

Abstract
Prostasin, a trypsinlike serine peptidase, is highly expressed in prostate, kidney, and lung epithelia, where it is bound to the cell surface, secreted, or both. Prostasin activates the epithelial sodium channel (ENaC) and suppresses invasion of prostate and breast cancer cells. The studies reported here establish mechanisms of membrane anchoring and secretion in kidney and lung epithelial cells and demonstrate a critical role for prostasin in regulating epithelial monolayer function. We report that endogenous mouse prostasin is glycosylphosphatidylinositol (GPI) anchored to the cell surface and is constitutively secreted from the apical surface of kidney cortical collecting duct cells. Using site-directed mutagenesis, detergent phase separation, and RNA interference approaches, we show that prostasin secretion depends on GPI anchor cleavage by endogenous GPI-specific phospholipase D1 (Gpld1). Secretion of prostasin by kidney and lung epithelial cells, in contrast to prostate epithelium, does not depend on COOH-terminal processing at conserved Arg322. Using stably transfected M-1 cells expressing wild-type, catalytically inactive, or chimeric transmembrane (not GPI)-anchored prostasins we establish that prostasin regulates transepithelial resistance, current, and paracellular permeability by GPI anchor- and protease activity-dependent mechanisms. These studies demonstrate a novel role for prostasin in regulating epithelial monolayer resistance and permeability in kidney epithelial cells and, furthermore, show specifically that prostasin is a critical regulator of transepithelial ion transport in M-1 cells. These functions depend on the GPI anchor as well as the peptidase activity of prostasin. These studies suggest that cell-specific Gpld1- or peptidase-dependent pathways for prostasin secretion may control prostasin functions in a tissue-specific manner.
Keywords: serine protease, epithelial sodium channel, glycosylphosphatidylinositol anchor, transepithelial resistance, tight junction
The human genome project has identified many previously unknown serine peptidase genes, including membrane peptidases that regulate key cellular functions such as ion transport, receptor activation, cell motility, and angiogenesis (1, 23, 27, 40, 46). Regulation of peptidase function occurs at multiple levels including transcription, activation, compartmentalization and secretion, and local balance between active peptidases and their inhibitors. Prostasin is a trypsinlike serine peptidase originally purified as an active, soluble enzyme from human seminal fluid and is highly expressed in prostate, bronchus, lung, and kidney in mouse and human (41, 43, 52). Prostasin (PRSS8) belongs to a distinct family of genes on syntenic regions of human chromosome 16p13.3/11.2 and mouse chromosomes 7 and 17 that includes tryptase-γ (TPSG), tryptase-ε (PRSS22), pancreasin (MPN), testisin (TEST1), and distal intestinal serine protease (Disp) (2, 4, 15, 32, 41, 47, 49, 53). In addition to being secreted, prostasin is expressed as a glycosylphosphatidylinositol (GPI)-anchored membrane protein in prostate epithelial cells (7). Soluble prostasin, purified from human seminal fluid, terminates at Arg323, suggesting that prostasin may be secreted via tryptic cleavage of its COOH-terminal hydrophobic domain (52). However, the posttranslational processing steps that lead to prostasin secretion have not been determined experimentally. Prostasin is not avidly secreted from cultured airway epithelial cells (36), suggesting that there may be cell-specific differences in prostasin processing.
Prostasin is also known as channel-activating protease (CAP)-1 and is the first of several membrane serine peptidases found to activate the epithelial sodium channel (ENaC) (27, 38). ENaC function is tightly regulated and is critical for maintaining salt and fluid balance in the lung and kidney (28). Hypertension and elevated levels of urinary prostasin develop in rats transiently overexpressing prostasin (44). Increased urinary prostasin excretion also occurs in patients with hypertension from primary hyperaldosteronism and is stimulated by saline infusion or mineralocorticoids (22, 25). Intrapulmonary administration of aprotinin, a broad-spectrum serine peptidase inhibitor, attenuates alveolar fluid clearance (26, 34). Recent studies show that prostasin is highly expressed in cystic fibrosis airways and is a strong basal activator of ENaC in cystic fibrosis airway epithelial cells (11, 36). These observations predict that prostasin has a critical role in regulating epithelial sodium transport in normal and pathological conditions in the kidney and lung. On this basis, prostasin has been proposed as a target for therapeutic inhibition in cystic fibrosis. In addition to regulating ENaC activity, prostasin inhibits prostate and breast cancer cell invasion in vitro, suggesting an additional role as a suppressor of tumor invasion (5, 6).
The physiological substrates of prostasin and the direct mechanism for ENaC activation by serine peptidases remain unknown. Patch-clamp experiments suggest that prostasin increases open probability of ENaC, but there is no evidence that prostasin directly cleaves ENaC α-, β-, or γ-subunits (38). In a Xenopus oocyte expression system full-length, but not COOH-terminally truncated, soluble prostasin/CAP-1 activated ENaC (39). However, other investigators found that soluble, active prostasin can augment ENaC activity in M-1 kidney cortical collecting duct cells (22). Additionally, in vitro invasion studies showed that overexpression of wild-type prostasin, but not a truncated secreted form of prostasin, inhibits cell invasion (5, 6). These observations suggest that prostasin function may be regulated by membrane localization or secretion. However, the specific roles of the GPI anchor and COOH-terminal domain have not been studied.
We hypothesize that regulation of prostasin activity at the cell surface may be an important mechanism for controlling its function in different cells and organs. To address this hypothesis we use mutagenesis, small interfering RNA (siRNA), and stable overexpression of prostasin mutants to identify mechanisms for GPI anchoring and secretion of prostasin in kidney and lung epithelial cells and to study the role of the COOH-terminal domain in prostasin function. In these studies, we demonstrate that secretion of prostasin by these cells is polarized and depends on release of GPI-anchored prostasin by endogenous GPI-specific phospholipase D1 (Gpld1) rather than on proteolytic cleavage of the COOH-terminal hydrophobic domain at Arg322. Furthermore, we report the novel observation that prostasin regulates transepithelial resistance (Rte) and paracellular permeability in addition to regulating amiloride-sensitive transepithelial current and that these prostasin functions depend on the COOH-terminal GPI anchor and peptidase activity.
Experimental Procedures
Materials
Cell culture media were obtained from GIBCO (Carlsbad, CA). Transwell polycarbonate cell culture membranes (0.4-μm pore, 1 cm2) were from Corning (Corning, NY). Fetal calf serum (FCS), Lipofectamine 2000, Superscript II reverse transcriptase, and pcDNA3.1 and pcDNA4-V5His-B expression vectors were from Invitrogen (Carlsbad, CA). G-418 was from Calbiochem (Pasadena, CA). Oligonucleotide primers, plasmid DNA, and RNA purification kits were obtained from Qiagen (Valencia, CA). AmplitaqGold DNA polymerase was from Perkin Elmer Bioscience (Foster City, CA). Restriction endonucleases, peptide N-glycosidase F (PNGase F), Deep Vent DNA polymerase, and T4 DNA ligase were from New England Biolabs (Beverly, MA). Escherichia coli DH5α and the QuikChange II mutagenesis kit were from Stratagene (La Jolla, CA). Anti-human prostasin monoclonal antibody was obtained from BD Biosciences (Palo Alto, CA). Anti-FLAG M2-horseradish peroxidase (HRP)-conjugated antibody and M2-agarose were from Sigma (St. Louis, MO). Anti-V5-HRP-conjugated antibody was from Invitrogen. Secondary antibodies were from Zymed Laboratories (South San Francisco, CA). Recombinant Bacillus thuringiensis phosphatidylinositol-specific phospholipase C (PI-PLC) and CyQuant cell enumeration assay were from Molecular Probes (Eugene, OR). Immobilon P polyvinylidene difluoride membranes and Microcon YM-10 centrifugal ultrafiltration concentrators were from Millipore (Bedford, MA). Super Signal West Pico reagents for enhanced chemiluminescence, bicinchoninic acid protein assay, and EZ-Link sulfo-N-hydroxysuccinimide (NHS)-biotin were obtained from Pierce (Rockford, IL). siRNA was purchased from Ambion (Austin, TX). All other reagents were from Sigma or Fisher Scientific (Suwannee, GA).
Cell culture
M-1 and MLE-12 cells were obtained from American Type Culture Collection (Manassas, VA). M-1 cells were cultured in a 50:50 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 medium supplemented with 5% FCS, 100 nM dexamethasone, penicillin, and streptomycin. MLE-12 cells were cultured in DMEM supplemented with insulin, transferrin, selenium, 10% FCS, and antibiotics. Cells were transfected with Lipofectamine 2000 (2 μl/μg DNA) in OptiMem per manufacturer's directions. For stable transfection, cells were selected for 10–14 days with 0.5 mg/ml G-418 to establish polyclonal cultures. Cells were cultured at 37°C with 5% CO2.
Detergent phase separation, deglycosylation, and GPI anchor cleavage
Secretion, membrane anchoring, and posttranslational modifications were studied in M-1 mouse cortical collecting duct cells, which express prostasin endogenously. Expression of prostasin was assessed by SDS-PAGE and immunoblotting. Immunoblots were probed with 1:1,000 anti-human prostasin monoclonal antibody and 1:1,000 goat anti-mouse IgG2a-HRP conjugate in blotting buffer (150 mM NaCl, 50 mM Tris pH 7.5, 0.5% Tween 20, 5% nonfat dry milk) and detected by enhanced chemiluminescence using SuperSignal West Pico substrate.
Secretion of endogenous prostasin by M-1 cells was assayed by immunoblotting of concentrated conditioned medium. Medium was concentrated by centrifugal ultrafiltration with Microcon YM-10 concentrators. To study apical and basolateral secretion, M-1 cells were cultured on Transwell membranes until monolayers developed high Rte (>1,000 Ω · cm2). Whole cell lysates were made in 150 mM NaCl-20 mM Tris pH 7.4 (Tris-buffered saline, TBS) with 1% Triton X-100 on ice. Triton X-114 detergent phase separation was done to isolate membrane and soluble proteins and to solubilize GPI-anchored proteins more efficiently (3). Briefly, cells were washed with ice-cold TBS, resuspended in TBS with 2% Triton X-114, and incubated on ice for 30–60 min and then at 37°C for 5 min to induce phase separation. The detergent phase was diluted with TBS to a final concentration of 2% Triton X-114, and proteins were precipitated from aqueous and detergent phases with 15% trichloroacetic acid (TCA).
To assess GPI anchoring, M-1 cells were treated with PI-PLC (0.5–1.0 U/ml) in PBS or control medium at 37°C for 60 min. PI-PLC conditioned medium was collected, concentrated, and assayed by immunoblot for prostasin. For deglycosylation experiments, GPI-anchored proteins were released from transiently transfected M-1 cells overexpressing prostasin by treatment with PI-PLC. Concentrated PI-PLC-conditioned medium was denatured by boiling for 10 min, incubated with PNGase F or buffer alone at 37°C for 18 h, and assayed by immunoblot for prostasin.
