

Introduction
Cirrhosis is defined as the histological development of regenerative nodules surrounded by fibrous bands in response to chronic liver injury, that leads to portal hypertension and end stage liver disease. Recent advances in the understanding of the natural history and pathophysiology of cirrhosis, and in treatment of its complications, resulting in improved management, quality of life and life expectancy of cirrhotic patients. At present, liver transplantation remains the only curative option for a selected group of patients, but pharmacological therapies that can halt progression to decompensated cirrhosis or even reverse cirrhosis are currently being developed. This concise overview focuses on diagnosis, complications and management of cirrhosis, and novel clinical and scientific developments.
Pathogenesis and pathophysiology of cirrhosis
Fibrosis describes encapsulation or replacement of injured tissue by a collagenous scar. Liver fibrosis results from the perpetuation of the normal wound healing response resulting in an abnormal continuation of fibrogenesis (connective tissue production and deposition). Fibrosis progresses at variable rates depending on the cause of liver disease, environmental and host factors (1-3). Cirrhosis is an advanced stage of liver fibrosis that is accompanied by distortion of the hepatic vasculature. It leads to shunting of the portal and arterial blood supply directly into the hepatic outflow (central veins), compromising exchange between hepatic sinusoids and the adjacent liver parenchyma, i.e., hepatocytes. The hepatic sinusoids are lined by fenestrated endothelia which rest on a sheet of permeable connective tissue (the space of Disse) which contains hepatic stellate cells (HSC) and some mononuclear cells. The other side of the space of Disse is lined by hepatocytes which execute most of the known liver functions. In cirrhosis the space of Disse is filled with scar tissue and endothelial fenestrations are lost, a process termed sinusoidal capillarization (4). Histologically, cirrhosis is characterized by vascularized fibrotic septa that link portal tracts with each other and with central veins, leading to hepatocyte islands that are surrounded by fibrotic septa and which are devoid of a central vein (Figure 1). The major clinical consequences of cirrhosis are impaired hepatocyte (liver) function, an increased intrahepatic resistance (portal hypertension) and the development of hepatocellular carcinoma (HCC). The general circulatory abnormalities in cirrhosis (splanchnic vasodilation, vasoconstriction and hypoperfusion of kidneys, water and salt retention, increased cardiac output) are intimately linked to the hepatic vascular alterations and the resulting portal hypertension. Cirrhosis and its associated vascular distortion are traditionally considered to be irreversible but recent data suggest that cirrhosis regression or even reversal is possible (5,6).
Fig.1. Vascular and architectural alterations in cirrhosis.
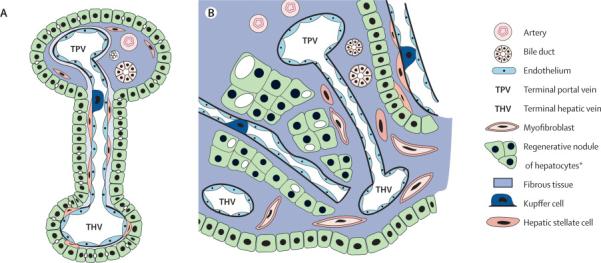
Mesenteric blood flows via the portal vein and the hepatic artery that extend branches into terminal portal tracts. A, normal liver: Terminal portal tract blood runs through the hepatic sinusoids where fenestrated sinusoidal endothelium which rest on loose connective tissue (space of Disse’) allow for extensive metabolic exchange with the lobular hepatocytes; sinusoidal blood is collected by terminal hepatic venules which disembogue into one of the 3 hepatic veins and finally the caval vein. B, cirrhosis: Activated myofibroblasts that derive from perisinusoidal hepatic stellate cells and portal or central vein fibroblasts proliferate and produce excess extracellular matrix (ECM). This leads to fibrous portal tract expansion, central vein fibrosis and capillarization of the sinusoids, characterized by loss of endothelial fenestrations, congestion of the space of Disse’ with ECM, and separation/encasement of perisinusoidal hepatocyte islands from sinusoidal blood flow by collagenous septa. Blood is directly shunted from terminal portal veins and arteries to central veins, with consequent (intrahepatic) portal hypertension and compromised liver synthetic function.
Epidemiology
The exact prevalence of cirrhosis worldwide is unknown. Cirrhosis prevalence was estimated at 0.15% or 400,000 in the USA (7), where it accounted for more than 25,000 deaths and 373,000 hospital discharges in 1998 (8). This may be an underestimation as we recognize the high prevalence of undiagnosed cirrhosis in both NASH and hepatitis C. Similar numbers have been reported from Europe, and numbers are even higher in most Asian and African countries where chronic viral hepatitis B or C are frequent. Since compensated cirrhosis often goes undetected for prolonged periods of time, a reasonable estimate is that up to 1% of populations may have histological cirrhosis.
Etiology of cirrhosis
The etiology of cirrhosis can usually be identified by the patient’s history combined with serologic and histologic evaluation (Table 1). Alcoholic liver disease and hepatitis C are the most common causes in the Western world, while hepatitis B prevails in most parts of Asia and sub-Saharan Africa. After the identification of the hepatitis C virus in 1989 and of nonalcoholic steatohepatitis (NASH) in obese and diabetic subjects, the diagnosis of cirrhosis without an apparent cause (cryptogenic cirrhosis) is rarely made. It is important to know the etiology of cirrhosis, since it can predict complications and direct treatment decisions. It also allows the discussion of preventive measures, e.g., with family members of patients with alcoholic cirrhosis or chronic viral hepatitis, and consideration of (genetic) testing and preventive advice for relatives of patients with genetic diseases, such as hemochromatosis or Wilson’s disease. Frequently multiple etiological factors contribute to the development of cirrhosis, as exemplified in epidemiological studies that identified regular (moderate) alcohol consumption, age above 50 years, and male gender as risk factors in chronic hepatitis C (9-11), or older age obesity, insulin resistance/type 2 diabetes, hypertension and hyperlipidemia (all features of the metabolic syndrome) in NASH (12,13).
Table 1.
Clinical Features of Cirrhosis
| GENERAL FINDINGS a | DESCRIPTION | ETIOLOGY | REFs |
|---|---|---|---|
| Jaundice | Yellow discoloration of skin, cornea and mucous membranes | Compromised hepatocyte excretory function, occurs when serum bilirubin >2mg/dl | 1-3 |
| Spider angiomata | Central arteriole with tiny radiating vessels, mainly on trunk and face | Elevated estradiol, decreased estradiol degradation in liver | 16,17 |
| Nodular liver | Irregular, hard surface on palpation | Fibrosis, irregular regeneration | 2 |
| Splenomegaly | Enlarged on palpation or in ultrasound | Portal hypertension, splenic congestion | 2 |
| Ascites | Proteinaceous fluid in abdominal cavity, clinically detected when ≥1.5 L | Portal hypertension | 1-3, 18 |
| Caput medusae | Prominent veins radiating from umbilicus | Portal hypertension, reopening of the umbilical vein that shunts blood from the portal vein | 2 |
| Cruveilhier Baumgarten syndrome | Epigastric vascular murmur | Shunts from portal vein to umbilical vein branches, can be present without Caput medusae | 19 |
| Palmar erythema | Erythema sparing the central portion of the palm | Elevated estradiol, decreased estradiol degradation in liver | 1-3 |
| White nails | Horizontal white bands and/or proximal white nail plate | Hypoalbuminemia | 20 |
| Hypertrophic osteoarthropathy/Finger clubbing | Painful proliferative osteoarthropathy of long bones | Hypoxemia due to right-to-left shunting, porto-pulmonary hypertension | 21 |
| Dupuytren’s contracture | Fibrosis and contraction of the palmar fascia | Enhanced oxidative stress, elevated hypoxanthine (alcohol exposure or diabetes) | 22 |
| Gynecomastia, loss of male hair pattern | Benign proliferation of glandular male breast tissue | Enhanced conversion of androstenedione to estrone and estradiol, decreased estradiol degradation in liver | 23 |
| Hypogonadism | Mainly in alcoholic cirrhosis and hemochromatosis | Direct toxic effect of alcohol or iron | 1-3 |
| Flapping tremor (asterixis) | Asynchronous flapping motions of dorsiflexed hands | Hepatic encephalopathy, disinhibition of motor neurons | 1-3 |
| Foetor hepaticus | Sweet, pungent smell | Volatile dimethylsulfide, especially in portosystemic shunting and liver failure | 24 |
| Anorexia, fatigue, weight loss, muscle wasting | Occurs in >50% of cirrhotics | Catabolic metabolism by diseased liver, secondary to anorexia | 1-3 |
| Type 2 diabetes | Occurs in 15-30% of cirrhotics | Disturbed glucose utilization and/or decreased insulin removal by the liver | 1-3 |
Clinical presentation
Cirrhosis is frequently indolent, asymptomatic and unsuspected until complications of liver disease present. A sizable proportion of these patients never come to clinical attention, and previously undiagnosed cirrhosis is still frequently found at autopsy (14). The diagnosis of asymptomatic cirrhosis is usually made when incidental screening tests such as liver transaminases or radiologic findings suggest liver disease and patients undergo further evaluation and liver biopsy. The recognition that 20% of HCV patients and perhaps as many as 10% of patients with NASH may progress to cirrhosis has led to the frequent use of biopsy in these high risk groups prior to development of clinical signs of cirrhosis. However, initial clinical presentation of patients with decompensated cirrhosis is still common and is characterized by the presence of dramatic and life-threatening complications, such as variceal hemorrhage, ascites, spontaneous bacterial peritonitis, or hepatic encephalopathy.
General physical and laboratory signs that are frequently found in cirrhosis are summarized in tables 1 and 2.
Table 2.
