

Abstract
The Dab1 docking protein is required for the proper organization of brain laminae and for a signal transduction pathway initiated by Reelin binding to the ApoER2 and VLDLR receptors on the cell surface of neurons. Dab1 physically interacts with APP, however, it is not known whether the APP gene influences Dab1 function. Here we demonstrate a genetic interaction between Dab1 and APP. Dab1-hypomorphic animals have neuronal ectopias in the neocortex and reduced cerebellar volume, possibly a consequence of Purkinje cell misplacement. These phenotypes are exacerbated in transgenic animals overexpressing a mutant form of APP, APPswe, which is characterized by increased processing at the β-secretase site. The Dab1-hypomorphic phenotype is improved in the cerebellum of animals that are deficient for APP. Together this suggests that APP expression constrains the consequences of Dab1 activity during brain development.
Keywords: Dab1, Reelin, APP, brain development, cerebellum, cortical lamination
Introduction
The Dab1 docking protein is an essential component of the Reelin signal transduction pathway, which is required for the appropriate developmental placement of neurons (Howell et al., 1999a; Rice et al., 1998). Mutant mice that lack Dab1 have pronounced neuronal ectopias in the cerebral cortex, hippocampus, and cerebellum (Gonzalez et al., 1997; Howell et al., 1997; Sheldon et al., 1997; Sweet et al., 1996; Ware et al., 1997). The cerebellar phenotype is particularly profound. Cerebellar Purkinje cells, which normally express Dab1, fail to migrate to their normal position, proximal to the granule cell precursors in the Dab1-null mutants. The proliferation of granule cell precursors, which is promoted by Shh, a short-range acting molecule secreted by Purkinje cells, is reduced when proximal Purkinje cells are missing or displaced (Smeyne et al., 1995; Wechsler-Reya and Scott, 1999). The ultimate consequence is a smaller and disorganized cerebellum (Gallagher et al., 1998; Yang et al., 2002).
Mutations in other genes that encode components of the Reelin signaling pathway have very similar phenotypes (D'Arcangelo et al., 1995; Goffinet, 1997; Ogawa et al., 1995; Trommsdorff et al., 1999). These include the genes encoding the secreted ligand Reelin, and the partially redundant transmembrane Reelin receptors, ApoER2 and VLDLR. Reelin binding to the receptors induces Dab1-dependent activation of the Src family kinases, Src, Fyn and Yes (SFY), and a SFY-dependent increase in Dab1 tyrosine phosphorylation (Arnaud et al., 2003). Mice with mutation in two Src family kinase members share phenotypic similarities with the other components of the signal transduction pathway, supporting a role for these kinases in the signaling pathway (Kuo et al., 2005).
Dab1 interacts with a C-terminal motif in the ApoER2 and VLDLR receptors that is centered around the peptide sequence N P X Y. Interestingly the Dab1 PTB/PI domain which mediates this interaction has also been shown to bind to a similar motif in the C-terminus of APP (Howell et al., 1999b). Confirmation of the legitimacy of the interactions has been provided by resolution of crystal structures of complexes of the Dab1 PTB/PI domain and the cognate peptides from ApoER2 (TKSMNFDNPVYRKT) and APP (NGYENPTYK) (Stolt et al., 2005; Yun et al., 2003). The APP mutants have no resemblance to the Dab1 mutant phenotype (Muller et al., 1994; Zheng et al., 1995), and the role for the Dab1-APP interaction is unresolved.
We examined whether APP expression influences aspects of brain development regulated by the Reelin pathway using mice that carry a novel Dab1 allele that expresses approximately 15% of normal levels and causes mild abnormalities in the cerebral cortex, hippocampus, and cerebellum. Overexpression of a disease-causing allele of APP augments phenotypes associated with the Dab1-hypomorphic allele. In contrast, loss of APP function partially rescues the hypoplastic cerebellar phenotype. These effects support a model in which a consequence of APP expression is to oppose or down-regulate Dab1 function during brain development. In addition our findings show that overexpression of mutant APP may cause anomalous brain development when Reelin-Dab1 signaling is suboptimal.
Results
Generation of the Dab1-conditional allele
We generated mice with a targeted insertion of a floxed Dab1 expression cassette in place of the Dab1 exon that encodes residues 23 to 69 of Dab1 p80 (Figure 1). The Dab1-conditional knock-in (cKI) places loxP sites upstream and downstream of a Dab1 expression cassette, comprised of the endogenous splice acceptor, Dab1 p80 coding sequence from residue 23 to residue 555, and a triple polyadenylation sequence. Insertion of a wild-type expression cassette, excluding the loxP sites, has previously been successful in supporting normal brain development (Howell et al., 2000). This targeting vector also includes a splice acceptor preceding a β-galactosidase reporter, which was introduced to indicate successful Cre-mediated excision in cells with active Dab1 locus transcription.
Figure 1.

Generation of a conditional allele of the Dab1 gene. Exon III of the Dab1 locus (encoding residues 23-69) was selected for replacement by a Dab1 expression cassette flanked by loxP sites (triangles). The targeting vector includes the floxed Dab1 expression cassette comprised of the endogenous splice acceptor, Dab1 p80 cDNA sequences from residue 23-555, some 3' untranslated region, and a triple repeated poly(A) signal. A splice acceptor β-galactosidase reporter was placed downstream of the Dab1 expression cassette to indicate Cre-mediated recombination. The PGK-neo drug selectable marker was flanked by frt sites (circles). The PGK promoter driving diphteria toxin expression (PGK-DTA) was placed outside of the targeted region to provide negative selection against non-homologous recombinants. Arrowheads represent primers used to identify homologous recombinants, and arrows denote primers used for genotyping of Dab1 cKIneo mice. S- SacI, S2-SacII unique site, E- EcoRI, X-XbaI.
