

Abstract
Loss-of-function mutations in the DJ-1 gene account for an autosomal recessive form of Parkinson’s disease (PD). To investigate the physiological functions of DJ-1 in vivo, we generated DJ-1 knockout (DJ-1-/-) mice. Younger (< 1year) DJ-1 -/- mice were hypoactive and had mild gait abnormalities. Older DJ-1-/-, however, showed decreased bodyweight and grip strength, and more severe gait irregularities compared to wild-type littermates. The basal level of extracellular dopamine, evoked dopamine release and dopamine receptor D2 sensitivity appeared normal in the striatum of DJ-1-/- mice, which was consistent with similar results between DJ-1-/- and controls in behavioral paradigms specific for the dopaminergic system. An examination of spinal cord, nerve and muscle tissues failed to identify any pathological changes that were consistent with the noted motor deficits. Taken together, our findings suggest that loss of DJ-1 leads to progressive behavioral changes without significant alterations in nigrostriatal dopaminergic and spinal motor systems.
Keywords: DJ-1, knockout mouse, Parkinson’s disease, dopamine, striatum, spinal cord, muscle, motor behavior
INTRODUCTION
Parkinson’s disease (PD) is the 2nd most common neurodegenerative disease with clinical features of resting tremor, rigidity, and bradykinesia, and pathological hallmarks of depletion of dopaminergic neurons in the substantia nigra and formation of intraneuronal Lewy bodies (Dauer and Przedborski, 2003). Although the majority of cases are sporadic, dominantly inherited mutations in α-synuclein and LRRK2, and recessively inherited mutations in parkin, DJ-1, and PINK1 have been linked to rare familial forms of PD. DJ-1-associated PD likely manifests via a loss of function mechanism as the first reported mutations were either a large genomic deletion resulting in an absence of protein and mRNA, or a L166P missense mutation that resulted in rapid protein degradation (Bonifati et al., 2003; Miller et al., 2003). The presence of DJ-1 protein within tau inclusions in post-mortem tissue taken from patients diagnosed with tauopathies suggests that DJ-1 loss may lead to a heterogenous catalogue of behavioral dysfunctions (Rizzu et al., 2004).
Four independent reports using different lines of DJ-1 knockout (DJ-1-/-) mice have suggested novel roles for DJ-1 in the modulation of dopaminergic neurons. DJ-1-/- mice were hypoactive in the open field, more susceptible to neuronal death from treatment of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), contained lower levels of presynaptic dopamine transporter (DAT) and had impaired dopamine receptor D2-mediated activities and long term depression (LTD) in acute striatal slices (Chen et al., 2005; Goldberg et al., 2005; Kim et al., 2005; Manning-Bog et al., 2007). These reports, however, are not entirely consistent with each other regarding either behavioral dysfunction or dopaminergic transmission, and only include data on the mice up to 12 months of age. A single recent report on aged DJ-1 mouse cohorts suggests that, with the exception of hypoactivity, no changes in basic motor activity are present in DJ-1-/- animals, and nigral pathology is normal (Yamaguchi and Shen, 2007).
To clarify the role of DJ-1 in neurotransmission, and identify any pathways linking the protein to PD, we generated DJ-1-/- mice by deleting the first coding exon (exon 2), and characterized the behavior and pathology until end stage (28 months). We chose behavioral paradigms suited to detecting alterations in both motor and dopaminergic function (Fleming and Chesselet, 2006), and extended previous studies by examining dopaminergic transmission in vivo using microdialysis. In addition to DJ-1-/- mice, other mouse models generated with genetic manipulations in parkin and α-synuclein have identified behavioral deficits consistent with alterations in the nigrostriatal pathway without accompanying nigral cell loss (Fleming et al., 2004; Goldberg et al., 2003). We now report that DJ-1 deficiency leads to progressive motor deficits without any obvious pathological changes in either the nigrostriatal system or spinal motor system and muscles.
MATERIALS AND METHODS
Generation of DJ-1-/- Mice
DNA fragments spanning exons 1-7 of DJ-1 were isolated from a mouse genomic phage library (Stratagene, CA). Targeting vectors were constructed through replacement of the second exon of DJ-1 with a neomycin resistance gene. The neomycin resistance gene was flanked by a 1.7 kb EcoRI-BamH1 left arm fragment and a 5.0 kb right arm consisting of a 3.3 kb Nhe1-EcoRI fragment joined to a 1.7 kb EcoRI-SacI fragment. Linearized targeting vector was transfected by electroporation into mouse ES cells derived from the 129/SvJ strain. G418-resistant colonies were selected and screened by Southern blot for homologous recombination with a 5′ and 3′ external probe. Positive cells were injected into C57BL6/J blastocysts to generate chimeras which were then mated with C57BL6/J wild-type mice to confirm germline transmission. All mice used in this study were F1 or F2 mice bred from founders with the hybrid background. Mice were genotyped by PCR using genomic DNA extracted from tail biopsy. The multi-primer system was designed with an intron 1 (5′-GCTGAAACTCTGCCATGTGA-3′) forward primer, an exon 2 (5′-CCATCTCCTCTGCTCCTTTG-3′) reverse primer and a neomycin (5′-GTTATCTGGGCGCTTGTCAT-3′) reverse primer. Mice were housed in a 12 h light/dark cycle and fed a regular diet ad libitum. The experimental protocols used are in accordance with guidelines set by each Insitutional Animal Care and Use Committee at The Johns Hopkins University, the National Institute on Drug Abuse, and the National Insititute on Aging.
Immunocytochemistry
Mice anaesthetized with an intraperitoneal injection of 15% chloral hydrate were sacrificed by transcardial perfusion with PBS (pH 7.4), followed by 4% paraformaldehyde in PBS (pH 7.4). Brains were removed and postfixed in the same fixative overnight before transferring to 30% sucrose for cryopreservation. Brains were then frozen and 30 μm coronal sections were obtained using a sliding microtome.
