

Abstract
Translation is often repressed in cell lines that are exposed to hypoxic conditions (0.5% – 1.5% O2) but this repression requires prolonged exposure (> 16 h). We report here that prolonged exposure to hypoxia results in the depletion of glucose from the media and that the loss of glucose correlates with the shut down in translation. Furthermore, we show that the addition of glucose or reoxygenation restores translation in hypoxic PC3 cells. This indicates that both glucose depletion and hypoxia are required for translational repression. We also show that eIF2α phosphorylation is reversed by glucose addition. Moreover, we present data that strongly indicate that eIF2α phosphorylation as well as the translational inhibition that occurs when cells are grown under conditions of glucose depletion and hypoxia is pancreatic eIF2α kinase (PERK) independent. We believe this is the first report to show that glucose depletion is required for translational repression under hypoxic conditions and that this explains why prolonged exposure to hypoxia is required for this inhibition. Since the physiological conditions that lead to tumor hypoxia would also likely lead to reduced glucose levels, understanding the interplay of glucose and hypoxia in regulating tumor metabolism will provide important information on the growth and development of solid tumors.
Keywords: hypoxia, glucose, translation, eIF2α, PERK
INTRODUCTION
Low oxygen environments have been shown to repress translation in a variety of cell lines. The impact on cellular translation is dependent on both the severity and duration of the low oxygen stress (Kraggerud et al. 1995; Stein et al. 1995; Tinton and Buc-Calderon 1999; Lang et al. 2002; Koritzinsky et al. 2005, 2006; Liu et al. 2006; Thomas and Johannes 2007). Under anoxic conditions (oxygen levels below 0.02%) translation is rapidly inhibited (Blais et al. 2004; Bi et al. 2005; Koritzinsky et al. 2005), while prolonged exposure (> 16 h) is required for translational repression under hypoxic (0.5% – 1.5% O2) conditions (Connolly et al. 2006; Liu et al. 2006). Although the reason(s) for this delay in the translational response is unclear, it appears that both anoxia and hypoxia effect translation by primarily inhibiting the initiation step of translation (Wouters et al. 2005; van den Beucken et al. 2006; Thomas and Johannes 2007).
The initiation of translation is the rate-limiting step in protein synthesis, and as such translational regulation is often mediated at this step (Pain 1996; Kozak 1999). To begin translation the 43S preinitiation complex, which consists of the 40S ribosomal subunit, the ternary complex eukaryotic translation initiation factor 3 (eIF3), and other eIFs, binds to the 5′-cap of an mRNA through interactions with the cap-binding complex (eIF4F) (Gingras et al. 1999b). eIF4F consists of three subunits (eIF4E, eIF4G, and eIF4A) and binds the 5′-cap of an mRNA via direct interactions with eIF4E (Kozak 1992; Pain 1996; Aronica et al. 1997). Reduced oxygen levels appear to regulate translation initiation at two major steps: (1) by inhibiting the activity of the cap-binding complex, via reducing the availability of eIF4E (Arsham et al. 2003; Connolly et al. 2006), and (2) by blocking the recycling of the ternary complex by inhibiting eIF2 function (Koumenis et al. 2002; Park et al. 2005; Blais et al. 2006). Acute anoxia primarily blocks recycling of the ternary complex, while chronic hypoxia appears to inhibit the activity of the cap-binding complex in addition to blocking recycling of the ternary complex (Wouters et al. 2005; Koritzinsky et al. 2006; van den Beucken et al. 2006).
Anoxia's rapid affect on global translation seems to be mediated by the unfolded protein response through activation of the pancreatic eIF2α kinase (PERK) (Harding et al. 2000a; Koumenis et al. 2002; Ron 2002). PERK phosphorylates eIF2α, which blocks the ability of eIF2 to exchange GDP with GTP. In the absence of eIF2-GTP the ternary complex cannot be recycled and translation is inhibited (Pain 1996; Bi et al. 2005). Unlike anoxia, short-term hypoxia has no effect on translation while prolonged exposure (> 16 h) represses translation in some but not all cell lines. Additionally, the magnitude of this repression is quite variable. In some cell lines translation is only inhibited by < 50% even after 48 h (Tinton and Buc-Calderon 1999; Arsham et al. 2003; Connolly et al. 2006), while other cell lines show 80% – 90% inhibition of translation after 20 h of exposure to hypoxic conditions (Thomas and Johannes 2007). The precise mechanism required for this translational repression under chronic hypoxia remains elusive, but several reports have implicated mTOR inactivation as well as eIF2α phosphorylation (McCay et al. 1935; Connolly et al. 2006).
There are two mTOR complexes in the cell, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Kim et al. 2002; Sarbassov et al. 2004). mTORC1 is rapamycin sensitive and plays an important role in regulating the availability of the cap-binding complex for translation by phosphorylating and modulating the activity of the eIF4E binding proteins (4EBPs) (Gingras et al. 1999a; Hay and Sonenberg 2004). Under growth stimulatory conditions, mTORC1 is active and phosphorylates the 4EBPs. Hyperphosphorylated 4EBPs do not interact with eIF4E and thus allow eIF4E to participate in the cap-binding complex, promoting translation. When mTORC1 is inactivated, as has been reported to occur during hypoxia, the 4EBPs become hypophosphorylated, which promotes their interaction with eIF4E. The binding of eIF4E to the 4EBPs sequesters eIF4E and blocks its ability to participate in the cap-binding complex, resulting in translational repression. In addition to this global effect on translation, mTORC1 activity is also required for the efficient translation of a subset of mRNAs that contain a 5′ terminal oligo-pyrimidine tract (5′ TOP) (Gingras et al. 2004; Wouters et al. 2005). Since most ribosomal protein mRNAs (rpmRNAs) are 5′ TOP containing mRNAs (Levy et al. 1991), hypoxia, through the inactivation of mTORC1, likely results in the reduction in ribosomal protein production. mTORC1 also phosphorylates and activates ribosomal protein S6 kinase (rpS6K), which stimulates cell growth and ribosomal biosynthesis. In contrast, mTORC2 is not rapamycin sensitive and does not phosphorylate rpS6K or the 4EBPs and is not directly involved with regulating cap-dependent translation.
In addition to inhibiting translation initiation, chronic hypoxia has also been reported to result in the phosphorylation of translational elongation factor 2 (EF2) (Connolly et al. 2006; Liu et al. 2006). Phosphorylation of EF2 prevents it from participating of translation elongation, resulting in the reduction in translation (Browne and Proud 2002). However the impact of this phosphorylation on translation remains unclear because in HEK293 and rh30 cells EF2 phosphorylation does not correlate with translational repression (Liu et al. 2006) while in MCF10A cells EF2 phosphorylation is required for full inhibition of translation under chronic hypoxia (Connolly et al. 2006).