Chimeric founder mice were generated through standard embryo transplant procedures with ES cells harboring the Dab1 cKI allele and retaining the PGK-neo sequence (Dab1 cKIneo). In the homozygous state Dab1 cKIneo did not cause an overt behavioral phenotype, in contrast to the Dab1-1 null or Dab1 5F alleles (Howell et al., 1997; Howell et al., 2000; data not shown). This suggested that the Dab1 cKIneo allele is at least partially functional before Cre-mediated excision of the Dab1 expression cassette.
Removal of the Dab1 expression cassette by Cre-mediated recombination in germ cells produced an excised Dab1 allele, Dab1 exKIneo, which behaves like the Dab1-null allele. Animals that are homozygous for Dab1 exKIneo have the typical ataxic behavior and developmental neurological disease characteristic of Dab1-null animals (Supplementary Figure S1 E and data not shown). This demonstrates that the Dab1 cKIneo allele is inactivated by Cre expression, a feature that will be useful for studies into the role of Dab1 in late development or adulthood.
The Dab1 cKIneo allele is hypomorphic and leads to a dosage sensitive phenotype
Other conditional alleles, especially in the form that retain the drug selectable marker, express less efficiently than their wild-type counterparts and cause hypomorphic phenotypes. This feature has been used to generate allelic series, which are useful in the study of gene dosage effects on brain development (Hirotsune et al., 1998). The Dab1 cKIneo mice have reduced Dab1 expression, which was approximately 15% of wild-type levels in postnatal day 1 (P1) animal brains (Supplementary Figure S2) as compared to Dab1-1 heterozygous animals that express approximately 50% of wild-type levels and do not demonstrate defects in brain lamination (Howell et al., 1997).
Examination of the brains of mice that are homozygous for the Dab1 cKIneo allele revealed mild ectopias and reduced cerebellar size (Supplementary Figure S1 C and data not shown). The most dramatic neuronal ectopia was identified in the cerebellum where Purkinje cells are displaced from the typical monolayer in several folia (Supplementary Figure S1). It is apparent that the cerebellar phenotype correlates with Dab1 gene dose. For instance, animals with one wild type allele have improved foliation and cerebellar size compared with Dab1 cKI homozygotes, where as animals with one null allele have more a severe cerebellar phenotype (Supplementary Figure S1 B, C, D). Purkinje cell ectopias are worse in mice with combined Dab1 cKIneo and Dab1-null alleles than in Dab1 cKIneo homozygous animals (data not shown). Further reduction in Dab1 expression, such as in Dab1 exKI homozygous animals, results in an even more severe phenotype (Supplementary Figure S1 E). Since the cerebellar phenotype is sensitive to Dab1 gene dose, the Dab1 cKIneo allele is potentially valuable for evaluating genetic interactions. We reasoned, therefore, that genetic modifiers that influenced Dab1 function might enhance or suppress the cerebellar phenotype.
APP expression spatially and temporally overlaps with Reelin signaling molecules
As a first step in analyzing whether APP influences Reelin-signaling, we investigated APP expression in mouse brain by immunohistochemistry, at P4. We also examined APP overexpression in age matched APPswe transgenic animals. APP is expressed in the cerebellum, cerebral cortex, and hippocampus of neonatal animals (Figure 2 A, C, and data not shown). In the cerebellum, APP expression is highest in Purkinje cells, but it is also present in other cells, particularly in the internal granule cell layer (IGL; Figure 2 A). In the cortex, APP is expressed in cortical plate cells (Figure 2 C), and in the hippocampus it is highest in the neuronal cell layer (data not shown). This expression pattern is supported by in situ hybridizations with anti-sense probes to APP performed by the Allen Brain Project (http://www.brainatlas.org/aba/). The APP 695 isoform in particular is highly expressed in the nervous system during this time. Dab1 is expressed in the projection neurons in the neocortex, the pyramidal neurons in the hippocampus and the Purkinje cells in the cerebellum (Gallagher et al., 1998; Rice et al., 1998). This indicates that APP is expressed during the developmental time frame and in the brain regions that are regulated by Reelin signaling.
Figure 2.

APP expression in the developing cerebellum and cortical plate. Immunostaining P4 brain of wild-type (wt; A) and APPswe transgenic animals (B) with anti-APP (green) and anti-Calbindin (red) showed similar levels of APP protein expression in Purkinje cells (yellow). However we observed increased expression of APP in the IGL of the APPswe cerebellum. APP immunostaining was present in the cortical plate of P4 neocortex in wt (C) and APPswe transgenic animals (D) had more expression in this region. DAPI fluorescence (blue) shows cell nuclei. Scale bar: 60 μm.
APPswe transgenic animals have higher APP expression than their wild-type counterparts in cerebellar Purkinje cells and cortical plate neurons in the neocortex at P4 (Figure 2 B, D). In the cerebellum the most marked increase in expression was observed in cells located in the IGL, which are likely granule cell neurons. Expression from the APPswe transgene overlaps with but is broader than endogenous expression and may impact neuronal processes that are regulated by Reelin signaling.
