

Abstract
The exit of lymphocytes from the interstitium of the lung, across the bronchial epithelium and into the airway lumen, is known as egression, or luminal clearance. Egression is important for immune surveillance and the resolution of inflammation, but the mechanisms involved are unknown. We show that egression of human T cells across the bronchial epithelium is a multistep process, driven in part by a polarized transepithelial gradient of CXCL11 that is up-regulated in patients with chronic obstructive airways disease. Previous studies have shown that T cells can migrate across a disrupted bronchial epithelium, but we provide evidence that egression does not require epithelial injury, and can take place across an intact epithelial barrier. After negotiating the extracellular matrix, the T cell adheres to the basal surface of the bronchial epithelial cell using α4 and leukocyte function associated-1 integrins before crossing the epithelium in an leukocyte function associated-1-dependent way. We demonstrate an egression-dependent decrease in transepithelial resistance across the epithelium without gross alteration in tight-junction proteins. The process of egression has been relatively overlooked when considering the control of leukocyte trafficking in the lung and other epithelial organs. This study highlights the role of the respiratory epithelium in the trafficking of T lymphocytes from the pulmonary interstitium and into the large airways, during the onset and resolution of pulmonary inflammation.
The lung comprises three distinct compartments: the bronchi, the alveoli, and the lung interstitium. The bronchi, lined with ciliated columnar epithelium, bring air to the alveoli; these are lined with a thin cuboidal epithelium, which is susceptible to damage or infection by inhaled allergens and pathogens. Lung disease is the single greatest cause of death world-wide (1), due to the high mortality associated with respiratory infections (2) and with chronic obstructive pulmonary disease (COPD)5 (3), a common but neglected disease of the airways caused by smoke and other pollutants (4, 5). Because of the risk of pulmonary infection, there is a need for constant immune surveillance and T lymphocytes migrate continuously from the interstitium into the bronchoalveolar airspaces (6, 7). A population of T cells remains within the bronchial epithelium and lumen (8) to provide immediate defense against airway pathogens (7, 9), and this response is reinforced by recruitment of effector T cells, activated by foreign Ags in regional lymph nodes.
Leukocytes are essential to fight infection, but in excessive numbers or over prolonged periods they can cause host damage. Accumulation of leukocytes in the lung interstitium and the alveolar airspace is especially detrimental. The bronchial T cells associated with COPD, tuberculosis, and viral disease are of the Th1/Tc1 phenotype (10-13) and express high levels of CXCR3 (10, 13, 14). These effector Th1/Tc1 cells may contribute to the immune-mediated lung damage seen in these and other diseases (10, 15-17). To understand pulmonary inflammation, we need to understand effector T cell traffic into and out of the lung.
For hollow organs, such as the lung and the gut, tissue accumulation of leukocytes is determined by recruitment of leukocytes from the blood; survival of tissue leukocytes; and migration of leukocytes from the interstitial space, either to the lymphatics or into the lumen of the organ, so called egression (18). After the leukocytes have crossed the epithelial barrier into the airway, they can be carried upwards on the mucociliary escalator to the pharynx for removal (19, 20). The physiological importance of this luminal clearance of inflammatory cells from the lung, was demonstrated by Corry and colleagues who showed that preventing egression of peribronchial leukocytes in a murine model of bronchial inflammation was fatal (21, 22). The benefit of clearing cells across the bronchial epithelium is in sharp contrast to the potentially deleterious effect of clearing leukocytes across the alveolar epithelium; the alveolar epithelium has no mucociliary escalator and the inflammatory cells that egress into the alveolar air-space remain there, interfering with gas exchange, and increasing mortality in animal studies of alveolar disease (23).
The chemokine receptor CCR7 directs the migration of CCR7+ lymphocytes from peripheral tissues via afferent lymphatics to the lymph nodes (24, 25), but the factors regulating the egression of effector CXCR3+ T cells from the lung interstitium, across the bronchial epithelium, and into the airway lumen are unknown. The ligands for CXCR3 are CXCL9, CXCL10, and CXCL11, and these chemokines also have defensin-like antimicrobial activity (26). CXCL9, CXCL10, and CXCL11 are produced by stimulated bronchial epithelium in vitro (27), and are up-regulated on infection of the epithelium with respiratory viruses (28) or tuberculosis (27) and in patients with COPD (14). These ligands for CXCR3 are also produced by leukocytes, including T lymphocytes and IFN-γ-stimulated mononuclear cells (26), both of which may be resident in the airways. This suggests a role for these ligands of antimicrobial defense coupled to the initial trafficking of CXCR3+ lymphocytes. Further lymphocyte egression might then follow in response to chemokines produced by inflammatory cells that are resident in, or have trafficked into, the airway lumen. This is supported by the observation that mice deficient in CXCR3 have a defect in egression of lymphocytes following bleomycin injury, and an increased mortality compared with wild-type controls (29).
Despite the importance of lymphocyte migration across bronchial epithelium, the immuno-biology underlying this process has not been well studied. For efficient egression, lymphocytes must move from the basal to the apical (luminal) surface of the epithelium, opposite to that for exit from the blood vessel across the endothelium, and must negotiate the intercellular junctions. Data indicate that lymphocytes may move across bronchial epithelial monolayers in the basal-to-apical direction when the epithelium is stimulated with IFN-γ or TNF-α plus IFN-γ (30, 31). This process requires leukocyte function associated-1 (LFA-1) on the lymphocyte to interact with ICAM-1 on the epithelial cell (30). However, both TNF-α and IFN-γ are known to disrupt the barrier function of the epithelium (32). In contrast, the movement of T lymphocytes across an epithelial barrier in which the junctions are intact, but across which a chemotactic gradient exists has not been investigated.
In this study, we demonstrate that the polarized production of CXCL11 by stimulated human bronchial epithelial cells results in a basal-to-apical transepithelial chemokine gradient. We show that human effector T cells are able to migrate across an intact bronchial epithelial barrier in response to such a gradient, and we examine the consequences of such egression on the epithelial barrier function. In addition, we demonstrate that CXCL11 is found in a polarized distribution in the human bronchial epithelium in vivo and that this gradient is increased in patients with COPD. We show that there are at least two discrete steps in the egression process, adhesion and diapedesis, each of which requires distinct adhesion molecules.
Materials and Methods
Abs and reagents
mAbs 38 (LFA-1 -subunit, function blocking), 15.2 (ICAM-1 blocking), 7C2/10 (IgG1 control), and 52U (IgG1 control) were gifts from Nancy Hogg (Cancer Research U.K., London). P5D2 (β1-subunit function-blocking Ab) was a gift from Fiona Watt (Cancer Research U.K., London). HP1/2 (α4-subunit blocking) was purchased from Serotec. MAb1C6 (CXCR3 blocking) and mAb FIB27 (β7-subunit blocking) were purchased from BD Biosciences and MC5 (CCR5) and 150503 (CCR7) were purchased from R & D Systems. Anti-CC chemokine receptor 5 (CCR5), MC-5 (33) was provided by Dr. Matthias Mack (Medizinische Poliklinik, Ludwig-Maximilians-University of Munich, Munich, Germany). The blocking Ab against VCAM-1 was Ig11 and this was purchased from AMS Biotechnology (Europe). Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary Abs were from Molecular Probes.