Site-directed mutagenesis and prostasin expression vector construction
The mouse prostasin (GenBank accession no. AF378085) coding sequence was amplified by PCR using primers 5′-CCCAAGCTTG GGCCCTTGTC CTAGGCCATG GCC and 5′-CCGGAATTCC GGCGAGTGGG CTCAAAGATC AAGTCG that encode HindIII and EcoRI linkers. PCR was done with AmplitaqGold and thermal cycling parameters of 95°C for 10 min for 1 cycle, 94°C for 30 s, 60°C for 30 s, 72°C for 90 s for 35 cycles, and 72°C for 3 min for 1 cycle. PCR products were purified, digested with HindIII and EcoRI, ligated into pcDNA3.1, and verified by sequencing to generate plasmid pcDNA3.1-Prostasin. Mutations were introduced into pcDNA3.1-Prostasin by site-directed mutagenesis with the Stratagene QuikChange II kit per manufacturer's instructions. Mutagenesis primers were designed with a web-based application (http://labtools.stratagene/QC). Prostasin mutants were generated by PCR using the primers listed in Table 1.
Table 1. Mouse prostasin mutagenesis primers.
| Mutation | Forward Primer | Reverse Primer |
|---|---|---|
| R322A | AGCCCCGAAATTGTTAGCGCCCGTACTTTTCCTGC | GCAGGAAAAGTACGGGCGCTAACAATTTCGGGGCT |
| S238A | CCAGGGTGACGCTGGGGGCCCAC | GTGGGCCCCCAGCGTCACCCTGG |
| S313N-S314L | CATCATCCTGTCTTCAACTTAGCGGCAGCCC | GGGCTGCCGCTAAGTTGAAGACAGGATGATG |
| K319A | GCGGCAGCCCCGGCATTGTTAAGGCCCGT | ACGGGCCTTAACAATGCCGGGGCTGCCGC |
| K319A-R322A | GCGGCAGCCCCGGCATTGTTAGCGCCCGT | ACGGGCGCTAACAATGCCGGGGCTGCCGC |
| S313N-S314L-R322A | Sequential use of S313N-S314L and R322A primers | |
| S313N-S314L-A315P | GTCTTCAACTTACCGGCAGCCCCGA | TCGGGGCTGCCGGTAAGTTGAAGAC |
| S313N-S314L-A315P-R322A | Sequential use of S313N-S314L-A315P and R322A primers | |
| S313N-S314L-A315P-K319A-R322A | Sequential use of S313N-S314L-A315P and R322A primers |
A chimera of mouse prostasin that replaces the GPI anchor signal with the COOH-terminal transmembrane peptide domain of mouse tryptase-γ, a member of the prostasin gene family (48), was constructed by overlap extension PCR to generate a transmembrane but not GPI-anchored form of prostasin. Prostasin cDNA (amino acids 1–287) was amplified with primers MPRF1 (5′-CCCAAGCTTG GGCCCTTGTC CTAGGCCATG GCC) and MPR-TPSGR1 (5′-AGCCCCCTGC CTCTGCCACA TGGTGGTG), which add a 5′ HindIII linker and a 3′ 10-base pair tryptase-γ sequence overlapping the 5′ end of the tryptase-γ amplimer. The tryptase-γ COOH-terminal hydrophobic domain (amino acids 264–311) was amplified with primers MPR-TPSGF2 (5′-GCAGGGGGCT CAGGAATG) and MPR-TPSGR2 (5′-GGGGGGTCTA GAATTTAAGA AAGACACTTG GGTTGG), which add a 3′ XbaI linker. Amplimers were gel purified and used as the template for a subsequent PCR using primers mPRF1 and mPR-TPSGR2. PCR products were gel purified, digested with HindIII and XbaI, repurified, and ligated into pcDNA3.1 to generate plasmid pcDNA3.1-mProstasin-Tpsg. Internally epitope-tagged prostasin, pcDNA3.1-mProstasin-FLAG, was generated by overlap extension PCR to insert FLAG epitope between prostasin amino acids 310 and 311 adjacent to the predicted site of GPI anchor attachment at Ser313. First-step PCRs were done with primer pairs MPRFLAG-F1/R1 (5′-CCCAAGCTTG GGCCCTTGTC CTAGGCCATG GCC/5′-CTTGTCATCG TCGTCCTTGT AATCAGGATG ATGGTTGCAG AGGTGGC) and MPRFLAG-F1/R2 (5′-GTCTTCAGCT CAGCGGCAGCC/5′-CCGGAATTCC GGCGAGTGGG CTCAAAGATC AAGTCG), using pcDNA3.1-mProstasin as template. First-step PCR products were purified, combined, and amplified, using primers MPRFLAG-F1 and MPRFLAG-R2 to generate full-length mProstasin-FLAG cDNA. Schematic diagrams of these constructs are shown in Fig. 5. pcDNA4-prostasin-Tpsg-V5 was constructed by subcloning prostasin-Tpsg into pcDNA4-V5His-B to attach V5 epitope to the COOH terminus of prostasin-Tpsg. All constructs were verified by DNA sequencing.
Fig. 5.
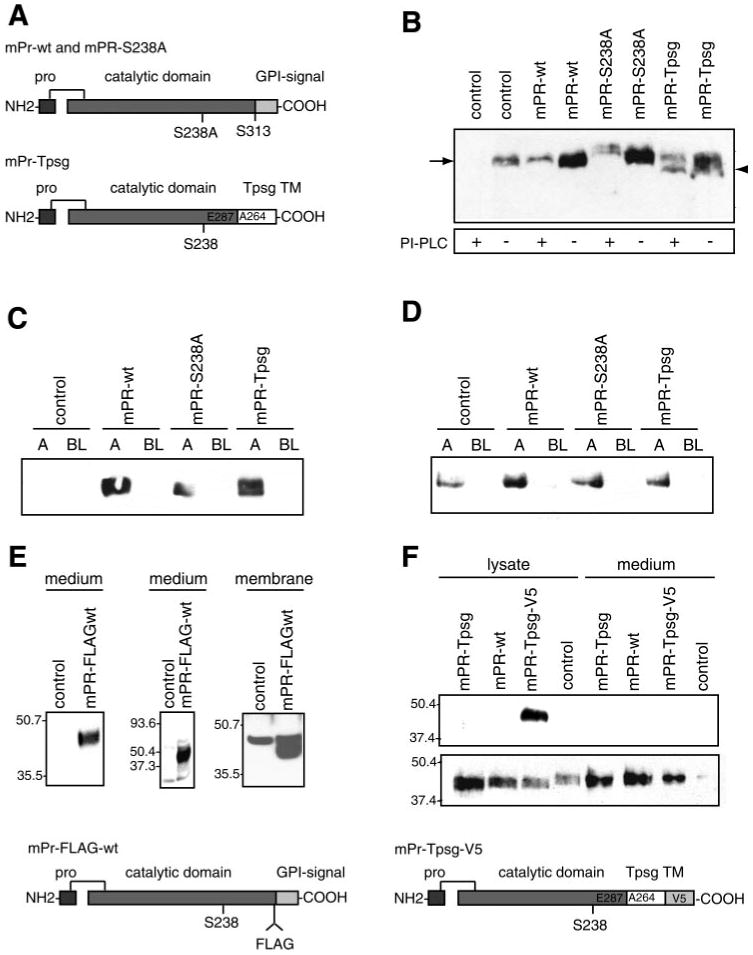
Characterization of stably transfected M-1 cell lines. M-1 cells were stably transfected with pcDNA3.1 empty vector (control), wild-type prostasin (mPR-wt), catalytically inactive prostasin (mPR-S238A), or chimeric transmembrane (TM), non-GPI-anchored prostasin (mPR-Tpsg). Prostasin was assayed by immunoblotting. Samples were normalized to cell number for conditioned media and protein concentration for cell lysates. A: schematic diagrams of prostasin expression constructs. B: membrane association and GPI anchoring of prostasin variants. Cells were treated with PI-PLC or buffer alone, extracted in 60 mM n-octylglucoside-1% Triton X-100 in TBS on ice, resolved by SDS-PAGE, and assayed for prostasin by immunoblotting. mPR-Tpsg (arrowhead) is resistant to shedding by PI-PLC and migrates slightly faster than prostasin (arrow). C: apical (A) and basolateral (BL) secretion of prostasin variants from stably transfected, polyclonal M-1 cell lines cultured on 0.4-μm-pore filters. D: domain-selective cell surface biotinylation. M-1 cells were grown on 0.4-μm-pore Transwell filters until stable transepithelial resistance (Rte) developed. Cells were incubated with sulfo-NHS-biotin (0.5 mg/ml) or PBS control (data not shown) in apical or basolateral chamber. Biotinylated proteins were captured with streptavidin-agarose, resolved by 10% SDS-PAGE, and assayed for prostasin by immunoblotting. E: M-1 cells were transfected with pcDNA3.1-mPR-FLAG (diagram of construct at bottom of figure) or control vector. Conditioned medium was assayed for prostasin by immunoblot with anti-prostasin antibody (left), immunoprecipitation with anti-FLAG M2 antibody followed by immunoblot with prostasin antibody (middle), or immunoblot with M2 antibody after cell surface proteins were isolated by biotinylation and streptavidin-agarose affinity capture (right). F: M-1 cells were transfected with pcDNA3.1-mPR-Tpsg, pcDNA3.1-mPR-wt, pcDNA4-mPR-Tpsg-V5 (diagram of construct at bottom of figure), or control. Cell lysates and conditioned media were assayed by immunoblot using V5 antibody (top) or prostasin antibody (bottom).
Secretion, membrane association, and GPI anchoring of prostasin mutants
M-1 mouse cortical collecting duct and MLE-12 mouse lung alveolar type II cells were transiently transfected with mouse prostasin mutants to identify specific residues involved in membrane anchoring and secretion of prostasin. Cells were transfected in six-well plates with 4 μg of plasmid and 8 μl of Lipofectamine 2000. For secretion studies conditioned medium was collected 48–72 h after transfection and concentrated by centrifugal ultrafiltration as above. Samples were normalized to load equivalent volumes of concentrated conditioned medium for analysis by 12% SDS-PAGE and immunoblotting. For pilot experiments, samples were also normalized to cell number measured with the CyQuant cell proliferation assay per the manufacturer's instructions. Results were identical when normalized to volume or cell number. Therefore, samples were normalized to load equivalent volume (data not shown).
For studies of membrane association, cellular proteins were extracted from transiently transfected cells by Triton X-114 detergent phase separation and concentrated by TCA precipitation. Samples were normalized to cell number for immunoblotting. For studies of GPI anchoring, transiently transfected M-1 cells were treated with PI-PLC (0.5 U/ml) in PBS. PI-PLC-conditioned medium was concentrated, normalized for cell number, and assayed by immunoblot. Immunoblots were quantitated by densitometry with ImageQuant5.0 (Molecular Dynamics, Sunnyvale, CA). Secretion of prostasin mutants was expressed as the ratio of densitometric units relative to wild-type prostasin. Secretion of mutant prostasins from stably transfected M-1 cell lines was characterized in immunoblots of apical and basolateral conditioned medium. Membrane association of mutant prostasins from these cell lines was characterized in immunoblots of proteins extracted in TBS, 1% Triton X-100, and 60 mM n-octylglucoside on ice.