Neither Dab1 protein levels nor tyrosine phosphorylation are affected by APP expression
If APP influences Dab1 function directly, we might expect to observe changes in Dab1 phosphorylation or protein levels in mice that overexpress APP. In brains of P1 animals, expression of APPswe does not alter Dab1 protein expression in mice either wild-type or hypomorphic for Dab1 (Supplementary Figure S2). It was difficult to detect Dab1 tyrosine phosphorylation in the Dab1-hypomorphic animals at P1, a time when phosphorylation is known to be low. We therefore examined Dab1 tyrosine phosphorylation in E16 mice of the same genotypes. APPswe expression does not alter Dab1 tyrosine phosphorylation in animals at this age (data not shown). We previously demonstrated that cultured neurons derived from APPswe transgenic mice have reduced levels of Dab1 tyrosine phosphorylation in response to Reelin, compared to wild-type neurons treated in the same manner (Pramatarova et al., 2006). The acute nature of Reelin stimulation in culture may put more demands on the pathway that reveal consequences of APPswe overexpression on Dab1 tyrosine phosphorylation, which are not apparent in biochemical analysis of whole brain.
It has recently been shown that Dab1 expression is not altered in mice that carry combined deficiencies for all three APP family members (Herms et al., 2004). In addition Dab1 protein or tyrosine phosphorylation levels are not altered in APP null animals (data not shown). Despite this lack of evidence for alterations in Dab1 expression or tyrosine phosphorylation in animals that have higher or lower levels of APP, APP could directly regulate Dab1; for example, by altering its subcellular localization, or access to key binding partners.
APPswe expression exacerbates the developmental phenotype of Dab1 hypomorphs
Previous studies examining APP transgenic animals have not identified lamination defects in the brains of animals with wild-type levels of Dab1. We confirmed this in the strain background used for our genetic tests by comparing the cerebella of wild-type and APPswe mutant animals. We did not observe changes in the overall size, and appearance of the folia, or Purkinje cell ectopia in the cerebellum. The organization of neuronal lamina in the cerebral cortex or hippocampus also appeared unchanged (Figure 3 and data not shown). This suggests that the overexpression of APPswe in animals with wild-type levels of Dab1 does not cause obvious lamination defects in the cerebellum, cerebral cortex, or hippocampus.
Figure 3.

A genetic interaction between Dab1 and APP influenced the development of the cerebellum. Midline sagittal sections through the cerebellum of wild type (wt; A, E), APPswe transgenic (B, F), homozygous Dab1 cKIneo (cKI/cKI; C, G, n=6) and homozygous Dab1 cKIneo and APPswe transgenic (cKI/cKI, APPswe; D, H, n=6) mice were stained with anti-Calbindin (brown) and hematoxylin (blue). Cerebella of Dab1-hypomorphic animals that carried the APPswe transgenic appeared smaller and occasionally the division between folia was lost (arrow; D). Purkinje cell ectopia was apparent in all Dab1 cKIneo homozygous animals (arrowheads; G, H), and Purkinje cell dendrites are apparent in white matter and IGL in Dab1 cKIneo homozygous animals carrying the APPswe transgene (brown stain in IGF; D, H). Scale bar is 100 μm in D, 20 μm in H.
Since Reelin signaling may be robust enough to mask subtle effects of APP overexpression in wild-type animals, we investigated whether an effect could be detected in mice with compromised Dab1 function. Ectopic Purkinje cells are clearly apparent in Dab1 cKIneo homozygous animals, both with and without the APPswe transgene (arrowheads; Figure 3 G, H), but the extent of the ectopia varies from one histological section to another. The cerebellar size as judged from sagittal sections near the midline is consistently smaller in Dab1 cKIneo homozygous animals that carry the APPswe transgene than those that are wild-type for APP (Figure 3 C, D) and a loss of the division between some folia is apparent (arrow; Figure 3 D). On the basis of these findings we chose to quantify cerebellar volumes to assess genetic interactions between Dab1 and APP.
To quantify the effects of hypomorphic levels of Dab1 on brain development and examine whether APP overexpression affected it, we compared vermal volumes that were normalized by the whole brain volume (Figure 4 A, B). The homozygous Dab1 cKIneo mice have smaller vermis to whole brain ratios than their wild-type siblings, which is consistent with our histological data. Mice that carry the APPswe transgene, and are wild-type for Dab1, have vermis to whole brain ratios that are similar to wild-type animals. In contrast, when the APPswe transgene was introduced into the Dab1 cKIneo homozygous background the vermis to whole brain ratios were significantly reduced compared to their Dab1 cKIneo littermates. This indicates that when Dab1 expression is decreased, APPswe overexpression can interfere with brain development.
Figure 4.