The requirement for prolonged exposure to hypoxia to induce translational inhibition may reflect changes in cellular metabolism or gene expression that require long periods of time to manifest themselves or may reflect changes in the media over time. This study shows that glucose depletion from the media is required for translational repression under hypoxic conditions in PC3 cells. We also show that glucose is utilized at an accelerated rate during hypoxia. Additionally our data strongly suggest that the phosphorylation of eIF2α under chronic hypoxia is PERK independent. Together this report provides important new information on the role of glucose in regulating translation and explains the delayed translational repression under hypoxic conditions. Since the conditions that result in hypoxia in tumors would also lead to reduced glucose levels, understanding the interplay of glucose and oxygen deprivation on cellular metabolism will likely provide new insights on the role of translation in regulating tumor growth.
RESULTS
Conditioned media from hypoxic cells accelerates translational repression during hypoxia
One of the curious observations about the impact of hypoxia on translation in PC3 cells is that it takes > 16 h of exposure to impact translation. In order to evaluate if this prolonged exposure is required for cellular adaptation to hypoxia or is due to alterations in the media, we analyzed the impact of fresh media (FM) and hypoxia conditioned media (HM) on translation. As shown in Figure 1, the addition of FM to normoxic (N) cells had no impact on translation. Exposure of cells to hypoxia for 20 h (H-20) resulted in a threefold reduction in translational activity, as has been previously reported (Thomas and Johannes 2007). When fresh media was added back to the hypoxic cells (H-20 FM), translation was restored to a level that was significantly higher (168%) than that found in normoxic cells. As expected there was no impact on translation when cells were exposed to hypoxia for only 3 h (H-3). However, when cells were exposed to hypoxia for 3 h in the presence of HM, translation was inhibited fivefold. These data show that HM markedly accelerates (from 20 h to 3 h) the translational inhibition observed under hypoxia and that FM can restore translation to hypoxic cells. Together these data indicate that a compound is either consumed from the media or secreted into the media that is directly involved with the translational inhibition that occurs during hypoxia.
FIGURE 1.

Fresh complete media reverses the hypoxia-mediated translational repression in PC3 cells. PC3 cells were maintained in normoxic conditions (N) or placed in a hypoxic chamber for 3 h (H-3) or 20 h (H-20). One hour before labeling, complete media (FM) was added to the cells. Conditioned hypoxic media (HM) was added to the cells immediately before placing into hypoxia for 3 h (H-3 HM). The cells were labeled with 35S-met/cys and the cellular translational activity was determined (%TCA/Total). Standard error bars are shown.
Glucose restores translation to hypoxic cells
Previous reports have implied that translational repression may be mediated through the loss of ATP and/or increases in the ADP/ATP ratio under prolonged hypoxic exposure (Liu et al. 2006). There are several sources of energy found in tissue culture media, including glucose (glc), glutamine (gln), and pyruvate (pyr). In order to evaluate the impact of these energy sources on translation we added each one of these compounds to translationally repressed hypoxic PC3 cells. As shown in Figure 2, exposure of cells to hypoxia for 20 h resulted in the inhibition of translation. When glucose was added to hypoxic cells, translation was restored (180% of normoxic) to a similar extent as occurs when fresh media was added (Fig. 1). The addition of glutamine or pyruvate had no effect on the translational repression in hypoxic cells. Reoxygenating the hypoxic cells by returning them to normoxic conditions for 1 h (Reoxy) fully restored translation. This latter observation is very interesting because translation fully recovers even though glucose is not added to the media.
FIGURE 2.

Glucose reverses the hypoxia-mediated translational repression in PC3 cells. PC3 cells were maintained in normoxic conditions (N) or placed in a hypoxic chamber for 20 h (H-20). One hour before harvesting the cells, glucose (glc), glutamine (gln), or pyruvate (pyr) was added to the media. A plate of hypoxic cells was returned to normoxic conditions 1 h prior to harvesting (Reoxy). Standard deviation bars are shown.
Glucose is utilized at an accelerated rate in hypoxic cells
If glucose depletion is required for translational repression under hypoxic conditions, then there should be lower levels of glucose in the media of hypoxic cells, as compared to normoxic cells. As shown in Figure 3, glucose began to be utilized at an accelerated rate after 4 h of exposure to hypoxic conditions, and by 15 h only 2% (1.9 mg/100 mL) of the glucose remained. In contrast, 75% (62 mg/100 mL) of the glucose remained in the media from normoxic cells after 15 h. These data demonstrate that hypoxic cells utilize glucose at an accelerated rate, as compared to normoxic cells. Even more interesting was the finding that glucose became nondetectable (< 1.0 mg/100 mL) coincidentally with translational repression (16 h).
FIGURE 3.

Glucose utilization is accelerated during hypoxia. PC3 cells were grown in hypoxic (△) or normoxic conditions (▲) and the media analyzed for glucose content at the times (hr) indicated. The amount of glucose present in the media at 0 h was set to 100%. Standard error bars are shown.
Glucose depletion does not impact translation under normoxic conditions
In order to establish if glucose depletion is solely responsible for translational repression under hypoxic conditions, we analyzed the impact of glucose depletion on normoxic cells. As shown in Figure 4A, glucose depletion had no effect on the translational activity of PC3 cells grown under normoxic conditions. However, when PC3 cells were grown in media lacking glucose, glutamine, and pyruvate, translation was repressed ∼50% (Fig. 4B). The addition of glucose or pyruvate to the media only had a marginal impact while addition of glutamine completely restored translation. Consistent with this finding, PC3 cells grown in the absence of glutamine were translationally repressed while glucose depletion had no effect (data not shown). Thus glucose depletion is required for translational repression under hypoxic conditions while glutamine depletion results in translational repression under normoxic conditions.
FIGURE 4.

Glucose does not significantly impair translation under normoxic conditions. (A) PC3 cells were grown in media containing 0, 0.1, 1.0, and 10 g/L glucose under normoxic conditions. Cells were harvested after 20 h and the translational activity was determined. As a control (C), cells were grown in media (containing 1 g/L glucose) for 48 h. (B) PC3 cells were grown under normoxic (N) conditions in complete media (+glc, +gln, +pyr) or media minus glucose, glutamine, and pyruvate (−glc, −gln, −pyr). After 20 h, glc, gln, or pyr was added back to the media 1 h prior to labeling. Cells were labeled and the translational activity (%TCA/Total) was determined. SEM bars are shown.