Laboratory Findings in Cirrhosis
| LABORATORY TEST a | DESCRIPTION | ETIOLOGY |
|---|---|---|
| AST and ALT | Often normal or moderately elevated | Leakage from damaged hepatocytes; AST to ALT ratio often above 1, especially in alcoholic cirrhosis (relative vitamin B6 deficiency) |
| ALP | Elevated <3-fold, except for PBC and PSC | Cholestasis |
| GGT | More specific for liver than ALP, high in active alcoholics | Cholestasis |
| Bilirubin | Elevated later than GGT and ALP, important predictor of mortality | Cholestasis, decreased hepatocyte and renal excretory function (exacerbated by systemic inflammation) |
| Albumin | Decreased in advanced cirrhosis | Decreased hepatic production, sequestration into ascites and interstitium (exacerbated in systemic inflammation), DD: malnutrition, protein losing enteropathy |
| Prothrombin time | Decreased in advanced cirrhosis | Decreased hepatic production of factor V/VII (While thrombin production is maintained), DD: vitamin K deficiency (e.g., due to mechanical biliary obstruction) |
| Immune globulins | Increased (mainly IgG) | Shunting of portal venous blood carrying (intestinal) antigens to lymph tissues with resultant stimulation of plasma cells (26) |
| Sodium imbalance | Hyponatremia | Unability to excrete free water via the kidneys due to increased activity of antidiuretic hormone (vasopressin 2 receptor effect) (27) |
| Anemia | Macro-, normo- or microcytic anemia | Folate deficiency, hypersplenism, direct toxicity (alcohol), gastrointestinal blood loss (e.g., via esophageal varices) |
| Thrombocytes and leukocytes | Thrombocytopenia (Leukopenia) | Hypersplenism, dysfibronogenemia, reduced hepatic thrombopoietin production (28) |
Imaging of cirrhosis
Ultrasonography, computerized tomography (CT) and magnetic resonance imaging (MRI) are not sensitive to detect cirrhosis, and final diagnosis still relies on histology. However, their specificity is high when an obvious cause is present and imaging reveals an inhomogeous hepatic texture or surface, rarefied hepatic central vein, an enlarged caudate lobe, splenomegaly or collateral veins (29). However, other etiologies such as portal vein thrombosis, parasitic diseases or hematological malignancies need to be excluded, and normal radiographic findings do not exclude compensated cirrhosis. The major role of radiography is for the detection and quantitation of complications of cirrhosis, i.e., ascites, HCC, and hepatic or portal vein thrombosis.
Ultrasonography provides important information on hepatic architecture, is cheap and widely available. Nodularity and increased echogenicity of the liver are often found in cirrhosis but are also present in steatosis (30,31). There is typically atrophy of the right lobe and hypertrophy of the left and especially caudate lobes. However, the width of the caudate relative to the right lobe is a poor predictor of cirrhosis (32). Ultrasonography and Doppler ultrasonography of portal and central vein diameters and velocities are useful screening tests for portal hypertension and vessel patency. Contrast ultrasonography examines the appearance of echogenic microbubbles in the hepatic vein. Their appearance after antecubital injection is correlated inversely with fibrosis (33,34). Ultrasonography is the first imaging modality for suspected HCC, but its sensitivity and specificity to detect HCC is below that of CT or MRI (35), and nodular lesions should be confirmed by helical CT and/or MRI. A high degree of suspicion, e.g., in patients with an alfa-fetoprotein above 200 μg/L, or pretransplant evaluation requires these more rigorous techniques even in the absence of ultrasonographic lesions. Contrast ultrasonography, harmonic imaging and power Doppler improve detection of HCC via sensitive visualization of abnormal vessels but are not yet generally available (36).
Conventional CT and MRI are not useful to define the severity of cirrhosis (37), while helical CT and MRI with contrast are the modalities of choice when HCC or vascular lesions are suspected (38). In a comparison MRI was superior to helical CT for detection of small HCC of 1-2cm size (39). MRI has also been shown to be effective in determining hepatic iron and fat content in hemochromatosis and liver steatosis, respectively (40,41).
Elasticity measurement (Fibroscan) is a promising technique based on the velocity of an elastic wave via an intercostally placed transmitter. Shear wave velocity is determined by pulse ultrasound and correlates with liver stiffness, i.e., fibrosis. The examination is limited by morbid obesity, ascites and small intercostal spaces. In a study of 327 patients with hepatitis C, histological cirrhosis was differentiated from milder stages of fibrosis with a receiver-operating characteristics (ROC) curve of 0.97 which is considered an almost ideal test (42). Elasticity scans have the ability to sample 1/500 of the liver and represent a useful, non-invasive test for diagnosing or excluding cirrhosis.
Liver biopsy
Biopsy is considered the gold standard for diagnosis of cirrhosis, and sequential histological grading of inflammation and staging of fibrosis can assess risk of progression. However, biopsy is prone to considerable sampling variability in all liver diseases (43-46). Thus when staging fibrosis in hepatitis C patients using the METAVIR system which is simple and uses only 4 stages (stage 4 being cirrhosis), one third of scores differed by at least one stage when a biopsy from the left liver lobe was compared to that from the right lobe, and with similar results for grading of inflammation (45). In hepatitis C, correct staging was only achieved for 65% and 75% of cases when biopsies were 15 mm and 25 mm in length, respectively (44), while in clinical practice only 16% of biopsies reach 25mm in length. Despite these shortcomings, biopsy is still required to confirm cirrhosis in patients with compensated liver function and to suggest its cause. Biopsy confirmation of cirrhosis is not necessary when clear signs of cirrhosis, such as ascites, coagulopathy, and a shrunken nodular appearing liver are present.
A liver biopsy is obtained by either a (radiographically-guided) percutaneous, a transjugular or laparoscopical route. A greater risk of bleeding following a biopsy has been observed with larger-diameter needles. In suspected cirrhosis a cutting is preferred over a suction needle, in order to prevent tissue fragmentation (47). 2 to 3 percent of patients require hospital admission for management of complications; pain or hypotension are the predominant causes. 60% of complications occur within two, and 96% within 24 hours after biopsy. Mortality, mainly due to severe bleeding is 1 in 10,000 to 12,000, and likely higher in cirrhosis (47). Blood products should be replaced when platelets are below 70,000/μL or prothrombin time is prolonged by more than four seconds, and/or a transjugular or laparoscopic approach chosen. Aspirin and other anti-platelet agents should be stopped at least a week before biopsy.
Natural History and Prognosis
The natural history of cirrhosis is dependent on both the etiology and treatment of the underlying cause. Annual rates of decompensation are 4% for HCV, 10% for HBV and the incidence of HCC is between 2 - 7% per year. Decompensation in alcoholic cirrhosis with continued alcohol use is even more rapid and often associated with alcoholic hepatitis on a background of cirrhosis. Once decompensation has occurred, mortality without transplant is as high as 85% over 5 years.
Numerous studies have attempted to develop a classification system that can both characterize the degree of liver injury and predict the prognosis of patients with cirrhosis based on clinical and laboratory parameters. Due to its low level of complexity and its fairly good predictive value, the Child-Pugh-Turcotte (CPT) classification is widely used (48, Table 4). One-year survival rates for patients with CPT A, B, and C cirrhosis are 100, 80, and 45 percent, respectively (49). CPT class predicts the development of complications, such as variceal hemorrhage and the response of patients to surgical interventions (50). More recently with the pressure in the allocation of scarce liver donors for transplantation, the Model for End Stage Liver Disease (MELD) has been developed to more precisely evaluate short term mortality (51). MELD best predicts 3 month survival of cirrhotics, irrespective of etiology. It is based on creatinine, bilirubin and INR, but lacks features of portal hypertension, such as ascites. It gives priority to patients who are most likely to die without a liver transplant, such as those with hepatorenal failure. In the USA the replacement of the former individualized system of organ allocation, which was heavily based on waiting time, by MELD reduced mortality on the waiting list without change in post-transplant outcome. The system is currently considered for further refinement, such as giving extra points to patients with HCC and hyponatremia <130mEq/mL (52). CPT and MELD scores can vary greatly when single parameters are modified by medical treatment, such as substitution of albumin, removal of ascites or diuretic treatment. Here, an increasing MELD score over time is a better predictor of cirrhosis severity and progression (53).
Table 4.
Child Pugh Turcotte (CPT) classification
| POINTS | 1 | 2 | 3 |
|---|---|---|---|
| Encephalopathy | absent | medically controlled | poorly controlled |
| Ascites | absent | controlled medically | poorly controlled |
| Bilirubin (mg/dL) | < 2 | 2-3 | > 3 |
| Albumin (g/dL) | < 3.5 | 2.8-3.5 | < 2.8 |
| INR | < 1.7 | 1.7-2.2 | > 2.2 |
| CPT A: 5-6 POINTS | CPT B: 7-9 POINTS | CPT C: 10-15 POINTS | |
|---|---|---|---|
| Life expectancy (years) | 15-20 | 4-14 | 1-3 |
| Perioperative mortality (abdominal surgery) (%) | 10 | 30 | 80 |
INR, international normalized ratio.
Treatment and Reversibility of cirrhosis
Causal
Elimination of the trigger(s) that lead to cirrhosis is likely to retard progression to a higher CPT class and to reduce the incidence of HCC. There is evidence that causal treatment may even reverse cirrhosis, although in some of the reports sampling variability cannot be excluded.
Patients with alcoholic cirrhosis must abstain, since continued alcohol consumption drives hepatitis which favours hepatic fibrogenesis and decompensation (54-56). Liver function often worsens in the first 2-3 weeks of withdrawal, since alcohol has an immunosuppressive effect (57).
Patients with compensated replicating HCV-cirrhosis benefit from interferon-based antiviral treatment. Viral eradication and a consequently lowered risk of hepatic decompensation and hepatocellular carcinoma can be achieved in up to 40 and 70% of patients with genotypes 1 and 2 or 3, respectively (58). In a recent meta-analysis 75 out of 153 biopsy-proven cirrhotics showed reversal of cirrhosis on biopsy after successful treatment (59), but results need conformation in view of biopsy sampling variability. How far maintenance interferon for 3-4 years can prevent hepatic decompensation or hepatocellular carcinoma in subjects with stage 3 or 4 fibrosis who did not respond to interferon-ribavirin therapy is currently evaluated in large prospective trials (HALT-C, EPIC-3 and COPILOT) (58).
Longterm treatment with oral nucleoside and nucleotide inhibitors of HBV polymerase may not only retard or reverse cirrhosis but were also shown to prevent complications of end stage liver disease. In a 3 year study of lamivudine for HBV, follow up liver biopsies suggested reversal of cirrhosis in 8/11 patients (73%) (60) and in 436/651 patients with HBV-cirrhosis treated with lamivudine for a mean of 32 months a >50% reduction of hard clinical endpoints, as defined by hepatic decompensation, hepatocellular carcinoma, spontaneous bacterial peritonitis, bleeding gastroesophageal varices, or death related to liver disease was attained (61). In replicating HBV-cirrhosis (>105 copies/mL) lamivudine treatment frequently resulted in clinical improvement, even after decompensation (62-64). The high rate of lamivudine resistance which reaches 56% and 70% after 3 and 4 years of treatment, respectively, is now of lesser concern, since equally well tolerable alternatives like adefovir (65), entecavir (66) or telbivudine (67), or their combinations are available which display lower rates of viral resistance and a different mutational profile. In one large study, adefovir treatment was successfully used in patients with lamivudine resistance pre-transplant, leading to suppression of HBV viral replication to undetectable levels in 76% of patients with either a stabilization or improvement in CTP score and a 90% survival (68).