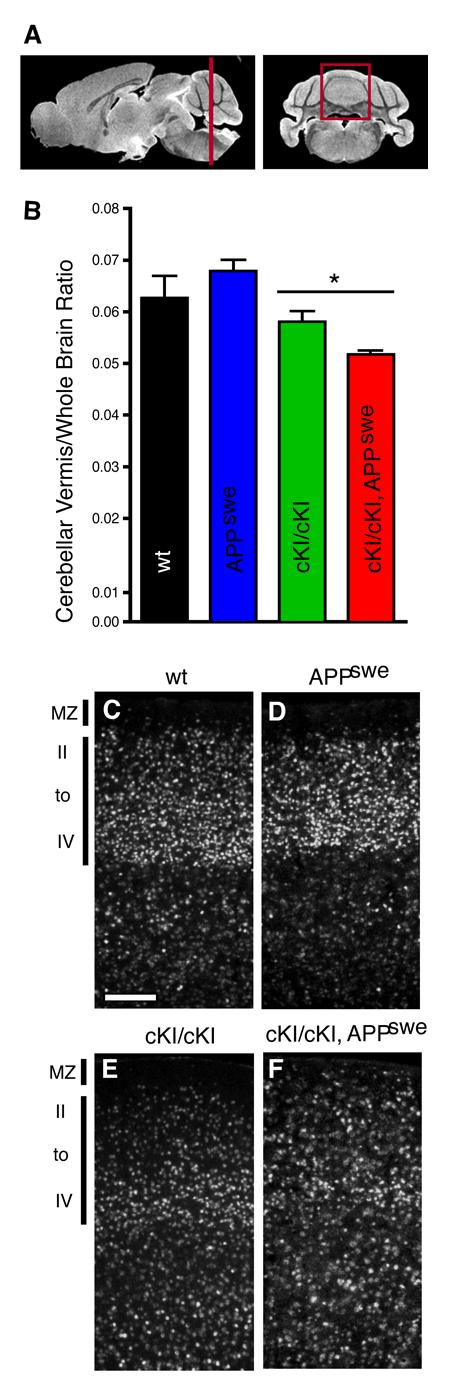
APPswe transgene expression enhanced the cerebellar and cortical phenotypes of Dab1-hypomorphic mice. Mouse brain images were collected by MRI (A) and areas of cross-sections were summed to determine the volume of various brain regions. The volume of the vermal area of the cerebellum (V; red box in A) was normalized by dividing by whole brain volume (W) to produce the V/W ratio (B). The V/W ratio of APPswe mice (blue bar) does not differ significantly from their wild type littermates (black). However the presence of the APPswe transgene on the Dab1 cKIneo hypomorphic background (red) resulted in a significant decrease in the V/W ratio compared to Dab1-hypomorphic animals expressing wild-type levels of APP (green). Coronal sections through the parietal cortex immunostained with an anti-CDP antibody (white; C-F, n=2) showed normal lamination for APPswe transgenic mice that express wild-type levels of Dab1 with most CDP positive cells in layers II-IV (compare D to C). However, Dab1 cKIneo hypomorphic animals expressing the APPswe transgene (F) had broader lamination defects in the neocortex than their Dab1 cKIneo littermates (E) with CDP positive neurons appearing in the marginal zone (MZ). Arrowheads indicate the pial surface. Scale bar is 60 μm. The data were compared by unpaired t-tests * P=0.0271, n=4.
The Reelin signaling pathway also regulates development of the neocortex and the hippocampus (Tissir and Goffinet, 2003). We therefore investigated whether these regions were also affected by APPswe expression. We examined the cortical lamination with antibodies against the transcription factors CDP and FoxP2, which are expressed in superficial layers (layers II-IV) and deep layers (layers V-VI), respectively. CDP positive neurons are more broadly distributed in Dab1-hypomorphic animals than animals expressing wild-type levels of Dab1, and they are evident in layers V and VI (Figure 4 C, E).
In the neocortex, APPswe expression in Dab1-hypomorphic mice was associated with a greater dispersion of CDP positive neurons (Figure 4 E versus F). In these APPswe expressing animals more CDP positive neurons are found in layers V and VI, and large numbers are also observed in the marginal zone (Figure 4 F). APPswe expression did not noticeably affect the position of CDP positive neurons in animals with a wild-type Dab1 genotype (Figure 4 D versus C). FoxP2 expressing neurons in the neocortex were appropriately positioned in layer V-VI in all genotypes, suggesting that early migration events, which form the deep layers, are not affected in these animals (data not shown). Since the more superficial layers are formed later, this suggests that Dab1 expression may be more critical at later times when Reelin levels are declining. APP expression may also have more impact later in development than it does at earlier stages. The hippocampal lamination in Dab1-hypomorphic animals is not altered by their APP genotype (data not shown). These observations suggest that expression of a form of APP that has been linked with Alzheimer's disease in humans can influence the development of the cerebellum and cerebral cortex in animals with compromised Dab1 function.
The effects of APPswe overexpression on brain development in mice that are deficient for Dab1 suggest that APPswe may directly interfere with Dab1 function or indirectly oppose a consequence of Dab1 activity. The mutations in APPswe are known to augment the production of Aβ,a peptide found in amyloid plaques in brains of Alzheimer's patients. The experiments above cannot distinguish whether the observed consequence of overexpressed APP are dependent upon the higher than normal production of Aβ peptides, increased levels of biologically active APP intracellular domain (AICD), or if expression of a wild-type version of APP would have the same activity.
APP loss-of-function partially rescues the cerebellar phenotype in Dab1-hypomorphic animals
To investigate if physiological levels of wild-type APP influence development of the cerebellum or the neocortex, we generated animals that have reductions in both Dab1 and APP gene expression. This approach circumvents general concerns associated with transgenic animals, where protein expression potentially exceeds physiological levels, spatial ranges or temporal limits. We examined APP null animals on a C57BL/6 congenic background and did not observe defects in lamination in the neocortex, cerebellum, or hippocampus (data not shown). In addition, the vermis to whole brain ratio in MR images are similar between APP null and wild-type animals (Figure 5).
Figure 5.

APP loss-of-function partially rescued the cerebellar phenotype of Dab1-hypomorphic mice. MRI quantification of brain volumes from mice with the APP loss-of-function allele showed no significant difference in the vermis to whole brain volume ratios between the wild type (wt; black) and APP null littermates (APP−/−; blue). However, both heterozygous and homozygous APP loss-of-function (APP −/+; yellow, and APP −/−; red respectively) resulted in increased V/W ratios in the Dab1 cKIneo hypomorphic animals (cKI) as compared to animals with wild-type APP levels (APP+/+; green). Data were compared by unpaired t-tests, *P=0.0434, **P=0.0123, n=3-4.