Muscle histology
Fresh skeletal muscle biopsies were obtained prior to fixation and frozen by immersion in isopentane cooled in liquid nitrogen. Sections (10μm) were cut and processed for NADH, ATPase (pH4.4 and 9.4), and acetlycholinesterase-silver using methods described previously (Dubowitz and Brooke, 1973; Sheehan and Hrapchak, 1987).
Electron Microscopy
Anaesthetized mice were perfused with 4% paraformaldehyde in 0.1M PBS (pH7.4). Ventral root, dorsal root and sciatic nerve were postfixed with 4% paraformaldehyde with 2% glutaraldehyde in 0.1M PBS (pH7.4). Subsequently, tissues were washed again in PBS, dehydrated and embedded in EPON. Thick (1 μm) and thin (100 nm) sections were obtained and stained respectively with toluidine blue and lead citrate/uranyl acetate.
Antibodies
Polyclonal antibodies for DJ-1 (Abcam, Cambridge, MA) and tyrosine hydroxylase (Pel Freeze, Rogers, AK) and monoclonal antibodies for ß-actin (Sigma, St. Louis, MO) and ubiquitin (Covance, Princeton, NJ) were used.
Stereology
Every fourth section in the substantia nigra, and every section containing locus coeruleus (LC) was processed for tyrosine hydroxylase (TH) immunohistochemically. The number of TH-positive cells was assessed using an unbiased stereological procedure with an optical fractionator (Stereo Investigator 6 from MicroBrightField).
Bodyweight
Mice were weighed every two weeks.
Behavioral Studies
Locomotor Activity
Mice were placed in a 40cm × 40cm × 38cm plexiglass cubicle, and spontaneous horizontal and vertical locomotor activity was measured by the Flex-Field activity system (San Diego Instruments, CA). Four 20W phosphorescent lamps shielded in a light box and placed in a dark room opposite of the testing area provided a diffuse source of light. Mice were habituated to the testing room and light level for 1 hour prior to testing. Flex-Field software was used to trace and quantify the movement of the mouse in the unit as number of beam breaks per second. The number of entries to the central zone of the open field was also recorded.
Rotarod
The mouse was placed onto a rotating rod with auto acceleration from 0 rpm to 40 rpm in 4 minutes (Rotor-Rod system, SD Instrument). The length of time the mouse was able to stay on the rotating rod was recorded. For the motor learning test, four trials were run for each mouse in a two-hour interval per day for three sequential days.
Plus Maze Task
An elevated (70cm) plus maze was used for testing anxiety in an environment similar to the locomotor activity task. Each subject was placed in the center of the apparatus and allowed to explore freely for 5 minutes. The number of arm entries made to the open and closed arms as well as time spent in each area was recorded.
Prepulse Inhibition
Prepulse inhibition of acoustic startle response was measured using the SR lab system (San Diego Instruments, San Diego, CA) as described by previously (Paylor and Crawley, 1997) following standard procedures (Holmes et al., 2001; Paylor and Crawley, 1997; Ralph-Williams et al., 2002). Briefly, for each test session a mouse was first placed in a cylindrical Plexiglas tube inside the chamber where it remained, undisturbed, for 5 minutes with 70 dB background noise prior to the start of the test session. The 70 dB white noise continued throughout the test session. Each test session consisted of seven trial types. One trial consisted of a 120 dB white noise burst lasting 40 ms. Five prepulse trials with varying prepulse stimulus levels (74, 78, 82, 86, and 90) lasting 20 ms followed 100 ms later by the onset of the 120 dB startle stimulus. The final trial type contained no stimulus sound, thereby providing a measure of baseline activity levels. Each session consisted of 6 blocks of seven trials in pseudorandom order, such that within every block each trial occurred only once. The intertrial interval range was 10-20 seconds averaging 15 seconds across the session. Startle responses were recorded for 65 ms (1 ms bins) beginning at the onset of the startle stimulus. Peak startle amplitude detected during the sampling window was used as the dependent measure.
Percent inhibition of acoustic startle response was calculated as follows: 100 - [(prepulse stimulus plus startle stimulus trials / acoustic startle stimulus only trials) × 100]. Consequently, a high percentage value indicates good suppression of the startle response, while a low percentage value indicates a deficit in startle response inhibition.
Pole Test
Animals were placed facing the top of a 50 cm length metal pole (1 cm diameter) wrapped in absorbent tape, with the base of the pole in the home cage, and the time taken for the mice to turn around the pole and move head first into the cage was measured. Mice were trained to learn the task for two days before a three trial experiment, and a cut-off time of 120 sec was used. Falls and slips were given a maximum score.
Gait Analysis
Gait movements were tracked on a 70 cm length/13 cm wide cardboard box with 10 cm walls lined with 3 mm white chromatography paper (Whatman). A single fluorescent light source was placed at the starting point of the walkway and the first 20 cm of the walkway was uncovered to encourage the mice to walk to the end of the track where the homecage was placed. Mice forepaws and hindpaws were dipped into non-toxic red and black tempera paints (Utrecht, Cranbury, NJ) respectively, and the mice were then placed on the track in the lit end, and allowed to traverse the walkway. Mice, age and weight-matched, were trained on the apparatus for several days until they were capable of performing the task without frequent stoppages or runs. Four major criteria were assessed in the test: (1) stride (the distance between two consecutive hindpaw footprints); (2) (3) hind-base and fore-base widths (separation distance between hind/forepaw footprints of the same stride); (4) matching (used to measure the uniformity of stride). The placement of hindprints on foreprints was used to assess stride uniformity. When the center of the hindprint perfectly overlapped with the center of the foreprint, a value of zero was recorded. Distances separating footprints that did not overlap were recorded from the center of the hindprint to the center of the foreprint. Weight matched cohorts of mice were used to avoid any weight bias on the test, and all the mice weighed less than 40 grams.