Addition of glucose to hypoxic cells results in the activation of mTORC1 and dephosphorylation of eIF2α
It has been reported that the translational repression that occurs during chronic hypoxia correlates with the inactivation of mTOR, phosphorylation of eIF2α, and/or phosphorylation of EF2 (Arsham et al. 2003; Wouters et al. 2005; Connolly et al. 2006; Liu et al. 2006; Thomas and Johannes 2007). Since glucose restores translation to hypoxic cells we wanted to examine the effects of glucose on these factors. To this end we maintained cells in complete media under hypoxic conditions for 20 h and then added glucose or glutamine to the media for 1 h. The cells were then harvested and the phosphorylation status of EF2, rpS6, and eIF2α was monitored. As a control, hypoxic cells (without added glc or gln) were returned to a normoxic environment for 1 h (Reoxy). Figure 5A shows that EF2 is phosphorylated under hypoxia in PC3 cells. Glucose, glutamine, and reoxygenation all reduced the level of phosphorylated EF2 relative to the hypoxic sample, but the degree of this reduction varied considerably among experiments (data not shown). The phosphorylation status of rpS6 was used as an indirect measurement of mTORC1 activity because this phosphorylation was completely rapamycin sensitive (Fig. 5B; Gingras et al. 2004; Wouters et al. 2005). Figure 5, A and B, shows that the amount of phosphorylated rpS6 was markedly reduced in hypoxic cells, and glucose addition or reoxygenation restored the amount of phosphorylated rpS6 to normoxic levels. Glutamine addition did not restore the phosphorylation of rpS6. Interestingly, the loss of phosphorylated rpS6 always correlated with a reduction in the amount of total rpS6. The amount of phosphorylated eIF2α was also increased under hypoxic conditions. Quantitative analysis of the data in Figure 5 revealed that the addition of glucose or reoxygenation reduced the amount of phosphorylated eIF2α relative to total eIF2α by 47% and 20%, respectively. In contrast, glutamine addition increased the ratio of phosphorylated eIF2α by 34%. In all cases the amount of phosphorylated eIF2α was significantly higher in hypoxic cells, as compared to normoxic cells. Taken together, the data in Figure 5 show that addition of glucose to hypoxic cells results in the activation of mTORC1 and the dephosphorylation of eIF2α.
FIGURE 5.

Glucose reverses the effect of hypoxia on the phosphorylation status on rpS6 and eIF2α. (A) Cells were maintained in complete media (*) under normoxic (N) conditions or placed in a hypoxic chamber for 20 h (H). One hour before harvesting, glucose (glc) or glutamine (gln) was added to the media. As a control, hypoxic cells were returned to a normoxic environment for 1 h (Reoxy). Cells were harvested and the phosphorylation status of eIF2α, rpS6, and EF2 was determined by Western analysis. Actin was used as a loading control. (B) Cells were treated with rapamycin and the effect on the phosphorylation of eIF2α, rpS6, and EF2 was analyzed in cells exposed to normoxia (N), hypoxia (H) for 20 h, or cells exposed to hypoxia for 20 h followed by glucose addition for 1 h. Additionally one set of hypoxic cells was returned to normoxic conditions for 1 h (O2). Actin was used as a loading control. (C) The translational activity was measured in PC3 cells grown under normoxic (N) and hypoxic (H-20) conditions in the presence or absence of rapamycin (20 ng/mL). After 20 h of hypoxic treatment, glucose was supplemented into the media (H-20 + glc) for 1 h or returned to normoxic conditions for 1 h (Reoxy) prior to analysis. The translational activity in the normoxic sample without rapamycin was set to 100%. The black bar represents rapamycin treatment (rap) and the gray bar represents untreated cells (control).
mTORC1 activity is not required for translational recovery of hypoxic cells
The activity of mTORC1 correlated well with the recovery of translation in hypoxic cells either by reoxygenation or by glucose addition. To determine whether mTORC1 activity was required for restoration of translation we added rapamycin, a potent inhibitor of mTORC1, 1 h prior to addition of glucose or reoxygenation. As shown in Figure 5C, rapamycin inhibited translation by ∼30% in normoxic cells, as previously reported (Thomas and Johannes 2007), and slightly enhanced the translational repression under hypoxic conditions. The addition of glucose or reoxygenation restored translation in the absence or presence of rapamycin (Fig. 5C) while completely blocking the phosphorylation of rpS6 (Fig. 5B). This strongly indicates that mTORC1 activity is not required for translational recovery.
The translational repression that occurs when cells are grown in glucose and glutamine depleted media under normoxic conditions does not require eIF2α phosphorylation
Analysis of eIF2α, rpS6, and EF2 during the translational repression that occurs when cells were grown under normoxic conditions in depleted media showed that EF2 is phosphorylated and rpS6 is dephosphorylated, while eIF2α phosphorylation was unaffected (Fig. 6). This indicates that eIF2α phosphorylation is not required for translational repression under these conditions. Restoration of mTORC1 activity, as measured by rpS6 phosphorylation, occurred with glucose or glutamine addition. Since translation is restored only when glutamine is added back to the depleted media indicates that mTORC1 activity is not required for translational recovery under these conditions. Since glucose addition results in the dephosphorylation of EF2 but not translational recovery suggests that phosphorylation of EF2 is not required for translational inhibition under these conditions. It was unexpected to find that eIF2α phosphorylation only occurred when glutamine was added and translation was restored because eIF2α phosphorylation usually correlates with translational repression. This suggests that this level of eIF2α phosphorylation does not inhibit translation under normoxic conditions. Overall these data indicate that the translational repression under hypoxic conditions utilizes a different mechanism than translational repression that occurs under depleted conditions in normoxic cells.
FIGURE 6.
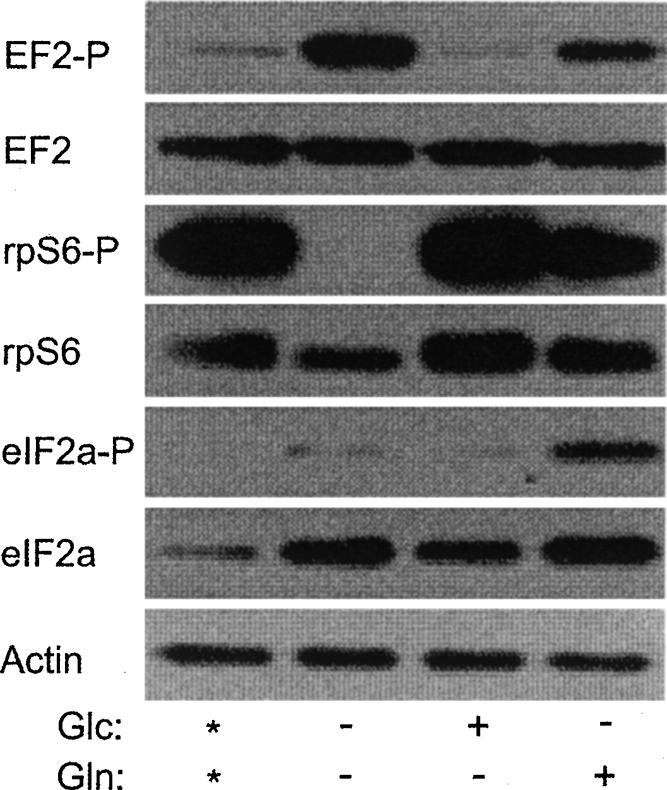
Glucose and glutamine depletion does alter the phosphorylation status of EF2, rpS6, and eIF2α. Cells were grown under normoxic conditions in complete media (*) or complete media minus glucose (glc), glutamine (gln), or both. Cells were harvested and the phosphorylation status of eIF2α, rpS6, and EF2 was determined by Western analysis. Actin was used as a loading control.