Cell culture
Primary normal human bronchial epithelial cells (NHBE; Clonetics, Cambrex Bio Science Wokingham) were cultured on flasks and membranes that had been precoated overnight at 4°C with 10% human placental collagen (Sigma-Aldrich). The medium used for growing the NHBE medium was bronchial epithelial basal medium (Clonetics). NHBE were used at passage 2–4. The SV40-transformed human bronchial epithelial cell line 16HBE14°−(16HBE) was a gift from Dr. Dieter C. Gruenert (Pacific Medical Center, San Francisco, CA). The 16HBE cells were cultured in DMEM plus 10% FCS on flasks or membranes that had been precoated overnight at 4°C with 30 μg/ml Vitrogen (Cohesion) 100 μg/ml BSA (Sigma-Aldrich), and 10 μg/ml fibronectin (Sigma-Aldrich) in PBS-A.
Preparation of lymphocytes
PBMC were prepared from single donor leukocyte buffy coats by centrifugation through Lymphoprep (Pharmacia Diagnostics AB). T cells were expanded from this population by culturing in RPMI 1640 plus 10% FCS (Invitrogen Life Technologies) in the presence of phytohaemagglutinin (Murex Diagnostics) at 1 μg/ml for 72 h as previously described (34). Cells were washed and maintained for 1–2 wk in medium supplemented with 20 ng/ml rIL-2 (EuroCetus). The cells, which were used between days 10 and 14, were a 99% CD3+ population, containing 65% CD8+ and 35% CD4+ cells. The population was negative for the NK cell marker CD56.
Flow cytometry of T lymphocytes
T cells were washed in 20 mM HEPES, 140 mM NaCl, and 2 mg/ml glucose (pH 7.4) (assay buffer), and 2 × 105 T cells in 50 μl assay buffer/0.1% BSA were added to flexiwell plate wells (Dynex Technologies), with appropriate concentrations of primary Abs. After a 60-min incubation on ice, T cells were washed three times and incubated with 3 μg/ml Alexa-488-conjugated goat anti-mouse IgG specific Ab for 20 min on ice. Fluorescence was detected using a FACScan flow cytometer (Becton Dickinson).
Immunohistochemistry
This study protocol was approved by the University College London Hospitals Medical Ethics Committees. Patients who were undergoing thoracic surgery with lung tissue removal gave informed consent for some of this tissue to be used for research purposes.
Formalin-fixed paraffin sections were deparaffinized, rehydrated, and had Ag retrieval performed using Epitope Retrieval Solution 2 (ER2) for 30 min (Vision BioSystems) on a Bond maX automated immunostaining system. The sections were then incubated with either a rabbit polyclonal Ab specific for CXCL11 (PeproTech), or rabbit polyclonal specific for histone H3 (negative control), or no primary Ab (for 20 min). The signal was amplified using the Vision BioSystems Bond Polymer Refine Detection system, visualized with diaminobenzidine and counterstained with hematoxylin.
Quantification of CXCL11 by ELISA
NHBE were grown to confluence on 0.4-μm pore size, 6.5 mm diameter polycarbonate filters (Costar, Corning). The growth medium was replaced with RPMI 1640/0.5% BSA before stimulating with cytokines at specified amounts for 18 h. Monolayers were stimulated on both apical and basal sides using cytokines diluted in RPMI 1640/0.5% BSA for specified time points. NHBE supernatant was collected from apical and basal sides of the monolayer and tested for CXCL11 concentration using a Quantikine ELISA kit (R & D Systems) according to the manufacturer's instructions. Total CXCL11 produced from either side of the monolayer was calculated using a standard calibration curve generated by a known amount of recombinant CXCL11.
Chemotaxis assays
Chemotaxis of T cells in response to various chemokines was investigated in 24-well Transwell chambers using 5-μm pore size, 6.5 mm diameter polycarbonate filters (Costar, Corning,). T cells were labeled with 50 nM 5-chloromethylfluorescein diacetate (Cell Tracker Green CMFDA; Molecular Probes, Invitrogen Life Technologies). Labeled T cells were washed and resuspended in RPMI 1640/0.5% BSA at 2 × 106/ml and 100 μl was added to the upper chamber. Chemokine was placed in the lower chamber in 600 μl of RPMI 1640/0.5% BSA. After 90 min, migrating T cells were retrieved from the lower chamber and counted as FL1 positive cells by FACS.
Transepithelial migration assay
The 16HBE (100 μl of cells at 1 × 106/m) were plated on 8-μm pore size, 6.5 mm diameter polycarbonate filters in 24-well Transwell chambers (Costar, Corning) and grown for 7–9 days. During this time, epithelial cells moved across the filter to grow on both sides of the filter. The monolayers were checked for their impermeability to fluid by their ability to maintain a fluid level difference between inner and outer wells. Cells were removed from one side of the monolayer with a cotton wool tip to leave a monolayer on either the top or bottom of the filter. The monolayers were left in culture for a further 24 h before recording their permeability to FITC-dextran 40 kDa (see above). Only monolayers with permeability of <0.5% of filter alone were used in subsequent assays. Monolayers were washed extensively to remove dextran. T cells and/or epithelial monolayers were preincubated with Abs for 30 min before beginning the assay. The epithelial monolayers were rinsed with RPMI 1640/0.5% BSA and 100 μl of labeled T cells at 4 × 106/ml were added to the monolayers, and the inserts were transferred to new wells (lower chambers) of a 24-well plate containing 0.6 ml of RPMI 1640/0.5% BSA and the indicated chemokine. The Transwells were then incubated at 37°C in 5% CO2. After 90 min, T cells, which had migrated through the epithelial monolayer into the lower chambers, were recovered. FL1 positive cells were counted on FACS and the percentage of migrated cells was calculated as above.
Real-time microscopy of transepithelial migration
Leukocyte/epithelial cell interactions were visualized in real time. The assay was set up as above, but the filters were moved into a four-chambered slide chamber. Microscopy was done with time-lapse photography. Nomarski differential interference contrast images were captured every 30 s and analyzed on QuickTime movies using ImageJ software.
Flow cytometry of bronchial epithelial cells
The 16HBE cells were passaged under gentle trypsinization in 0.02% trypsin/0.0016% EDTA with 0.02% EGTA and 1% polyvinylpyrrolidine solution (Sigma-Aldrich) in HEPES buffered saline (polyvinylpyrrolidine-EGTA-trypsin). After release, the cells were spun down for 5 min at 1,000 rpm, and allowed to recover at 37°C for 1 h in MEM with 10% FCS. Following recovery, cells were washed and 2 × 105 cells in 50 μl RPMI 1640/0.1% BSA were added to flexiwell plate wells (Dynex Technologies), with appropriate concentrations of primary Abs. After 60 min incubation on ice, epithelial cells were washed three times and incubated with an appropriate secondary Ab on ice for 30 min, before washing three times. Fluorescence was detected using a FACScan flow cytometer (Becton Dickinson).
Actin staining of bronchial epithelial cells
Bronchial epithelial cells were grown on coverslips until 80% confluent. The monolayers were stimulated with 100 ng/ml CXCL11 for various time points before being fixed with 4% formaldelhyde at 38°C for 30 min. Cell were then permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature and stained with tetramethylrhodamine isothiocyanate-phalloidin at 250 ng/ml for 10 min at room temperature. Cells were washed three times before mounting and viewing on a fluorescence microscope (Leica). Monolayers were viewed under identical fluorescence and camera exposure to allow comparison between conditions.