L-Dopa Responsive Gait Analysis
Mice were administered intraperitoneally with 12.5 mg/kg of benzeraside hydrochloride (Sigma, St. Louis, MO) followed by 50 mg/kg L-DOPA (Sigma, St. Louis, MO)) given with a twenty minute delay between injections. Thirty minutes after the second injection, gait indices were measured. Both drugs were dissolved in 0.9% saline.
Grip Strength
Forelimb grip strength was measured using a grip strength meter (Chatillon Instruments, Greensboro, NC). Mice were lowered by their tail and allowed to reach out to the metal bar on the meter. Mice were slowly pulled away from the meter at a horizontal plane until the forepaws released from the bar. The force applied to the bar at the point of release was recorded digitally by the meter. Three measurements were made per mouse and averaged to determine mouse grip strength.
Microdialysis
Mice were anesthetized with sodium pentobarbital (75 mg/kg i.p.) and placed in a stereotaxic frame adapted for the mouse (David Kopf, Topanga, CA). The skull was exposed and a small hole was drilled for insertion of a guide cannula (CMA 11, CMA microdialysis, North Chelmsford, MA) into the dorsal striatum according to an atlas (Slotnick and Leonard, 1975). The coordinates, relative to bregma, were: antero-posterior: +0.4 mm, lateral: -2.1 mm, ventral: -2.1 mm for the dorsal striatum. These coordinates were calculated for a 2 mm membrane length probe. The cannulae were fixed to the skull with cranioplastic cement (Geristore, Santa Maria, CA) and further secured with dental acrylic. After surgery, mice were individually housed in the colony room. Four days after surgery, the microdialysis probe (CMA/11, CMA microdialysis, North Chelmsford, MA) was connected to the dialysis system, flushed with artificial cerebrospinal fluid (aCSF: 145 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 0.25 mM ascorbic acid, and 5.4 mM D-glucose, pH 7.2), and slowly inserted into the guide cannula. The dialysis system consisted of FEP tubing (CMA microdialysis) that connected the probe to a 1 ml gastight syringe (Hamilton Co., Reno, NV) mounted on a microdialysis pump (CMA/102) through a quartz-lined, low resistance swivel (375/D/22QM, Instech, Plymouth Meeting, PA). After probe insertion, the mouse was placed in the dialysis chamber with food and water freely available, and the probe perfused overnight with aCSF at a flow rate of 0.6 μl/min. The next morning, the perfusion syringes were loaded with fresh aCSF and probes were allowed to equilibrate for an additional 1 hour prior to the commencement of experiments.
For no net flux experiments, five different concentrations of DA (0, 5, 10, 20 and 40 nM) in aCSF were perfused in random order through the dialysis probe. Each DA concentration was perfused for 30 minutes, and then 2×10 minute samples were collected. Following completion of no net flux experiments, normal aCSF was again perfused through the probe for 30 min. allowing for a period of equilibration. Five baseline samples were then collected followed by changing the perfusion buffer to aCSF containing 60 mM KCl (NaCl concentration was reduced accordingly to maintain osmolarity) and three 10 min. samples were obtained. Additional groups of animals were perfused with the D2 receptor agonist, quinpirole (0, 0.1, 0.3 and 1 μM) and dialysate samples were collected at 10 min. intervals.
After the experiment, mice were sacrificed by a sodium pentobarbital overdose and their brains were removed and frozen on dry ice. Brains were stored at -80° C until histological analysis. Probe location was assessed on 20 μm serial cryostat sections. Only the data from the animals with correct probe placements were used for subsequent statistical analysis.
DA was determined by HPLC coupled to electrochemical detection. The chromatographic system consisted of a CMA/200 refrigerated microinjector (CMA microdialysis, North Chelmsford, MA) a BAS PM-80 pump (BAS, West Lafayette, IN) and a BAS LC-4C amperometric detector. The mobile phase (0.15 M sodium phosphate, 2.24 mM sodium octanesulfonic acid, 0.94 mM EDTA and 11% methanol (vol/vol), adjusted to pH 5.0) was filtered through a 0.22 μm nylon filter and degassed by a BAS on-line degasser and pumped through the system at a flow rate of 0.6 ml/min. DA was separated in a BAS C18 column (100 mm × 2.0 mm × 3 μm) and detected on a glassy carbon working electrode at an oxidation potential of +700 mV vs. Ag/AgCl. Dialysate DA levels were quantified by external standard curve calibration, using peak heights for quantification. All the reagents used for the mobile phase were analytical grade. Under these conditions, retention time for DA was 3 min and the limit of detection was 0.25 nM.
Data Analysis
In the no net flux studies, the dialysate DA concentrations (DAout) obtained during the perfusion of the different DA concentrations (DAin) were used to construct a linear equation for each animal. The net flux of DA through the probe (DAin- DAout) was plotted against DAin. Extracellular DA levels (DAext) and the extraction fraction of the probe (Ed) were determined as DAext equals the DAin concentration at which there is no net flux of DA through the probe (DAin-DAout= 0). The extraction fraction (Ed) is the slope of the regression line and has been shown to provide an estimate of changes in transporter-mediated DA uptake (Justice, 1993; Smith et al., 1994). Significant differences between the two genotypes in the above parameters were assessed by a student t-test.
For subsequent experiments, the mean baseline dialysate DA level was determined for each animal and drug-evoked DA concentrations were transformed to percent of this baseline. Since the percent transformed data did not follow a normal distribution, non-parametric analyses were performed. The effect of the different treatments within each genotype was assessed by the Friedman repeated measures analysis of variance on ranks. Posthoc analysis was performed using the Student-Newman-Keuls test. Between genotypes comparisons were performed using a Mann-Whitney’s U-test.