Domain-selective cell surface labeling was done to examine polarized expression of prostasin variants with stably transfected M-1 cell lines cultured on 4.7-cm2, 0.4-μm-pore Transwells. After monolayers developed high Rte, cells were incubated on ice for 60 min with sulfo-NHS-biotin (0.5 mg/ml in PBS) or PBS alone applied to the apical or basolateral chamber and then quenched with cold serum-free medium for 5 min. After washing with PBS, membranes were excised from the Transwell inserts, and cells were homogenized in TBS-1% Triton X-100–60 mM n-octylglucoside lysis buffer. Biotinylated proteins were captured by incubating cell lysates with streptavidin-agarose beads overnight at 4°C. Bound proteins were eluted from streptavidin-agarose by boiling in reducing SDS-PAGE sample buffer, and samples were assayed for prostasin by immunoblotting.
RNA interference
Small interfering duplex RNA (siRNA) corresponding to mouse Gpld1 (GenBank accession no. NM_008156) and Silencer negative control no. 1 siRNAs were obtained from Ambion. Gpld1 sense and antisense siRNA sequences are 5′-GGCACAGUAU GUACUAACUTT and 5′-AGUUAGUACA UACUGUGCCTT, respectively. M-1 cells were transfected with 100 nM siRNA, using Lipofectamine 2000 as described above. Gpld1 expression was assayed by semiquantitative PCR using primers 5′-ACCCTAACCC AAGTCCTGCT and 5′-CAGGTCAGTC AGGTGCAGAA. Mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified with primers 5′-GTGTTCCTAC CCCCAATGTG and 5-AGGAGACAAC CTGGTCCTCA as a housekeeping gene control. Total RNA was purified from cells 4 days after transfection with siRNA. Two micrograms of RNA was reverse transcribed with Superscript II. Gpld1 and GAPDH cDNA were amplified by PCR using the following thermal cycling parameters: 94°C for 30 s, 57°C for 30 s, 72°C for 45 s for 35 cycles. Relative expression was assessed by 2% agarose gel electrophoresis and staining with ethidium bromide. Conditioned medium was collected 4, 6, and 8 days after transfection, concentrated, normalized to cell number, and assayed for prostasin by immunoblotting. Prostasin secretion was quantitated by densitometry, and data were analyzed with a two-sided t-test.
Measurement of Rte, current, and permeability
M-1 cells were seeded onto 0.4-μm-pore, 1-cm2 polycarbonate Transwell membranes (500,000 cells/cm2). Rte and potential difference (Vte) were measured with an epithelial voltohmeter (EVOM) using Ag:AgCl chopstick electrodes (World Precision Instruments, Sarasota, FL). Measurements were made every 24–72 h, and data were analyzed when monolayers showed stable Rte and Vte for at least 24 h, which occurred 15–18 days after filters were seeding. Equivalent short-circuit current (Ieq) was calculated by Ohm's law (Ieq = Vte/Rte) and normalized to surface area of the Transwell membrane as in other studies of M-1 cells (19). To avoid disturbing the monolayer and altering Rte, in studies requiring medium changes or addition of inhibitors or tracers before Rte measurements the medium was changed with a perfusion technique by withdrawing and gently replacing 50% of the medium volume five times. To assess sensitivity of Ieq to amiloride, cells were exposed to 10 μM amiloride in the apical media and measurements were done at 15-min intervals. Treatment of untransfected M-1 cells with aprotinin (20 μg/ml) or amiloride (10 μM) confirmed that transepithelial current is attenuated by aprotinin and amiloride in our cells (data not shown), similar to data previously reported by other investigators using M-1 cells (19). Experiments were done in triplicate wells and repeated three to five times (total of 9–15 filters per cell line). Data are presented as means (SE). Transepithelial permeability was assayed by measuring apical-to-basal flux of fluorescein isothiocyanate (FITC)-inulin, a passive tracer of paracellular permeability, by fluorescence (excitation 485 nm, emission 520 nm).
Real-time RT-PCR
Relative quantitative real-time PCR was done to compare expression of endogenous prostasin and the prostasin-Tpsg chimera in stably transfected M-1 cells. Primer pairs were designed to span the site of prostasin-Tpsg fusion in order to amplify specifically endogenous prostasin transcripts or prostasin-Tpsg transcripts. Endogenous prostasin-specific primers (5′-TACGGGCCTTAACAATTTCG and 5′-CTGAGTCTGGGGGACAACTC) yield a 157-bp amplimer. Prostasin-Tpsg-specific primers (5′-TACTTGGCAGGCATTGTGAG and 5′-GAGACCAGCAGCAGGAAGAG) yield a 197-bp amplimer. Two micrograms of total RNA from M-1 cells stably transfected with control vector or pcDNA3.1-prostasin-Tpsg was reverse transcribed and amplified with SYBR Green to detect products. Data are normalized to GAPDH expression and presented as fold change relative to control cells determined by the threshold cycle method. Real-time RT-PCR experiments were done in the Biomolecular Resource Facility at the University of Virginia with a ABI Prism 7900HT thermal cycler per manufacturer's instructions with SYBR Green to detect PCR products.
Cell proliferation
Cell proliferation was measured with a fluorescent DNA assay (CyQuant Cell Enumeration Assay; Molecular Probes). Stably transfected M-1 cell lines were plated into 96-well tissue culture plates (5,000 cells/well) and assayed after 24 h per manufacturer's instructions. Experiments were done five times with four or five wells per cell line, and data are expressed as means (SE).
Statistical analysis
Statistical analysis was done with SPSS 11 (SPSS, Chicago, IL). Analysis of variance was used to compare means between groups. For multiple-comparisons testing, the Student-Newman-Keuls test was applied to test for differences between groups and Dunnett's test was applied to test for differences compared with control. P < 0.05 was considered significant.
Results
Constitutive secretion, N-glycosylation, and GPI anchoring of prostasin in M-1 cells
Secretion and membrane anchoring of prostasin were studied in M-1 mouse cortical collecting duct cells. Immunoblots of concentrated conditioned medium showed that M-1 cells constitutively secrete ∼42-kDa prostasin (Fig. 1A). Detergent phase separation demonstrated that prostasin partitions exclusively into the detergent phase of Triton X-114 cell lysates, indicating that cellular prostasin resides in a membrane compartment and not in a soluble intracellular pool. In contrast, prostasin is detected only weakly when cells are extracted with 1% Triton X-100, indicating that prostasin localizes to Triton X-100-resistant membranes, most likely lipid rafts (3). Our findings differ from those of Narikiyo et al. (22), who reported that prostasin is secreted but not membrane bound in M-1 cells. This difference is probably due to cell extraction conditions in our study that solubilize GPI-anchored proteins more efficiently (3). Prostasin was secreted only from the apical surface of polarized M-1 cells grown on 0.4-μm filters (Fig. 1B). Treatment with B. thuringiensis PI-PLC released prostasin from the cell surface, and prostasin was nearly undetectable in the detergent phase after PI-PLC treatment, demonstrating that most cellular prostasin is GPI anchored (Fig. 1C). Incubation with PNGase F reduced prostasin migration by ∼10 kDa, to ∼33 kDa (expected molecular mass of catalytic domain Ile42-Ser313 is 29 kDa), consistent with N-glycosylation at both consensus sites (Fig. 1D) (41). The difference between observed and predicted molecular mass may be due to anomalous binding of SDS to the GPI anchor remnant associated with the protein after cleavage by PI-PLC (18).
Fig. 1.
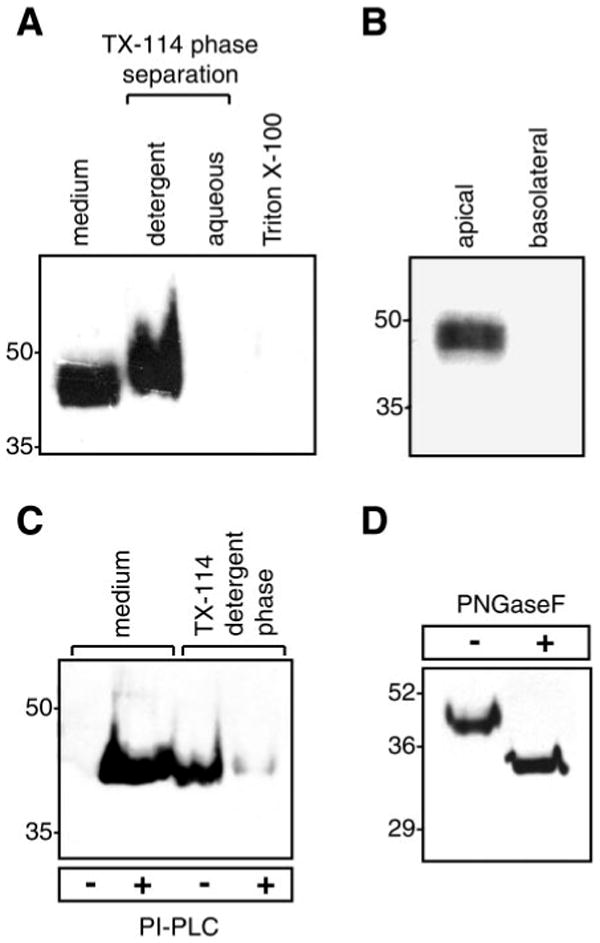
Endogenous expression and posttranslational modifications of prostasin in M-1 kidney epithelial cells. A: immunoblot of native prostasin. Proteins were extracted from M-1 cells in 1% Triton X-100 or by detergent phase separation using 2% Triton X-114 (TX-114). Samples were normalized for cell number and separated by SDS-PAGE. B: immunoblot for prostasin in apical and basolateral conditioned media from M-1 cells grown to confluence [transepithelial resistance (Rte) > 1,000 Ω × cm2] on 0.4-μm-pore filters. C: glycosylphosphatidylinositol (GPI) anchoring of prostasin. M-1 cells were treated with phosphatidylinositol-specific phospholipase C (PI-PLC) or PBS. Prostasin was assayed in conditioned medium and detergent phase cell lysates by immunoblotting. D: deglycosylation of prostasin. M-1 cells were treated with PI-PLC, concentrated conditioned medium was incubated with protein N-glycosidase F (PNGase F), and proteins were separated by SDS-PAGE and immunoblotted to detect prostasin.