The data on reversibility and stabilization of other causes of cirrhosis is less well defined. Cohort studies showed that some cirrhotic patients with autoimmune hepatitis showed regression after long-term treatment with corticosteroids (69,70), and venesection of patients with hereditary hemochromatosis could decrease the development of complications of portal hypertension (71).
Complications of Cirrhosis
A detailed review of the complications of cirrhosis is beyond the scope of this article. Major advances have been made in recent years to both prevent and treat the common complications of cirrhosis such as variceal bleeding, ascites, spontaneous bacterial peritonitis and encephalopathy (72-78, Table 5). It is important to note that bacterial infections are frequent, especially in decompensated cirrhotics, exacerbating hepatic dysfunction, encephalopathy and portal hypertension and underlining the need for vigilance and rigorous antibiotic treatment in cirrhosis. Enhanced bacterial translocation from the intestine, a compromised immune function and an excessive proinflammatory cytokine release have been implicated in the pathogenesis of the cirrhosis-associated systemic inflammatory syndrome (79). An example is the failure to control esophageal variceal bleeding with associated bacterial infection (80).
Table 5.
Complications of cirrhosis, their prevention and treatment
| COMPLICATION | PREVENTION | TREATMENT |
|---|---|---|
| Variceal bleeding (72-74) | Non selective beta blockers Variceal band ligation |
Acute: Resuscitation Vasocontrictors Sclerotherapy Band Ligation TIPSS Surgical Shunts |
|
Chronic: Variceal obliteration TIPS Surgical Shunts | ||
| Ascites (72,76) | Low Na diet | Low Na diet Diuretics Large volume paracentesis TIPSS (LeVeen / Denver shunts) |
| Renal failure (77) | Avoid hypovolemia | Discontinue diuretics Rehydration Albumin infusion |
|
Hepatorenal syndrome: Add Terlipressin or Midodrine (Noradrenaline) and Somatostatin (Octreotide) | ||
| Encephalopathy (78) | Avoid precipitants |
Treat precipitating factors: Infection Bleeding Electrolyte imbalance Sedatives High protein intake |
| Lactulose Neomycin, Metronidazole, Rifaximin | ||
| Spontaneous bacterial peritonitis (72) | Treat ascites | Early diagnosic paracentesis: Neutrophils >250/cc → antibiotics iv Secondary prophlaxis with a po antibiotic such as Levofloxacin |
TIPSS: Transjugular intrahepatic porto-systemic shunt; vasoconstrictors: vasopressin, octreotide/somatostatin, terlipressin; non-selective beta blockers: nadolol, propranolol
An important realization for the clinician is that once complications have developed, suitable patients should be referred to a Liver Center that specializes in both the care of patients with end stage liver disease and liver transplantation. Special attention has also to be paid to the circulatory and cardiac abnormalities in cirrhosis that can preclude transplant eligibility. The hepatopulmonary syndrome which occurs in 15-20% of cirrhotics is due to overproduction of NO and overexpression of the endothelin B receptor with consequent pulmonary arteriolar vasodilation and hypoxemia (81,82). It is largely reversible after transplantation. Portopulmonary hypertension is rare, but its prevalence rises to 16-20% of patients with refractory ascites. It is likely caused by an excess of pulmonary arteriolar vasoconstrictors and profibrogenic factors like TGFβ1 (83). The condition is considered irreversible and a pulmonary artery pressure >40 mmHg precludes liver transplantation (84). Cirrhotic cardiomyopathy is characterized by a blunted stress response of the heart, combined with hypertrophy (85). Severe forms increase postoperative mortality and preclude transplantation.
Hepatocellular Carcinoma
HCC is one of the commonest solid organ tumors worldwide and cirrhosis is the major risk factor for progression to HCC (86-88). Other risk factors are listed in Table 6. The pathogenic appears to be the development of regenerative nodules with small cell dysplasia and then invasive HCC. The mortality rate of HCC associated with cirrhosis is rising in most developed countries, whereas mortality from non-HCC complications of cirrhosis is decreasing (89). Cirrhosis due to HCV is associated with the highest HCC incidence in Japan compared to the West, followed by hereditary hemochromatosis (5-year cumulative incidence 17-30%). In cirrhosis due to HBV, the major cause for HCC-related deaths in the world, the 5-year cumulative incidence of HCC is 15% in high endemic areas and 10% in the West. 5-year HCC incidence is lower in alcoholic cirrhotics, or in patients with biliary cirrhosis (8% and 4%, respectively). HCC is increasing in the USA, where its incidence had increased from 1.8 per 100,000 to 2.5 per 100,000 over one decade, mainly attributable to HCV infection (90).
Table 6.
Risk factors for hepatocellular carcinoma
| Cirrhosis |
| Decompensated cirrhosis |
| Viral Hepatitis B and C |
| NASH |
| Type 2 diabetes |
| Aflatoxin exposure |
| Co-infection with multiple viruses; HBV, HCV and HIV (risk 2-6 fold) |
| Increasing Age |
| Male Sex |
| Positive family history of HCC |
| Associated secondary alcohol abuse (risk 2-4 fold) or NASH as a co-factor |
Screening for HCC is one of the most important tasks in following patients with cirrhosis. Current AASLD and EASL guidelines recommend at least one annual screening for HCC in patients with cirrhosis using imaging with ultrasound, triphasic CT scan or gadolinium enhanced MRI (86-88). Serum alfa-fetoprotein,which was an integral component of prior screening algorithms, is no longer recommended due to its poor sensitivity and specificity. Once HCC is detected, multiple treatment modalities are available that depend on tumor size, tumor number and local expertise. In the non-cirrhotic patient, surgical resection is an option and can be curative. However, most patients with cirrhosis will not tolerate liver resection or have microscopic satellite lesions, and the best option for cure is with liver transplantation. The Milan criteria have suggested that the mortality and recurrence rate of HCC is acceptable if liver transplant is performed for either a solitary tumor <5cm in diameter or no more than 3 tumors with the largest being <3cm in diameter. Alternative treatments for HCC patients who do not meet the criteria for surgical resection or transplant are radiofrequency ablation, chemoembolization, alcohol ablation and cyberknife radiotherapy (86-88). Selection of these modalities depends on local expertise, and randomized trials suggesting that they improve long term survival are scarce.
Liver transplantation
The ultimate therapy for cirrhosis and end stage liver disease is liver transplantation. Indications and contraindications for liver transplant are given in Table 5. The most recent survival data from the United Network of Organ Sharing (UNOS) indicates a 1 year survival of 83%, a 5 year survival of 70% and an 8 year survival of 61% (91). Survival is best in patients who are at home at the time of transplant compared to those who are in the hospital or in the ICU. A great advance in liver transplantation has been the improvement in immunosuppressive regimens so that allograft loss from rejection is now relatively rare (92,93). The major issues that remain in the care of the patient post liver transplantation are recurrent disease in the transplant, particularly HCV, and longterm consequences of immunosuppressive agents such as hypertension, hyperlipidemia and renal disease.
RECENT ADVANCES AND FUTURE DIRECTIONS
Molecular pathology of hepatic fibrosis and cirrhosis
The scar tissue in cirrhosis is composed of a complex assembly of different extracellular matrix (ECM) molecules, comprising the fibril forming interstitial collagens type I and III, basement membrane collagen type IV, noncollagenous glycoproteins like fibronectin and laminin, elastic fibers and glycosaminoglycans and proteoglycans among others (94). Toxins, viruses, cholestasis, or hypoxia can trigger a wound healing reaction termed fibrogenesis, i.e., the excess synthesis and deposition of ECM. Initially, fibrogenesis is counterbalanced by removal of excess ECM by proteolytic enzymes, such as certain matrix metalloproteinases (MMPs) (95). Chronic damage usually favours fibrogenesis over fibrolysis, with an upregulation of tissue inhibitors of MMPs (TIMPs) (79). The major hepatic ECM producing cells are myofibroblasts that either derive from activated hepatic stellate cells (HSC) or perivascular fibroblasts (96-98). Myofibroblast activation is mainly driven via fibrogenic cytokines and growth factors that are released by activated macrophages (Kupffer cells), other inflammatory cells and bile duct epithelia (Figure 2). The most prominent profibrogenic cytokine is transforming growth factor β which suppresses inflammation, but drives fibrogenic gene expression in these Myofibroblasts (96,98,99).
Fig.2. Initiation and maintenance of fibrogenesis.
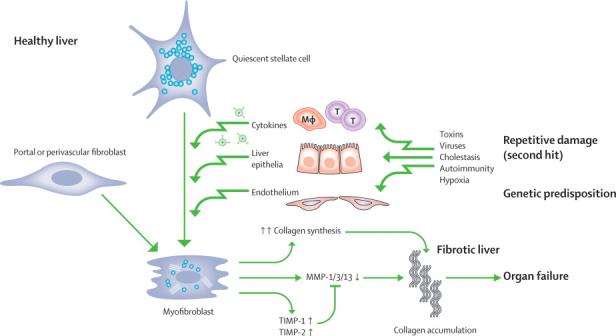
With continuous injury, primarily to hepatocytes or bile duct epithelia, and / or mechanical stress the normally quiescent hepatic stellate cells and portal/perivenular fibroblasts undergo activation and transdifferentiation to myofibroblasts. These myofibroblasts produce excessive amounts of collagens, downregulate certain matrix metalloproteinases (MMPs) and show an enhanced expression of the physiological inhibitors of the MMPs (TIMP-1 and -2). TIMP-1 can also promote myofibroblast proliferation and inhibit their apoptosis.
Genetic predisposition for cirrhosis
The variable rates of development of cirrhosis amongst individuals with similar risk factors such as HCV or alcohol abuse had long been unexplained. Recently, a growing number of functional genetic polymorphisms that likely increase the risk of fibrosis progression has been described. Implicated genes encode cytokines/chemokines and their receptors (100,101), molecules involved in fibrogenesis or fibrolysis (102), blood coagulation (103), antigen presentation (104), iron uptake (105), oxidative and antioxidative metabolism (106), detoxification (107) and polygenetic traits linked to the metabolic syndrome and NASH. In a recent gene association study 1,609 out of 24,882 single nucleotide polymorphisms (SNPs) were found to be associated with fibrosis progression in chronic hepatitis C, with the DDX5 gene having a high positive predictive value (108). Together with established extrinsic risk factors like excess alcohol consumption, obesity or advanced age these SNPs will allow the establishment of risk profiles for the individual patient (109).