The translational repression that occurs during chronic hypoxia appears to be PERK independent
It has been reported that PERK activity is induced, via the integrated stress response, during acute anoxia and phosphorylates eIF2α resulting in translational repression (Bi et al. 2005; Blais et al. 2006). In order to evaluate if PERK activity is required for the translational repression that occurs under chronic hypoxia, we used RNA interference to knock down PERK and evaluated the impact on translational repression. Additionally, we evaluated if HIF1 or HIF2 activity was required for this translational repression by using a siRNA directed against HIF1β. HIF1β is the binding partner of both HIF1α and HIF2α, and by knocking down HIF1β the formation of both of these transcription complexes is blocked. As shown in Figure 7A, siRNAs directed against the PERK and HIF1β mRNAs specifically reduced expression of their respective mRNAs by 84% and 88%, respectively. A siRNA targeting a scrambled sequence (scr) had no effect on either of these mRNAs. As shown in Figure 7B, hypoxic exposure of cells transfected with the SCR, PERK, or HIF1β siRNA reduced translation by 83%, 88%, and 67%, respectively.
FIGURE 7.

Knockdown of PERK does not impact the hypoxia-mediated translational repression. (A) PC3 cells were transfected with siRNAs targeting PERK, HIF1β, or a scrambled (scr) sequence. Four days after transfection RNA was harvested and the amount of PERK (open bars) or HIF1β (dark bars) mRNA present in the cells was determined by qRT-PCR. All data were normalized to the amount of cyclophilin A mRNA. The ratio of PERK/cyclophilin A and the ratio of HIF1β/cyclophilin A found in the cells transfected with a scrambled siRNA were set to 100% (relative mRNA conc.). A control (Cnt) using untransfected cells is also shown. (B) Cells were treated with siRNAs targeting PERK, HIF1β, or a scrambled (scr) sequence for 3 d. Cells were then transferred to a hypoxic environment for 20 h (dark bars) or maintained in a normoxic environment (open bars) and analyzed for translational activity. A separate plate of hypoxic cells was returned to normoxic conditions (Reoxy, gray bars) for 1 h prior to analysis. The translational activity of the cells under normoxic conditions was set to 100%. (C) PC3 cells were transfected with siRNAs targeting a scrambled (scr) sequence or the PERK mRNA and the amount of the ATF3 mRNA (open bars) and PERK mRNA (dark bars) present after 6 h of thapsigargin treatment is shown. (D) PC3 cells were transfected with siRNAs targeting a scramble (scr) sequence or the PERK mRNA and maintained in a normoxic environment (open bars) or exposed to hypoxia for 20 h (dark bars) and the relative amount of the ATF 3 mRNA (% ATF3 mRNA) is shown. The amount of ATF3 mRNA in the hypoxic cells transfected with the scr siRNA was set to 100%. (E) The relative amount of the PERK mRNA found in the PC3 cells transfected with the scramble or PERK siRNA used in D is shown. Standard deviation bars are shown. All RNAs were normalized to the cyclophilin A mRNA.
To verify that the PERK knockdown was sufficient to impact PERK-dependent physiological responses we analyzed the ATF3 mRNA, whose expression is dependent on PERK activity (Jiang et al. 2004), in thapsigargin-treated PC3 cells. Thapsigargin, through activation of the unfolded protein response, is a potent inducer of PERK. PERK stimulates the translational induction of the transcription factor ATF4 (Harding et al. 2000a,b), which in turn activates the transcription of ATF3. As shown in Figure 7C, cells treated with the PERK siRNA, but not with a scrambled siRNA, reduced the expression of ATF3 by fivefold in thapsigargin-treated PC3 cells, which demonstrates that the knockdown is sufficient to impact downstream events mediated by PERK. The fact that the PERK knockdown was sufficient to inhibit downstream effects mediated by PERK, i.e., ATF3 expression, in response to thapsigargin, but had very little, if any, impact on the translational repression under hypoxic conditions supports the hypothesis that PERK activity is not required for the translation repression that occurs during hypoxia. Consistent with this hypothesis, we found that the PERK knockdown had no impact on the induction of the ATF3 mRNA under conditions of hypoxia and glucose deprivation (Fig. 7D), which indicates that hypoxic treatment induces ATF3 in a PERK independent manner.
In order to verify that the knockdown of HIF1β was sufficient to impact downstream events modulated by HIF1β, we analyzed the expression of a HIF1α target mRNA, Adrenomedullin (ADM) (Kitamura et al. 1993; Udono et al. 2001). As shown in Figure 8A, cells treated with the HIF1β siRNA reduced the hypoxic expression of ADM by threefold, as compared to control cells. This effect was specific for the HIF1β siRNA because no effect on ADM expression was observed with the PERK siRNA. Thus the knockdown of HIF1β was sufficient to impact events modulated by HIF1β. Since translation continued to be reduced significantly in cells treated with the HIF1β siRNA we conclude that neither HIF1 nor HIF2 expression is required for the translational repression that occurs during hypoxia in PC3 cells. However, since the magnitude of the translational repression was reduced in the HIF1β knockdown we cannot rule out the possibility that HIF1 or HIF2 expression augments the translational repression that occurs during hypoxia.
FIGURE 8.

eIF2α phosphorylation and glucose utilization is not inhibited in cells treated with siRNAs targeting HIF1β or PERK. (A) PC3 cells were untransfected (cnt) or transfected with siRNAs targeting HIF1β or the PERK mRNA and exposed to hypoxia for 20 h. The amount of the ADM (open bars) and HIF1β (gray bars) mRNAs, relative to untransfected (cnt), are shown. (B) The phosphorylation status of eIF2α was analyzed in the Scr and PERK siRNA-treated cells under normoxic (N) and hypoxic (H) conditions. Actin was used as a loading control. (C) Glucose utilization during hypoxia was measured in control cells (◆) and cells treated with siRNAs directed against HIF1β (△), PERK (×), or a scrambled sequence (scr, □). Standard deviations bars are shown.
To determine if eIF2α continued to be phosphorylated under hypoxic conditions in the cells treated with the PERK siRNA, we analyzed the phosphorylation status of eIF2α. Quantitative analysis of the data shown in Figure 8B revealed that hypoxic treatment increased the amount of phosphorylated eIF2α relative to total eIF2α in the control, scr siRNA, and PERK siRNA-treated cells by 2.2-, 2.6-, and 2.4-fold, respectively. The data in Figures 7 and 8 strongly indicate that eIF2α is phosphorylated via a PERK independent mechanism during chronic hypoxia and is consistent with the hypothesis that eIF2α phosphorylation is required for translational repression under these conditions. Together these data indicate that hypoxia and glucose deprivation results in the phosphorylation eIF2α and induction of the ATF3 mRNA, similar to what occurs during the unfolded protein response. However, unlike the UPR, hypoxia and glucose deprivation appear to mediate these changes using a PERK independent mechanism.
As a control we analyzed the impact of the siRNAs on glucose utilization and found that the cells treated with the siRNAs targeting HIF1β and PERK showed slightly reduced glucose utilization at 4 h of hypoxic treatment, as compared to control cells or those transfected with a siRNA targeting a scrambled sequence (Fig. 8C). However by 16 h of hypoxic treatment all cells had equivalent levels of glucose that became undetectable by 20 h of hypoxic treatment.