Sequence determinants of prostasin secretion
We generated mouse prostasin mutants based on the following data and predictions regarding determinants of secretion and GPI anchoring (see schematic, Fig. 2). Soluble human prostasin, purified from seminal fluid, terminates in Arg323 (52). This Arg residue is conserved in mice (Arg322), rats, and humans, suggesting that prostasin may be secreted by cleavage of the COOH-terminal hydrophobic domain by a tryptic peptidase or by prostasin itself. In mouse prostasin another potential tryptic cleavage site, although not conserved in humans, lies nearby at Lys319. Using “big-PI Predictor” (http://mendel.imp.univie.ac.at/sat/gpi/gpi_server.html), a model for GPI anchor attachment site prediction, we identified Ser313 and Ser314 as the most likely sites for GPI anchor addition and Ala315 as a secondary site. To explore the role of COOH-terminal processing by prostasin or other peptidases we generated mutations of Arg322, Lys319, and the active site Ser238. To probe the role of the GPI anchor in prostasin membrane localization and secretion we generated mutations of the putative GPI anchor addition sites. The Ser313Asn/Ser314Leu double mutation was chosen to mimic rat prostasin, which has a weaker GPI anchor signal than that in mouse. Ala315Pro was selected because this mutation is predicted to abolish GPI anchoring and potentially generate non-GPI, transmembrane prostasin.
Fig. 2.
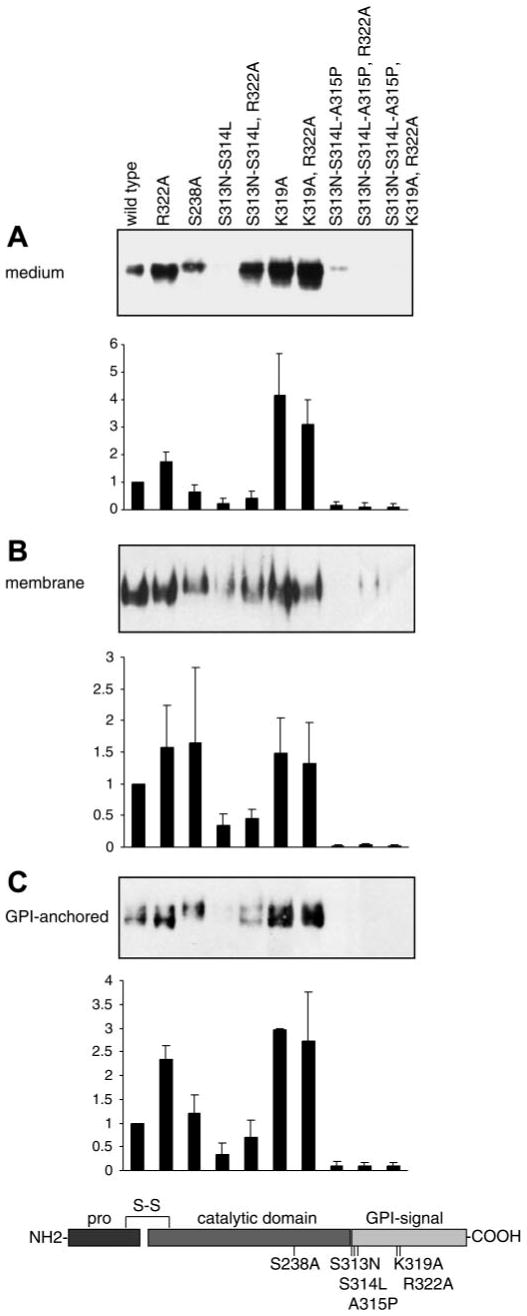
Determinants of secretion and GPI anchoring of mouse prostasin in M-1 cells. Specific amino acid mutations were introduced into prostasin to disrupt residues hypothesized to regulate GPI anchoring and secretion of prostasin as shown in the schematic diagram. Samples from M-1 cells transiently transfected with prostasin mutants were analyzed by immunoblotting and densitometry. A: conditioned medium was concentrated and normalized to load equivalent volumes; results were identical when loading was normalized to cell number. B: proteins were extracted from transfected M-1 cells by Triton X-114 detergent phase separation, precipitated with 15% trichloroacetic acid (TCA), and normalized to cell number for analysis. C: transiently transfected M-1 cells were treated with PI-PLC to release cell surface GPI-anchored proteins. Conditioned medium was concentrated, and gel loading was normalized to cell number. All experiments were done at least 3 times, and representative blots are shown. Prostasin in each compartment was quantified by densitometry and data is expressed as ratio of prostasin mutation to wild-type densitometric units. Data are expressed as means ± SD (n = 3–7).
Secretion of mutant prostasins varied strikingly (Fig. 2A). Mutation of potential tryptic cleavage sites Arg322 and Lys319, singly or in combination, significantly increased secretion compared with wild-type prostasin (P < 0.05). On the other hand, the Ser313Asn/Ser314Leu double mutant, lacking the preferred GPI attachment sites, was secreted much less efficiently than wild-type prostasin (P < 0.05). However, secretion of the triple mutant Ser313Asn/Ser314Leu/Arg322Ala, which alters both the preferred GPI attachment sites and the potential COOH-terminal tryptic cleavage site, was increased compared with the Ser313Asn/Ser314Leu double mutant (P < 0.05) and was not different from wild-type prostasin (P > 0.05). Mutating all predicted GPI anchor attachment sites with the Ser313Asn/Ser314Leu/Ala315Pro triple mutant completely blocked secretion, which is not rescued by the combined mutation Ser313Asn/Ser314Leu/Ala315Pro/Lys319Ala/Arg322Ala. The catalytically inactive prostasin mutant, Ser238Ala, was secreted with an efficiency similar to that of wild-type prostasin (P > 0.05). The active site mutant, Ser238Ala, has a slightly higher molecular mass by SDS-PAGE than wild-type prostasin. Endogenous prostasin is expressed in insufficient quantity to be visualized on these immunoblots but was visible on prolonged exposures and comigrated with prostasin expressed by transient transfection (data not shown).
Sequence determinants for GPI anchoring of prostasin
We used Triton X-114 detergent phase separation and PI-PLC treatment of transfected cells to assess effects of these mutants on cellular prostasin. Native and mutant prostasin partitioned into the detergent phase in proportion to the amount detected in conditioned medium (Fig. 2B). Incubation of transfected cells with PI-PLC released prostasin into medium in proportion to the amount of prostasin secreted into conditioned medium and in detergent phase of cell lysates for each mutation (Fig. 2C). As observed in the secretion studies, mutations of Arg322 and Lys319 increased PI-PLC-releasable prostasin compared with wild type (P < 0.05). After incubation with PI-PLC, the Ser313Asn/Ser314Leu mutant was weakly detectable in the medium but Ser313Asn/Ser314Leu/Ala315Pro was undetectable. Thus the Ser313Ser314Ala315 motif is necessary for GPI anchoring of prostasin. The combined mutation Ser313Asn/Ser314Leu/Arg322Ala partially restored susceptibility to PI-PLC compared with Ser313Asn/Ser314Leu (P < 0.05), as with prostasin secretion. These data are consistent with our predictions that Ser313 and Ser314 are preferred sites for GPI anchor attachment and that Ala315 may serve as an additional site for GPI anchor attachment. The Ser313/Ser314/Ala315 sequence is conserved in humans, suggesting that GPI anchor addition also occurs at these sites in human prostasin. Membrane association and GPI anchoring of catalytically inactive prostasin (Ser238Ala) were similar to wild type (P > 0.05). As in conditioned medium, the active site mutant Ser238Ala has a slightly higher molecular mass by SDS-PAGE than wild-type prostasin in detergent phase cell lysates and after PI-PLC treatment.
No differences were observed between M-1 kidney cells and MLE-12 lung cells (which do not express prostasin natively) in secretion and GPI anchoring of these prostasin mutants (Fig. 3). Thus mechanisms of prostasin membrane anchoring and secretion are likely to be similar in kidney collecting duct epithelium and lung alveolar type II cells.
Fig. 3.
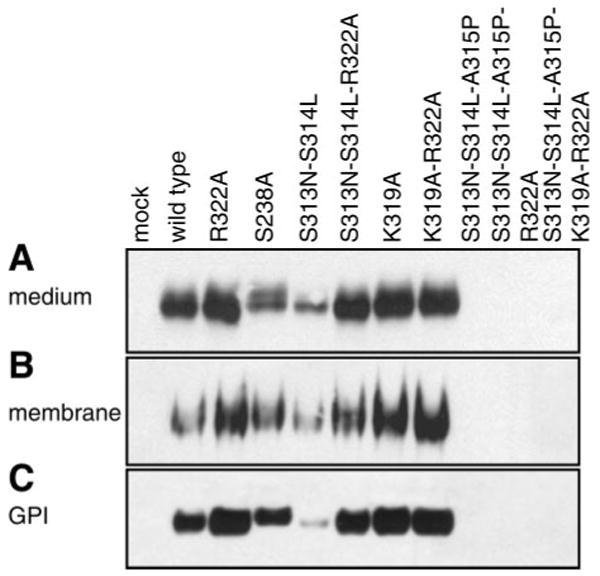
Determinants of secretion and GPI anchoring of prostasin in mouse lung epithelial cells. MLE-12 cells were transiently transfected with mutated prostasins, separated by SDS-PAGE, and immunoblotted for prostasin. A: conditioned medium was concentrated and normalized to load equivalent volumes; results were identical when loading was normalized to cell number. B: proteins were extracted from MLE-12 cells by Triton X-114 detergent phase separation and precipitated with 15% TCA for analysis. Samples were normalized to cell number for immunoblotting. C: MLE-12 cells were treated with PI-PLC to release cell surface GPI-anchored proteins. Conditioned medium was concentrated, and gel loading was normalized to cell number. All experiments were done at least 3 times, and representative blots are shown. Mock transfected MLE-12 cells (no plasmid) do not express prostasin.
Constitutive secretion of prostasin depends on Gpld1
Our mutagenesis studies demonstrate that prostasin secretion does not depend on potential tryptic cleavage sites Arg322 or Lys319 in the COOH-terminal domain. Furthermore, the relationship between membrane, GPI-anchored, and secreted prostasin is proportional, suggesting that secretion may occur through shedding of GPI-anchored prostasin. Mammalian Gpld1 is implicated in physiological shedding of some GPI-anchored proteins (21). We used RNA interference to assess the effect of Gpld1 inhibition on constitutive secretion of endogenous prostasin in M-1 cells. As shown in Fig. 4, transfection with Gpld1-specific siRNA, but not control siRNA, decreased secretion of endogenously expressed prostasin to 26 ± 18% of control [mean (SD), n = 3; P < 0.05] on day 6 after transfection. In addition, in M-1 cells stably transfected to overexpress prostasin or empty pcDNA3.1 vector, inhibiting Gpld1 with siRNA increased the amount of prostasin that is releasable by PI-PLC, consistent with accumulation of GPI-anchored prostasin at the cell surface in these cells (Fig. 4B). Semiquantitative PCR using GAPDH as a housekeeping gene control showed a similar time course in reduction of Gpld1 transcripts and demonstrated that the decline in prostasin secretion was not due to a general inhibition of transcription (Fig. 4C).
Fig. 4.