Feasibility of pharmacological reversal of cirrhosis
The observations that even cirrhosis can regress once the fibrogenic trigger is eliminated (5,6,59,60,69-71,110) can be explained by the dynamic processes of fibrogenesis and fibrolysis even in cirrhosis (6). While the central role of activated HSC (myofibroblasts) in fibrogenesis is unchallenged, other cells contribute. Thus macrophages/Kupffer cells retarded progression in early but promoted progression in advanced fibrosis (111). Furthermore, regression from macro- to micronodular cirrhosis and possible cirrhosis reversal depends on the degree of ECM crosslinking, which is catalyzed by enzymes such as tissue transglutaminase (112). The rapid progress in understanding the molecular mechanisms that lead to cirrhosis or its reversal have spawned the development of antifibrotic drugs. We can classify the therapeutic approaches to reversal of fibrosis as primary and secondary. Primary approaches focus on treatment of the underlying disease such as HBV and HCV that have convincingly been shown to result in regression of (compensated) cirrhosis (59,60,72). The secondary approach is to develop pharmacotherapy that is directly focused on the mechanism of fibrogenesis, intrinsic antifibrotic drugs, irrespective of the etiology of the liver disease.
The major obstacle to antifibrotic drug development has been the difficulty in defining validated endpoints for clinical trials. The combination of a slowly evolving disease (years to decades) and an established endpoint (liver biopsy) which has limited sensitivity and significant sampling variability represents a stumbling block for study design. In particular, without short term surrogate markers for liver fibrosis, exploratory studies are hampered by the need for significant sample size and high risk of failure.
Noninvasive markers of fibrogenesis and fibrolysis
Non-invasive serological markers to cross-sectionally stage liver fibrosis (113-123) have been extensively reviewed (124-126). Although showing potential, particularly for the diagnosis of cirrhosis, none meet the criteria for an ideal surrogate fibrosis marker (Table 8). A problem is the heterogeneity of liver diseases, with different stages being present in different areas of the liver, particularly between stages 1 to 3. These markers either reflect hepatic function (113-119) or turnover of the ECM (120-123) (Table 9). Combinations have been developed since no single biomarker has adequate sensitivity and specificity. Unfortunately, current ECM-derived serum markers correlate mainly with fibrosis stage, and to a lesser degree with fibrogenesis. We consider the performance of the majority of these biomarkers to be similar with a diagnostic accuracy approaching 80% for the differentiation between mild fibrosis (Metavir F0/1) and moderate to severe fibrosis (F2-4). However, the performance is consistently better at both spectrums of disease from no fibrosis to cirrhosis, and importantly, for predicting cirrhosis.
Table 8.
Desired characteristics of noninvasive markers of liver fibrosis
| 1.) Liver specific |
| 2.) Levels not influenced by alterations in liver, renal or reticuloendothelial function. |
| 3.) Exact measurement of one or more of the following processes: |
| ■ Stage of fibrosis |
| ■ Activity of matrix deposition (fibrogenesis) |
| ■ Activity of matrix removal (fibrolysis) |
| 4.) Easy and reproducible performance characteristics |
| 5.) Able to predict risk of disease progression or regression |
Table 9.
Differentiation of fibrosis stage F0-1 from F2-4 by serum markers and fibroscanN
| ETIOLOGY | AUROC | % CLASSIFIED | REF | ||
|---|---|---|---|---|---|
| Fibrotest a | 352 | HCV | 0.76 (0.03) | 46% | Poynard 2003 (113) |
| Fibrotest | 209 | HBV | 0.78 (0.04) | Myers 2003 (114) | |
| Forns Index b | 476 | HCV | 0.78 | 49% | Forns 2002 (115) |
| APRI c | 192 | HCV | 0.80 (0.06) | 51% | Wai 2003 (116) |
| APRI | 484 | HCV | 0.74 | 57% | Berg 2004 (117) |
| HA, TIMP-1, α2M | 696 | HCV | 0.831 | Patel 2003 (120) | |
| HA, PIIINP, TIMP-1, age | 921 | all liver diseases | 0.804 (0.02) | Rosenberg 2004 (121) | |
| HA, albumin, AST | 137 | HCV/HIV | 0.87 | Kelleher 2005 (122) | |
| Comparisons | |||||
| APRI vs Fibrotest | 323 | HCV | 0.74 (0.03) 0.83 (0.02) |
Le Calvez 2004 (118) | |
| APRI vs AST-ALT ratio | 239 | HCV | 0.773 0.820 |
Giannini 2003 (119) | |
| Fibroscan plus Fibrotest | 183 | HCV | 0.88 | Castera 2005 (126) | |
Algorithms of:
bilirubin, γ-GT, γ-globulin, haptoglobin, α2-macroglobulin, age
γ-GT, cholesterol, platelets, age.
AST to platelet ratio APRI: AST (ULN) / platelets (109/ L) ≤ 0.5 vs. > 1.5
α2M, α2-macroglobulin; matrix-derived markers: HA, hyaluronic acid; PIIINP, aminotermninal propeptide of procollagen III; TIMP-1, tissue inhibitor of matrix metalloproteinases 1.
Performance of the tests is better for differentiating F3-4 (F4 being cirrhosis) from F0-1.
Hepatic elasticity measurement (Fibroscan) (42,126,127) in combination with these serum indices may yield a better prediction of histological fibrosis than either test alone (126), and a recent study showed that Fibroscan was superior to Fibrotest in hepatitis C patients with persistently normal or low transaminases (127).
Several of these tests are now available for use in clinical practice and there is a clinical role for surrogate fibrosis markers. A simple algorithm for using biomarkers is given in Figure 2. The major focus for research is to identify new biomarkers that allow assessment of the dynamic processes of fibrogenesis and fibrolysis, in order to monitor the effect of antifibrotic therapies in patients. This may be achievable with serum proteomics or glycomics (128, 129), or novel imaging techniques for sensitive assessment of fibrogenesis representing the whole liver. Such techniques could be based on CT or MRI imaging with the use of contrast media that target activated HSC. Their validation likely requires parallel analysis of the liver transcriptome of patients with slow or rapid fibrosis progression (130), an approach that requires invasive sampling of liver tissue.
Pharmacological and cellular reversal of hepatic fibrosis and cirrhosis
Numerous agents with proven direct and indirect antifibrotic effects in experimental animals would merit clinical testing (98,131-134), and efficient reversal therapies likely require antifibrotic drug combinations (Table 10, Figure 4). Of note, many potential antifibrotics possess a reasonable safety profile, while their long-term safety in cirrhotic patients has to be proven. However, optimization of such treatment heavily relies on the availability of sensitive non-invasive markers or techniques that allow their rapid testing in low numbers of patients.
Table 10. Antifibrotic agents.
Antifibrotic drug candidates
| Inhibition of profibrogenic activation of HSC |
| Cytokines/ Cytokine antagonists |
| * Interferon-α/β/γ |
| TGFβ- and TGFβ-signaling antagonists (TGFβ antisense oligonucleotides, soluble TGFβ decoy receptors) |
| Inhibition of TGF-β activation: anti-Integrin αvβ6 (EMD409849) |
| Phosphodiesterase-inhibitors |
| * Pentoxifylline, Phosphodiesterase-3/4-inhibitors (Rolipram) |
| MMP-inducers |
| Halofuginone |
| Prostanoids |
| Prostaglandin E2 |
| Vasoactive modulators |
| Endothelin A receptor antagonists (LU135252) |
| * Angiotensin system inhibitors (Captopril, Enalapril, Pirindopril; Losartan, Irbesartan) |
| NO-donors (Pyrro-NO) |
| Histone deacetylase inhibitors |
| Trichostatin A, MS-275 |
| PPAR-α agonists |
| Fibrates (Bezafibrate, Fenofibrate) |
| PPAR-γ agonists |
| * Glitazones: Pioglitazone, Rosiglitazone, Troglitazone |
| * Plant derived, mainly antioxidants |
| Apigenin, Compound 861, FuZhengHuaYu, Glycyrrhicin, Inchin-ko-to (TJ-135), Quercetin, |
| Resveratrol, Rooibus, Salvia miltiorrhiza, Sho-saiko-to (TJ-9), Silymarin, |
| Farnesoid X receptor agonists |
| 6-ethyl chenodeoxycholic acid |
| Inhibition of HSC migration/proliferation |
| HMG-CoA-reductase inhibitors |
| Statins |
| Diuretics |
| Aldosterone (Spironolactone); Na+/H+ exchanger (Cariporide) |
| Immunosuppressants |
| Mycophenolate mofetil, Rapamycin |
| Angiogenesis inhibitors |
| VEGF-receptor 1&2 antagonists (PTK 787) |
| Anti-Integrin αvβ3 (EMD409915) |
| Other kinase inhibitors |
| Anti-PDGFβ receptor kinase: Imatinib, SU9518 |
| Hepatocyte maintenance/protection |
| Hepatocyte growth factor (HGF) |
| Insulin like growth factor I (IGF I) |
Antioxidants
Examples of drugs for which antifibrotic activity has been shown in suitable animal models of liver fibrosis, for which an in vivo antifibrotic effect can be anticipated from in vitro studies on activated hepatic stellate cells (HSC), or which have been or are currently being tested for their antifibrotic effect in patients (marked by an asterisk). This is a selection of published data, based on best scientific evidence, and further examples can be found in recent reviews (82,111-114). Most of these drugs suppress HSC activation directly, others prevent hepatocyte damage or loss, or halt proliferation of bile duct epithelial cells which via release of profibrogenic factors drive fibrogenesis. Drug effects can vary greatly between lobular vs. biliary fibrosis. This makes their preclinical testing in suitable animal models of lobular and biliary fibrosis obligatory. Once an antifibrotic effect has been proven (which largely depends on the development of better noninvasive markers of imaging of fibrosis progression or regression) these agents are likely to be used as combinations, either for long-term or interval therapy.
HSC, hepatic stellate cell/ myofibroblast; Integrin, receptor for matrix proteins or cell-adhesion molecules; MMP, matrix metalloproteinase; PDGF, platelet derived growth factor; PPAR, peroxosome proliferator activated receptor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.
Fig.4. Antifibrotic approaches and candidates for combination therapy.