DISCUSSION
Although it has been well documented that prolonged exposure to hypoxia is required for translational repression, the reason(s) for this delayed response has not been elucidated. Our data strongly support the hypothesis that glucose depletion is required for translational repression under hypoxic conditions and that the prolonged exposure time is needed for the glucose in the media to be consumed. Specifically we found that when glucose is present in the media hypoxia does not inhibit translation but when glucose is depleted hypoxia results in the dramatic inhibition of translation. We also observed that hypoxic PC3 cells utilize glucose at a greatly accelerated rate, as compared to normoxic cells, and that glucose reaches undetectable levels by 16 h of hypoxic treatment. This loss of glucose correlates with the translational repression that occurs during hypoxia (Thomas and Johannes 2007). These data, together with the fact that the addition of glucose restores translation in hypoxic cells, strongly support the conclusion that glucose depletion is required for translational repression during hypoxia in PC3 cells. This dependence on glucose depletion may be common and may help to explain why exposure to hypoxia for 16 h or longer is required for translational repression. It would be interesting to examine the rate of glucose consumption in cell lines that do not repress translation upon hypoxic exposure. Perhaps these cell lines do not consume glucose at an accelerated rate during hypoxia and therefore do not deplete glucose from the media, which would allow for translation to be maintained.
The increase in glucose consumption is likely explained by the hypoxia-mediated shift from oxidative phosphorylation to gylcolysis, often referred to as the Warburg effect (Warburg 1956; Shaw 2006). Hypoxia represses oxidative phosphorylation (Kim et al. 2006; Papandreou et al. 2006) and increases the expression of several glucose transporters, especially Glut1 and -3, which increases glucose transport into the cell (Semenza 2003; Schofield and Ratcliffe 2004; Brahimi-Horn et al. 2007; Thomas and Johannes 2007). This hypoxia-mediated increase in glucose utilization and potential glucose depletion is often overlooked when examining the impact of hypoxia on gene expression and cellular metabolism. It is possible that many of the cellular events attributed to chronic hypoxia may be a result of the combined stresses of hypoxia and glucose depletion.
Since it has been shown that glucose starvation can have a suppressive effect on translation under normoxic conditions in certain cell lines (Gomez et al. 2004; Vander Mierde et al. 2007) we wanted to determine if the translational repression that we observe under hypoxic conditions is merely a consequence of glucose depletion. Our data clearly show that short-term (20 h) glucose depletion under normoxic conditions has little impact on protein synthesis while dramatically inhibiting translation under hypoxic conditions. This strongly indicates that the translational repression that is observed during chronic hypoxia requires both glucose and oxygen depletion. Interestingly, glutamine, but not glucose, restores translation in normoxic cells that are energy starved while glucose, but not glutamine, restores translation under hypoxic conditions. This implies that the molecular mechanisms responsible for translational repression under hypoxia/glucose depletion and normoxia/glutamine depletion are distinct. Consistent with this hypothesis we find that eIF2α phosphorylation correlates with the translational repression in glucose depleted/chronically hypoxic cells but not in energy starved normoxic cells.
We find that chronic hypoxic treatment results in the loss of phosphorylated rpS6 (reflecting mTORC1 inactivation), the phosphorylation of eIF2α, and an increase in the phosphorylation status of EF2. Restoration of translation by glucose addition or reoxygenation correlated with mTORC1 activation and the reduction of phosphorylated eIF2α, while glutamine addition, which does not restore translation, did not. Together these data show that inactivation of mTORC1 and phosphorylation of eIF2α correlates with the hypoxia-mediated translational repression. However unlike previous reports (Arsham et al. 2003; Bernhardt et al. 2006; Connolly et al. 2006; Liu et al. 2006; Thomas and Johannes 2007) we show that both of these events are reversed by glucose addition as well as reoxygenation. The observation that mTORC1 inactivation occurs within a few hours of hypoxic treatment (Thomas and Johannes 2007) when glucose levels are still high (Fig. 3) indicates that mTORC1 activity is initially more responsive to oxygen levels than to glucose levels. However upon prolonged exposure to hypoxia, addition of glucose activates mTORC1. This suggests that the regulation of mTORC1 by hypoxia and glucose is altered when cells are exposed to chronic hypoxia. These observations and our previous studies (Thomas and Johannes 2007) strongly suggest that hypoxia alone can inactivate mTOR while eIF2α phosphorylation requires both hypoxia and glucose deprivation.
Although mTORC1 activity correlates well with the translational activity, several lines of evidence indicate that mTORC1 is not the key player in regulating global translation when cells are exposed to hypoxic and low glucose conditions. First, our previous work has shown that mTORC1 is inhibited by 2 h of hypoxic treatment while translational repression requires > 16 h of hypoxic treatment (Thomas and Johannes 2007); second, rapamycin treatment only has a modest effect on translation under normoxic conditions; and third, translational recovery by either glucose or reoxygenation is not affected by rapamycin treatment. Thus, we conclude that the inhibition of mTORC1 by hypoxia has only a modest impact on translation, most likely resulting from the specific translational repression of 5′ TOP mRNAs. Furthermore, its activity is not required for translational recovery by reoxygenation or glucose addition. These data appear to be in contrast to what has been reported to occur in the immortalized breast epithelium cell line MCF10A, in which mTOR inactivation and EF2 phosphorylation are responsible for the translational repression mediated by chronic hypoxia (Connolly et al. 2006). These authors also report that the more transformed breast cancer cell lines, HTB20 and CRL2324, had constitutively active mTOR and were refractory to the hypoxia-mediated translational repression (Connolly et al. 2006). Since PC3 cells are highly transformed they are more similar to the HTB20 and CRL2324 cells than to the MCF10A cells. Although mTORC1 is not constitutively activated in the PC3 cells, its inactivity is not required for translational repression under chronic hypoxia. Thus both prostate cancer and breast cancer cell lines uncouple regulation of mTORC1 from global translational repression. A similar phenomenon is also seen in HEK293 cells in which rpS6 is hypophosphorylated within 6 h of hypoxic treatment but translational repression requires greater than 16 h of exposure (Liu et al. 2006). Thus in these transformed cell lines mTORC1 activity is uncoupled from global translational repression. It would be interesting to determine if glucose consumption correlated with the translational repression in the MCF10A cell line and if glucose depletion in conjunction with hypoxia inhibits translation in the more transformed breast cancer cell lines. It is possible that the glucose dependency on translational repression during hypoxia is common in highly transformed cells and not in primary cells or weakly transformed cell lines.
Consistent with our previous work, our data indicate that the phosphorylation of eIF2α plays a central role in regulating translation when cells are grown in hypoxic and depleted glucose conditions. Similar observations have been reported to occur under acute anoxic conditions (Koumenis et al. 2002; Bi et al. 2005; Koritzinsky et al. 2006); however, the molecular mechanisms leading to the phosphorylation of eIF2α under the two conditions must be distinct. Under anoxic conditions the phosphorylation of eIF2α is rapid and PERK dependent while phosphorylation under chronic hypoxia is slow and appears to be PERK independent (Bernhardt et al. 2006; Blais et al. 2006; Connolly et al. 2006; Koritzinsky et al. 2006; van den Beucken et al. 2006). Since it has been shown that PERK can be activated by glucose deprivation under normoxic conditions (Elouil et al. 2007; Shang et al. 2007), it would be interesting to know if PERK activation during anoxia is influenced by glucose.