Epithelial permeability assay
Transepithelial electrical resistance (TER) was measured using a volt ohmmeter. Confluent monolayers were washed and placed in HBSS to equilibrate for 30 min at 37°C. The voltage across the monolayer was recorded and expressed as ohm/cm2. Permeability to FITC-dextran was measured by washing the monolayers into RPMI 1640/0.5% BSA, and adding 100 μl of FITC-Dextran m.w. 40 kDa to the upper chamber. After 30 min, 100 μl of medium were withdrawn from the lower chamber and fluorescence measured on a plate reader. Background fluorescence was subtracted from the readings. Fluorescence was linear for the concentration of FITC-dextran used and the permeability of the monolayer was expressed as percentage of permeability of the filter alone.
Confocal microscopy
An assay of transepithelial migration was performed as above, but using unlabeled T cells, and with blocking mAbs as appropriate. At the indicated time, the monolayers were washed and fixed with 4% formaldehyde for 20 min at room temperature. Monolayers to be permeabilized were incubated with 0.4% Triton X-100 for 10 min at room temperature. Once transepithelial migration was stopped and the monolayers had been fixed and permeabilized, the T cells were labeled with anti-LFA mAb followed by a goat anti-mouse secondary, and junctions were visualized with rabbit anti-ZO-1 and a goat anti-rabbit secondary. Confocal microscopy was performed using a Zeiss laser-scanning microscope LSM 510 equipped with a 60× oil immersion objective (Karl Zeiss). Images were collected as horizontal sections taken at intervals through whole cell volumes and were compiled for display from a projection of the complete Z-series, as a Y-Z display using ImageJ software. Because the holes in the filters take up fluorescence the sections that pass through the filter are easily identified, and the basal (next to the filter) and apical (furthest from the filter) surfaces can be distinguished (data not shown). In addition, staining of ZO-1 in the epithelial tight junctions allowed us to distinguish basal epithelium and migrating T cells that had not yet crossed the tight junctions from apical epithelium and migrating T cells that had crossed the tight junctions (data not shown). In some situations images were taken at 0.2 μm intervals extending 1 μm basal to the ZO-1 staining and for 1 μm apical to the ZO-1 staining and then projected as a composite attack of images. In addition, the ZO-1 staining from the same series has been analyzed frame by frame and the composite was imported into Adobe Illustrator and recreated as an overlay in orange to show the junctions without the confusion of the background staining.
Adhesion assay
The 16HBE cells were grown to confluence in 96-well tissue culture plates. At 3 days postconfluence, the plates were washed three times into RPMI 1640/0.5% BSA. T cells were washed and labeled with 2.5 μM acetoxymethyl ester of 2,7-bis(2-carboxyethyl)-5,6-carboxyfluorescein (BCEFAM; Calbiochem) and resuspended in RPMI 1640/0.5% BSA. T cells (2 × 105) were added to each well together with CXCL11 and mAbs as indicated to a final volume of 100 μl. Plates were spun at 1,000 rpm for 2 min and then incubated at 37°C for 30 min. Nonadherent cells were removed by washing three times with warmed RPMI 1640/0.5%BSA (150 μl/well). Adhesion was quantified by recording emission at 530 nm following excitation at 485 nm using a plate reader, and expressing the reading for each well as percentage of total emission for that well before incubation.
Results
Human T lymphocytes cultured in IL-2 have an effector phenotype
T cells from healthy volunteers, were cultured in IL-2, and then analyzed in a FACS assay. They expressed LFA-1 (Fig. 1C) but not other β2 integrins (data not shown), β1 (Fig. 1D), β7 (Fig. 1J), α4 (Fig. 1E), and α5 (Fig. 1F) integrin subunits. The lymphocytes were of a Th1/Tc1 effector phenotype expressing CXCR3 (Fig. 1G). In addition, there was a CCR5 negative and a CCR5 positive population (Fig. 1H) as well as CCR7 positive and CCR7 negative populations (Fig. 1I).
FIGURE 1.

Human T lymphocytes cultured in IL-2 have an effector cell Th1 phenotype and express CXCR3. FACS analysis of 14-day T lymphocytes shows no primary Ab (A), secondary goat anti-mouse immunoglobulins: alexa-488 alone (B); mouse mAb against LFA-1 (C), β1 integrins (D), α4 (E), and α5 integrin subunits (F) and the chemokine receptors CXCR3 (G), CCR5 (H), and CCR7 (I). J, Rat anti-human β7-integrin (filled histogram) with line overlay of goat anti-rat immunoglobulins: alexa-488.
CXCL11 secretion from primary human bronchial epithelium is polarized
To determine the polarity of CXCL11 production in vitro by human bronchial epithelium, a confluent polarized monolayer of primary human lung epithelial cells was established on a filter. CXCL11 production was monitored using an ELISA. As shown in Fig. 2, there was only a small amount of CXCL11 secretion from the unstimulated bronchial epithelial cells. There was no significant difference in the secretion of CXCL11 from the basal surface compared with the apical with secretion from both surfaces being close to the limitations of the assay (13.9 pg/ml). However, when the epithelial monolayer was stimulated (on both the apical and basal sufaces) with IFN-γ plus TNF-α, the amount of CXCL11 detected on both sides of the epithelium was increased, but with 45-fold greater amount of CXCL11 released from the apical surface (Fig. 2).
FIGURE 2.
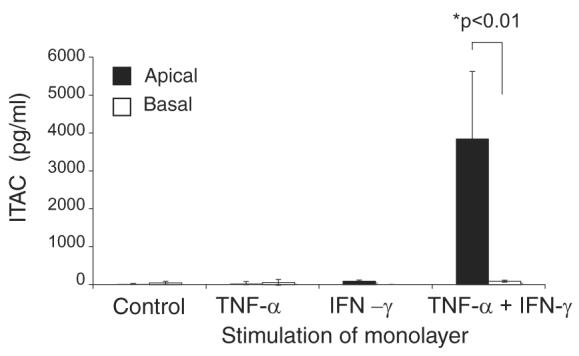
Polarised distribution of epithelial CXCL11 in vitro. Primary human lung epithelial cells were grown to confluence on a filter and left unstimulated or stimulated with IFN-γ and/or TNF-α. Samples of medium from either side of the monolayer (apical/luminal and basal/interstitial) were assayed by ELISA for CXCL11. The results show the combined results from three experiments performed in duplicate, and are expressed as mean ± SEM.
CXCL11 can stimulate transepithelial migration of T lymphocytes across intact bronchial epithelial monolayers in either direction (basal-to-apical and apical-to-basal)
We then investigated whether an epithelial gradient of CXCL11 could stimulate lymphocyte movement across the bronchial epithelium in a basal-to-luminal direction as occurs during lymphocyte egression. The bronchial epithelial cell line 16HBE was cultured either as standard or inverted monolayers on Transwell filters (Fig. 3A). The presence of tight junctions was confirmed using fluorescence microscopy for zona-occludens, (ZO-1; Fig. 3B). The 16HBE monolayers were fluid impermeable with TER >250 W/cm2 and permeability to 40 kDa fluorescent dextran of <0.5% relative to the filter alone. A total of <5% of cultured T cells added to the inner well moved across either an uncoated filter or a monolayer of 16HBE cells in the absence of a chemotactic gradient (Fig. 3C). In response to a gradient of CXCL11 (400 ng/ml), 8–16% of the cells moved across either the filter (chemotaxis; Fig. 3D) or the epithelial monolayer (transepithelial migration; Fig. 3E). Interexperimental variation reflected differences between individual T cell donors. Both chemotaxis and transepithelial migration were prevented when CXCL11 was added to the upper and lower chambers (Fig. 3, D and E), or when the T cells were preincubated with a blocking mAb against CXCR3 (Fig. 3F). CXCL11 in the upper well did not induce a haptotactic response in either a chemotaxis (data not shown) or a transepithelial migration assay (Fig. 4A). T cell migration could take place in either direction across the monolayer in response to CXCL11 (Fig. 4A). Transepithelial migration appeared more efficient in the physiological basal-to-apical direction (equivalent to moving from the interstitium of the lung into the airway lumen) when measured at time points up to 90 min (Fig. 4B), but at later time points there was no significant difference between the two directions (Fig. 4C).