Rotating Disk Electrode Voltammetry
Mice were sacrificed by decapitation and the brains rapidly removed. Using a brain matrix, the olfactory tubercles were removed to enable visualization of the diagonal bands and a blade placed at the level at which the diagonal bands intersect. The second blade was positioned 1 mm anterior. The section was placed on an ice-cold Petri dish and dissected to obtain the caudate-putamen. The average wet weight of tissue obtained was 4.7±0.3 mg (n=11). The dissected tissue was washed twice with 500 μl of ice-cold buffer (124mM NaCl, 3mM KCl, 1.24 mM KH2PO4, 1.30 mM MgSO4, 2.50 mM CaCl2, 26.0 mM NaHCO3, and 10 mM D-glucose, pH 7.4 gassed with 95% O2 / 5% CO2) and then pre-incubated in 500 μl of room temperature buffer for 4 min. The tissue was then minced with two scalpel blades and placed into the incubation chamber containing 300 μl of buffer. The incubation chamber consisted in a well with an auxiliary platinum electrode and a Ag/AgCl reference electrode, fitted in a metal block and kept at 37°C by a circulating water bath. The tissue was stirred by a rotating glassy carbon working electrode (Pine Instruments Inc., Grove City, PA), operating at a rotation rate of 4000-rpm, and continuously oxygenated by directing a stream of 95% O2/ 5% CO2 to the surface of the incubation buffer.
Voltammetric measurements were made using a potentiostat (LC 4B, Bioanalytical Systems Inc., West Lafayette, IN) with the time constant changed to 200 msec. The applied potential (Eapp) was +450 mV versus Ag-AgCl. Data acquisition and analysis were performed using Origin data acquisition software (MicroCal, Northampton, MA). After a 15 min stabilization period, DA [DA0] was added to the tissue suspension (6 μl addition volume).at a final concentration of 2 μM DA. The clearance of DA was monitored as the decay in the oxidation current. The initial velocity (v) of transport was estimated as the slope of the initial 20 seconds of the DA clearance curve (pmoles/sec), and corrected for the amount of tissue (grams wet weight) added to the incubation chamber. The average clearance rates for each genotype were compared with a Student’s t-test.
Other Statistical Analysis
All statistical analysis was performed with StatView 5.0.1. Unpaired two-tailed Student’s t-tests were used when genotype was the only grouping variable. Analysis of variance was used with multiple grouping variables, and Spearman rank correlations were used to assess the strength of the relationship between two variables. Prepulse inhibition data were analyzed using two-way ANOVAs with repeated measures. Post hoc comparisons, when required, were made using Newman-Keuls and simple effect tests.
RESULTS
Generation of DJ-1-/- Mice
Targeted deletion of mouse DJ-1 was accomplished by replacing exon 2 with a neomycin resistance gene cassette (Supplemental Fig 1A). Successful homologous recombination of the targeted exon 2 disruption was confirmed by Southern blot (Supplemental Fig 1B) and Western blot (Supplemental Fig 1C).
DJ-1-/- Mice are Hypoactive and Display Gait Abnormalities
All offspring from DJ-1 heterozygous mating were viable, fertile, and lacked obvious developmental abnormalities. From birth until 12 months, there were no significant differences in bodyweights between genotype (F (1, 64) = 0.412, p > 0.5). After 1 year of age, however, DJ-1-/- males had significantly lower bodyweight (F (1, 168) = 33.446, p < 0.0001) that was progressive with age (F (11, 104) = 3.54, p < 0.005) (Fig 1A). Similar changes of bodyweights were observed in female cohorts as well (data not shown). Since clinical symptoms of patients harboring DJ-1 mutations include bradykinesia and heightened anxiety, we examined spontaneous ambulatory activity in four age groups (2, 5, and 14, and 23 months) of mice in a motion sensitive Flex-Field activity system (San Diego Instruments). ANOVA test indicated a significant (genotype F (1, 47) = 8.287, p < 0.01) but non-progressive (genotype × age F (1, 47) = 0.596, p > 0.4) hypokinesia in DJ-1-/- mice at five and fourteen months of age (Figure 1B) that was independent of any anxiety deficits detectable by either the elevated plus maze (n=11, p > 0.1) or thigmotaxis associated with the activity system (data not shown). Because altered postural gait and shortened stride are prominent characteristics of many neurological disorders including PD, we analyzed the stride patterns of age and weight matched male mice, and measured the displacement of the hind paws and the uniformity of strides (matching). Mouse cohorts were chosen with matching weights to discount the normal stride differences that would associate with alterations in bodyweight. Two month-old DJ-1-/- mice displayed changes in both stride uniformity (DJ-1+/+: 0.66 ± 0.05 cm, n = 10; DJ-1-/-: 0.92 ± 0.07 cm, n = 8; p < 0.005) and hind base displacement (DJ-1+/+: 2.35 ± 0.08 cm; DJ-1-/-: 2.60 ± 0.05 cm; p < 0.02), but showed similar stride lengths (DJ-1+/+: 5.65 ± 0.19 cm, n = 8; DJ-1-/-: 5.35 ± 0.20 cm, n = 8; p > 0.2) (Figure 2A). Strikingly, twenty-four month old DJ-1-/- showed a significant decrease in stride length (DJ-1+/+: 5.90 ± 0.16 cm, n = 8; DJ-1-/-: 5.35 ± 0.18 cm, n = 10; p < 0.05) in addition to deficits in stride uniformity (DJ-1+/+: 1.04 ± 0.08 cm; DJ-1-/-: 1.40 ± 0.12 cm; p < 0.02) and hind base displacement (DJ-1+/+: 2.91 ± 0.06 cm; DJ-1-/-: 3.18 ± 0.07 cm; p < 0.01, Figures 2B-2C). ANOVA reveals an age times genotype correlation at the limit of significance (F (3, 367) = 3.739, p = 0.05) for stride, indicating that the motor deficit was progressive. While both activity and gait tests were performed with only male mice, nearly identical results were observed with similarly aged female mice (data not shown), suggesting that the phenotypes were not sex-specific.