Although our knockdown data strongly support the hypothesis that the phosphorylation of eIF2α is PERK independent, we cannot completely rule out the possibility that the trace amounts of PERK remaining in the cells is sufficient to phosphorylate eIF2α when cells are grown under hypoxic and depleted glucose conditions. However, the fact that the PERK knockdown is sufficient to inhibit the expression of ATF3 in thapsigargin-treated cells, but not in hypoxic-treated cells, strongly suggests that if PERK were involved with the hypoxia-mediated translational repression and eIF2α phosphorylation we would have detected it in our system. If the phosphorylation of eIF2α is PERK independent, as our data indicate, it is likely that one of the other eIF2α kinases, such dsRNA-activated protein kinase (PKR) (Chong et al. 1992), general control of nitrogen metabolism kinase 2 (GCN2) (Dever et al. 1992), or the heme-regulated inhibitor kinase (HRI) (Chen and London 1995), might be involved with increasing the phosphorylation of eIF2α (Kaufman 2004). Conversely, chronic hypoxia and glucose deprivation might inactivate phosphatase PP1, which would reduce the dephosphorylation of eIF2α, resulting in a net increase in phosphorylated eIF2α (Vander Mierde et al. 2007). We are in the process of investigating the molecular mechanism involved with the increased phosphorylation of eIF2α during glucose depletion and chronic hypoxia.
Under these conditions in which mTORC1 is inactivated and eIF2α is phosphorylated it has been reported that certain mRNAs continue to be translated (Blais et al. 2004; Koritzinsky et al. 2006; Thomas and Johannes 2007). It is likely that these mRNAs have the ability to recruit ribosomes when cap-dependent translation is suppressed and there is a reduced level of ternary complex. One such mechanism is utilized by the ATF4 mRNA where the presence of short upstream open reading frames (uORFs) in its 5′ UTR blocks the ability of ribosomes to reinitiate at the ATF4 AUG when ternary complex is abundant (Lu et al. 2004). When the ternary complex becomes limited, such as when eIF2α is phosphorylated, ribosome bypass the second uORF and reinitiate translation at the ATF4 AUG stimulating translation of this protein. Another mechanism that may be involved is internal initiation of translation, in which internal ribosome entry sites (IRESs) directly recruit ribosomes to the mRNAs containing these elements (Hellen and Sarnow 2001; Komar and Hatzoglou 2005). IRES-mediated translation has been implicated in the translation of several mRNAs that continue to be translated during hypoxia such as HIF1α (Lang et al. 2002) and VEGFA (Miller et al. 1998) and is by definition cap independent and thus would not be impeded by a reduction in cap-binding complex mediated by mTORC1 inactivation. Additionally, the cricket paralysis-like virus (CrPV) intergenic IRES functions by mimicking the initiating tRNA and thus is not dependent on eIF2 activity or cap-binding complex (Wilson et al. 2000a,b). Although no cellular IRES has been shown to use a similar mechanism, the cationic amino acid transporter (cat-1), platelet derived growth factor, and VEGF IRESs have been reported to be stimulated when eIF2α is phosphorylated (Fernandez et al. 2002; Gerlitz et al. 2002). Thus it is possible that certain cellular IRESs may be able to bypass the need for both eIF4F and the ternary complex. Although it appears that certain mRNAs are refractory to the translational repression that occurs during hypoxia (Thomas and Johannes 2007), the precise mechanism(s) that is used by these mRNAs to recruit ribosomes remains to be elucidated. It is also important to note that although translation is severely reduced it is not completely blocked under these conditions, and therefore mRNAs can still recruit ribosomes but much less efficiently. Therefore, the loss of protein production that results from the suppression of translation can likely be overcome by increasing the abundance of the mRNA, without the need for an alternative mechanism of translational initiation.
Our work shows that the translational inhibition mediated by chronic hypoxia requires both a low oxygen environment and depleted glucose. This glucose dependency is likely mediated through changes in the ADP/ATP ratio or ATP levels, which are key signals that regulate cellular metabolism in response to energy stress (Liu et al. 2006). Moreover, our work strongly indicates that mTORC1 activity is rapidly inactivated by hypoxia alone but is not involved with the inhibition of global translation under conditions of chronic hypoxia and glucose depletion. In contrast, eIF2α phosphorylation appears to require both low glucose and low oxygen conditions and correlates well with the translational repression. These conditions are likely to be found in the hypoxic regions of tumors where inadequate blood flow would result in both reduced oxygen and glucose delivery. Under these conditions it is likely the translation is severely impaired. This inhibition of translation may have important consequences in tumor cell survival as well as sensitivity to chemo or radiation therapy. Therefore understanding the translational status of hypoxic tumor cells may provide important insights into the growth and treatment of tumors.
MATERIALS AND METHODS
Tissue culture and hypoxic treatment
PC3 (ATCC # CRL-1435) cells were grown in DMEM media (Mediatech) supplemented with 10% FBS (Mediatech) and Pen/Strep at 37°C in 5% CO2. For hypoxic treatment, cells were plated under normoxic conditions and grown for 16 – 20 h and then placed into a hypoxic chamber (Coy Laboratory) preequilibrated to 1.0% O2, 5% CO2 at 37°C. Oxygen levels were verified using an independent oxygen sensor (Drager, Inc.). Rapamycin (Alexis Biochemicals) was used at 2 μg/mL. For the glucose depletion experiments DMEM media without glucose was supplemented with the appropriate amount of glucose. For the reoxygenation experiments hypoxic cells were retuned to a normoxic environment (air, 5% CO2, 37°C) for 1 h before assaying or labeling. Cells were treated with 100 μM thapsigargin for 6 h prior to isolating RNA.
Metabolic labeling
Cells were labeled for 30 min by adding 70 μCi/mL of Pro-mix (Pro-mix L-35S, Amersham Bioscience) to the media (1 mL) in 12 well plates. After labeling, cycloheximide was added to 100 μg/mL and incubated for 5 min (to block further protein synthesis) and the amount of label incorporated into TCA perceptible (ppt) counts was performed as previously described (Thomas and Johannes 2007). Translational activity was determined by normalizing the TCA ppt counts to total label incorporation into the cells.
Glucose assays
The glucose concentration in the media was determined using the Glucose Assay Kit (Biovision Research Products) following the manufacturer's instructions. Media was changed 2 h before placing the cells in the hypoxic chamber and the 0 time point was set to 100%.