FIGURE 3.

A model system for transepithelial migration demonstrates that similar numbers of T cells migrate across a filter (chemotaxis) or epithelial monolayer in response to CXCL11, despite 200-fold lower permeability of the epithelial monolayer. A, Human bronchial epithelial cells (HBE14°−) can be grown on either side of a Costar transwell filter, and T cell migration can be measured in the apical-to-basal direction (epithelial cells grown on the topside of the filter) and in the more physiological basal-to-apical direction (epithelial cells are grown on the underside of the filter). B, Epithelial cells grown on a filter polarize with the TJ marker ZO-1 correctly positioned; ZO-1 is in red on the Z series and ZX series, the porous filter is stained green. CXCL11 (400 ng/ml) stimulates chemotaxis of T cells across a 5 μM pore-sized filter (C) and a tight epithelial monolayer (D). E, Preincubation of the T cells with Abs blocking CXCR3 abolishes T cell movement in the transepithelial assay. Data are expressed as mean ± SD of triplicates, and one representative experiment of three is shown. The p values were calculated using a two-tailed Student's t test. ***, p < 0.001.
FIGURE 4.
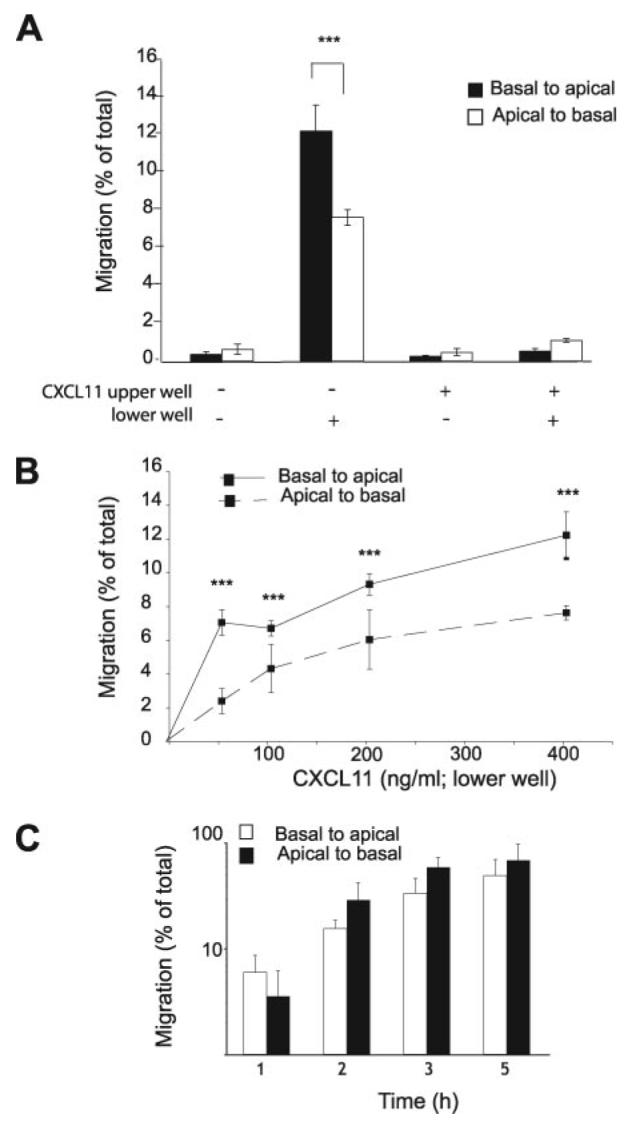
T lymphocytes migrate across a tight epithelial monolayer in either direction in response to CXCL11. A, CXCL11 (400 ng/ml) stimulates migration of T cells across a tight epithelial monolayer in either direction. Abolition of the CXCL11 gradient inhibits transepithelial migration, and there is no transepithelial migration seen when CXCL11 is placed in the upper well of the assay. B, T cell migration in response to CXCL11 is enhanced in the physiological basal-to-apical direction at early time points (<90 min) (C), but there is no significant difference in the two directions at later time points. Data are expressed as mean ± SD of triplicates, and one representative experiment of three is shown. The p values were calculated using a two-tailed Student's t test. ***, p < 0.001.
Video microscopy of transepithelial migration
We used video microscopy of transepithelial migration to examine the route and time taken to cross the epithelium for the T cells (see Supplementary Video 1).6 The time course of transepithelial migration was slower than that for chemotaxis. Chemotaxis was complete by 90 min (data not shown), but T cells were still migrating several hours after the beginning of a transepithelial migration assay (Fig. 5A). As illustrated in Fig. 5, B–E, the video microscope was focused on the basal surface of the monolayer, with the filter above the plane of focus so that the holes in the filters appear out of focus in Fig. 5B (open black arrows and numbered 1 and 2 in white). The T cells can be seen coming into the field of view at the start of the assay (Fig. 5B; white arrows), by 60 min the cells were aligned on the basal surface along, what appear to be, intercellular junctions (Fig. 5C; thin black arrows). At 71 min, one T cell has passed through the intercellular junction and moved out of the plane of focus (Fig. 5D; thick black arrow). Over the next 20 min, three more T cells pass through the intercellular junctions (Fig. 5E), while one T cell remains on the basal surface apparently resting on a junction (Fig. 5E).
FIGURE 5.
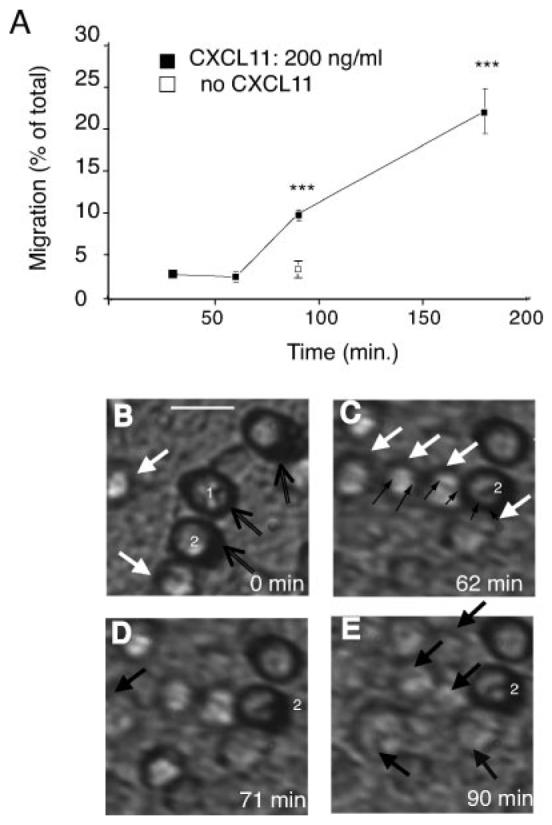
Live cell imaging and fixed cell analysis of the different stages of transepithelial migration. Data are expressed as mean ± SD of triplicates, and one representative experiment of three is shown. The p values were calculated using a two-tailed Student's t test; ***, p < 0.001. A, Time course of transepithelial migration across an inverted epithelial monolayer. B–E, Time course video imaging of T lymphocytes during transepithelial migration. B, White arrows mark T cells on the basal surface of the epithelium. Open black arrows mark the 8-μM pores in the filter that are out of focus and through which the T cells have already passed. Two of these pores have been labeled “1” and “2” in white. C, The epithelial junctions can be seen and one section is marked with small black arrows. D and E, T cells that have successfully undergone transepithelial migration move out of the plane of focus and are labeled with closed black arrows. Scale bar, 10 μm. Complete set of images is available in Supplementary Video 1 online.