Figure 1. Bodyweight changes and hypoactivity in DJ-1-/- mice.

(A) After 12 months, DJ-1-/- mice fail to gain weight like their wild-type littermates. (B) DJ-1-/- exhibit a non-progressive hypoactivity at five and fourteen months. All data are means ± SEM. * represents p < 0.05.
Figure 2. Progressive gait abnormalities in DJ-1-/- mice.

(A) Stride patterns of typical 24-month old mice with hind paws (black) and fore paws (red) shown. (B) At 2 months, DJ-1-/- show a greater hind base displacement, and more stride matching differences. (C) At 24 months, DJ-1-/- mice have shorter strides, greater displacement between hind paws, and greater stride matching differences. * represents p < 0.05.
Older DJ-1-/- Mice Show Decreased Grip Strength but Enhanced Motor Learning
Since muscle wasting and peripheral nerve damage accompanies weight loss in several different mouse models for motor neuron disease and muscular dystrophy (Gabellini et al., 2006; Wong et al., 1995), and two mutations in DJ-1 co-segregate with motor neuron disease (Annesi et al., 2005), we examined spinal motor system and muscles, and tested the grip strength of the forelimbs of mice. At 16 months, DJ-1-/- mice showed a significant decrease in foregrip strength (DJ-1+/+: 0.55 ± 0.027 N; DJ-1-/-: 0.47 ± 0.027 N; n = 16 each; p < 0.05) that was absent in juvenile mice (DJ-1+/+: 0.55 ± 0.049 N; DJ-1-/-: 0.56 ± 0.027 N; n = 9 each; p > 0.5) (Figure 3A) despite an absence of gross pathological differences between skeletal muscle and peripheral nerves (Figures 3B-3C). The number of motor neurons remaining in 24-month-old mice was also similar between the DJ-1-/- mice and their control littermates (data not shown). We then used the accelerating rotarod to determine whether the gait abnormalities in the DJ-1-/- mice were linked to deficits in motor coordination. No differences were detected between mouse genotypes with one-day rotarod testing (ANOVA: F (1, 29) = 0.087, p > 0.7). Similarly, multiple trial testing conducted over three days on the rotarod, which is a cerebellar and striatum-dependent motor learning task (Lalonde et al., 1995) revealed no differences between genotypes (Figure 3D: F(3, 58) = 0.817, p > 0.3). Surprisingly, DJ-1-/- (F (1, 28) = 5.362, p < 0.03) display enhanced motor learning over the 3-day test period relative to wild-type littermates (F (1, 30) = 3.012, p > 0.09). The wild-type mice show only a 34.6% increase from the first testing day to the third, compared to the 62.3% increase detected in the DJ-1-/- mice. To establish if bodyweight changes may influence the differences seen in the grip strength and the motor learning we used a Spearman rank correlation, and found no significant correlation between bodyweight on grip strength (p > 0.08, ρ=0.313) or rotarod latency (p > 0.3, ρ= -0.16)
Figure 3. Grip strength loss and enhanced motor learning.

(A) A loss in forearm grip strength was observed in DJ-1-/- mice at 16 months that were absent at 2 months of age. (B) No differences were observed in gross muscle pathology with NADH, esterase, or hematoxylin and eosin (H & E) staining. (C) No evidence of peripheral nerve degeneration. (D) Enhanced motor learning in the DJ-1-/- mice over a three day set of trials. * represents p < 0.05.
DJ-1-/- Mice Show Altered Response to L-DOPA administration
As the dopamine synthesis precursor L-DOPA remains the standard treatment of both sporadic and familial PD, we hypothesized that the stride deficit in DJ-1-/- mice may be rescued through L-DOPA administration. 24-month old mice were given two injections of drug (12.5 mg/kg bodyweight benzeraside hydrochloride followed by 50 mg/kg bodyweight L-DOPA) with the injections spaced 20 minutes apart. Twenty minutes after the second injection, gait indices were measured again for both groups of mice (Figure 4). While DJ-1+/+ showed a significant increase in stride length (n =12; F (1, 172) = 10.820, p < 0.005) after drug treatment, the DJ-1-/- mice did not (n =10; F (1, 174) = 0.986, p > 0.3), so while the stride deficit was not rescued, the acute L-DOPA injection appears to result in differential responses between DJ-1+/+ and DJ-1-/- mice.
Figure 4. DJ-1-/- mice show a lower response to L-DOPA administration.
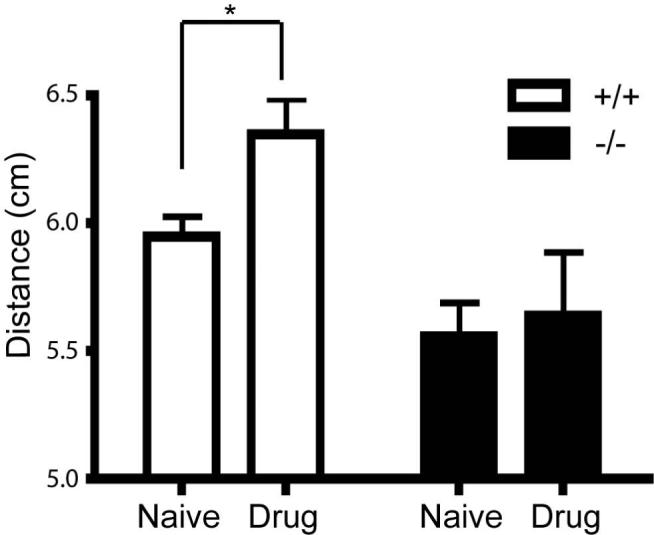
Gait indices of DJ-1-/- and wild-type control mice at 24 months were measured following injection of L-DOPA. All data are means ± SEM. * represents p < 0.05.