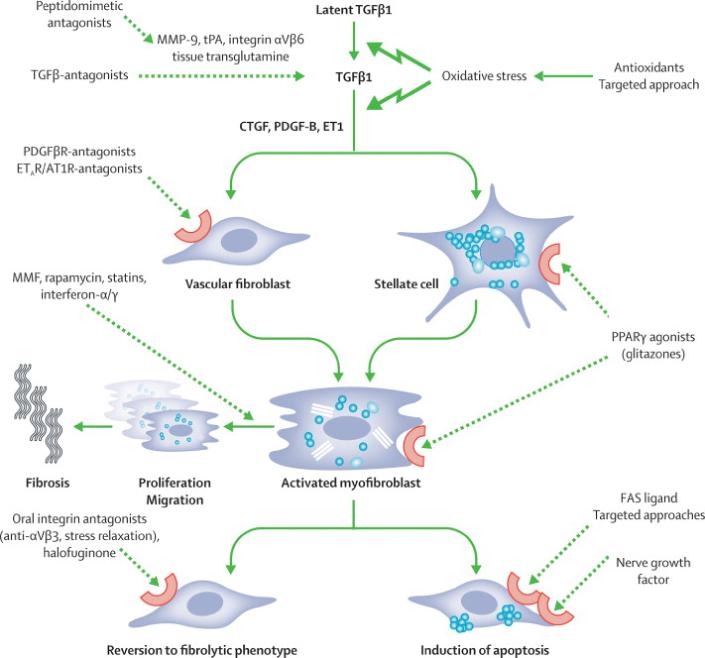
An important principle is inhibition of TGFβ, either by blocking molecules that induce its proteolytic activation from latent TGFβ, or by its direct inhibition. However, this has to be a targeted approach, since complete abrogation of TGFβ leads to cellular dedifferentiation and severe (intestinal) inflammation. AT, angiotensin; AT1R, angiotensin 1 receptor; CTGF, connective tissue growth factor; ET-1, endothelin-1; ETAR, endothelin A receptor; FASL, FAS ligand; MMF, mycophenolate mofetil; MMP, matrix metalloproteinase; NGF, nerve growth factor; PDGF, platelet derived growth factor; TGF, transforming growth factor; tPA tissue plasminogen activator; PPAR, peroxisome proliferator activated receptor.
In order to achieve quick restitution of the functional parenchymal mass in concert with reversal of cirrhosis, the combination of antifibrotic therapy and hepatocyte renewal is attractive (135-137). Thus hepatocyte transplantation improved liver function (138,139) and ameliorated or even reversed advanced fibrosis (140,141). Hepatocyte engraftment was increased by oxidative preconditioning and HSC activation (142-143), and infusion of HGF, a potent hepatocyte mitogen, improved liver function (144). The isolation and in vitro expansion of hepatocyte stem or progenitor cells for cell transplantation may hold promise for an unlimited donor pool (145,146). Reports that infusion of bone marrow stem cells replenished hepatocytes, either by hepatocytic transdifferentiation (147), fusion with hepatocytes (148,149), or indirectly by hepatotrophic growth factors released from stem cells engrafted in the hepatic vasculature (150) sparked much enthusiasm. However, efficiency of stem or progenitor cell engraftment is generally low (151) and the manipulations that are currently needed to allow for sufficient engraftment in humans would incur great risks for patients with cirrhosis and liver failure, necessitating considerable refinement before this techniques can be applied to patients. Similarly, the observation that genetic restitution of telomerase, an enzyme that abrogates cellular ageing by preventing chromosomal telomer shortening, can accelerate hepatic regeneration and ameliorate experimental liver fibrosis has evoked much interest (152). However, increased telomerase activity also favours hepatocarcinogenesis which dampens the enthusiasm for this approach (153).
Summary
Many advances have occurred in the clinical care of patients with cirrhosis and the complications of end stage liver disease. The majority of these have focused on treatment of the underlying cause of cirrhosis and management of complications of portal hypertension. The next 10 years may see us focus on the primary prevention and treatment of cirrhosis. Examples are the use of non-invasive tests to screen for earlier stages of fibrosis and to monitor antifibrotic drug effects, and pharmacological targeting of fibrogenesis pathways. Stem cell or hepatocyte transplantation aiming at reconstitution of liver function may become a clinical reality. Continued basic and clinical research is critical to be able to finally remove cirrhosis as an irreversible condition and a major contributor to morbidity and mortality in our patients.
Fig.3. Utilization of biomarkers for staging liver fibrosis and diagnosis of cirrhosis.
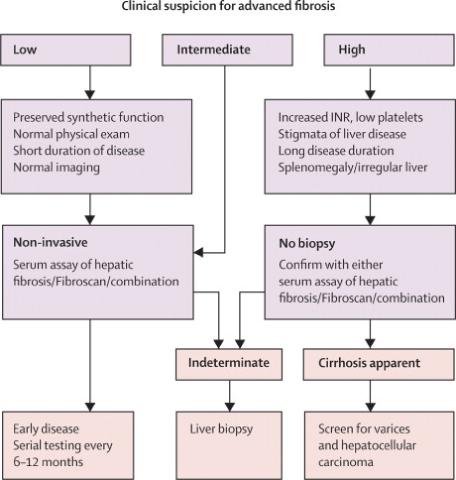
Table 3.
Diagnostic tests in chronic liver disease
| ETIOLOGY | SPECIFIC PHYSICAL ASSOCIATIONS | DIAGNOSTIC (LABORATORY) PARAMETERS | VALUE OF LIVER BIOPSY |
|---|---|---|---|
| HBV | Arthritis | HBsAg, (HBeAg), anti-HBc, HBV-DNA | + |
| HCV | Cryoglobulinemia, | anti-HCV, HBV-RNA | + |
| HDV | (HBsAg), anti-HDV, HDV-RNA | ++ (HDAg) | |
| Alcoholic | AST/ALT ≥2, CDT↑, γGT↑ | ++ (Mallory bodies, steatosis, granulocytes >hepatocyte ballooning) | |
| NASH | Overweight/obesity, metabolic syndrome, type 2 diabetes | Uric acid, fasting glucose/insulin/triglycerides, | ++ (Mallory bodies, steatosis, hepatocyte ballooning>granulocytes) |
| Autoimmune | Autoantibodies (ANA, anti-LKM, anti-SLA), γ-globulins↑↑ | +++ (bridging necrosis) | |
| PBC | Sicca-syndrome, xanthelasma | AMA, ALP/γGT↑, cholesterol↑ | ++ (cholangitis, paucity of bile ducts, granuloma, ductopenia) |
| PSC | Ulcerative colitis (90%) | Anti-pANCA (70%), ALP/γGT↑ imaging: beaded intra- (and extra-) hepatic bile ducts |
+++ (concentric peri-bile ductular fibrosis, ductopenia) |
| Hemochromatosis | Arthritis, myocarditis, diabetes | Fasting transferrin saturation >60% (♂), >50% (♀), ferritin↑↑, HFE mutation | ++ (periportal Fe- loaded hepatocytes, quant. liver Fe |
| Wilson’s | Neurological | Coeruloplasmin↓, urinary Cu (24h) ↑, slit-lamp: corneal Cu deposits | +++ (quant. liver Cu) |
| α1-Antitrypsin | Pulmonary fibrosis | α1-AT ↓, α1-AT subtyping |
+++ (α1-AT-loaded hepatocytes) |
| Congenital | +++ (bile ductular plate malformations etc.) |
A1-AT, α1 antitrypsin; AMA, anti-mitochondrial antibodies; ANA, anti-nuclear antibodies; CDT, carbohydrate deficient transferrin; Cu, copper; Fe, iron; HBV/ HCV/HDV, hepatitis B/C/D virus; HBc/HBe/HBs Ag, hepatitis B core/envelope/surface antigen; LKM, liver kidney membrane; SLA, soluble liver antigen; pANCA, perinuclear neutrophil cytoplasmic antigen.
Table 7.
Indications and contraindications for orthotopic liver transplantion
| INDICATIONS |
| Advanced Chronic Liver Failure |
| - CPT score > 7 |
| - Qualifying MELD Score for Organ Allocation |
| Acute Liver Failure |
| Unresectable Hepatic Malignancy |
| Inherited Metabolic Disorders |
| General |
| No alternative form of therapy |
| No absolute contraindications |
| Willingness to comply with follow up care |
| Ability to provide for costs of OLT |
| CONTRAINDICATIONS |
| Relative |
| HIV seropositivity |
| Methadone dependence |
| Stage 3 HCC * |
| Absolute |
| Extrahepatic malignancy |
| AIDS |
| Cholangiocarcinoma |
| Severe, uncontrolled systemic infection |
| Multiorgan failure |
| Advanced cardiopulmonary disease |
| Active substance abuse |
not fulfilling the Milan criteria (see text).
Acknowledgements
We thank the Espinosa Liver Fibrosis Fund at Beth Israel Deaconess Medical Center and the LIFER Foundation, Boston, MA, for research support.