To determine whether loss of APP modified the cerebellar phenotype of the Dab1-hypomorphic animals, we compared mutants that were homozygous for Dab1 and were wild-type for APP or had lost one or both copies of the APP gene. Reduction in APP levels are correlated with significant improvement of cerebellar size in Dab1-hypomorphic animals (Figure 5). Both heterozygous and homozygous APP mutant animals show partial improvement of the cerebellar volume phenotype. This suggests that physiological levels of APP regulate brain development through genetic influences on Dab1 function.
We investigated whether loss of APP improved the neocortical phenotype of the Dab1-hypomorphic animals, since APP overexpression made it worse. The organization of CDP positive neurons in the neocortex in Dab1-hypomorphic mice was not improved by loss of APP function (data not shown). The cerebellar phenotype may be more sensitive to APP levels than the neocortical phenotype, or perhaps other APP family members have a more critical role in the development of the neocortex. However, it seems probable that Dab1 function is influenced by APP expression in the neocortex, since APPswe exacerbated the phenotype of this brain region in Dab1 hypomorphs.
Discussion
In this study we revealed a genetic interaction between Dab1 and APP, which highlights a role for APP in brain development. Previously, our understanding of APP function in development has been limited since APP overexpression does not cause overt developmental phenotypes (Hsiao et al., 1995; Mucke et al., 2000) and APP loss-of-function results in only subtle phenotypes (Zheng et al., 1995). Disruption APP and two paralogs APLP1 and APLP2 does produce a profound phenotype, however, and results in neonatal death and type II cobblestone lissencephaly (Herms et al., 2004). In the triple mutant animals the lamination of the brain is relatively normal outside of the areas of focal heterotopias. This is distinct from the Reelin pathway mutant phenotypes and suggests that the APP family members do not play an essential role in promoting Dab1 function downstream of Reelin signaling. The demonstration here that APPswe overexpression enhances, and APP loss-of-function suppresses a Dab1-hypomorphic phenotype suggests that APP antagonizes Dab1 function during brain development.
Dab1 is not the only PTB/PI domain containing protein to bind the YENPTY motif in the cytoplasmic domain of APP. The docking proteins X11/Mint, Fe65 and related adaptors bind to the same sequence (Borg et al., 1996; Fiore et al., 1995; Rogelj et al., 2006). Interestingly, combined mutations in Fe65 and Fe65L1 lead to phenotypes with leptomeningeal eruptions and focal dysplasias very similar to the triple APP family deficiency phenotype (Guenette et al., 2006). Notably, the phenotypes of Fe65 and APP homologs in C. elegans, Feh-1 and Apl, share a similar pharyngeal pumping defect suggesting that a common function for APP and Fe65 family members may be evolutionarily conserved (Zambrano et al., 2002). It has been demonstrated by a number of groups that Fe65 and the AICD fragment, the product of γ-secretase activity on APP, function together to promote transcription (Cao and Sudhof, 2001; Chang and Suh, 2005). In support of the concept that Fe65 and the AICD have a role in development, mutations in Psen1, a component of γ-secretase that produces AICD, result in leptomeningeal eruptions similar to those mentioned above (Hartmann et al., 1999). Since the Dab1-null and -hypomorphic animals have distinct phenotypes it appears that Dab1 does not participate in a pathway with APP and Fe65 family members to promote the integrity of the external limiting membrane of the brain.
Instead, the genetic interactions reported here suggest that APP opposes Dab1 function in some developmental events at least. We have observed a similar antagonism between Drosophila APP-like and Dab1 in a Drosophila eye model system (Pramatarova et al., 2006). In aged mice it has recently been shown that Reelin expression is reduced in an APP- expressing Alzheimer's animal model, with a significant reduction in Reelin-expressing pyramidal neurons in the entorhinal cortex, suggesting that pathological forms of APP may influence the Reelin transcription or stability (Chin et al., 2007). The mechanism of APP influence over Dab1 remains a mystery. We did not detect a change in Dab1 tyrosine phosphorylation, which was anticipated based on our previous study showing APPswe overexpression reduced Reelin-induced Dab1 tyrosine phosphorylation (Pramatarova et al., 2006). The physical interaction between the Dab1 PTB/PI domain and APP may be significant. The interaction motif in the APP C-terminus, YENPTY, is conserved in all family members including homologs from Drosophila and C. elegans (Luo et al., 1990) and is not altered in the APPswe mutant. APP may compete with the Reelin receptors for Dab1 binding, and recruit it to different signaling complexes or subcellular compartments. Alternatively, APP may have an indirect effect on Dab1 function and Reelin signaling by regulating a common downstream phenomenon. For instance, both APP and Reelin signaling have been reported to influence cell adhesion and intracellular trafficking (Gotthardt et al., 2000; Hoe et al., 2006; Kins et al., 2006; Morimura et al., 2005; Sabo et al., 2001; Sanada et al., 2004). It will be interesting to determine if APP and Dab1 have opposing influences on these cell biological properties.
In animals with hypomorphic levels of Dab1, overexpression of APPswe resulted in increased severity of phenotypes in the cerebellum and cerebral cortex. Previous work with various models of Alzheimer's disease, including those that overexpress mutant forms of APP alone or in combination with mutations of Psen1 and Tau, have not revealed anomalies in brain development (Hsiao et al., 1995; Oddo et al., 2003). Interestingly in humans, cerebellar hypoplasia has been observed in trisomy 21 or Down syndrome, where the APP gene dose is 50% higher than normal (Pinter et al., 2001). It remains to be determined, however, if this phenotype is dependent upon APP overexpression in the presence of the amplification of other genes on chromosome 21. A prospective longitudinal study in nuns demonstrated that there is a correlation between linguistic skills early in life and the development of Alzheimer's disease later in life (Riley et al., 2005). One interpretation of this is that genetic predisposition to Alzheimer's disease also causes a mild developmental phenotype that negatively influences language proficiency. Together these studies leave open the possibility that genetic susceptibility to Alzheimer's disease causes a mild developmental disorder that becomes more apparent in the presence of secondary genetic factors.