Basal dopamine levels and D2 autoreceptor sensitivity is normal
To investigate in vivo dynamics of dopaminergic transmission in the mice, we first measured extracellular dopamine (DA) function in the dorsal striatum of six-month old male DJ-1-/- mice and their wild-type littermates using the no-net flux microdialysis technique in vivo (Chefer et al., 2006; Parsons and Justice, 1994). No differences between genotypes were observed in the extracellular DA concentrations (DJ-1+/+: 15.4 ± 2.4 nM, n = 6; DJ-1-/-: 15.5 ± 3.4 nM, n = 6) (Figure 5A). In addition, the in vivo DA extraction fraction (determined as the slope of the net flux regression line) did not differ between genotypes, suggesting that DA uptake was not altered in DJ-1-/- mice (Figure 5A). This was confirmed in striatal tissue in vitro by rotating disk electrode voltammetry (Figure 5B). We next investigated whether DJ-1-/- would show alterations in evoked DA release. Depolarizing concentrations of KCl (60 mM) were perfused through the microdialysis probe implanted in the dorsal striatum and DA overflow was quantified. No significant genotype differences were observed (Figure 5C). Finally we evaluated whether there was an alteration in DA autoreceptor sensitivity by perfusing increasing concentrations of the D2 receptor agonist quinpirole through the microdialysis probe. No significant differences were observed in quinpirole-induced decrease of dialysate DA between DJ-1-/- and wild-type mice (p = 0.33), indicating a rather normal DA autoreceptor activity in DJ-1-/- mice (Figure 5D).
Figure 5. Intact nigrostriatal dopaminergic systems in DJ-1-/- mice.

(A) No differences observed in the basal level of extracellular dopamine estimated using no-net flux microdialysis. (B) No changes in dopamine uptake using rotating disk voltammetry. (C) No significant differences observed in evoked dopamine release using depolarizing concentrations (60mM) of potassium chloride. (D) No alterations in D2 autoreceptor sensitivity using D2 agonist quinpirole.
We then chose to test whether mice exhibited any changes in prepulse inhibition (PPI) or in performance of the pole test, which can both detect fluctuations in dopaminergic function (Matsuura et al., 1997; Ralph-Williams et al., 2002; Ralph et al., 1999). Neither behavioral task showed any significant differences between genotypes when tested at 16 months of age for the PPI and 20 months of age in the pole test (Supplemental Figures 2A-B; p > 0.1), which was consistent with the microdialysis data. Brain sections from 13 month, 22 month and 28 month old mice (n = 6 per genotype) were also probed with a polyclonal rabbit anti-tyrosine hydroxylase (TH) to determine whether any dopaminergic cell loss was evident. No differences were detected in TH staining between genotypes in the substantia nigra (DJ-1+/+: 10,800 ± 300 TH+ neurons; DJ-1-/-: 10,200 ± 300 TH+ neurons; n = 5 each; p > 0.3) or the locus coeruleus (data not shown) at any of the ages examined using unbiased stereology. Collectively, any dopaminergic dysfunction in the DJ-1-/- mice that may exist appears in the attempted L-DOPA rescue of gait abnormalities. By contrast, basal DA dynamics and D2 autoreceptor function are not impaired.
DISCUSSION
Recessive mutations in DJ-1 are a rare cause of early-onset Parkinson’s disease. To better understand the role the predicted loss of DJ-1 contributes to the pathophysiology of disease, we generated DJ-1 knockout (DJ-1-/-) mice, and observed behavioral and pathological changes over the entire lifespan of the mice. We have demonstrated that DJ-1-/- mice develop progressive behavioral abnormalities in gaits and forearm grip strength, and are hypoactive at both juvenile and adult ages.
Previously, alterations in ambulatory activity were detected in both dopamine D1 and D2 receptor deficient mice, suggesting a link between motor activity and dopamine receptor function. Additionally, mice with only a single copy of Nurr1, a transcription factor essential for the development of dopaminergic neurons, show a progressive loss of striatal dopamine and hypoactivity in the open field (Jiang et al., 2005). Goldberg and colleagues (2005), who observed hypoactivity in 3-month old DJ-1-/- mice, concluded that solely D2 receptors were perturbed in DJ-1-/- mice based on findings of intact long-term potentiation (LTP), and impaired long term-depression (LTD) that was restored by D2 and not D1 receptor agonists in striatal slices of 1-month old mice. These authors also observed increased stimulation-evoked re-uptake of dopamine in striatal slices obtained from 3-month old DJ-1-/- mice. A recent report characterizing a separate and distinct line of DJ-1-/- mice suggests that these changes in dopamine re-uptake may be due to alterations in presynaptic dopamine transporter (DAT) levels (Manning-Bog et al., 2007). Chen and colleagues (2005), who observed hypoactivity in DJ-1-/- mice at 11-months of age, used fast-scan cyclic voltammetry in vitro to measure functional alterations in striatal tissue obtained from 4-month old mice, and found a higher stimulated release of dopamine in the dorsal striatum of DJ-1-/-. Based on these findings, we decided to extend the study of the dopaminergic system to an in vivo setting using microdialysis probes placed in the dorsal striatum of 6-month old DJ-1+/+ and DJ-1-/- mice. With this paradigm, we did not detect any differences in either evoked dopamine release using depolarizing concentrations of KCl or in dopamine autoreceptor sensitivity using the D2 agonist quinpirole. Our in vivo measurements of extracellular dopamine showed no differences in the DJ-1-/- mice. Similarily, no alterations in basal DA dynamics or re-uptake were observed in quantitative microdialysis and rotating disk voltammetry studies, respectively. The sensitivity and time resolution of DA detection could be different between the in vitro and in vivo paradigms, which may explain why we could not detect any obvious difference in DA transmission in DJ-1-/- mice. However, there is a possibility that similar experiments performed in aged DJ-1-/- mouse cohorts may be capable of detecting nigrostriatal changes, as some of the behavioral phenotypes were observed in the older animals.