CXCL11 causes increased actin polmerization in human bronchial epithelial cells which is blocked by mAbs to CXCR3, but this is not essential for efficient transepithelial migration
FACS analysis showed that 16HBE cells express high levels of CXCR3, even more than seen on T lymphocytes (Fig. 6A), and this led us to investigate whether CXCL11 has an autocrine effect on the bronchial epithelium that produces it. Stimulation of the bronchial epithelium for 1 h, with concentrations of CXCL11 between 100 and 400 ng/ml led to similar increases in epithelial actin polymerization (data not shown); and so the lower concentration of 100 ng/ml CXCL11 was used for subsequent experiments (Fig. 6). The increase in epithelial cell actin polymerization, was apparent at 5 min (Fig. 6C) but maximal at 30 min (Fig. 6D) and maintained at 60 min (Fig. 6E). This actin polymerization was inhibited by preincubation of the epithelium with a blocking mAb to CXCR3 (Fig. 6G). These experiments confirmed that CXCR3 is functional on the bronchial epithelium. To investigate the relative contributions of CXCR3 on the epithelium and on the T lymphocyte during transepithelial migration, we conducted transepithelial migration assays after preincubation of the epithelium or of the lymphocytes with a blocking mAb to CXCR3. Preincubation of the bronchial epithelium with mAb against CXCR3 had no effect on transepithelial migration (Fig. 6H), in contrast to the inhibitory effect when T cells were preincubated with the same mAb (Fig. 6H). This demonstrated that the autocrine effect of CXCL11 on the epithelium was not essential for efficient lymphocyte transepithelial migration.
FIGURE 6.
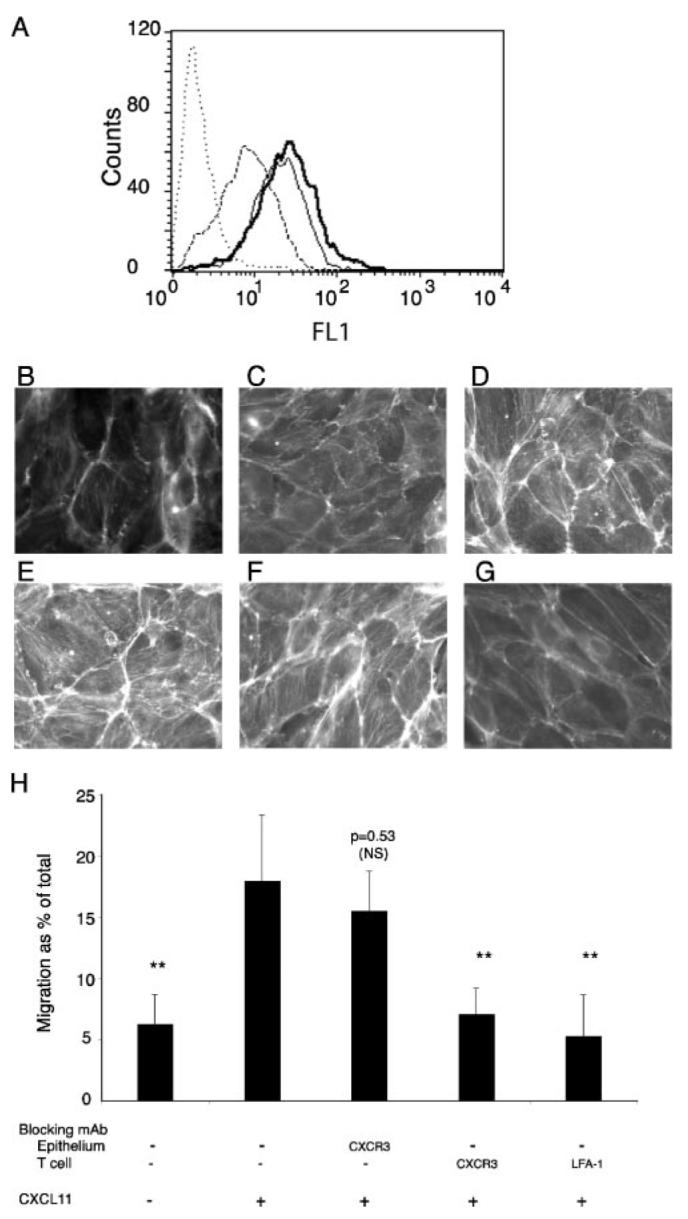
Bronchial epithelial cells express CXCR3 and undergo actin rearrangements in response to CXCL11. A, FACS analysis of CXCR3 expression on bronchial epithelial cells released with EGTA (thin line) or polyvinylpyrrolidine-EGTA-trypsin (thick line), and on T cells (dashed line). Bronchial epithelial cells incubated with secondary alone are shown as dotted control. Results are representative of four different experiments. B–G, Incubation of bronchial epithelium with 100 ng/ml CXCL11 at various time points 0 min (B), 5 min (C), 15 min (D), 30 min (E and G), and 60 min (F) causes an increase in actin polymerization and stress fiber formation that is inhibited by preincubation of the epithelium with a blocking mAb against CXCR3 (G). Scale bar, 10 μm. One representative experiment of three is shown. H, Transepithelial migration is inhibited by blocking mAbs against CXCR3 on the T cell, but not against CXCR3 on the epithelial cell. Transepithelial migration assay after 120 min in response to 400 ng/ml CXCL11. T cells or epithelial monolayers were preincubated with Abs to CXCR3 or LFA-1 before being washed and used in a migration assay. Data are expressed as mean ± SD of triplicates, and one representative experiment of three is shown. The p values were calculated using a Student's t test. **, p < 0.01.
T cell adhesion to, and migration across, the epithelial monolayer causes localized decreases in barrier function
The observation that T cells were able to move across epithelial junctions in large numbers (2 × 105/cm2 of epithelium, equivalent to a ratio of 2:1 T cell to epithelial cell over 90 min) in response to CXCL11, raised the possibility that transepithelial migration might compromise the barrier function of the epithelium. To investigate tight junction integrity, we measured TER and permeability to 40 kDa fluorescent dextran, before and after transepithelial migration. For a control of barrier disruption, we used Cytochalasin D at 2 μM, which is the lowest dose that prevents the epithelial monolayer from maintaining a fluid level difference between the inner and outer wells. Cytochalsain D inhibits actin polymerization and disrupting the interepithelial junctions.