Immuno-blot analysis
Whole cell extracts were prepared by lysis in RIPA buffer containing HALTS Protease Inhibitor Cocktail (Pierce) using standard procedures. Protein concentration was determined using the Bio-Rad DC Kit. Thirty-five micrograms of protein were separated by 10% SDS-PAGE and transferred to Immobilon-P membrane (Millipore). Transfer efficiency was evaluated by Ponceu S staining of the membrane. Antibodies for RPS6 (5G10), phosphorylated RPS6 (ser 235/236), EF2, and phosphorylated EF2 (Thr-56), eIF2α, and phosphorylated eIF-2α (Ser 51) were purchased from Cell Signaling Technology. The blot was developed using the ECL Plus Detection Kit (Amersham Biosciences). After detection the blot was stripped and reprobed using anti-actin (Sigma). Quantitation was performed by determining the density of the bands from the resulting autoradiogram using the Quantity One software (Bio-Rad).
siRNA transfection
PC3 cells were transfected with siRNA (200 nM) directed against PERK (ID: 137,754), ARNT (ID: 106,533), or a scrambled siRNA. All siRNAs were purchased from Ambion. Transfections were performed using Oligofectamine (Invitrogen) according to the manufacturer's protocol. Forty-eight hours after transfection cells were subjected to hypoxic (1% O2) or normoxic conditions for 20 h. Cells were then metabolically labeled or RNA was isolated. The effectiveness of the knockdown was determined by qRT-PCR.
Quantitative real-time PCR (qRT-PCR)
RNA templates for qRT-PCR consisted of total RNA isolated using the RNA easy kit (Qiagen). Primer/probe sets were purchased from ABI and the gene and catalog number are as follows: GAPDH, Hs99999905-m1; PERK (EIF2AK3), Hs00178128_m; HIF1β, Hs00231048_m1; ADM, Hs00181605_m1; ATF3, Hs00231069_m1. For RNA analysis, cyclophilin A (Hs99999904_m1) was used as a loading control. Analysis was performed using the MxPro Software (Stratagene) using the MX 3000 and the Brilliant qRT-PCR system according to the manufacturer's instructions. All probes were tested in no RT control reactions, and all reactions were performed in duplicate or triplicate.
ACKNOWLEDGMENTS
We thank Dr. Shelly Waggoner for helpful discussions and critical reading and editing of the manuscript and Dr. Peter Sarnow for insights contributing to this work. This work is supported by the Public Health Service grant CA112086 from the National Cancer Institute.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.857308.
REFERENCES
- Aronica, S.M., Gingras, A.C., Sonenberg, N., Cooper, S., Hague, N., Broxmeyer, H.E. Macrophage inflammatory protein-1α and interferon-inducible protein 10 inhibit synergistically induced growth factor stimulation of MAP kinase activity and suppress phosphorylation of eukaryotic initiation factor 4E and 4E binding protein 1. Blood. 1997;89:3582–3595. [PubMed] [Google Scholar]
- Arsham, A.M., Howell, J.J., Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- Bernhardt, W.M., Schmitt, R., Rosenberger, C., Munchenhagen, P.M., Grone, H.J., Frei, U., Warnecke, C., Bachmann, S., Wiesener, M.S., Willam, C., et al. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006;69:114–122. doi: 10.1038/sj.ki.5000062. [DOI] [PubMed] [Google Scholar]
- Bi, M., Naczki, C., Koritzinsky, M., Fels, D., Blais, J., Hu, N., Harding, H., Novoa, I., Varia, M., Raleigh, J., et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–3481. doi: 10.1038/sj.emboj.7600777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais, J.D., Filipenko, V., Bi, M., Harding, H.P., Ron, D., Koumenis, C., Wouters, B.G., Bell, J.C. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 2004;24:7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais, J.D., Addison, C.L., Edge, R., Falls, T., Zhao, H., Wary, K., Koumenis, C., Harding, H.P., Ron, D., Holcik, M., et al. PERK-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006;26:9517–9532. doi: 10.1128/MCB.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahimi-Horn, M.C., Chiche, J., Pouyssegur, J. Hypoxia signaling controls metabolic demand. Curr. Opin. Cell Biol. 2007;19:223–229. doi: 10.1016/j.ceb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Browne, G.J., Proud, C.G. Regulation of peptide-chain elongation in mammalian cells. Eur. J. Biochem. 2002;269:5360–5368. doi: 10.1046/j.1432-1033.2002.03290.x. [DOI] [PubMed] [Google Scholar]
- Chen, J.J., London, I.M. Regulation of protein synthesis by heme-regulated eIF-2 α kinase. Trends Biochem. Sci. 1995;20:105–108. doi: 10.1016/s0968-0004(00)88975-6. [DOI] [PubMed] [Google Scholar]
- Chong, K.L., Feng, L., Schappert, K., Meurs, E., Donahue, T.F., Friesen, J.D., Hovanessian, A.G., Williams, B.R. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, E., Braunstein, S., Formenti, S., Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006;26:3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever, T.E., Feng, L., Wek, R.C., Cigan, A.M., Donahue, T.F., Hinnebusch, A.G. Phosphorylation of initiation factor 2 α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- Elouil, H., Bensellam, M., Guiot, Y., Vander Mierde, D., Pascal, S.M., Schuit, F.C., Jonas, J.C. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia. 2007;50:1442–1452. doi: 10.1007/s00125-007-0674-4. [DOI] [PubMed] [Google Scholar]
- Fernandez, J., Yaman, I., Sarnow, P., Snider, M.D., Hatzoglou, M. Regulation of internal ribosomal entry site-mediated translation by phosphorylation of the translation initiation factor eIF2α. J. Biol. Chem. 2002;277:19198–19205. doi: 10.1074/jbc.M201052200. [DOI] [PubMed] [Google Scholar]
- Gerlitz, G., Jagus, R., Elroy-Stein, O. Phosphorylation of initiation factor-2 α is required for activation of internal translation initiation during cell differentiation. Eur. J. Biochem. 2002;269:2810–2819. doi: 10.1046/j.1432-1033.2002.02974.x. [DOI] [PubMed] [Google Scholar]
- Gingras, A.C., Gygi, S.P., Raught, B., Polakiewicz, R.D., Abraham, R.T., Hoekstra, M.F., Aebersold, R., Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes & Dev. 1999a;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras, A.C., Raught, B., Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999b;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Gingras, A.C., Raught, B., Sonenberg, N. mTOR signaling to translation. Curr. Top. Microbiol. Immunol. 2004;279:169–197. doi: 10.1007/978-3-642-18930-2_11. [DOI] [PubMed] [Google Scholar]