Several reports suggest that DJ-1 may act as an anti-oxidant, which confers protection to cells from oxidative stresses associated with challenges from the well characterized mitochondrial complex I inhibitors 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone, or paraquat (Menzies, 2005; Meulener, 2005; Park, 2005; Martinat, 2004; Pisani, 2006). Flies lacking both human DJ-1 fly orthologues DJ-1α and DJ-1β show selective vulnerability to exposure from either rotenone or paraquat that can be rescued with expression of either or both homologues (Meulener, 2005). Additionally, knockdown of DJ-1 in neuroblastoma lines with siRNA accelerates cell death induced by MPTP metabolite MPP+ and hydrogen peroxide (Taira, 2004). Notably, acidic isoforms of DJ-1, representing oxidized products, accumulate in brains from patients with idiopathic PD and in both dopaminergic cell lines and brains from rats exposed to rotenone, suggesting a direct link between oxidative stress and disease (Choi, 2006; Betarbet, 2006). Two independent reports observe that administration of MPTP leads to increased hypoactivity in the open field, and increased loss of striatal DA and metabolites in DJ-1-/- mice compared with wild-type controls (Kim et al., 2005; Manning-Bog et al., 2007). A recent report indicates that exposure to paraquat leads to both motoric deficits and selective striatal dopamine losses in DJ-1-/- mice that are absent in controls, supporting a neuroprotective functional role for DJ-1 (Yang et al., 2007). Despite challenging the DJ-1 mouse cohort with two different concentrations of paraquat known to result in premature lethality in both mice lacking in superoxide dismutase 1 (SOD1) (Wong et al., 2000) and mice deficient in familial ALS gene ALS2 (Cai et al., 2005), we found no evidence that DJ-1-/- mice are more susceptible to oxidative stressors, in terms of lifespan, than their wild-type littermates (our unpublished observations), in agreement with Goldberg and colleagues (2005), though we did not undertake a more stringent examination of the mouse tissue post injection as Yang and colleagues (Yang et al., 2007).
Due to the prominent shuffling and postural gait alterations that are clinical symptoms of PD, we used footprint patterns to assess whether any movement changes were evident in our DJ-1-/- mice. Previously, mice treated with MPTP, a drug selectively toxic to dopaminergic neurons, showed significantly shorter stride lengths and greater interstride variability (Amende et al., 2005). Both mice carrying a human Huntington’s disease mutation (Carter et al., 1999) and those modeling Down’s syndrome (Hampton et al., 2004) exhibit shortened stride patterns in the footprint test, showing that the paradigm is well suited to detecting irregularities in gait in a variety of neurologically relevant diseases. Several groups have measured gait indices in mice over-expressing either wild-type (Fleming et al., 2004), mutant A53T (Gispert et al., 2003) or A30P human α-synuclein (Gomez-Isla et al., 2003). Only transgenic mice expressing mutant A53T human α-synuclein display shortened stride similar to our DJ-1-/- mice, and in these mice, deficits in grip strength are also noted (Gispert et al., 2003). The other gait abnormalities we observed in the mice were reminiscent of mice expressing the mutant human Huntington’s disease gene (Carter et al., 1999). Interestingly, DJ-1-/- mice failed to show stride differences at 3-4 months of age in a recent report, supporting our results that these footprint differences are age related (Manning-Bog et al., 2007). Our attempts to rescue the stride deficits with exogenously applied L-DOPA suggest that normal conversion of L-DOPA to dopamine may be compromised, or dopamine is catabolized more quickly in DJ-1-/-. The latter possibility seems unlikely as we failed to detect increases in dopamine metabolites DOPAC or HVA by HPLC (data not shown) in striatal tissue dissected from uninjected mice in agreement with other groups, and saw no changes in extracellular dopamine (Goldberg et al., 2005; Manning-Bog et al., 2007). Our observations that the DJ-1-/- mice have decreased bodyweight gain and decreased grip strength are interesting in that these phenotypes appear in mice expressing mutant human α-synuclein (Gispert et al., 2003) and in parkin-deficient mice (Palacino et al., 2004) suggesting that there may be a pathway linking these familial gene products. While each of these other PD mouse models also have been reported to have mild nigrostriatal deficits, selective dopaminergic cell death is absent. Perhaps, the presence of prominent nigrostriatal DA neuron loss requires longer lifespan than the typical 2-2.5 years afforded by mice. For example, genetically manipulated mice that have approximately only 5% of residual vesicular monoamine transporter 2 (VMAT2) protein levels show no gross changes in the number of nigrostriatal DA neurons until they reach 18 months of age despite prominent decreases in striatal dopamine in young mutants, early motor coordinaton deficits in the rotarod that were rescued with L-DOPA treatment, and progressive motor dysfunction (Caudle et al., 2007; Colebrooke et al., 2006). The clear phenotypes present in these mice are helpful in establishing a baseline, and it will certainly be interesting to see whether specific pharmacological treatments can attenuate the hypoactive behavior or gait deficits in these mice.
Supplementary Material
(A) Strategy of generating DJ-1-/- targeted allele. (B) Southern blot of HindIII digested genomic DNA extracted from DJ-1+/+, DJ-1+/-, and DJ-1-/- mice with a 5′ probe. (C) Western blot on whole brain tissue dissected from littermate mice.
(A) No statistical differences noted (n=16, p > 0.1) between 16-month old male DJ-1+/+ and DJ-1-/- for percent prepulse inhibition at different prepulse stimulus levels given. (B) No statistical differences (n=11, p > 0.1) noted between 20-month old female DJ-1+/+ and DJ-1-/- mice in trial task times in the pole test paradigm.