Transepithelial migration increased monolayer permeability without disruption of ZO-1 and β-catenin
CXCL11 alone had no effect on either TER or dextran permeability (Fig. 7, A and B). However, T cells alone, in the absence of an CXCL11 gradient caused a consistent but not always significant decrease in TER, but with no change in dextran permeability (Fig. 7, A and B). T cells migrating across the epithelium in response to an exogenous CXCL11 gradient caused a significant decrease in TER and an increase in dextran permeability (Fig. 7, A and B). Cytochalasin D, in contrast, caused a marked decrease in TER and an increase in dextran permeability (Fig. 7, A and B). Analysis of junctional components revealed no disruption of either ZO-1 (Fig. 7D) or β-catenin (Fig. 7G) during transepithelial migration as compared with the control monlayers (Fig. 7, C and F). In contrast, monolayers treated with Cytochalasin D showed disruption of these tight junction molecules (Fig. 7, E and H). In summary, CXCL11 alone does not affect epithelial permeability, but T cell migration across the epithelial monolayer causes localized decreases in barrier function, which appears proportional to the number of migrating cells.
FIGURE 7.

Epithelial permeability is increased by adherent and migrating T cells, without alteration of junctional proteins. A, The permeability of the monolayer to FITC-dextran 40 kDa or the transepithelial resistance of the monolayer (B) were measured as described in the presence of CXCL11, T cells, and both CXCL11 and T cells. Cytochalasin D was included as a positive control of barrier disruption. Readings were made from triplicate wells. Only wells with TER of >150 ohm/cm2 were used. Data were taken from triplicate samples and are expressed as mean ± SD and p values were calculated using a Student's t test. C–E, ZO-1 staining, and β-catenin staining of monolayers (F–H), after 1 h of transepithelial migration. (The small white rings indicate the underlying pores in the filter on which the epithelial cell are growing). C and F, Control monolayer. D and G, T cells on apical surface and CXCL11 on basal surface. E and H, Cytochalasin D 2 μM treatment for 30 min. Scale bar, 10 μm.
A polarized distribution of CXCL11 is seen in vitro and in vivo in healthy human bronchial epithelium, and this chemotactic gradient is increased in patients with COPD
To examine the localization of CXCL11 in human bronchial epithelium in vivo, we stained lung biopsies for CXCL11. Multiple samples of pulmonary epithelium from eight patients with COPD (representative samples shown in Fig. 8, A–C, E, and F) were stained positively for CXCL11 throughout the epithelium, with increased staining on the apical surface (Fig. 8, C, E, and F); the staining was not seen with either secondary Ab alone (Fig. 8A) or an isotype-matched control (Fig. 8B). Normal lung from three control patients showed only low-level staining for CXCL11 (Fig. 8D) which had the same polarized distribution.
FIGURE 8.

Human lung samples show a polarized distribution of epithelial CXCL11 in vitro, which is increased in patients with COPD. A–F, Lung tissue from patients stained for human CXCL11. Epithelium from patients with COPD (A–C, E, and F), or control patients (D), stained with secondary Ab alone (A), isotype-matched control Ab (B), or anti-CXCL11 Ab (C and F). The airway lumen is marked with *. Scale bar, 10 μm.
During transepithelial migration, α4 and LFA-1 integrins are required for T cell adhesion to the basal epithelial surface and LFA-1 is required for diapedesis
To investigate the mechanisms involved in transepithelial migration of T cells, we made use of mAbs to specific adhesion molecules. Transepithelial migration was inhibited when either α4 integrins or the β2 integrin LFA-1 on the T cells were blocked (Fig. 9A). The block was more complete with LFA-1 inhibition, and under these conditions, an α4-inhibiting Ab had no additional effect (Fig. 9A). A blocking Ab to the LFA-1 ligand, ICAM-1, also inhibited transepithelial migration (Fig. 9A). Inhibition of β1 and β7 integrins on the T cells significantly reduced transepithelial migration, and combined inhibition was equivalent to inhibition against α4 integrins, indicating involvement of both α4β1 and α4β7 on the T cells (data not shown). The 16HBE cells express a small amount of VCAM-1, which is a ligand for both α4β1 and α4β7 and blocking VCAM-1 on the epithelial cells consistently inhibited transepithelial migration, but to a lesser extent than blocking ICAM-1 (Fig. 9, A and B).
FIGURE 9.
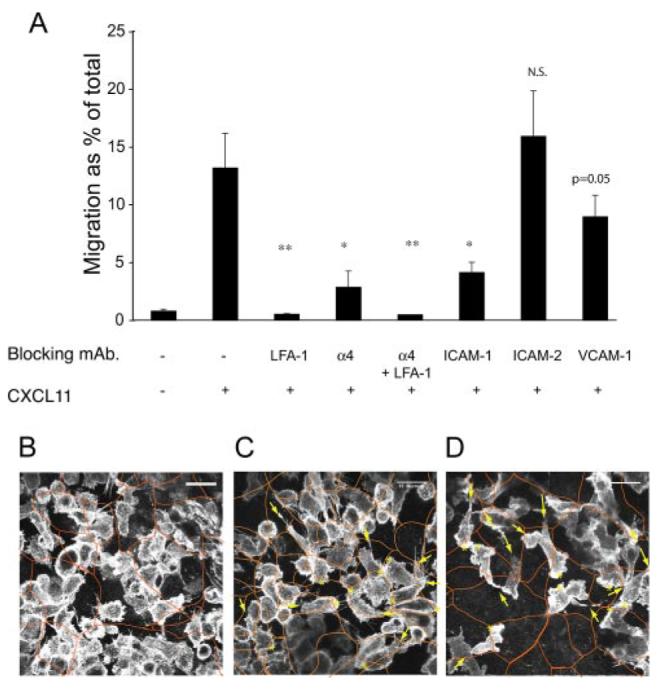
Transepithelial migration is inhibited by blocking mAbs against the T cell integrins α4 and LFA-1, and to the epithelial ligands ICAM-1 and VCAM-1. A, Transepithelial migration is inhibited by blocking mAbs against the T cell integrins α4 and LFA-1, and to the epithelial ligands ICAM-1 and VCAM-1. Transepithelial migration assay after 90 min in response to 400 ng/ml CXCL11. T cells were preincubated with Abs to α4 and/or LFA-1, bronchial epithelial monolayers were preincubated with Abs against ICAM-1, and VCAM-1. Data are expressed as mean ± SD of triplicates, and one representative experiment of three is shown. The p values were calculated using a Student's t test. *, p = 0.05; **, p < 0.01; ***, p < 0.001. B–D, Following T cell adhesion, LFA-1 is required for T cell migration across the epithelium. B–D, Laser scanning confocal microscopy of the basal surface of the epithelium after I hour of transepithelial migration showing migrating T cells (labeled with anti-LFA-1), and epithelium (labeled with anti-ZO-1; orange overlay). Scale bar, 15 μm (B). No CXCL11 (C and D) CXCL11 gradient; leading edge (*) and a trailing uropod (arrows) shown for eight cells. D, T cells preincubated with an LFA-1 integrin blocking mAb.