Abbreviations
- CPT
Child-Pugh Turcotte
- HCC
hepatocellular carcinoma
- HSC
hepatic stellate cell
- NASH
nonalcoholic steatohepatitis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. 2nd Edition Oxford University Press; 1999. [Google Scholar]
- 2.Sherlock S, Dooley J, editors. Diseases of the Liver and Biliary System. 11th Edition Blackwell Science; Oxford, UK; Malden, MA: 2002. [Google Scholar]
- 3.Schiff ER, Sorrell MF, Maddrey EC, editors. Schiff’s Diseases of the Liver. 9th Edition Lippincott, Williams & Wilkins; Philadelphia: 2003. [Google Scholar]
- 4.Schaffner H, Popper H. Capillarization of the sinusoids. Gastroenterology. 1963;44:339–42. [Google Scholar]
- 5.Desmet VJ, Roskams T. Cirrhosis reversal: a duel between dogma and myth. J Hepatol. 2004;40:860–7. doi: 10.1016/j.jhep.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599–607. doi: 10.5858/2000-124-1599-ROHC. [DOI] [PubMed] [Google Scholar]
- 7.Digestive diseases in the United States: Epidemiology and Impact. NIDDK; Bethesda, MD: 1994. NIH Publication No. 94-1447. [Google Scholar]
- 8.National Center for Health Statistics. US Department of Health and Human Services, Centers for Disease Control and Prevention; Hyattsville, MD: 2005. (Series 13). [Google Scholar]
- 9.Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet. 1997;349:825–32. doi: 10.1016/s0140-6736(96)07642-8. [DOI] [PubMed] [Google Scholar]
- 10.Bellentani S, Pozzato G, Saccoccio G, et al. Clinical course and risk factors of hepatitis C virus related liver disease in the general population: report from the Dionysos study. Gut. 1999;44:874–80. doi: 10.1136/gut.44.6.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellentani S, Saccoccio G, Costa G, et al. The Dionysos Study Group Drinking habits as cofactors of risk for alcohol induced liver damage. Gut. 1997;41:845–50. doi: 10.1136/gut.41.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(3 Suppl 1):S5–10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 13.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl 1):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 14.Conn H, Atterbury C. Cirrhosis. In: Schiff L, Schiff E, editors. Diseases of the Liver. 7th edition Lippencott Company, Philadelphia; Philadelphia: 1993. pp. 875–934. [Google Scholar]
- 15.Groszmann RJ, Abraldes JG. Portal hypertension. From bedside to bench. J Clin Gastroenterol. 2005;39(Suppl 2):S125–30. doi: 10.1097/01.mcg.0000155552.14396.3d. [DOI] [PubMed] [Google Scholar]
- 16.Pirovino M, Linder R, Boss C, Kochli HP, Mahler F. Cutaneous spider nevi in liver cirrhosis: Capillary microscopical and hormonal investigations. Klin Wochenschr. 1988;66:298–302. doi: 10.1007/BF01727516. [DOI] [PubMed] [Google Scholar]
- 17.Foutch PG, Sullivan JA, Gaines JA, Sanowski RA. Cutaneous vascular spiders in cirrhotic patients: correlation with hemorrhage from esophageal varices. Am J Gastroenterol. 1988;83:723–26. [PubMed] [Google Scholar]
- 18.Cattau E, Benjamin SB, Knuff TE, Castell DO. The accuracy of the physical exam in the diagnosis of suspected ascites. JAMA. 1982;247:1164–66. [PubMed] [Google Scholar]
- 19.Erlinger S, Benhamou J. Cirrhosis: Clinical aspects. In: Mcintyre N, Benhamou J, Rizzetto M, Rodes J, editors. Oxford Textbook of Clinical Hepatology. University Press; Oxford: 1991. p. 380. [Google Scholar]
- 20.Muercke RC. The finger-nails in hypoalbumenemia: a new physical sign. Br Med J. 1956;4979:1327–28. doi: 10.1136/bmj.1.4979.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epstein O, Adukiewicz AB, Dick R, Sherlock S. Hypertrophic hepatic osteoarthropathy. Clinical roentgenologic, biochemical hormonal and cardiorespiratory studies, and review of the literature. Am J Med. 1979;67:88–97. doi: 10.1016/0002-9343(79)90078-0. [DOI] [PubMed] [Google Scholar]
- 22.Attali P, Ink O, Pelletier G, et al. Dupuytren’s contracture, alcohol consumption, and chronic liver disease. Arch Intern Med. 1987;147:1065–67. [PubMed] [Google Scholar]
- 23.Van Thiel DH, Gavaler JS, Schade RR. Liver disease and the hypothalamic pituitary gonadal axis. Semin Liver Dis. 1985;5:35–45. doi: 10.1055/s-2008-1041756. [DOI] [PubMed] [Google Scholar]
- 24.Tangerman A, Meuwese-Arends MT, Jansen JB. Cause and composition of foetor hepaticus. Lancet. 1994;343:483. doi: 10.1016/s0140-6736(94)92729-4. [DOI] [PubMed] [Google Scholar]
- 25.Pratt D, Kaplan M. Evaluation of the Liver A: Laboratory Tests. In: Schiff E, Sorrell M, Maddrey W, editors. Schiff’s Diseases of the Liver. Eighth Edition Lippincott Williams Wilkens; Philadelphia: 1999. p. 205. [Google Scholar]
- 26.Triger DR, Wright R. Hyperglobulinaemia in liver disease. Lancet. 1973;1:1494–96. doi: 10.1016/s0140-6736(73)91827-8. [DOI] [PubMed] [Google Scholar]
- 27.Papadakis MA, Fraser CL, Arieff AI. Hyponatraemia in patients with cirrhosis. Q J Med. 1990;76:675–88. [PubMed] [Google Scholar]
- 28.Peck-Radosavljevic M, Wichlas M, Zacherl J, et al. Thrombopoietin induces rapid resolution of thrombocytopenia after orthotopic liver transplantation through increased platelet production. Blood. 2000;95:795–801. [PubMed] [Google Scholar]
- 29.Martinez-Noguera A, Montserrat E, Torrubia S, Villalba J. Doppler in hepatic cirrhosis and chronic hepatitis. Semin Ultrasound CT MR. 2002;23:19–36. doi: 10.1016/s0887-2171(02)90027-2. [DOI] [PubMed] [Google Scholar]
- 30.Di Lelio A, Cestari C, Lomazzi A, Beretta L. Cirrhosis: Diagnosis with sonographic study of the liver surface. Radiology. 1989;172:389–92. doi: 10.1148/radiology.172.2.2526349. [DOI] [PubMed] [Google Scholar]
- 31.Tchelepi H, Ralls PW, Radin R, Grant E. Sonography of diffuse liver disease. J Ultrasound Med. 2002;21:1023–32. doi: 10.7863/jum.2002.21.9.1023. [DOI] [PubMed] [Google Scholar]
- 32.Awaya H, Mitchell DG, Kamishima T, Holland G, Ito K, Matsumoto T. Cirrhosis: modified caudate-right lobe ratio. Radiology. 2002;224:769–74. doi: 10.1148/radiol.2243011495. [DOI] [PubMed] [Google Scholar]
- 33.Albrecht T, Blomley MJ, Cosgrove DO, et al. Non-invasive diagnosis of hepatic cirrhosis by transit-time analysis of an ultrasound contrast agent. Lancet. 1999;353:1579–83. doi: 10.1016/S0140-6736(98)06373-9. [DOI] [PubMed] [Google Scholar]
- 34.Blomley MJ, Lim AK, Harvey CJ, et al. Liver microbubble transit time compared with histology and Child-Pugh score in diffuse liver disease: a cross sectional study. Gut. 2003;52:1188–93. doi: 10.1136/gut.52.8.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim CK, Lim JH, Lee WJ. Detection of hepatocellular carcinomas and dysplastic nodules in cirrhotic liver: accuracy of ultrasonography in transplant patients. J Ultrasound Med. 2001;20:99–104. doi: 10.7863/jum.2001.20.2.99. [DOI] [PubMed] [Google Scholar]
- 36.Lencioni R, Cioni D, Bartolozzi C. Tissue harmonic and contrast-specific imaging: back to gray scale in ultrasound. Eur Radiol. 2002;12:151–65. doi: 10.1007/s003300101022. [DOI] [PubMed] [Google Scholar]
- 37.Ito K, Mitchell DG, Hann HW, et al. Viral-induced cirrhosis: Grading of severity using MR imaging. AJR Am J Roentgenol. 1999;173:591–96. doi: 10.2214/ajr.173.3.10470885. [DOI] [PubMed] [Google Scholar]
- 38.Choi D, Kim SH, Lim JH, et al. Detection of hepatocellular carcinoma: combined T2-weighted and dynamic gadolinium-enhanced MRI versus combined CT during arterial portography and CT hepatic arteriography. J Comput Assist Tomogr. 2001;25:777–85. doi: 10.1097/00004728-200109000-00018. [DOI] [PubMed] [Google Scholar]
- 39.Burrel M, Llovet JM, Ayuso C, et al. MRI angiography is superior to helical CT for detection of HCC prior to liver transplantation: an explant correlation. Hepatology. 2003;38:1034–42. doi: 10.1053/jhep.2003.50409. [DOI] [PubMed] [Google Scholar]
- 40.Bonkovsky HL, Rubin RB, Cable EE, et al. Hepatic iron concentration: Noninvasive estimation by means of MR imaging techniques. Radiology. 1999;212:227–34. doi: 10.1148/radiology.212.1.r99jl35227. [DOI] [PubMed] [Google Scholar]
- 41.Qayyum A, Goh JS, Kakar S, et al. Accuracy of liver fat quantification at MR imaging: comparison of out-of-phase gradient-echo and fat-saturated fast spin-echo techniques - initial experience. Radiology. 2005;237:507–11. doi: 10.1148/radiol.2372040539. [DOI] [PubMed] [Google Scholar]
- 42.Ziol M, Handra-Luca A, Kettaneh A, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology. 2005;41:48–54. doi: 10.1002/hep.20506. [DOI] [PubMed] [Google Scholar]
- 43.Abdi W, Millan JC, Mezey E. Sampling variability on percutaneous liver biopsy. Arch Intern Med. 1979;139:667–69. [PubMed] [Google Scholar]
- 44.Bedossa P, Dargere D, Paradis V. Sampling variability of liver fibrosis in chronic hepatitis C. Hepatology. 2003;38:1449–57. doi: 10.1016/j.hep.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 45.Regev A, Berho M, Jeffers LJ, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol. 2002;97:2614–18. doi: 10.1111/j.1572-0241.2002.06038.x. [DOI] [PubMed] [Google Scholar]
- 46.Ratziu V, Charlotte F, Heurtier A, et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128:1898–906. doi: 10.1053/j.gastro.2005.03.084. [DOI] [PubMed] [Google Scholar]
- 47.Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med. 2001;344:495–500. doi: 10.1056/NEJM200102153440706. [DOI] [PubMed] [Google Scholar]
- 48.Pugh RN, Murray-Lyon IM, Dawson JL, et al. Transection of the oesophagus for bleeding esophageal varicies. Br J Surg. 1973;60:646–49. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 49.Infante-Rivard C, Esnaola S, Villeneuve JP. Clinical and statistical validity of conventional prognostic factors in predicting short-term survival among cirrhotics. Hepatology. 1987;7:660–64. doi: 10.1002/hep.1840070408. [DOI] [PubMed] [Google Scholar]
- 50.de Franchis R, Primignani M. Why do varices bleed? Gastroenterol Clin North Am. 1992;21:85–101. [PubMed] [Google Scholar]
- 51.Wiesner R, Edwards E, Freeman R, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology. 2003;124:91–96. doi: 10.1053/gast.2003.50016. [DOI] [PubMed] [Google Scholar]
- 52.Wiesner RH. Evidence-based evolution of the MELD/PELD liver allocation policy. Liver Transpl. 2005;11:261–63. doi: 10.1002/lt.20362. [DOI] [PubMed] [Google Scholar]
