakamura T. Am J Respir Cell Mol Biol. 2002;26:525–533. doi: 10.1165/ajrcmb.26.5.4714. [DOI] [PubMed] [Google Scholar]
- 11.Yaekashiwa M, Nakayama S, Ohnuma K, Sakai T, Abe T, Satoh K, Matsumoto K, Nakamura T, Takahashi T, Nukiwa T. Am J Respir Crit Care Med. 1997;156:1937–1944. doi: 10.1164/ajrccm.156.6.9611057. [DOI] [PubMed] [Google Scholar]
- 12.Ito W, Kanehiro A, Matsumoto K, Hirano A, Ono K, Maruyama H, Kataoka M, Nakamura T, Gelfand EW, Tanimoto M. Am J Respir Cell Mol Biol. 2005;32:268–280. doi: 10.1165/rcmb.2004-0058OC. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura T, Mizuno S, Matsumoto K, Sawa Y, Matsuda H. J Clin Investig. 2000;106:1511–1519. doi: 10.1172/JCI10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ono M, Sawa Y, Mizuno S, Fukushima N, Ichikawa H, Bessho K, Nakamura T, Matsuda H. Circulation. 2004;110:2896–2902. doi: 10.1161/01.CIR.0000146342.30470.30. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann G, Naldini L, Weidner KM, Sachs M, Vigna E, Comoglio PM, Birchmeier W. Proc Natl Acad Sci U S A. 1992;89:11574–11578. doi: 10.1073/pnas.89.23.11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto K, Nakamura T. Cancer Sci. 2003;94:321–327. doi: 10.1111/j.1349-7006.2003.tb01440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. FEBS Lett. 1997;420:1–6. doi: 10.1016/s0014-5793(97)01475-0. [DOI] [PubMed] [Google Scholar]
- 18.Fehlner-Gardiner CC, Cao H, Jackson-Boeters L, Nakamura T, Elliott BE, Uniyal S, Chan BM. Differentiation. 1999;65:27–42. doi: 10.1046/j.1432-0436.1999.6510027.x. [DOI] [PubMed] [Google Scholar]
- 19.Kinoshita M, Miyamoto T, Ohashi N, Sasayama S, Matsumori A. Circulation. 2002;106:3133–3138. doi: 10.1161/01.cir.0000039344.98537.be. [DOI] [PubMed] [Google Scholar]
- 20.Serafin WE, Sullivan TP, Conder GA, Ebrahimi A, Marcham P, Johnson SS, Austen KF, Reynolds DS. J Biol Chem. 1991;266:1934–1941. [PubMed] [Google Scholar]
- 21.Caughey GH, Raymond WW, Wolters PJ. Biochim Biophys Acta. 2000;1480:245–257. doi: 10.1016/s0167-4838(00)00076-5. [DOI] [PubMed] [Google Scholar]
- 22.Tchougounova E, Pejler G, Abrink M. J Exp Med. 2003;198:423–431. doi: 10.1084/jem.20030671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polanowska J, Krokoszynska I, Czapinska H, Watorek W, Dadlez M, Otlewski J. Biochim Biophys Acta. 1998;1386:189–198. doi: 10.1016/s0167-4838(98)00085-5. [DOI] [PubMed] [Google Scholar]
- 24.Irani AM, Bradford TR, Kepley CL, Schechter NM, Schwartz LB. J Histochem Cytochem. 1989;37:1509–1515. doi: 10.1177/37.10.2674273. [DOI] [PubMed] [Google Scholar]
- 25.Matin R, Tam EK, Nadel JA, Caughey GH. J Histochem Cytochem. 1992;40:781–786. doi: 10.1177/40.6.1588024. [DOI] [PubMed] [Google Scholar]
- 26.Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. J Cell Biol. 1995;131:775–789. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell EJ, Campbell MA, Boukedes SS, Owen CA. J Clin Investig. 1999;104:337–344. doi: 10.1172/JCI6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartmann T, Ruoss SJ, Raymond WW, Seuwen K, Caughey GH. Am J Physiol. 1992;262:L528–L534. doi: 10.1152/ajplung.1992.262.5.L528. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto K, Kataoka H, Date K, Nakamura T. J Biol Chem. 1998;273:22913–22920. doi: 10.1074/jbc.273.36.22913. [DOI] [PubMed] [Google Scholar]
- 30.Raymond WW, Waugh Ruggles S, Craik CS, Caughey GH. J Biol Chem. 2003;278:34517–34524. doi: 10.1074/jbc.M304087200. [DOI] [PubMed] [Google Scholar]
- 31.Powers JC, Tanaka T, Harper JW, Minematsu Y, Barker L, Lincoln D, Crumley KV, Fraki JE, Schechter NM, Lazarus GG, Nakajima K, Nakashino K, Neurath H, Woodbury RG. Biochemistry. 1985;24:2048–2058. doi: 10.1021/bi00329a037. [DOI] [PubMed] [Google Scholar]
- 32.Sanker S, Chandrasekharan UM, Wilk D, Glynias MJ, Karnik SS, Husain A. J Biol Chem. 1997;272:2963–2968. doi: 10.1074/jbc.272.5.2963. [DOI] [PubMed] [Google Scholar]
- 33.Saarinen J, Kalkkinen N, Welgus HG, Kovanen PT. J Biol Chem. 1994;269:18134–18140. [PubMed] [Google Scholar]
- 34.Kofford MW, Schwartz LB, Schechter NM, Yager DR, Diegelmann RF, Graham MF. J Biol Chem. 1997;272:7127–7131. doi: 10.1074/jbc.272.11.7127. [DOI] [PubMed] [Google Scholar]
- 35.Saito K, Muto T, Tomimori Y, Imajo S, Maruoka H, Tanaka T, Yamashiro K, Fukuda Y. Biochem Biophys Res Commun. 2003;302:773–777. doi: 10.1016/s0006-291x(03)00263-8. [DOI] [PubMed] [Google Scholar]
- 36.Caughey GH. Mol Immunol. 2002;38:1353–1357. doi: 10.1016/s0161-5890(02)00087-1. [DOI] [PubMed] [Google Scholar]
- 37.Kunori Y, Koizumi M, Masegi T, Kasai H, Kawabata H, Yamazaki Y, Fukamizu A. Eur J Biochem. 2002;269:5921–5930. doi: 10.1046/j.1432-1033.2002.03316.x. [DOI] [PubMed] [Google Scholar]
- 38.Karlson U, Pejler G, Tomasini-Johansson B, Hellman L. J Biol Chem. 2003;278:39625–39631. doi: 10.1074/jbc.M301512200. [DOI] [PubMed] [Google Scholar]
- 39.Yano K, Nakao K, Sayama K, Hamasaki K, Kato Y, Nakata K, Ishii N, Butter-field JH, Galli SJ. Biochem Biophys Res Commun. 1997;239:740–745. doi: 10.1006/bbrc.1997.7546. [DOI] [PubMed] [Google Scholar]
- 40.Chan AM, Rubin JS, Bottaro DP, Hirschfield DW, Chedid M, Aaronson SA. Science. 1991;254:1382–1385. doi: 10.1126/science.1720571. [DOI] [PubMed] [Google Scholar]