The role of LFA-1 in CXCL11 induced transepithelial migration
Next, using confocal microscopy, we investigated the appearance of the T cells as they migrated across the bronchial epithelium. Fig. 9, B–D, shows the basal surface of the epithelial monolayer, which has been fixed 1 h after addition of T cells. The T cells have been labeled with anti-LFA-1 and the epithelial cells with anti-ZO-1 (outlined in red in Fig. 9, B–D). In the absence of a CXCL11 gradient, control T cells adhered to the basal surface of the epithelium; these cells were relatively unpolarized (Fig. 9B). In the presence of a CXCL11 gradient, a similar number of T cells were adherent to the epithelium but many of the T cells were polarized, with a trailing uropod and a leading edge; in many cases, this leading edge was seen at a junction between two or three epithelial cells (Fig. 9C). When LFA-1 on the T cells was blocked, there were fewer adherent T cells. However, the cells that were adherent were polarized normally (Fig. 9D), but these adherent cells were still unable to complete the transmigration across the epithelium (Fig. 9A). When α4 was blocked, <50% of the added T cells are able to cross the filter alone in a chemotaxis assay (data not shown), and this may account for some of the inhibition seen in a transepithelial migration assay (Fig. 9A). In summary, α4 integrins are required for the T cell to cross the filter to come into contact with the basal surface of the epithelium. The T cell then adheres to the basal epithelium using LFA-1 and α4 integrins. A further postadhesive step involving LFA-1, and potentially other integrins, is then required for successful completion of transepithelial migration.
Because of the dependence of transepithelial migration on LFA-1:ICAM-1 interactions, we looked at the effect of CXCL11 on bronchial epithelial expression of adhesion receptors and found an up-regulation of ICAM-1 (Fig. 10, A and B), but not of ICAM-2 and VCAM-1 (data not shown). There was more than a 2-fold increase in epithelial ICAM-1, and this was maximal at 30–60 min depending on the concentration of CXCL11 used to stimulate the epithelium (Fig. 10C). The increase in ICAM-1 was maintained for at least 120 min (Fig. 10C). However, despite this increase in epithelial cell expression of ICAM-1, there was no resultant increase in LFA-1:ICAM-1 dependent adhesion of lymphocytes to the bronchial epithelium after stimulation with CXCL11 (Fig. 10D). These results indicated that epithelial levels of ICAM-1 are not a limiting factor for T cells adhesion to the unstimulated epithelium and suggests the up-regulation of ICAM-1 by CXCL11 does not increase the efficiency of transepithelial migration.
FIGURE 10.

CXCL11 up-regulates ICAM-1 on bronchial epithelial cells but does not increase LFA-1:ICAM-1 mediated adhesion of lymphocytes to the bronchial epithelium. A and B, FACS analysis of ICAM-1 expression on bronchial epithelial cells after stimulation with CXCL11 for 30 min (A) and 120 min (B). Dotted lines show epithelial cells labeled with secondary Ab alone. Thick line, no CXCL11; thin line, 100 ng/ml; dashed line, 400 ng/ml. One experiment representative of three similar experiments is shown. C, FACS analysis of ICAM-1 expression on bronchial epithelial cells, after stimulation with CXCL11, and normalized to unstimulated control cells: no CXCL11 white bars, 100 ng/ml gray bars, and 400 ng/ml black bars. The results are shown as the average and SD from three similar FACS experiments. The p values were calculated using a Student's t test. *, p = 0.05; **, p < 0.01. D, Adhesion of T lymphocytes to the bronchial epithelium is LFA-1: ICAM-1 dependent but is not increased by various concentrations of CXCL11. T cells adhesion to the bronchial epithelium was measured in the presence of increasing amounts of CXCL11 and various blocking mAbs directed against LFA-1 on the lymphocyte, or ICAM-1, ICAM-2 and VCAM-1 on the epithelium: no CXCL11, white bars; 100 ng/ml, gray bars; and 400 ng/ml, black bars. The results are shown as the average and SD from three similar experiments. The p values were calculated using a Student's t test. *, p = 0.05; ***, p < 0.005.
Discussion
This study demonstrates that egression of human effector T cells from the lung can take place across an intact bronchial epithelium in response to a chemotactic gradient of CXCL11 and dissects key components of the mechanism regulating such migration. Bronchial epithelial cells are known to secrete CXCL11, but our data demonstrate a basal-to-apical gradient of CXCL11 across the epithelium both in vitro and in vivo. This gradient is present to some degree in normal lung tissue, but markedly increased in lung specimens from patients with COPD. Findings from cultured epithelial monolayers indicate that the gradient is established in part by the stimulated epithelial cells, although additional factors such as production of CXCL11 by IFN-γ stimulation of luminal alveolar macrophages (26) or other inflammatory cells, with subsequent retention of CXCL11 at the apical epithelium, may also be involved. Our results do not distinguish whether the basal secretion detected when the bronchial epithelium is stimulated is true basal secretion or reflects movement of CXCL11 across the disrupted barrier from apical to basal side.
In our cellular model, transepithelial migration in response to a gradient of CXCL11 is a true chemotactic response that can operate in both the basal-to-apical and apical-to-basal direction. Data from pigs (35, 36) and mice (25) show that lymphocytes can move from the airway lumen back into the lung, emphasizing that the chemotactic gradient may operate in both directions across the epithelium depending on the underlying pathophysiology. Although we focus on CXCL11, an exclusive T cell chemoattractant, it is likely that there are other chemotactic gradients across the respiratory epithelium that are established by respiratory epithelial or other luminal cells. Consistent with this, another T cell attractant CCL5 was found on the apical side of the bronchial epithelium in patients with asthma (30). In addition, multiple chemokine receptors, including CXCR3 and CCR7 have been implicated in different phases of the asthmatic response including the migration of lymphocytes to allergen challenged airways (37). Thus, the specific chemoattractant-dependent leukocyte migration may depend on the underlying disease stimulus and vary as the inflammatory responses evolves.
Theoretically, there are several ways in which apical CXCL11 on the apical side of the epithelium can be sensed by lymphocytes on the basal side, even when the epithelium is intact: diffusion of the chemokine across the tight junctions; transcytosis of the chemokine from apical to basal surfaces; sampling of the apical milieu by T lymphocytes extending processes across the tight junctions as has been shown for dendritic cells in the gut (38); or movement in the plane of the membrane. We have shown that 3kD and 10kD dextran can move slowly across the tight junctions (data not shown), and it is probable that CXCL11 (molecular mass 8.3 KDa) can do the same. Alternatively, both endothelial and epithelial cells transcytose proteins and present them on the opposite surface. For example, endothelial cells transcytose IL-8 and present it apically (39), and epithelial cells have been shown to transcytose luminal cobalumin and present basally (40). It is possible that epithelial cells actively transport apical CXCL11 for basal presentation.
Next, we investigated the route taken by the T cell during transepithelial migration. It appears that many, if not all, T cells cross the epithelium by a para-cellular route across the tight junctions. It has been well described that leukocytes may cross the endothelium by a para-cellular route largely regulated by the interendotheial cell:cell junctions. During transendothelial migration, adhesion molecules (such as ICAM-1 and VCAM-1) on the endothelium not only play a role in capturing leukocytes, but induce signaling on integrin ligation, which mediate endothelial junctional remodelling (41-44). This junctional remodelling may lead to transient increases in endothelial permeability and facilitates leukocyte transendothelial migration (42, 45, 46). We therefore examined the effect of lymphocyte adhesion and migration on the integrity of the epithelial monolayer. Adhesion of T cells to the basal surface of the epithelial cells and migration across the monolayer caused small, localized increases in the permeability of the epithelium. The decrease in TER was greater than the increase in permeability to dextran, implying local discontinuous disruptions of the monolayer. A similar process of rapid and reversible increase in epithelial permeability, without loss or redistribution of tight junction proteins, has been described when neutrophils adhere to and cross the intestinal epithelium (47). Cross-linking of ICAM-1 has been linked to permeability increases in the lung epithelium (48), but we found no evidence for this in our assay. Our results do not determine whether the small but consistent decrease in TER, in the absence of CXCL11, is induced by adhesion of T cells to the basal epithelium independent of transepithelial migration, or by the low level of constitutive transepithelial migration observed, or by both. One possibility is that this small, constitutive, but significant decrease in TER when T cells interact with the basal epithelium may allow CXCL11 from the apical surface to be detected by basal lymphocytes, and stimulate further egression and a further increase in epithelial permeability. A surprising observation is that, in response to a given concentration of CXCL11, a similar number of T cells migrate across a filter as migrate across a tight epithelial monolayer. The epithelium is 200 times “tighter” that the filter alone, and is not dramatically disrupted during transepithelial migration. This indicates that the epithelial cells are not just bystanders, but they play an active role in the egression process.