- 53.Huo TI, Wu JC, Lin HC, et al. Evaluation of the increase in model for end-stage liver disease (DeltaMELD) score over time as a prognostic predictor in patients with advanced cirrhosis: risk factor analysis and comparison with initial MELD and Child-Turcotte-Pugh score. J Hepatol. 2005;42:826–32. doi: 10.1016/j.jhep.2005.01.019. Epub 2005 Mar 31. [DOI] [PubMed] [Google Scholar]
- 54.Powell WJ, Jr, Klatskin G. Duration of survival in patients with Laennec’s cirrhosis. Influence of alcohol withdrawal, and possible effects of recent changes in general management of the disease. Am J Med. 1968;44:406–20. doi: 10.1016/0002-9343(68)90111-3. [DOI] [PubMed] [Google Scholar]
- 55.Orrego H, Blake JE, Blendis LM, Medline A. Prognosis of alcoholic cirrhosis in the presence and absence of alcoholic hepatitis. Gastroenterology. 1987;92:208–14. doi: 10.1016/0016-5085(87)90861-4. [DOI] [PubMed] [Google Scholar]
- 56.Runyon BA. Historical aspects of treatment of patients with cirrhosis and ascites. Semin Liver Dis. 1997;17:163–73. doi: 10.1055/s-2007-1007195. [DOI] [PubMed] [Google Scholar]
- 57.Stickel F, Schuppan D, Hahn EG, Seitz HK. Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut. 2002;51:132–39. doi: 10.1136/gut.51.1.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Everson GT. Management of cirrhosis due to chronic hepatitis C. J Hepatol. 2005;42(Suppl1):S65–74. doi: 10.1016/j.jhep.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 59.Poynard T, McHutchison J, Manns M, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122:1303–13. doi: 10.1053/gast.2002.33023. [DOI] [PubMed] [Google Scholar]
- 60.Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124:105–17. doi: 10.1053/gast.2003.50013. [DOI] [PubMed] [Google Scholar]
- 61.Liaw YF, Sung JJ, Chow WC, et al. Cirrhosis Asian Lamivudine Multicentre Study Group Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521–31. doi: 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- 62.Lok AS, McMahon BJ. Practice Guidelines Committee, American Association for the Study of Liver Diseases (AASLD). Chronic hepatitis B: update of recommendations. Hepatology. 2004;39:857–61. doi: 10.1002/hep.20110. [DOI] [PubMed] [Google Scholar]
- 63.Villeneuve JP, Condreay LD, Willems B, et al. Lamivudine treatment for decompensated cirrhosis resulting from chronic hepatitis B. Hepatology. 2000;31:207–10. doi: 10.1002/hep.510310130. [DOI] [PubMed] [Google Scholar]
- 64.Fontana RJ, Hann HW, Perrillo RP, et al. Determinants of early mortality in patients with decompensated chronic hepatitis B treated with antiviral therapy. Gastroenterology. 2002;123:719–27. doi: 10.1053/gast.2002.35352. [DOI] [PubMed] [Google Scholar]
- 65.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Adefovir Dipivoxil 438 Study Group Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B. N Engl J Med. 2005;352:2673–81. doi: 10.1056/NEJMoa042957. [DOI] [PubMed] [Google Scholar]
- 66.Chang TT, Gish RG, Hadziyannis SJ, et al. BEHoLD Study Group A dose-ranging study of the efficacy and tolerability of entecavir in Lamivudine-refractory chronic hepatitis B patients. Gastroenterology. 2005;129:1198–209. doi: 10.1053/j.gastro.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 67.Lai CL, Leung N, Teo EK, et al. Telbivudine Phase II Investigator Group A 1-year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen-positive chronic hepatitis B. Gastroenterology. 2005;129:528–36. doi: 10.1016/j.gastro.2005.05.053. [DOI] [PubMed] [Google Scholar]
- 68.Schiff ER, Lai CL, Hadziyannis S, et al. Adefovir Dipovoxil Study 435 International Investigators Group Adefovir dipivoxil therapy for lamivudine-resistant hepatitis B in pre- and post-liver transplantation patients. Hepatology. 2003;38:1419–27. doi: 10.1016/j.hep.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 69.Roberts SK, Therneau TM, Czaja AJ. Prognosis of histological cirrhosis in type 1 autoimmune hepatitis. Gastroenterology. 1996;110:848–57. doi: 10.1053/gast.1996.v110.pm8608895. [DOI] [PubMed] [Google Scholar]
- 70.Dufour JF, DeLellis R, Kaplan MM. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann Intern Med. 1997;127:981–85. doi: 10.7326/0003-4819-127-11-199712010-00006. [DOI] [PubMed] [Google Scholar]
- 71.Fracanzani AL, Fargion S, Romano R, et al. Portal hypertension and iron depletion in patients with genetic hemochromatosis. Hepatology. 1995;22:1127–31. doi: 10.1002/hep.1840220417. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Tsao G. Current management of the complications of cirrhosis and portal hypertension: variceal hemorrhage, ascites, and spontaneous bacterial peritonitis. Gastroenterology. 2001;120:726–48. doi: 10.1053/gast.2001.22580. [DOI] [PubMed] [Google Scholar]
- 73.Bosch J, Garcia-Pagan JC. Prevention of variceal rebleeding. Lancet. 2003;361:952–4. doi: 10.1016/S0140-6736(03)12778-X. [DOI] [PubMed] [Google Scholar]
- 74.de Franchis R, Dell’Era A, Iannuzzi F. Diagnosis and treatment of portal hypertension. Dig Liver Dis. 2004;36:787–98. doi: 10.1016/j.dld.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 75.Boyer TD. Transjugular intrahepatic portosystemic shunt: current status. Gastroenterology. 2003;124:1700–10. doi: 10.1016/s0016-5085(03)00377-9. [DOI] [PubMed] [Google Scholar]
- 76.Gines P, Cardenas A, Arroyo V, Rodes J. Management of cirrhosis and ascites. N Engl J Med. 2004;350:1646–54. doi: 10.1056/NEJMra035021. [DOI] [PubMed] [Google Scholar]
- 77.Gines P, Guevara M, Arroyo V, Rodes J. Hepatorenal syndrome. Lancet. 2003;362:1819–27. doi: 10.1016/S0140-6736(03)14903-3. [DOI] [PubMed] [Google Scholar]
- 78.Butterworth RF. Complications of cirrhosis III. Hepatic encephalopathy. J Hepatol. 2000;32(1 Suppl):171–80. doi: 10.1016/s0168-8278(00)80424-9. [DOI] [PubMed] [Google Scholar]
- 79.Riordan SM, Williams R. The intestinal flora and bacterial infection in cirrhosis. J Hepatol. 2006;45:744–57. doi: 10.1016/j.jhep.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 80.Papatheodoridis GV, Patch D, Webster GJ, Brooker J, Barnes E, Burroughs AK. Infection and hemostasis in decompensated cirrhosis: a prospective study using thrombelastography. Hepatology. 1999;29:1085–90. doi: 10.1002/hep.510290437. [DOI] [PubMed] [Google Scholar]
- 81.Arguedas MR, Abrams GA, Krowka MJ, Fallon MB. Prospective evaluation of outcomes and predictors of mortality in patients with hepatopulmonary syndrome undergoing liver transplantation. Hepatology. 2003;37:192–7. doi: 10.1053/jhep.2003.50023. [DOI] [PubMed] [Google Scholar]
- 82.Fallon MB. Mechanisms of pulmonary vascular complications of liver disease: hepatopulmonary syndrome. J Clin Gastroenterol. 2005;39(4 Suppl 2):S138–42. [PubMed] [Google Scholar]
- 83.Blendis L, Wong F. Portopulmonary hypertension: an increasingly important complication of cirrhosis. Gastroenterology. 2003;125:622–4. doi: 10.1016/s0016-5085(03)00969-7. [DOI] [PubMed] [Google Scholar]
- 84.Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003;37:401–9. doi: 10.1053/jhep.2003.50060. [DOI] [PubMed] [Google Scholar]
- 85.Gaskari SA, Honar H, Lee SS. Therapy insight: Cirrhotic cardiomyopathy. Nat Clin Pract Gastroenterol Hepatol. 2006;3:329–37. doi: 10.1038/ncpgasthep0498. [DOI] [PubMed] [Google Scholar]
- 86.Bruix J, Sherman M, Llovet JM, et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol. 2001;35:421–30. doi: 10.1016/s0168-8278(01)00130-1. [DOI] [PubMed] [Google Scholar]
- 87.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–17. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 88.Sherman M, Klein A. AASLD single-topic research conference on hepatocellular carcinoma: Conference proceedings. Hepatology. 2004;40:1465–73. doi: 10.1002/hep.20528. [DOI] [PubMed] [Google Scholar]
- 89.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology. 2004;127(5 Suppl 1):S35–50. doi: 10.1053/j.gastro.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 90.El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36:S74–83. doi: 10.1053/jhep.2002.36807. [DOI] [PubMed] [Google Scholar]
- 91.Roberts MS, Angus DC, Bryce CL, Valenta Z, Weissfeld L. Survival after liver transplantation in the United States: a disease-specific analysis of the UNOS database. Liver Transpl. 2004;10:886–97. doi: 10.1002/lt.20137. [DOI] [PubMed] [Google Scholar]
- 92.Fung J, Kelly D, Kadry Z, Patel-Tom K, Eghtesad B. Immunosuppression in liver transplantation: beyond calcineurin inhibitors. Liver Transpl. 2005;11:267–80. doi: 10.1002/lt.20373. [DOI] [PubMed] [Google Scholar]
- 93.Perry I, Neuberger J. Immunosuppression: towards a logical approach in liver transplantation. Clin Exp Immunol. 2005;139:2–10. doi: 10.1111/j.1365-2249.2005.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Sem Liver Dis. 2001;21:351–72. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 95.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–84. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 96.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–50. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 97.Knittel T, Kobold D, Saile B, et al. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology. 1999;117:1205–21. doi: 10.1016/s0016-5085(99)70407-5. [DOI] [PubMed] [Google Scholar]
- 98.Schuppan D, Krebs A, Bauer M, Hahn EG. Hepatitis C and liver fibrosis. Cell Death Differ. 2003;10(Suppl 1):S59–67. doi: 10.1038/sj.cdd.4401163. [DOI] [PubMed] [Google Scholar]
- 99.Bissell DM, Roulot D, George J. Transforming growth factor β and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 100.Muhlbauer M, Bosserhoff AK, Hartmann A, et al. A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology. 2003;125:1085–93. doi: 10.1016/s0016-5085(03)01213-7. [DOI] [PubMed] [Google Scholar]
- 101.Hellier S, Frodsham AJ, Hennig BJ, et al. Association of genetic variants of the chemokine receptor CCR5 and its ligands, RANTES and MCP-2, with outcome of HCV infection. Hepatology. 2003;38:1468–76. doi: 10.1016/j.hep.2003.09.027. [DOI] [PubMed] [Google Scholar]
- 102.Satsangi J, Chapman RW, Haldar N, et al. A functional polymorphism of the stromelysin gene (MMP-3) influences susceptibility to primary sclerosing cholangitis. Gastroenterology. 2001;121:124–30. doi: 10.1053/gast.2001.25527. [DOI] [PubMed] [Google Scholar]