Dab1 expression and Reelin presentation have been shown to regulate APP processing by enhancing the α cleavage, of APP (Hoe et al., 2006), which is mutually exclusive of the β cleavage the first step in the production of the pathogenic Aβ peptide. It will therefore be interesting to determine whether loss of Dab1 function augments phenotypes, such as hyperphosphorylation of the microtubule protein Tau (Mapt) and Aβ deposition in mouse models of Alzheimer's disease. Along this line, on certain strain backgrounds Dab1-null mice show hyperphosphorylated Mapt in the hippocampus (Brich et al., 2003). Hyperphosphorylated Mapt is a hallmark of Alzheimer's disease and has been correlated with the development of neurofibrillary tangles (Blurton-Jones and Laferla, 2006). A greater understanding of how these molecules regulate development should aid the resolution of APP function and the pathological consequences that lead to the development of Alzheimer's disease.
Experimental Methods
Mouse mutants
The targeting vector design was based on that previously described for the Dab1 wild-type (p80WTKI) and 5F (p805FKI) knock-in alleles, which substitutes exon III with the Dab1 p80 coding sequence from residue 23 to 555 (Howell et al., 2000). To generate the Dab1-conditional allele (Dab1 cKIneo) loxP sites were added both 5' and 3' to the Dab1 expression cassette, which includes the upstream endogenous splice acceptor and a downstream triple poly(A) signal (detailed cloning strategy available upon request). A splice acceptor β-galactosidase reporter gene (Friedrich and Soriano, 1991) and a neomycin drug resistance expression cassette flanked by frt sites were cloned 3' to the floxed Dab1 sequences (Yamaguchi et al., 1994). The vector was designed such that excision of the Dab1 expression cassette will turn on the β-galactosidase reporter in cells with active Dab1 locus transcription. A PGK-diphtheria toxin negative selection marker, introduced to select against cells where non-homologous recombination had occurred, was cloned outside of the targeting region of the vector and 5' to the short homologous arm (Figure 1). The construct was confirmed by sequencing.
Embryonic stem cells (129Sv/Sor) were electroporated with the Dab1 cKIneo targeting vector after linearization at a unique Sac II site downstream of the targeting sequences, as previously described (Howell et al., 2000). Homologous recombinants were selected by PCR using primers 5'- TAG GAG GAG AAT GGG CTG AC -3' (located in mouse genomic sequence upstream of the short arm) and 5'- CTT GAA GAC GAA AGG GCC T -3' (located in vector sequence between 5' loxP site and Dab1 cDNA cassette), which generate a 1kb product. Chimeric animals were generated using standard protocols (Ramirez-Solis et al., 1993). The Dab1 cKIneo line was maintained on a 129/C57Bl6 mixed background. Animals were genotyped by PCR using primers in the Dab1 locus 5'-TGA TGC TAT CCC TAG CAA GAC - 3' (intron II) and 5'- GTG GCT TCG CTG CGA TCC TGA C -3' (exon III), which amplify a wild type allele of 356 bp and a larger Dab1 cKI allele of 597 bp due addition of loxP and flanking sequence.
The APPswe (B6;SLJ) and APP-null (C57BL/6) mice have been described previously (Hsiao et al., 1995; Zheng et al., 1995) and were obtained from Taconic and Jackson labs respectively. To minimize the effect of genetic modifiers the animals compared were either offspring from the same crosses or crosses of siblings set up in parallel. Previously genetic background effects have been shown to cause premature death and Tau hyperphosphorylation in Dab1 mutants (Brich et al., 2003).Animals were analyzed between 1-4 months of age. All mice used in this study were handled under the animal care and use guidelines of the NIH.
Immunoblotting
The cerebral hemispheres of P1 mice were homogenized in RIPA buffer (0.15M NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 10mM sodium phosphate pH 7.0, 2 mM EDTA, 50 mM NaF, 1 mM phenylarsine oxide, 14mM 2-mercaptoethanol, and protease inhibitors [complete mini, EDTA free; Roche]) as previously described (Howell et al., 1999a).
Histology and immunostaining
Anesthetized animals were perfused with ice-cold phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA). The brains were fixed in 4% PFA for 16 h and infiltrated with 30% sucrose and frozen in Tissue-Tek OCT compound (Sakura Finetechnical Co.) prior to cryosectioning. Immunostaining was performed on 30μm thick floating sections using the following antibodies: mouse anti-Calbindin-D-28K 1:1000 (CB-955, Sigma), rabbit anti-CDP 1:100 (M-222, Santa Cruz Biotechnology), rabbit anti-FoxP2 1:4000 (ab16046, Abcam), rabbit anti-Amyloid Precursor Protein 1:1000 (A8717, Sigma). Colorimetric detection was done with DAB substrate kit (Vectastain ABC Elite), then counterstained with Hematoxylin QS (Vector Laboratories). Bright field images were acquired on a Leica stereo-microscope equipped with a color Spot camera (Diagnostic Instruments Inc.). Immunoflorescent labeling was visualized using a DeltaVision microscope (Applied Precision).