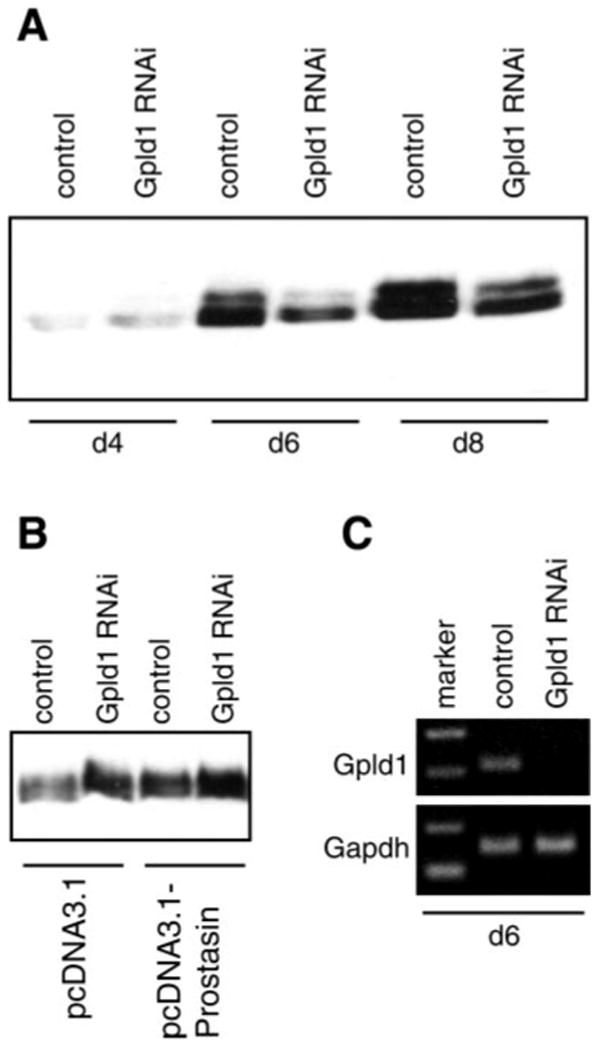
GPI-specific phospholipase D (Gpld1)-dependent secretion of prostasin. M-1 cells were transfected with Gpld1-specific small interfering RNA (siRNA) or control siRNA. A: conditioned medium was collected at 48-h intervals, concentrated, normalized to cell number, and assayed for prostasin by immunoblotting. There was no difference in cell proliferation between Gpld1- and control siRNA-transfected cells (data not shown). Relative levels of prostasin in conditioned medium were quantified by densitometry and expressed as means ± SD. Transfection with Gpld1 siRNA reduced prostasin in conditioned medium by 74% (P < 0.05; n = 3). B: M-1 cells stably transfected to express prostasin or empty vector (see Experimental Procedures and Fig. 5 for details) were transfected with Gpld1 or control siRNA and treated with PI-PLC. Medium was assayed for prostasin by immunoblotting. Inhibition of Gpld1 increased PI-PLC-releasable prostasin. C: expression of Gpld1 was assessed by semiquantitative RT-PCR relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control 6 days after transfection with Gpld1 and control siRNA. d4, d6, d8, 4, 6, and 8 days after transfection.
Characterization of stably transfected M-1 cell lines
M-1 cells express prostasin and have a phenotype consistent with serine peptidase regulation of ENaC (19). To investigate specific effects of prostasin activity and the COOH-terminal domain on epithelial monolayer function, we stably transfected M-1 cells to generate polyclonal lines overexpressing empty pcDNA3.1 vector as control, wild-type prostasin (mPR-wt), active site mutant prostasin-Ser238Ala (mPR-S238A), or chimeric prostasin-Tpsg (mPR-Tpsg). In the prostasin-Tpsg chimera, the COOH-terminal domain of prostasin (amino acids 288–339) including the putative GPI anchor signal (amino acids 313–339) was replaced with the COOH-terminal transmembrane peptide domain (amino acids 264–311) of mouse tryptase-γ (also termed transmembrane tryptase; gene name Tpsg) (Fig. 5A). Prostasin-Tpsg is predicted to generate transmembrane but not GPI-anchored prostasin based on analysis of mouse tryptase-γ, which is expressed as a transmembrane protein in mast cells and is not known to be secreted (48).
Immunoblots of cell lysates confirmed that stably transfected cell lines overexpress prostasins compared with the empty vector-transfected control cell line. mPR-wt and mPR-S238A are sensitive to shedding by PI-PLC (Fig. 5B). mPR-Tpsg is detergent extractable, migrates slightly faster on SDS-PAGE than endogenous prostasin, and resists shedding by PI-PLC (Fig. 5B). Immunoblots of apical and basal medium show that prostasin is secreted apically but not basolaterally in mPR-wt, mPR-S238A, and mPR-Tpsg cell lines (Fig. 5C). Endogenous prostasin was not detected in medium in these samples because of insufficient quantity. Apical or basolateral domain-selective cell surface labeling with biotin confirmed polarized expression of membrane-associated prostasin and its variants at the apical surface of M-1 cells (Fig. 5D). Prostasin was not detectable in control samples from cells exposed to PBS instead of sulfo-NHS-biotin (data not shown). Levels of prostasin expression were similar in media and cell lysates in stably transfected cell lines overexpressing prostasin variants. Cell proliferation was greater in mPR-wt cells but not in the other cell lines compared with control (142% of control; P < 0.05).
We unexpectedly observed increased secretion of immunoreactive prostasin after transfection with pcDNA3.1-prostasin-Tpsg. Prostasin secreted from prostasin-Tpsg-expressing cells migrates similarly to wild-type prostasin on SDS-PAGE (Fig. 5), suggesting that secretion may occur in M-1 cells by processing within the Tpsg domain to remove the transmembrane peptide or, alternatively, that endogenous prostasin expression or secretion is increased. To exclude the possibility that prostasin-Tpsg induces native prostasin, we performed quantitative RT-PCR using primers specific for endogenous prostasin transcripts or prostasin-Tpsg transcripts. In prostasin-Tpsg-expressing cells, there was no difference in endogenous prostasin transcripts [0.90 (SE 0.06)-fold change compared with control vector-transfected cells; n = 3] and a large increase in prostasin-Tpsg transcripts [754 (SE 25)-fold change compared with control vector-transfected cells; n = 3]. To further assess processing of prostasin-Tpsg and wild-type prostasin, we transfected M-1 cells with pcDNA3.1-prostasin-FLAG (internal epitope tag) and pcDNA4-prostasin-Tpsg-V5 (COOH-terminal epitope tag) plasmids. In M-1 cells transfected with pcDNA3.1-prostasin-FLAG, immunoblotting of conditioned medium for prostasin or immunoprecipitation of conditioned medium with anti-FLAG M2 antibody and immunoblotting for prostasin revealed a single prostasin band (mPR-FLAG-wt) (Fig. 5E). Cell surface biotinylation and immunoblotting with M2 antibody also revealed mPR-FLAG-wt expression, as well as a background band that was also present in control vector-transfected cells. The presence of FLAG epitope in both membrane and secreted prostasin-FLAG protein indicates that the COOH terminal of secreted prostasin lies on the carboxy side of amino acid 310. This observation demonstrates that prostasin secretion occurs via processing between residues 311 and 339 (e.g., within the GPI signal sequence). Immunoblots demonstrate that the COOH-terminal V5 epitope is intact in cell lysates, but not in the secreted prostasin, from M-1 cells overexpressing prostasin-Tpsg-V5 protein (mPR-Tpsg-V5) (Fig. 5F). Secreted prostasin-Tpsg-V5 migrates similarly to prostasin-Tpsg and wild-type prostasin on SDS-PAGE. Finally, immunoblots of medium from M-1 cells endogenously secreting or overexpressing wild-type prostasin do not reveal any smaller bands that could suggest that an alternative biological pathway for prostasin secretion exists in these cells. In aggregate, these data are consistent with secretion of prostasin-Tpsg through processing within the Tpsg domain, rather than an alternative biologically relevant pathway for secretion of wild-type prostasin. Because prostasin was secreted similarly into the apical compartment in both mPR-wt and mPR-Tpsg cells, these cell lines should be a useful model to study the effect of the type of membrane anchor on prostasin function.
Regulation of transepithelial resistance and current by prostasin: role of GPI anchoring
Stable overexpression of prostasin and prostasin mutants in M-1 cells resulted in significant differences in monolayer bioelectric properties as shown in Fig. 6. Vector control, mPR-S238A, and mPR-Tpsg cell lines developed high Rte and were impermeable to FITC-inulin by day 7 of culture (Fig. 6, A and C). In contrast, at day 7 mPR-wt cell monolayers exhibited significantly lower Rte than other cell lines and were permeable to inulin. In all of these stably transfected cell lines, Rte and Vte reached plateaus at days 15–18, and at this time mPR-wt cell monolayers had also become impermeable to FITC-inulin. However, maximal Rte remained significantly different between cell lines. Overexpression of wild-type prostasin resulted in significantly lower-Rte monolayers compared with the other cell lines. In contrast, mPR-Tpsg cells exhibited markedly higher Rte than the other cell lines. Rte was not different between mPR-S238A and vector control cell monolayers.
Fig. 6.