- 103.Wright M, Goldin R, Hellier S, et al. Factor V Leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut. 2003;52:1206–10. doi: 10.1136/gut.52.8.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshizawa K, Ota M, Saito S, et al. Long-term follow-up of hepatitis C virus infection: HLA class II loci influences the natural history of the disease. Tissue Antigens. 2003;61:159–65. doi: 10.1034/j.1399-0039.2003.00015.x. [DOI] [PubMed] [Google Scholar]
- 105.Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38:335–42. doi: 10.1016/s0168-8278(02)00415-4. [DOI] [PubMed] [Google Scholar]
- 106.Silvestri L, Sonzogni L, De Silvestri A, et al. CYP enzyme polymorphisms and susceptibility to HCV-related chronic liver disease and liver cancer. Int J Cancer. 2003;104:310–7. doi: 10.1002/ijc.10937. [DOI] [PubMed] [Google Scholar]
- 107.Stickel F, Osterreicher CH, Datz C, et al. Prediction of progression to cirrhosis by a glutathione S-transferase P1 polymorphism in subjects with hereditary hemochromatosis. Arch Intern Med. 2005;165:1835–40. doi: 10.1001/archinte.165.16.1835. [DOI] [PubMed] [Google Scholar]
- 108.Huang H, Shiffman ML, Cheung RC, et al. Identification of two gene variants associated with risk of advanced fibrosis in patients with chronic hepatitis C. Gastroenterology. 2006;130:1679–87. doi: 10.1053/j.gastro.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 109.Bataller R, North KE, Brenner DA. Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology. 2003;37:493–503. doi: 10.1053/jhep.2003.50127. [DOI] [PubMed] [Google Scholar]
- 110.Issa R, Williams E, Trim N, et al. Apoptosis of hepatic stellate cells: involvement in resolution of biliary fibrosis and regulation by soluble growth factors. Gut. 2001;48:548–57. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Issa R, Zhou X, Constandinou CM, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 113.Poynard T, McHutchison J, Manns M, Myers RP, Albrecht J. Biochemical surrogate markers of liver fibrosis and activity in a randomized trial of peginterferon alfa-2b and ribavirin. Hepatology. 2003;38:481–92. doi: 10.1053/jhep.2003.50319. [DOI] [PubMed] [Google Scholar]
- 114.Myers RP, Tenturier MH, Ratziu V, et al. Prediction of liver histological lesions with biochemical markers in patients with chronic hepatitis B. J Hepatol. 2003;39:222–30. doi: 10.1016/s0168-8278(03)00171-5. [DOI] [PubMed] [Google Scholar]
- 115.Forns X, Ampurdanes S, Llovet JM, et al. Identification of chronic hepatitis C patients without fibrosis by a simple predictive model. Hepatology. 2002;36:986–92. doi: 10.1053/jhep.2002.36128. [DOI] [PubMed] [Google Scholar]
- 116.Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38:518–26. doi: 10.1053/jhep.2003.50346. [DOI] [PubMed] [Google Scholar]
- 117.Berg T, Sarrazin C, Hinrichsen H, et al. Does noninvasive staging of fibrosis challenge liver biopsy as a gold standard in chronic hepatitis C? Hepatology. 2004;39:1456–57. doi: 10.1002/hep.20226. [DOI] [PubMed] [Google Scholar]
- 118.Le Calvez S, Thabut D, Messous D, et al. The predictive value of Fibrotest vs. APRI for the diagnosis of fibrosis in chronic hepatitis C. Hepatology. 2004;39:862–63. doi: 10.1002/hep.20099. [DOI] [PubMed] [Google Scholar]
- 119.Giannini E, Testa R. Noninvasive diagnosis of fibrosis: the truth is rarely pure and never simple. Hepatology. 2003;38:1312–3. doi: 10.1053/jhep.2003.50500. [DOI] [PubMed] [Google Scholar]
- 120.Patel K, Gordon SC, Jacobson I, et al. Evaluation of a panel of non-invasive serum markers to differentiate mild from moderate-to-advanced liver fibrosis in chronic hepatitis C patients. J Hepatol. 2004;41:935–42. doi: 10.1016/j.jhep.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 121.Rosenberg WM, Voelker M, Thiel R, et al. Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology. 2004;127:1704–13. doi: 10.1053/j.gastro.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 122.Kelleher TB, Mehta SH, Bhaskar R, et al. Prediction of hepatic fibrosis in HIV/HCV co-infected patients using serum fibrosis markers: the SHASTA index. J Hepatol. 2005;43:78–84. doi: 10.1016/j.jhep.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 123.Rosenberg WM. Rating fibrosis progression in chronic liver diseases. J Hepatol. 2003;38:357–60. doi: 10.1016/s0168-8278(03)00010-2. [DOI] [PubMed] [Google Scholar]
- 124.Afdhal NH, Nunes D. Evaluation of liver fibrosis: a concise review. Am J Gastroenterol. 2004;99:1160–74. doi: 10.1111/j.1572-0241.2004.30110.x. [DOI] [PubMed] [Google Scholar]
- 125.Parkes J, Guha IN, Roderick P, Rosenberg W. Performance of serum marker panels for liver fibrosis in chronic hepatitis C. J Hepatol. 2006;44:462–74. doi: 10.1016/j.jhep.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 126.Castera L, Vergniol J, Foucher J, et al. Prospective comparison of transient elastography, Fibrotest, APRI, and liver biopsy for the assessment of fibrosis in chronic hepatitis C. Gastroenterology. 2005;128:343–50. doi: 10.1053/j.gastro.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 127.Colletta C, Smirne C, Fabris C, et al. Value of two noninvasive methods to detect progression of fibrosis among HCV carriers with normal aminotransferases. Hepatology. 2005;42:838–45. doi: 10.1002/hep.20814. [DOI] [PubMed] [Google Scholar]
- 128.Low TY, Leow CK, Salto-Tellez M, Chung MC. A proteomic analysis of thioacetamide-induced hepatotoxicity and cirrhosis in rat livers. Proteomics. 2004;4:3960–74. doi: 10.1002/pmic.200400852. [DOI] [PubMed] [Google Scholar]
- 129.Callewaert N, Van Vlierberghe H, Van Hecke A, et al. Noninvasive diagnosis of liver cirrhosis using DNA sequencer-based total serum protein glycomics. Nat Med. 2004;10:429–34. doi: 10.1038/nm1006. [DOI] [PubMed] [Google Scholar]
- 130.Asselah T, Bieche I, Laurendeau I, et al. Liver Gene Expression Signature of Mild Fibrosis in Patients With Chronic Hepatitis C. Gastroenterology. 2005;129:2064–2075. doi: 10.1053/j.gastro.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 131.Friedman SL. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]
- 132.Pinzani M, Rombouts K, Colagrande S. Fibrosis in chronic liver diseases: diagnosis and management. J Hepatol. 2005;42(Suppl1):S22–36. doi: 10.1016/j.jhep.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 133.Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3:95–107. doi: 10.1016/s1542-3565(04)00445-8. [DOI] [PubMed] [Google Scholar]
- 134.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fausto N. Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477–87. doi: 10.1002/hep.20214. [DOI] [PubMed] [Google Scholar]
- 136.Gupta S, Chowdhury JR. Therapeutic potential of hepatocyte transplantation. Semin Cell Dev Biol. 2002;13:439–46. doi: 10.1016/s1084952102001325. [DOI] [PubMed] [Google Scholar]
- 137.Strom S, Fisher R. Hepatocyte transplantation: new possibilities for therapy. Gastroenterology. 2003;124:568–71. doi: 10.1053/gast.2003.50072. [DOI] [PubMed] [Google Scholar]
- 138.Kobayashi N, Ito M, Nakamura J, et al. Hepatocyte transplantation in rats with decompensated cirrhosis. Hepatology. 2000;31:851–7. doi: 10.1053/he.2000.5636. [DOI] [PubMed] [Google Scholar]
- 139.Ahmad TA, Eguchi S, Yanaga K, et al. Role of intrasplenic hepatocyte transplantation in improving survival and liver regeneration after hepatic resection in cirrhotic rats. Cell Transplant. 2002;11:399–402. [PubMed] [Google Scholar]
- 140.Cai J, Ito M, Nagata H, et al. Treatment of liver failure in rats with end-stage cirrhosis by transplantation of immortalized hepatocytes. Hepatology. 2002;36:386–94. doi: 10.1053/jhep.2002.34614. [DOI] [PubMed] [Google Scholar]
- 141.Nagata H, Ito M, et al. Treatment of cirrhosis and liver failure in rats by hepatocyte xenotransplantation. Gastroenterology. 2003;124:422–31. doi: 10.1053/gast.2003.50065. [DOI] [PubMed] [Google Scholar]
- 142.Malhi H, Gorla GR, Irani AN, Annamaneni P, Gupta S. Cell transplantation after oxidative hepatic preconditioning with radiation and ischemia-reperfusion leads to extensive liver repopulation. Proc Natl Acad Sci U S A. 2002;99:13114–19. doi: 10.1073/pnas.192365499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Benten D, Kumaran V, Joseph B, et al. Hepatocyte transplantation activates hepatic stellate cells with beneficial modulation of cell engraftment in the rat. Hepatology. 2005;42:1072–81. doi: 10.1002/hep.20889. [DOI] [PubMed] [Google Scholar]
- 144.Matsuno Y, Iwata H, Umeda Y, et al. Hepatocyte growth factor gene transfer into the liver via the portal vein using electroporation attenuates rat liver cirrhosis. Gene Ther. 2003;10:1559–66. doi: 10.1038/sj.gt.3302052. [DOI] [PubMed] [Google Scholar]
- 145.Malhi H, Irani AN, Gagandeep S, Gupta S. Isolation of human progenitor liver epithelial cells with extensive replication capacity and differentiation into mature hepatocytes. J Cell Sci. 2002;115:2679–88. doi: 10.1242/jcs.115.13.2679. [DOI] [PubMed] [Google Scholar]
- 146.Nowak G, Ericzon BG, Nava S, et al. Identification of expandable human hepatic progenitors which differentiate into mature hepatic cells in vivo. Gut. 2005;54:972–9. doi: 10.1136/gut.2005.064477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6:1229–34. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 148.Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422:901–4. doi: 10.1038/nature01539. [DOI] [PubMed] [Google Scholar]
- 149.Willenbring H, Bailey AS, Foster M, et al. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat Med. 2004;10:744–8. doi: 10.1038/nm1062. [DOI] [PubMed] [Google Scholar]
- 150.Sakaida I, Terai S, Yamamoto N, et al. Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology. 2004;40:1304–11. doi: 10.1002/hep.20452. [DOI] [PubMed] [Google Scholar]
- 151.Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology. 2006;43:2–8. doi: 10.1002/hep.21015. [DOI] [PubMed] [Google Scholar]
- 152.Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science. 2000;287:1253–8. doi: 10.1126/science.287.5456.1253. [DOI] [PubMed] [Google Scholar]
- 153.Edwards JT, Macdonald GA. Hepatocellular carcinoma. Curr Opin Gastroenterol. 2000;16:275–281. doi: 10.1097/00001574-200005000-00011. [DOI] [PubMed] [Google Scholar]