- Gomez, E., Powell, M.L., Greenman, I.C., Herbert, T.P. Glucose-stimulated protein synthesis in pancreatic β-cells parallels an increase in the availability of the translational ternary complex (eIF2-GTP·Met-tRNAi) and the dephosphorylation of eIF2 α. J. Biol. Chem. 2004;279:53937–53946. doi: 10.1074/jbc.M408682200. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000a;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., Zhang, Y., Bertolotti, A., Zeng, H., Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000b;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Hay, N., Sonenberg, N. Upstream and downstream of mTOR. Genes & Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hellen, C.U., Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes & Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- Jiang, H.Y., Wek, S.A., McGrath, B.C., Lu, D., Hai, T., Harding, H.P., Wang, X., Ron, D., Cavener, D.R., Wek, R.C. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, R.J. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem. Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kim, D.H., Sarbassov, D.D., Ali, S.M., King, J.E., Latek, R.R., Erdjument-Bromage, H., Tempst, P., Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim, J.W., Tchernyshyov, I., Semenza, G.L., Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kitamura, K., Sakata, J., Kangawa, K., Kojima, M., Matsuo, H., Eto, T. Cloning and characterization of cDNA encoding a precursor for human adrenomedullin. Biochem. Biophys. Res. Commun. 1993;194:720–725. doi: 10.1006/bbrc.1993.1881. [DOI] [PubMed] [Google Scholar]
- Komar, A.A., Hatzoglou, M. Internal ribosome entry sites in cellular mRNAs: Mystery of their existence. J. Biol. Chem. 2005;280:23425–23428. doi: 10.1074/jbc.R400041200. [DOI] [PubMed] [Google Scholar]
- Koritzinsky, M., Seigneuric, R., Magagnin, M.G., van den Beucken, T., Lambin, P., Wouters, B.G. The hypoxic proteome is influenced by gene-specific changes in mRNA translation. Radiother. Oncol. 2005;76:177–186. doi: 10.1016/j.radonc.2005.06.036. [DOI] [PubMed] [Google Scholar]
- Koritzinsky, M., Magagnin, M.G., van den Beucken, T., Seigneuric, R., Savelkouls, K., Dostie, J., Pyronnet, S., Kaufman, R.J., Weppler, S.A., Voncken, J.W., et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. 2006;25:1114–1125. doi: 10.1038/sj.emboj.7600998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis, C., Naczki, C., Koritzinsky, M., Rastani, S., Diehl, A., Sonenberg, N., Koromilas, A., Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak, M. Regulation of translation in eukaryotic systems. Annu. Rev. Cell Biol. 1992;8:197–225. doi: 10.1146/annurev.cb.08.110192.001213. [DOI] [PubMed] [Google Scholar]
- Kozak, M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Kraggerud, S.M., Sandvik, J.A., Pettersen, E.O. Regulation of protein synthesis in human cells exposed to extreme hypoxia. Anticancer Res. 1995;15:683–686. [PubMed] [Google Scholar]
- Lang, K.J., Kappel, A., Goodall, G.J. Hypoxia-inducible factor-1α mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol. Biol. Cell. 2002;13:1792–1801. doi: 10.1091/mbc.02-02-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, S., Avni, D., Hariharan, N., Perry, R.P., Meyuhas, O. Oligopyrimidine tract at the 5′ end of mammalian ribosomal protein mRNAs is required for their translational control. Proc. Natl. Acad. Sci. 1991;88:3319–3323. doi: 10.1073/pnas.88.8.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L., Cash, T.P., Jones, R.G., Keith, B., Thompson, C.B., Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, P.D., Harding, H.P., Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCay, C.M., Crowell, M.F., Maynard, L.A. The effect of retarded growth upon the length of lifespan and upon the ultimate body size. J. Nutr. 1935;10:62–79. [PubMed] [Google Scholar]
- Miller, D.L., Dibbens, J.A., Damert, A., Risau, W., Vadas, M.A., Goodall, G.J. The vascular endothelial growth factor mRNA contains an internal ribosome entry site. FEBS Lett. 1998;434:417–420. doi: 10.1016/s0014-5793(98)01025-4. [DOI] [PubMed] [Google Scholar]
- Pain, V.M. Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem. 1996;236:747–771. doi: 10.1111/j.1432-1033.1996.00747.x. [DOI] [PubMed] [Google Scholar]
- Papandreou, I., Cairns, R.A., Fontana, L., Lim, A.L., Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Park, E.H., Lee, J.M., Blais, J.D., Bell, J.C., Pelletier, J. Internal translation initiation mediated by the angiogenic factor Tie2. J. Biol. Chem. 2005;280:20945–20953. doi: 10.1074/jbc.M412744200. [DOI] [PubMed] [Google Scholar]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov, D.D., Ali, S.M., Kim, D.H., Guertin, D.A., Latek, R.R., Erdjument-Bromage, H., Tempst, P., Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Schofield, C.J., Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Shang, J., Gao, N., Kaufman, R.J., Ron, D., Harding, H.P., Lehrman, M.A. Translation attenuation by PERK balances ER glycoprotein synthesis with lipid-linked oligosaccharide flux. J. Cell Biol. 2007;176:605–616. doi: 10.1083/jcb.200607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Stein, I., Neeman, M., Shweiki, D., Itin, A., Keshet, E. Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Mol. Cell. Biol. 1995;15:5363–5368. doi: 10.1128/mcb.15.10.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J.D., Johannes, G.J. Identification of mRNAs that continue to associate with polysomes during hypoxia. RNA. 2007;13:1116–1131. doi: 10.1261/rna.534807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinton, S.A., Buc-Calderon, P.M. Hypoxia increases the association of 4E-binding protein 1 with the initiation factor 4E in isolated rat hepatocytes. FEBS Lett. 1999;446:55–59. doi: 10.1016/s0014-5793(99)00185-4. [DOI] [PubMed] [Google Scholar]
- Udono, T., Takahashi, K., Nakayama, M., Yoshinoya, A., Totsune, K., Murakami, O., Durlu, Y.K., Tamai, M., Shibahara, S. Induction of adrenomedullin by hypoxia in cultured retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2001;42:1080–1086. [PubMed] [Google Scholar]
- van den Beucken, T., Koritzinsky, M., Wouters, B.G. Translational control of gene expression during hypoxia. Cancer Biol. Ther. 2006;5:749–755. doi: 10.4161/cbt.5.7.2972. [DOI] [PubMed] [Google Scholar]
- Vander Mierde, D., Scheuner, D., Quintens, R., Patel, R., Song, B., Tsukamoto, K., Beullens, M., Kaufman, R.J., Bollen, M., Schuit, F.C. Glucose activates a protein phosphatase-1-mediated signaling pathway to enhance overall translation in pancreatic β-cells. Endocrinology. 2007;148:609–617. doi: 10.1210/en.2006-1012. [DOI] [PubMed] [Google Scholar]
- Warburg, O. Origin of cancer cells. Oncologia. 1956;9:75–83. [PubMed] [Google Scholar]
- Wilson, J.E., Pestova, T.V., Hellen, C.U., Sarnow, P. Initiation of protein synthesis from the A site of the ribosome. Cell. 2000a;102:511–520. doi: 10.1016/s0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- Wilson, J.E., Powell, M.J., Hoover, S.E., Sarnow, P. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol. Cell. Biol. 2000b;20:4990–4999. doi: 10.1128/mcb.20.14.4990-4999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters, B.G., van den Beucken, T., Magagnin, M.G., Koritzinsky, M., Fels, D., Koumenis, C. Control of the hypoxic response through regulation of mRNA translation. Semin. Cell Dev. Biol. 2005;16:487–501. doi: 10.1016/j.semcdb.2005.03.009. [DOI] [PubMed] [Google Scholar]