Volumetric measurements of mouse cerebellum
Brains were dissected from 13-16 weeks old littermates perfused as described above. Three dimensional MRI images were acquired using a volumetric spin-echo pulse sequence on a 7 Tesla Bruker Biospin NMR spectrometer with a microimaging apparatus including a Micro 2.5 gradient insert (Bruker Biospin, Inc). A 15 mm RF coil insert was used to image the specimen, which was immersed in Fomblin LC-08 (Solvey Solexis) a per-fluronated poly-ether designed to suppress image artifacts from the air-tissue interface. The acquisition parameters for the MSME sequence were: Repetition time (TR) = 1500 ms, Echo Time (TE) = 30 ms, FOV = 17 × 12 × 10 mm, with two averages for a total acquisitions time of 16h:25m. The spatial resolution of the acquired data was 78 microns isotropic with a final data resolution of 47 microns isotropic after zero-filling prior to Fourier transformation. Images were imported into ImageJ (Research Services Branch, NIH) and manually segmented. Volumes were measured using ImageJ and the data was presented as the ratio of the volume of the vermal area of the cerebellum divided by the volume of the whole brain excluding the olfactory bulbs.
Supplementary Material
The Dab1 cKIneo allele is hypomorphic. Midline sagittal sections through the cerebellum of mice that are wild type (wt/wt; A), heterozygous Dab1 cKIneo (cKI/wt; B), homozygous Dab1 cKIneo (cKI/cKI; C), hemizygous Dab1 cKIneo/Dab1-1 (cKI/null; D), or homozygous for the excised Dab1-conditional allele (exKI/exKI; E) were stained with anti-calbindin and hematoxylin to indicate the size and foliation. Scale bar is 100 μm.
Dab1 expression was reduced in Dab1 cKIneo mutant mice but is not altered by the presence of an APP transgene. Brain lysates from one day old Dab1 wild-type (wt; lanes 1, 6,7) , Dab1-1/wt (null/wt; lane 2) or Dab1 cKIneo homozygous (cKIneo/cKIneo; lanes 3-5) animals were compared for Dab1 expression by Western blotting with a C-terminal anti-Dab1 antibody. Dab1 expression in Dab1-1 heterozygous animals was approximately 50% of normal (lane 2), while expression in Dab1 cKIneo homozygous mice was approximately 15% the wild-type levels (lanes 3, 5). Dab1 expression was not affected by the presence of the APPswe transgene in mice that were wild-type or homozygous for Dab1 cKIneo (lanes 4 and 7).
Acknowledgements
We would like to thank Amy Chen for generating chimeric mice, Mark Henkemeyer and Philippe Soriano for the gift of vectors, Heinz Arnheiter, Huaibin Cai, Bill Rebeck, Monique Dubois-Dalcq, and Kenneth Fischbeck for stimulating discussions, and Alan Koretsky and Doug Morris for support with the MRI analysis. This work was financed in part by intramural funds from NIH/NINDS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnaud L, Ballif BA, Förster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of Disabled-1 during brain development. Curr Biol. 2003;13:9–17. doi: 10.1016/s0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Laferla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res. 2006;3:437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brich J, Shie FS, Howell BW, Li R, Tus K, Wakeland EK, Jin LW, Mumby M, Churchill G, Herz J, Cooper JA. Genetic modulation of tau phosphorylation in the mouse. J Neurosci. 2003;23:187–192. doi: 10.1523/JNEUROSCI.23-01-00187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Chang KA, Suh YH. Pathophysiological roles of amyloidogenic carboxy-terminal fragments of the beta-amyloid precursor protein in Alzheimer's disease. J Pharmacol Sci. 2005;97:461–471. doi: 10.1254/jphs.cr0050014. [DOI] [PubMed] [Google Scholar]
- Chin J, Massaro CM, Palop JJ, Thwin MT, Yu GQ, Bien-Ly N, Bender A, Mucke L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer's disease. J Neurosci. 2007;27:2727–2733. doi: 10.1523/JNEUROSCI.3758-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Howell BW, Soriano P, Cooper JA, Hawkes R. Cerebellar abnormalities in the disabled (mdab1-1) mouse. J Comp Neurol. 1998;402:238–251. [PubMed] [Google Scholar]
- Goffinet AM. Unscrambling a disabled brain. Nature. 1997;389:668–669. doi: 10.1038/39453. [DOI] [PubMed] [Google Scholar]
- Gonzalez JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA. Birthdate and cell marker analysis of scrambler: a novel mutation affecting cortical development with a reeler-like phenotype. J. Neurosci. 1997;17:9204–9211. doi: 10.1523/JNEUROSCI.17-23-09204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotthardt M, Trommsdorff M, Nevitt MF, Shelton J, Richardson JA, Stockinger W, Nimpf J, Herz J. Interactions of the Low Density Lipoprotein Receptor Gene Family with Cytosolic Adaptor and Scaffold Proteins Suggest Diverse Biological Functions in Cellular Communication and Signal Transduction. J Biol Chem. 2000;275:25616–25624. doi: 10.1074/jbc.M000955200. [DOI] [PubMed] [Google Scholar]
- Guenette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. Embo J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann D, De Strooper B, Saftig P. Presenilin-1 deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasia similar to human type 2 lissencephaly. Curr Biol. 1999;9:719–727. doi: 10.1016/s0960-9822(99)80331-5. [DOI] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. Embo J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S, Fleck M, Gambello M, Bix G, Chen A, Clark G, Ledbetter D, McBain C, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem. 2006;281:35176–35185. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999a;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–885. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol. 1999b;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Kins S, Lauther N, Szodorai A, Beyreuther K. Subcellular trafficking of the amyloid precursor protein gene family and its pathogenic role in Alzheimer's disease. Neurodegener Dis. 2006;3:218–226. doi: 10.1159/000095259. [DOI] [PubMed] [Google Scholar]