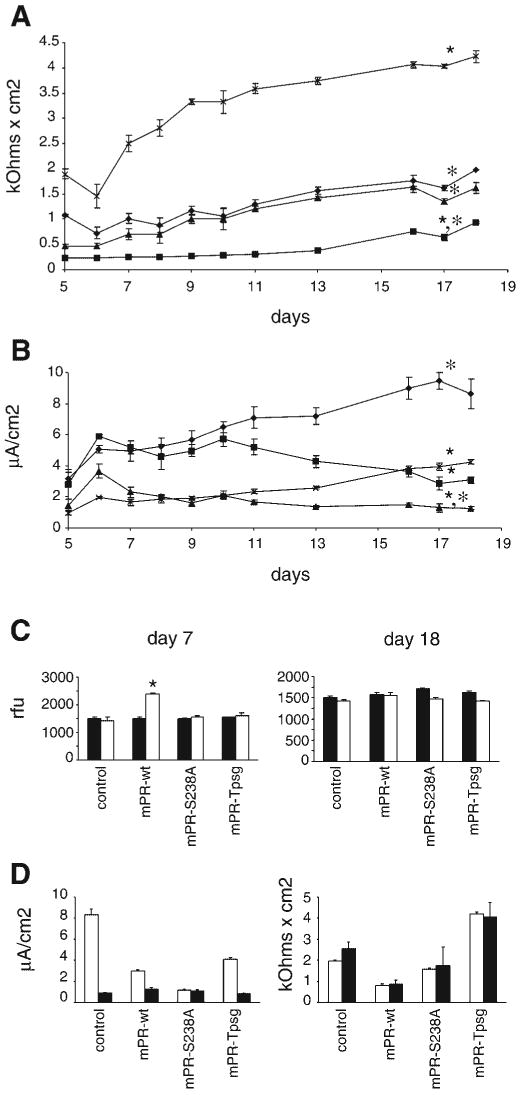
Effect of GPI anchoring and catalytic activity of prostasin on transepithelial resistance, current, and permeability. Stably transfected M-1 cell lines (described in Results and Fig. 5) were grown on 0.4-μm polycarbonate filters. Rte and potential difference were measured with an epithelial voltohmeter, and equivalent short-circuit current (Ieq) was calculated by Ohm's law. Data are expressed as means ± SE from 5 independent experiments with triplicate wells. Data were analyzed when resistance and potential difference were stable and monolayers of all cell lines were impermeable to inulin (data in C). A: time course of Rte in stable transfected M-1 cells overexpressing prostasin variants (♦, mPR-ctrl; ■, mPR-wt; ▲, mPR-S238A; ×, mPR-Tpsg). ★P < 0.05 compared with mPR-ctrl; *P < 0.05 compared with mPR-Tpsg. B: time course of Ieq in stably transfected M-1 cell lines. ★P < 0.05 compared with mPR-ctrl; *P < 0.05 compared with mPR-Tpsg. C: paracellular permeability of M-1 cell lines at days 7 and 18 of culture was assayed by measuring apical-to-basal flux of FITC-inulin (20 μg/ml) over 24 h. Data are expressed as means ± SE relative fluorescence units (rfu) in basal medium at 0 (filled bars) and 24 (open bars) h. Mean apical and basal fluorescence at time 0 were ∼23,000 and ∼1,600 rfu, respectively. ★P < 0.05 for comparison between 0 and 24 h. D: effect of amiloride on Ieq (left) and Rte (right) in stably transfected M-1 cell lines. Ten micromolar amiloride was applied to the apical surface of cell monolayers on days 18–19 of culture. Ieq and Rte were measured 45–60 min after exposure to amiloride. Open and filled bars depict baseline and postamiloride measurements, respectively. Data are expressed as means ± SE from 5 independent experiments with triplicate wells. Exposure to amiloride abolished the differences in Ieq between cell lines and reduced Ieq to similarly low current (P > 0.05). There was no difference in baseline and postamiloride Rte in any of these cell lines (P > 0.05).
In control M-1 cells (stably transfected with empty vector), Ieq was quantitatively similar to Ieq reported by Helms et al. (13) in untransfected M-1 cells grown under similar culture conditions. Similar to data reported in untransfected M-1 cells, treatment of the control cell line with 10 μM amiloride applied to the apical surface to inhibit ENaC reduced Ieq to 11% of baseline without significantly altering Rte (Fig. 6D). Ieq was compared in the stably transfected M-1 cell lines when Rte and Vte had reached stable plateaus and monolayers were impermeable to inulin. Ieq was significantly lower in mPR-S238A than control cell monolayers (Fig. 6B). In mPR-wt and mPR-Tpsg monolayers, Ieq did not differ but were less than Ieq of vector-transfected control cells. As shown in Fig. 6D, exposure to 10 μM amiloride decreased Ieq markedly to 0.8 –1.2 μA/cm2, and Ieq did not differ between the stably transfected cell lines (P > 0.05). Amiloride did not affect Rte, and the differences in Rte between cell lines persisted after treatment with amiloride (P < 0.05).
Discussion
Membrane serine peptidases activate ENaC in kidney and lung epithelial cells and thereby may provide an important extracellular pathway for physiologically and pharmacologically regulating ion and water transport in these organs. Several reports have modeled the effects of serine peptidases on ENaC function using broad-spectrum peptidases and inhibitors such as trypsin and aprotinin (19, 26, 34), but few studies have specifically addressed the roles of the peptidases that are hypothesized to activate ENaC in vivo. In these studies, we report the novel observation that prostasin specifically regulates transepithelial resistance, current, and permeability through mechanisms that depend on the GPI anchor as well as peptidase activity. Furthermore, we demonstrate that constitutive secretion of prostasin depends on Gpld1 in these cells, rather than on proteolytic cleavage of the COOH-terminal domain. These findings suggest that prostasin function may be regulated through cell-specific variations in prostasin localization and secretion that are coordinated by the COOH-terminal domain and GPI anchor.
In human prostate epithelial cells, prostasin is GPI anchored and constitutively secreted (7, 52). Sequencing of human prostasin from seminal fluid suggests that soluble prostasin is generated by cleavage at Arg323 (Arg322 in mouse prostasin) by tryptic peptidases to remove the COOH-terminal hydrophobic domain (52). No alternative mRNA transcripts have been identified to explain termination at Arg323 (41, 43, 51). In contrast to prostate, cultured airway epithelial cells do not secrete prostasin efficiently (36), suggesting that mechanisms for prostasin secretion may be cell specific. Using site-directed mutagenesis in M-1 kidney and MLE-12 lung epithelial cells, we found that changes in secreted, membrane-associated, and GPI-anchored prostasin are proportional for each prostasin mutant (Figs. 2 and 3). Inactivation of predicted sites for GPI anchor addition (Ser313, Ser314, Ala315) abolished prostasin secretion. In contrast, mutations of basic residues in the COOH-terminal domain (Lys319, Arg322) that are putative targets for cleavage by tryptic peptidases did not block secretion of prostasin. These observations suggest that shedding of GPI-anchored prostasin is the major pathway for prostasin secretion in these cells, rather than tryptic proteolysis of the COOH-terminal domain at Lys319 or Arg322 as may occur in prostate.
To investigate this possibility further we specifically inhibited Gpld1 expression by siRNA in M-1 cells, which endogenously express and secrete prostasin. Inhibiting Gpld1 markedly reduced constitutive secretion of prostasin, leading to accumulation of GPI-anchored prostasin on the cell membrane (Fig. 4). These independent, complementary data from mutagenesis and Gpld1 inhibition support our hypothesis that GPI anchor addition and subsequent processing of the GPI anchor by Gpld1 are the primary mechanism for secretion of prostasin in M-1 kidney epithelial cells. In these cells, prostasin secretion did not require basic amino acids in the COOH-terminal domain that could be targeted by trypsinlike peptidases. Although Iwashita et al. (16) reported that broad-spectrum serine peptidase inhibitors (aprotinin and nafamostat) diminish prostasin secretion in M-1 cells, our mutagenesis data suggest that secretion of prostasin is not directly due to serine peptidase cleavage at Arg322 or Lys319 in these cells. The effect of aprotinin and nafamostat on prostasin secretion could be due to cleavage at another site or to an indirect mechanism. GPI transamidation at Ser313, Ser314, or Ala315 (as may occur in kidney cells) would remove the COOH-terminal peptide of prostasin including Arg322, whereas proteolytic cleavage at Arg322 (as may occur in prostate cells) would remove the hydrophobic COOH-terminal sequence and inactivate the GPI anchor signal (37). Currently a unique example, folate receptor-β is expressed as a GPI-anchored protein but is secreted by proteolysis of the COOH-terminal GPI anchor signal without GPI modification (45). A similar pathway could explain differences in prostasin processing between M-1 kidney cells and prostate epithelial cells. Alternatively, these differences could be due to variation between mouse and human or possibly as a phenomenon of immortalized or transfected cells. However, these alternatives are considered less likely based on the high sequence and functional homology of prostasin across species and our data implicating Gpld1 in secretion of endogenous prostasin in M-1 cells.
We observed a marked decrease in cellular prostasin in the Ser313Ser314Ala315 mutations. This finding suggests that disruption of sites of GPI addition in prostasin (despite leaving the COOH-terminal hydrophobic domain intact) may lead to degradation rather than secretion or transmembrane peptide anchoring. Mutagenesis studies of Q7b and acetylcholinesterase report that mutations of the ω-site or other sites in the GPI signal peptide that prevent cleavage lead to retention of unprocessed protein in the endoplasmic reticulum (ER) and rapid degradation (8, 10). A recent study reports that the ER translocon can differentiate between hydrophobic sequences of a GPI signal and a transmembrane peptide such that ineffectual GPI addition results in degradation rather than transmembrane anchoring (9). Thus our observations are consistent with recent studies of GPI anchor signal requirements and extend the generalizability of these conclusions. Interestingly, mutation of Lys319 and/or Arg322 increased GPI-anchored prostasin and partially rescued GPI anchoring and secretion of the ω-site mutations. This finding was not predicted from big-PI scores for these mutations (wild-type 9.74, Arg322Ala 9.12, Lys319Ala 10.23, Lys319Ala/Arg322Ala 7.85, Ser313Asn/Ser314Leu −2.09, Ser313Asn/Ser314Leu/Arg322Ala −1.49, and Ser313Asn/Ser314Leu/Lys319Ala/Arg322Ala −2.93; positive score predicts GPI anchoring). Variations in big-PI score were attributable to differences in spacer hydrophobicity score (data not shown). In this predictive model, thresholds for spacer hydrophobicity are not highly validated at present because of the paucity of experimental data from various GPI-anchored proteins (12).
The active site mutant (Ser238Ala) has a slightly higher molecular mass by SDS-PAGE than wild-type prostasin (Fig. 2). This difference persists after treatment with PI-PLC, in which the COOH termini should be identical, suggesting that the difference is due to retention of the ∼1.5-kDa NH2-terminal propeptide. This observation suggests the possibility that prostasin may autoactivate. However, other investigators report that recombinant, soluble proprostasin could not be activated with active, soluble (COOH-terminally truncated) prostasin (33). This result predicts that autoactivation, if it occurs, may require prostasin in a membrane-bound rather than soluble form.
Existing data on regulation of ENaC by prostasin conflict regarding the contributions of membrane-anchored and secreted prostasin to this function, leading some investigators to speculate that membrane localization is critical and others to suggest that prostasin is secreted in order to exert paracrine effects on ENaC (22, 39, 43). In addition, most studies have used soluble trypsin, a broad-spectrum serine peptidase that is nonphysiological in the kidney and lung, to model peptidase activation of ENaC in cell culture or in vivo (19, 26, 34). The specific effect of GPI anchoring on prostasin function has not been reported previously. To study the effects of prostasin more specifically, we generated stably transfected M-1 cell lines overexpressing empty vector as a control, wild-type prostasin, prostasin active site mutant (prostasin-Ser238Ala), or chimeric transmembrane (not GPI)-anchored prostasin-Tpsg and assessed effects on monolayer function. As shown in Figs. 5 and 6, these cell lines express prostasin at the apical surface, develop high Rte, and exhibit predominantly amiloride-sensitive Ieq. The prostasin-Tpsg chimera is resistant to PI-PLC, consistent with our prediction that it would be a transmembrane protein rather than GPI anchored. Unexpectedly, prostasin-Tpsg was also secreted and migrated similarly to wild-type prostasin on SDS-PAGE (Fig. 5). Additional control experiments using the prostasin-FLAG construct demonstrate that secretion of wild-type prostasin occurs through processing of the COOH-terminal carboxyl to residue 310 (e.g., within the GPI anchor signal sequence). Immunoblots of detergent cell lysates and medium from M-1 cells expressing COOH-terminally tagged prostasin-Tpsg-V5 show that the COOH terminal of prostasin-Tpsg is intact in the membrane form but removed from the secreted protein. Predicted molecular masses of the proprostasin domain (amino acids 31–287) and the Tpsg domain of prostasin-Tpsg chimera are 27,584 and 5,043 Da, respectively; soluble prostasin migrates near its predicted molecular mass at ∼33,000 Da after deglycosylation (Fig. 1). Thus, if immunoreactive prostasin secreted from mPR-Tpsg cells were generated by cleavage of chimeric molecule within the prostasin domain, then the prostasin band would migrate at least ∼5,000 Da smaller than wild-type prostasin. Similar migration of prostasins secreted from control, mPR-wt, and mPR-Tpsg cells suggests that secreted prostasin-Tpsg is generated by processing within the Tspg domain of the chimera, rather than through an alternative, biological pathway for prostasin secretion. Since prostasin membrane expression and apical secretion were similar in mPR-wt, mPR-S238A, and mPR-Tpsg cells, these cell lines should serve as a useful model to study the effect of the type of membrane anchor on prostasin function.