Acknowledgment
This work is supported by the intramural programs of National Institute on Aging, National Institute of Drug Abuse, and National Institute of Mental Health. We thank the Transgenic Core Facility of the Johns Hopkins University School of Medicine for the blastocyst injection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amende I, et al. Gait dynamics in mouse models of Parkinson’s disease and Huntington’s disease. J Neuroengineering Rehabil. 2005;2:20. doi: 10.1186/1743-0003-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annesi G, et al. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58:803–7. doi: 10.1002/ana.20666. [DOI] [PubMed] [Google Scholar]
- Bonifati V, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Cai H, et al. Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J Neurosci. 2005;25:7567–74. doi: 10.1523/JNEUROSCI.1645-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19:3248–57. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–48. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, et al. Quantitative no-net-flux microdialysis permits detection of increases and decreases in dopamine uptake in mouse nucleus accumbens. J Neurosci Methods. 2006;155:187–93. doi: 10.1016/j.jneumeth.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Chen L, et al. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–26. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- Choi J, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–24. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colebrooke RE, et al. Age-related decline in striatal dopamine content and motor performance occurs in the absence of nigral cell loss in a genetic mouse model of Parkinson’s disease. Eur J Neurosci. 2006;24:2622–30. doi: 10.1111/j.1460-9568.2006.05143.x. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dubowitz V, Brooke M. Muscle Biopsy: A Modern Approach. WB Saunders Co., LTD; London: 1973. [Google Scholar]
- Fleming SM, Chesselet MF. Behavioral phenotypes and pharmacology in genetic mouse models of Parkinsonism. Behav Pharmacol. 2006;17:383–91. doi: 10.1097/00008877-200609000-00004. [DOI] [PubMed] [Google Scholar]
- Fleming SM, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–40. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabellini D, et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–7. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- Gispert S, et al. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–29. doi: 10.1016/s1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–35. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–96. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2003;24:245–58. doi: 10.1016/s0197-4580(02)00091-x. [DOI] [PubMed] [Google Scholar]
- Hampton TG, et al. Gait dynamics in trisomic mice: quantitative neurological traits of Down syndrome. Physiol Behav. 2004;82:381–9. doi: 10.1016/j.physbeh.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Holmes A, et al. Behavioral characterization of dopamine D5 receptor null mutant mice. Behav Neurosci. 2001;115:1129–44. [PubMed] [Google Scholar]
- Hwang DY, et al. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res Mol Brain Res. 2003;114:123–31. doi: 10.1016/s0169-328x(03)00162-1. [DOI] [PubMed] [Google Scholar]
- Jiang C, et al. Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp Neurol. 2005;191:154–62. doi: 10.1016/j.expneurol.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Kim RH, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102:5215–20. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, et al. Rotorod sensorimotor learning in cerebellar mutant mice. Neurosci Res. 1995;22:423–6. doi: 10.1016/0168-0102(95)00916-h. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, et al. Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol Dis. 2007;27:141–150. doi: 10.1016/j.nbd.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Martinat C, et al. Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES- derived cell model of primary Parkinsonism. PLoS Biol. 2004;2:e327. doi: 10.1371/journal.pbio.0020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, et al. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods. 1997;73:45–8. doi: 10.1016/s0165-0270(96)02211-x. [DOI] [PubMed] [Google Scholar]
- Menzies FM, et al. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr Biol. 2005;15:1578–82. doi: 10.1016/j.cub.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Meulener M, et al. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol. 2005;15:1572–7. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- Miller DW, et al. L166P mutant DJ-1, causative for recessive Parkinson’s disease, is degraded through the ubiquitin-proteasome system. J Biol Chem. 2003;278:36588–95. doi: 10.1074/jbc.M304272200. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Park J, et al. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361:133–9. doi: 10.1016/j.gene.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Parsons LH, Justice JB., Jr. Quantitative approaches to in vivo brain microdialysis. Crit Rev Neurobiol. 1994;8:189–220. [PubMed] [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132:169–80. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Ralph-Williams RJ, et al. Differential effects of direct and indirect dopamine agonists on prepulse inhibition: a study in D1 and D2 receptor knock-out mice. J Neurosci. 2002;22:9604–11. doi: 10.1523/JNEUROSCI.22-21-09604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph RJ, et al. The dopamine D2, but not D3 or D4, receptor subtype is essential for the disruption of prepulse inhibition produced by amphetamine in mice. J Neurosci. 1999;19:4627–33. doi: 10.1523/JNEUROSCI.19-11-04627.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzu P, et al. DJ-1 colocalizes with tau inclusions: a link between parkinsonism and dementia. Ann Neurol. 2004;55:113–8. doi: 10.1002/ana.10782. [DOI] [PubMed] [Google Scholar]
- Sheehan D, Hrapchak B. Theory and Practice of Histotechnology. Batelle Memorial Institute; 1987. [Google Scholar]
- Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–16. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wong PC, et al. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci U S A. 2000;97:2886–91. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Shen J. Absence of dopaminergic neuronal degeneration and oxidative damage in aged DJ-1-deficient mice. Mol Neurodegener. 2007;2:10. doi: 10.1186/1750-1326-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, et al. Paraquat induces dopaminergic dysfunction and proteasome impairment in DJ-1-deficient mice. Hum Mol Genet. 2007 doi: 10.1093/hmg/ddm249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Strategy of generating DJ-1-/- targeted allele. (B) Southern blot of HindIII digested genomic DNA extracted from DJ-1+/+, DJ-1+/-, and DJ-1-/- mice with a 5′ probe. (C) Western blot on whole brain tissue dissected from littermate mice.
(A) No statistical differences noted (n=16, p > 0.1) between 16-month old male DJ-1+/+ and DJ-1-/- for percent prepulse inhibition at different prepulse stimulus levels given. (B) No statistical differences (n=11, p > 0.1) noted between 20-month old female DJ-1+/+ and DJ-1-/- mice in trial task times in the pole test paradigm.