- Kuo G, Arnaud L, Kronstad-O'Brien P, Cooper JA. Absence of Fyn and Src causes a reeler-like phenotype. J Neurosci. 2005;25:8578–8586. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo LQ, Martin-Morris LE, White K. Identification, secretion, and neural expression of APPL, a Drosophila protein similar to human amyloid protein precursor. J Neurosci. 1990;10:3849–3861. doi: 10.1523/JNEUROSCI.10-12-03849.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura T, Hattori M, Ogawa M, Mikoshiba K. Disabled1 regulates the intracellular trafficking of reelin receptors. J Biol Chem. 2005;280:16901–16908. doi: 10.1074/jbc.M409048200. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Cristina N, Li ZW, Wolfer DP, Lipp HP, Rulicke T, Brandner S, Aguzzi A, Weissmann C. Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell. 1994;79:755–765. doi: 10.1016/0092-8674(94)90066-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Pinter JD, Eliez S, Schmitt JE, Capone GT, Reiss AL. Neuroanatomy of Down's syndrome: a high-resolution MRI study. Am J Psychiatry. 2001;158:1659–1665. doi: 10.1176/appi.ajp.158.10.1659. [DOI] [PubMed] [Google Scholar]
- Pramatarova A, Ochalski PG, Lee CH, Howell BW. Mouse disabled 1 regulates the nuclear position of neurons in a Drosophila eye model. Mol Cell Biol. 2006;26:1510–1517. doi: 10.1128/MCB.26.4.1510-1517.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Solis R, Davis AC, Bradley A. Gene targeting in embryonic stem cells. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D'Arcangelo G, Nakajima K, Goldowitz D, Curran T. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development. 1998;125:3719–3729. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- Riley KP, Snowdon DA, Desrosiers MF, Markesbery WR. Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun Study. Neurobiol Aging. 2005;26:341–347. doi: 10.1016/j.neurobiolaging.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Rogelj B, Mitchell JC, Miller CC, McLoughlin DM. The X11/Mint family of adaptor proteins. Brain Res Rev. 2006;52:305–315. doi: 10.1016/j.brainresrev.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D'Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Chu T, Lewin A, Bian F, S CS, Kunsch C, Lira SA, Oberdick J. Local control of granule cell generation by cerebellar Purkinje cells. Mol. Cell. Neurosci. 1995;6:230–251. doi: 10.1006/mcne.1995.1019. [DOI] [PubMed] [Google Scholar]
- Stolt PC, Chen Y, Liu P, Bock HH, Blacklow SC, Herz J. Phosphoinositide binding by the disabled-1 PTB domain is necessary for membrane localization and Reelin signal transduction. J Biol Chem. 2005;280:9671–9677. doi: 10.1074/jbc.M413356200. [DOI] [PubMed] [Google Scholar]
- Sweet HO, Bronson RT, Johnson KR, Cook SA, Davisson MT. Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm Genome. 1996;7:798–802. doi: 10.1007/s003359900240. [DOI] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor-2. Cell. 1999:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Ware ML, Fox JW, Gonzalez JL, Davis NM, Lambert C, Russo CJ, Chua SC, Jr, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994;8:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]
- Yang H, Jensen P, Goldowitz D. The community effect and Purkinje cell migration in the cerebellar cortex: analysis of scrambler chimeric mice. J Neurosci. 2002;22:464–470. doi: 10.1523/JNEUROSCI.22-02-00464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M, Keshvara L, Park CG, Zhang YM, Dickerson JB, Zheng J, Rock CO, Curran T, Park HW. Crystal structures of the Dab homology domains of mouse disabled 1 and 2. J Biol Chem. 2003;278:36572–36581. doi: 10.1074/jbc.M304384200. [DOI] [PubMed] [Google Scholar]
- Zambrano N, Bimonte M, Arbucci S, Gianni D, Russo T, Bazzicalupo P. feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J Cell Sci. 2002;115:1411–1422. doi: 10.1242/jcs.115.7.1411. [DOI] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJS, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisodia SS, Chen HY, van der Ploeg LHT. Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Dab1 cKIneo allele is hypomorphic. Midline sagittal sections through the cerebellum of mice that are wild type (wt/wt; A), heterozygous Dab1 cKIneo (cKI/wt; B), homozygous Dab1 cKIneo (cKI/cKI; C), hemizygous Dab1 cKIneo/Dab1-1 (cKI/null; D), or homozygous for the excised Dab1-conditional allele (exKI/exKI; E) were stained with anti-calbindin and hematoxylin to indicate the size and foliation. Scale bar is 100 μm.
Dab1 expression was reduced in Dab1 cKIneo mutant mice but is not altered by the presence of an APP transgene. Brain lysates from one day old Dab1 wild-type (wt; lanes 1, 6,7) , Dab1-1/wt (null/wt; lane 2) or Dab1 cKIneo homozygous (cKIneo/cKIneo; lanes 3-5) animals were compared for Dab1 expression by Western blotting with a C-terminal anti-Dab1 antibody. Dab1 expression in Dab1-1 heterozygous animals was approximately 50% of normal (lane 2), while expression in Dab1 cKIneo homozygous mice was approximately 15% the wild-type levels (lanes 3, 5). Dab1 expression was not affected by the presence of the APPswe transgene in mice that were wild-type or homozygous for Dab1 cKIneo (lanes 4 and 7).