In EVOM studies of monolayer bioelectric properties, we observed striking differences in transepithelial current and resistance between M-1 cells stably overexpressing prostasin variants (Fig. 6). Prior studies in cortical collecting duct cells reported that treatment with exogenous trypsin, without prior exposure to serine peptidase inhibitor, does not activate ENaC, suggesting that activation of ENaC by trypsinlike peptidases is maximal under basal conditions (19, 43). In our studies, overexpression of active site mutant prostasin-Ser238Ala reduced Ieq by 78% compared with control, without altering Rte. This finding is similar to inhibition of ENaC with amiloride and thus is consistent with a dominant-negative effect of the prostasin active site mutant on ENaC activation. The magnitude of this effect is also comparable to the effect of apical aprotinin (a broad-spectrum serine peptidase inhibitor) on M-1 cells (60% reduction in Ieq) (19) and to prostasin knockdown in cystic fibrosis airway epithelial cells (74% reduction in Ieq) (36). Although other ENaC-activating serine peptidases have been identified in cortical collecting duct cells (42), the quantitatively similar effects of prostasin-Ser238Ala and aprotinin suggest that prostasin activity is critical for activating basal sodium transport in M-1 cells. These data support the idea that prostasin could be a modifier gene or a novel target for therapeutic inhibition in salt-sensitive hypertension. Although we expected that prostasin overexpression would not further augment Ieq, we found that stable overexpression of prostasin decreased Ieq compared with control cells line stably transfected with empty vector. Since EVOM studies measure net transepithelial current, these data suggest the possibility that long-term overexpression of prostasin in polarized M-1 cells could downregulate ENaC, modify expression or function of other regulators of ENaC, or alter expression or function of other transcellular or paracellular channels and thereby decrease Ieq. Ieq was reduced to similarly low currents in all the prostasin-variant M-1 cell lines after exposure to amiloride, indicating that most of the transepithelial current in these cell lines is sensitive to amiloride and suggesting that observed differences in Ieq do reflect differences in ENaC activity. It is unlikely that the differences in Ieq between M-1 cell lines are simply an artifact of transfection since we used polyclonal cell lines to minimize random effects of integration, verified that vector-transfected control cells had Ieq and Rte similar to published data in untransfected M-1 cells, and observed a dominant-negative phenotype on amiloride-sensitive current in cells overexpressing the active site mutation of prostasin. Additional studies are required to identify the specific molecular mechanisms through which prostasin overexpression leads to these differences in Ieq.
Interestingly, Rte was markedly lower in mPR-wt cells compared with vector-transfected control cells, whereas Rte was significantly increased in mPR-Tpsg cells. Furthermore, development of impermeability to inulin was delayed in the mPR-wt cell monolayers. This difference in Rte is not explained solely by differences in ENaC channel activity since mPR-wt and mPR-Tpsg monolayers have similar amiloride-sensitive Ieq and treatment with amiloride did not significantly alter Rte (Fig. 6). Since amiloride nearly abolishes Ieq without altering Rte in M-1 cells, paracellular resistance must exceed transcellular resistance in these cells. The large opposing effects of wild-type prostasin and prostasin-Tpsg overexpression on monolayer resistance and differences in inulin permeability suggest that prostasin may regulate paracellular permeability and resistance through a mechanism that depends on the GPI anchor or COOH-terminal domain. Liu et al. (19) reported that apical aprotinin treatment reduces Rte by ∼60% in M-1 cells, and Swystun et al. (34) reported that apical trypsin treatment increases Rte in airway epithelial cells. In both studies the authors speculated that apical serine peptidases regulate paracellular resistance and tight junction function, in addition to regulating sodium channels.
Our observations of the effects of prostasin on Rte and inulin permeability provide more specific evidence that prostasin regulates tight junction function, in addition to its reported role in regulating ENaC activity. Since amiloride diminished Ieq to similar levels without affecting Rte in our M-1 cell lines, prostasin regulation of tight junctions may be independent from prostasin effects on ENaC function. Using skin-specific prostasin gene inactivation in mice, Leyvraz and colleagues (17) recently reported that skin permeability is increased in mice deficient in prostasin. Rte in cultured keratinocytes from these mice was not reported in this study. The observations of Leyvraz et al. are consistent with the effect on paracellular permeability and Rte that we observed with prostasin-Tpsg (transmembrane, not GPI, anchored) and are opposite to the effect of wild-type (GPI anchored) prostasin. Potential explanations for this difference could be cell-specific differences in COOH-terminal posttranslational processing of prostasin or differences in tight junction components between epidermal cells and kidney cortical collecting duct cells. Recent studies have demonstrated that tight junctions, the primary determinant of paracellular resistance, form functionally heterogeneous, ion-selective paracellular channels (29, 35). Tight junction function, including ion selectivity and paracellular resistance, depends on type and combination of subunits such as claudins (a family of >24 members) and can be altered by changes in subunit assembly or posttranslational modifications, including by serine peptidases (29, 30, 35, 50). The different effects of wild-type prostasin and prostasin-Tpsg on Rte and paracellular permeability in M-1 cells indicate that GPI anchoring is critical to prostasin function. Since in both mPR-wt and mPR-Tpsg cells prostasin was secreted similarly into the apical compartment, it is likely that these differences in Rte are due to membrane-anchored prostasin rather than soluble prostasin. Alternatively, differences in the COOH-terminal of soluble wild-type prostasin and prostasin-Tpsg could alter prostasin activity since cleavage of GPI anchor can lead to conformational changes affecting protein activity or immunogenicity (31). In summary, our data on Rte and Ieq in these prostasin-variant M-1 cell lines suggest that prostasin function depends on the GPI anchor in addition to peptidase activity and that prostasin plays a key role in regulating epithelial tight junction function in addition to activating ENaC.
These studies suggest that GPI anchoring regulates prostasin function at the cell surface, perhaps by controlling access to prostasin substrates and inhibitors. GPI anchoring directs proteins to restricted membrane microdomains (lipid rafts) that may promote clustering of proteins into functional complexes (20). In addition, the GPI anchor and local lipid ordering may induce conformational changes that can affect protein function (31). The tight junction proteins occludin and ZO-1 and a fraction of ENaC channels have been localized to lipid rafts (14, 24). The present studies report the novel observations that membrane localization and secretion of prostasin depend on the GPI anchor in kidney and lung epithelial cells, that prostasin is an important regulator of tight junction and ENaC function in M-1 cells, and that GPI anchoring is critical for prostasin regulation of ENaC and tight junctions. These findings suggest that prostasin function could be dynamically regulated in a cell-specific manner through Gpld1-mediated GPI anchor cleavage and secretion. Similar pathways may regulate function of other GPI-anchored serine peptidases. Further studies are necessary to identify the substrates of prostasin that lead to modulation of ENaC activity and tight junction function.
Acknowledgments
The authors gratefully acknowledge Dr. Hans Folkesson for helpful discussions and thoughtful reading of this manuscript and Dr. Yongde Bao in the Biomolecular Resource Facility at the University of Virginia for assistance with quantitative PCR.
Grants
These studies were supported by National Heart, Lung, and Blood Institute Grants HL-67920 (G. M. Verghese) and HL-024136 (G. H. Caughey), the Department of Veterans Affairs, and the Northern California Institute for Research and Education.
References
- 1.Bauvois B. Transmembrane proteases in cell growth and invasion: new contributors to angiogenesis? Oncogene. 2004;23:317–329. doi: 10.1038/sj.onc.1207124. [DOI] [PubMed] [Google Scholar]
- 2.Bhagwandin V, Hau L, Clair JMS, Wolters P, Caughey G. Structure and activity of human pancreasin, a novel tryptic serine peptidase expressed primarily by the pancreas. J Biol Chem. 2003;278:3363–3371. doi: 10.1074/jbc.M209353200. [DOI] [PubMed] [Google Scholar]
- 3.Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 4.Caughey GH, Raymond WW, Blount JL, Hau LW, Pallaoro M, Wolters PJ, Verghese GM. Characterization of human gamma-tryptases, novel members of the chromosome 16p mast cell tryptase and prostasin gene families. J Immunol. 2000;164:6566–6575. doi: 10.4049/jimmunol.164.12.6566. [DOI] [PubMed] [Google Scholar]
- 5.Chen LM, Chai KX. Prostasin serine protease inhibits breast cancer invasiveness and is transcriptionally regulated by promoter DNA methylation. Int J Cancer. 2002;97:323–329. doi: 10.1002/ijc.1601. [DOI] [PubMed] [Google Scholar]
- 6.Chen LM, Hodge GB, Guarda LA, Welch JL, Greenberg NM, Chai KX. Down-regulation of prostasin serine protease: a potential invasion suppressor in prostate cancer. Prostate. 2001;48:93–103. doi: 10.1002/pros.1085. [DOI] [PubMed] [Google Scholar]
- 7.Chen LM, Skinner ML, Kauffman SW, Chao J, Chao L, Thaler CD, Chai KX. Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. J Biol Chem. 2001;276:21434–21442. doi: 10.1074/jbc.M011423200. [DOI] [PubMed] [Google Scholar]
- 8.Coussen F, Ayon A, Le Goff A, Leroy J, Massoulie J, Bon S. Addition of a glycophosphatidylinositol to acetylcholinesterase. Processing, degradation, and secretion. J Biol Chem. 2001;276:27881–27892. doi: 10.1074/jbc.M010817200. [DOI] [PubMed] [Google Scholar]
- 9.Dalley JA, Bulleid NJ. The endoplasmic reticulum (ER) translocon can differentiate between hydrophobic sequences allowing signals for glycosylphosphatidylinositol anchor addition to be fully translocated into the ER lumen. J Biol Chem. 2003;278:51749–51757. doi: 10.1074/jbc.M303978200. [DOI] [PubMed] [Google Scholar]
- 10.Delahunty MD, Stafford FJ, Yuan LC, Shaz D, Bonifacino JS. Uncleaved signals for glycosylphosphatidylinositol anchoring cause retention of precursor proteins in the endoplasmic reticulum. J Biol Chem. 1993;268:12017–12027. [PubMed] [Google Scholar]
- 11.Donaldson SH, Hirsh A, Li DC, Holloway G, Chao J, Boucher RC, Gabriel SE. Regulation of the epithelial sodium channel by serine proteases in human airways. J Biol Chem. 2002;277:8338–8345. doi: 10.1074/jbc.M105044200. [DOI] [PubMed] [Google Scholar]
- 12.Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292:741–758. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- 13.Helms MN, Fejes-Toth G, Naray-Fejes-Toth A. Hormone-regulated transepithelial Na+ transport in mammalian CCD cells requires SGK1 expression. Am J Physiol Renal Physiol. 2003;284:F480–F487. doi: 10.1152/ajprenal.00299.2002. [DOI] [PubMed] [Google Scholar]
- 14.Hill WG, An B, Johnson JP. Endogenously expressed epithelial sodium channel is present in lipid rafts in A6 cells. J Biol Chem. 2002;277:33541–33544. doi: 10.1074/jbc.C200309200. [DOI] [PubMed] [Google Scholar]
- 15.Hooper J, Bowen N, Marshall H, Cullen L, Sood R, Daniels R, Stuttgen M, Normyle J, Higgs D, Kastner D, Ogbourne S, Pera M, Jazwinska E, Antalis T. Localization, expression and genomic structure of the gene encoding the human serine protease testisin. Biochim Biophys Acta. 2000;1492:63–71. doi: 10.1016/s0167-4781(00)00071-3. [DOI] [PubMed] [Google Scholar]
- 16.Iwashita K, Kitamura K, Narikiyo T, Adachi M, Shiraishi N, Miyoshi T, Nagano J, Tuyen do G, Nonoguchi H, Tomita K. Inhibition of prostasin secretion by serine protease inhibitors in the kidney. J Am Soc Nephrol. 2003;14:11–16. doi: 10.1097/01.asn.0000043900.39397.48. [DOI] [PubMed] [Google Scholar]
- 17.Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, Breiden B, Sandhoff K, Hummler E. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170:487–496. doi: 10.1083/jcb.200501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Littlewood GM, Hooper NM, Turner AJ. Ectoenzymes of the kidney microvillar membrane. Affinity purification, characterization and localization of the phospholipase C-solubilized form of renal dipeptidase. Biochem J. 1989;257:361–367. doi: 10.1042/bj2570361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Hering-Smith KS, Schiro FR, Hamm LL. Serine protease activity in m-1 cortical collecting duct cells. Hypertension. 2002;39:860–864. doi: 10.1161/01.hyp.0000013055.48885.8d. [DOI] [PubMed] [Google Scholar]
- 20.Lucero H, Robbins P. Lipid rafts-protein association and the regulation of protein activity. Arch Biochem Biophys. 2004;426:208–224. doi: 10.1016/j.abb.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 21.Metz CN, Brunner G, Choi-Muira NH, Nguyen H, Gabrilove J, Caras IW, Altszuler N, Rifkin DB, Wilson EL, Davitz MA. Release of GPI-anchored membrane proteins by a cell-associated GPI-specific phospholipase D. EMBO J. 1994;13:1741–1751. doi: 10.1002/j.1460-2075.1994.tb06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, Shiraishi N, Nonoguchi H, Chen LM, Chai KX, Chao J, Tomita K. Regulation of prostasin by aldosterone in the kidney. J Clin Invest. 2002;109:401–408. doi: 10.1172/JCI13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH, Antalis TM. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22:237–258. doi: 10.1023/a:1023003616848. [DOI] [PubMed] [Google Scholar]
- 24.Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113:1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- 25.Oliviero O, Castagna A, Guarini P, Chiecchi L, Gherardo S, Pizzolo F, Corrocher R, Righetti P. Urinary prostasin: a candidate marker of epithelial sodium channel activation in humans. Hypertension. 2005;46:683–688. doi: 10.1161/01.HYP.0000184108.12155.6b. [DOI] [PubMed] [Google Scholar]
- 26.Planes C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay M, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1099–L1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- 27.Rossier BC. The epithelial sodium channel: activation by membrane-bound serine proteases. Proc Am Thorac Soc. 2004;1:4–9. doi: 10.1513/pats.2306007. [DOI] [PubMed] [Google Scholar]
- 28.Schild L, Kellenberger S. Structure function relationships of ENaC and its role in sodium handling. Adv Exp Med Biol. 2001;502:305–314. doi: 10.1007/978-1-4757-3401-0_20. [DOI] [PubMed] [Google Scholar]
- 29.Schneeberger E, Lynch R. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 30.Schneeberger EE. Claudins form ion-selective channels in the paracellular pathway. Focus on “Claudin extracellular domains determine paracellular charge selectively and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol. 2003;284:C1331–C1333. doi: 10.1152/ajpcell.00037.2003. [DOI] [PubMed] [Google Scholar]
- 31.Sharom F, Radeva G. GPI-anchored protein cleavage in the regulation of transmembrane signals. Subcell Biochem. 2004;37:285–315. doi: 10.1007/978-1-4757-5806-1_9. [DOI] [PubMed] [Google Scholar]
- 32.Shaw-Smith CJ, Coffey AJ, Leversha M, Freeman TC, Bentley DR, Walters JR. Characterisation of a novel murine intestinal serine protease, DISP. Biochim Biophys Acta. 2000;1490:131–136. doi: 10.1016/s0167-4781(99)00226-2. [DOI] [PubMed] [Google Scholar]
- 33.Shipway A, Danahay H, Williams JA, Tully DC, Backes BJ, Harris JL. Biochemical characterization of prostasin, a channel activating protease. Biochem Biophys Res Commun. 2004;324:953–963. doi: 10.1016/j.bbrc.2004.09.123. [DOI] [PubMed] [Google Scholar]
- 34.Swystun V, Chen L, Factor P, Siroky B, Bell PD, Matalon S. Apical trypsin increases ion transport and resistance by a phospholipase C-dependent rise of Ca2+ Am J Physiol Lung Cell Mol Physiol. 2005;288:L820–L830. doi: 10.1152/ajplung.00396.2004. [DOI] [PubMed] [Google Scholar]
- 35.Tang V, Goodenough D. Paracellular ion channel at the tight junction. Biophys J. 2003;84:1660–1673. doi: 10.1016/S0006-3495(03)74975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong Z, Illek B, Bhagwandin VJ, Verghese GM, Caughey GH. Prostasin, a membrane-anchored serine peptidase, regulates sodium currents in JME/CF15 cells, a cystic fibrosis airway epithelial cell line. Am J Physiol Lung Cell Mol Physiol. 2004;287:L928–L935. doi: 10.1152/ajplung.00160.2004. [DOI] [PubMed] [Google Scholar]
- 37.Udenfriend S, Kodukula K. How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu Rev Biochem. 1995;64:563–591. doi: 10.1146/annurev.bi.64.070195.003023. [DOI] [PubMed] [Google Scholar]
- 38.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 39.Vallet V, Pfister C, Loffing J, Rossier BC. Cell-surface expression of the channel activating protease xCAP-1 is required for activation of ENaC in the Xenopus oocyte. J Am Soc Nephrol. 2002;13:588–594. doi: 10.1681/ASN.V133588. [DOI] [PubMed] [Google Scholar]
- 40.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 41.Verghese GM, Tong ZY, Bhagwandin V, Caughey GH. Mouse prostasin gene structure, promoter analysis, and restricted expression in lung and kidney. Am J Respir Cell Mol Biol. 2004;30:519–529. doi: 10.1165/rcmb.2003-0251OC. [DOI] [PubMed] [Google Scholar]
- 42.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J Gen Physiol. 2002;120:191–201. doi: 10.1085/jgp.20028598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vuagniaux G, Vallet V, Jaeger NF, Pfister C, Bens M, Farman N, Courtois-Coutry N, Vandewalle A, Rossier BC, Hummler E. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J Am Soc Nephrol. 2000;11:828–834. doi: 10.1681/ASN.V115828. [DOI] [PubMed] [Google Scholar]
- 44.Wang C, Chao J, Chao L. Adenovirus-mediated human prostasin gene delivery is linked to increased aldosterone production and hypertension in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1031–R1036. doi: 10.1152/ajpregu.00660.2002. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Shen F, Yan W, Wu M, Ratnam M. Proteolysis of the carboxyl-terminal GPI signal independent of GPI modification as a mechanism for selective protein secretion. Biochemistry. 1997;36:14583–14592. doi: 10.1021/bi970845w. [DOI] [PubMed] [Google Scholar]
- 46.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 47.Wong G, Li L, Madhusudhan M, Krilis S, Gurish M, Rothenberg M, Sali A, Stevens R. Tryptase 4, a new member of the chromosome 17 family of mouse serine proteases. J Biol Chem. 2001;276:20648–20658. doi: 10.1074/jbc.M010422200. [DOI] [PubMed] [Google Scholar]
- 48.Wong GW, Foster PS, Yasuda S, Qi JC, Mahalingam S, Mellor EA, Katsoulotos G, Li L, Boyce JA, Krilis SA, Stevens RL. Biochemical and functional characterization of human transmembrane tryptase (TMT)/tryptase gamma. TMT is an exocytosed mast cell protease that induces airway hyperresponsiveness in vivo via an IL-13/IL-4Ralpha/STAT6-dependent pathway. J Biol Chem. 2002;277:41906–41915. doi: 10.1074/jbc.M205868200. [DOI] [PubMed] [Google Scholar]
- 49.Wong GW, Yasuda S, Madhusudhan MS, Li L, Yang Y, Krilis SA, Sali A, Stevens RL. Human tryptase epsilon (PRSS22), a new member of the chromosome 16p13.3 family of human serine proteases expressed in airway epithelial cells. J Biol Chem. 2001;276:49169–49182. doi: 10.1074/jbc.M108677200. [DOI] [PubMed] [Google Scholar]
- 50.Yu AS. Claudins and epithelial paracellular transport: the end of the beginning. Curr Opin Nephrol Hypertens. 2003;12:503–509. doi: 10.1097/00041552-200309000-00004. [DOI] [PubMed] [Google Scholar]
- 51.Yu JX, Chao L, Chao J. Molecular cloning, tissue-specific expression, and cellular localization of human prostasin mRNA. J Biol Chem. 1995;270:13483–13489. doi: 10.1074/jbc.270.22.13483. [DOI] [PubMed] [Google Scholar]
- 52.Yu JX, Chao L, Chao J. Prostasin is a novel human serine proteinase from seminal fluid. Purification, tissue distribution, and localization in prostate gland. J Biol Chem. 1994;269:18843–18848. [PubMed] [Google Scholar]
- 53.Yu JX, Chao L, Ward DC, Chao J. Structure and chromosomal localization of the human prostasin (PRSS8) gene. Genomics. 1996;32:334–340. doi: 10.1006/geno.1996.0127. [DOI] [PubMed] [Google Scholar]