Given the active role of epithelial cells in the process of transepithelial migration, it is possible that CXCL11, as well as being a chemoattractant for T cells, also has an autocrine effect on the epithelium. There has been some debate in the literature on the presence of CXCR3 on bronchial epithelium, which has been found by some researchers (49-51), but not by others (52). CXCR7 can also function as a receptor for CXCL11 (53), but it is not found on the surface of lung epithelial cells (53). We demonstrate high levels of functional trypsin-resistant CXCR3 on the bronchial epithelium, and have shown that CXCL11 stimulation of the bronchial epithelium cells leads to actin rearrangements and stress fiber formation, similar to that shown for CXCL10 (50). These effects of CXCL11 are prevented when the epithelium is preincubated with a blocking mAb against CXCR3. However, we show that these autocrine effects of CXCL11 on the epithelium are not essential for egression, as a blocking mAb against epithelial CXCR3 does not inhibit transepithelial migration.
We then examined the molecular mechanisms involved in transepithelial migration. Egressing T cells first come into contact with extracellular matrix on the filter, before binding to the basal surface of the epithelium and moving across the junctions. In the control situation, with no CXCL11 gradient, T cells adhere well to the basal surface of the epithelium. Such adhesion was blocked with a combination of blocking Abs against LFA-1 and α4 integrins. These integrins are not constitutively active on T cells and require an activation step (54). The nature of the activating stimulus is unknown, but our data show that activation of integrins for adhesion does not require CXCL11. CXCL11 is required for polarization of the T cells. In the absence of CXCL11, although the majority of adherent T cells are rounded, some are polarized with a uropod and a leading edge. In the presence of a CXCL11 gradient, the T cells on the basal epithelial surface became more spread and more obviously polarized, with a trailing uropod and a leading edge.
The quantification of leukocyte adhesion to the basal surface of the epithelium is difficult. However, we found that migration across the extracellular matrix requires α4-mediated adhesion and can be reduced with Abs against α4. The majority of lymphocytes that manage to cross the extracellular matrix during α4-block appear to bind to the epithelial cell basal surface normally, polarize in response to CXCL11, and move across the monolayer (data not shown).
Blocking Abs against β1 integrins inhibit chemotaxis across a filter by ∼50% but had a less and variable effect on transepithelial migration. This may be because the bronchial epithelial cells also express β1 integrins that contribute to the integrity of the monolayer. In contrast, inhibition of β7 integrins on the T cells had no effect on chemotaxis but did reduce transepithelial migration by ∼50%; this demonstrates that both α4β1 and α4β7 integrins can contribute to initial leukocyte-epithelial adhesion. Bronchial epithelial cells express VCAM-1, a surface ligand for both α4β1 and α4β7, and blocking VCAM-1 on the epithelium has a small but consistent effect on transepithelial migration.
Inhibition of LFA-1 reduces the binding to T cells to the epithelium, but T cells can still adhere to the epithelium in reasonable numbers. However, despite the ability of the T cells to adhere to the epithelium, they are unable to complete the migration across the epithelium. The observation that blocking LFA-1 completely prevents transepithelial migration, even though cells are still adherent to the epithelium, indicates an essential postadhesion step that is LFA-1-dependent, analogous to the β2-integrin steps that have been described for transendothelial migration (55-57). There are several theoretical possibilities on the nature of this LFA-1-dependent step that we are currently investigating: LFA-1 may be required for the T cell to move to the intercellular junctions, perhaps in response to a combination of ICAM-1 at the junctions and a chemotactic gradient across the junctions. Alternatively, binding of lamellipodial LFA-1 to ICAM-1 at the junctions may allow release of the T cell uropod, allowing the T cell to progress on ICAM-1 (58, 59). Interestingly, although CXCL11 up-regulates ICAM-1 on the surface of the epithelium, this does not result in increased LFA-1: ICAM-1-dependent adhesion of the T cells to the epithelium. These results suggest that epithelial expression of ICAM-1 is not a limiting factor for initial lymphocyte adhesion to the epithelium; but they do not exclude a requirement for up-regulation of epithelial ICAM-1 in subsequent postadhesion events that may be necessary for successful transepithelial migration.
On arrival at the luminal side of the epithelium, the T cells adhere briefly to the epithelial cell before detaching. Intratracheal transfer studies have shown that bronchial effector T cells down-regulate LFA-1 on arrival in the airway (60), which may allow their removal on the mucociliary escalator (12), and prevent excessive IFN-γ-dependent amplification of CXCR3+ lymphocyte recruitment (61). Down-regulation of LFA-1 may also prevent T cells moving back across the epithelium (apical-to-basal) into the lung interstitium.
In conclusion, we have shown that CXCL11, known to play a role in transendothelial migration (62), is a potent inducer of T cell migration across a tight epithelial barrier, and we have examined the molecular processes by which human T lymphocytes can migrate out of the lung and into the airway lumen in response to a chemotactic gradient. We have shown that epithelial CXCL11 is found in a polarized distribution in the bronchial epithelium in normal lung, and that staining for CXCL11 is increased in lungs from patients with COPD but always in the same polarized distribution. We have identified key steps in the CXCL11 gradient-induced egression of effector Th1/Tc1 cells across a bronchial epithelial monolayer and have shown that T cell movement across the epithelium is a multistep process, analogous to that across the endothelium, with sequential and essential involvement of different adhesion receptors. In particular, although α4 integrins and LFA-1 both contribute to adhesion to the epithelium, there is an LFA-1-dependent step, after initial adhesion, which is essential for successful egression. We have demonstrated that lymphocytes are able to migrate across intact, undamaged epithelium; that luminal lymphocytes are not simply a non-specific result of epithelial disruption; and that the bronchial epithelial cells play an active role in egression. A wealth of knowledge concerning leukocyte extravasation has led to targeted therapies to prevent and treat inflammation (63) and a similar understanding of leukocyte egression may prove valuable in the therapeutic approach to inflammatory diseases of the lung and other epithelial organs.
Supplementary Material
Acknowledgments
The idea to investigate lymphocyte transepithelial migration in the lung came from discussions with Nancy Hogg while J.P. was in her laboratory. We also acknowledge Nancy Hogg and members of her laboratory for gifts of reagents and preparation of T lymphocytes. We thank Dieter Gruenert for providing the 16HBE cell line. Thanks to Jon Friedland, Alison Lloyd, Martin Raff, and members of Alan Hall's Laboratory for helpful discussions and careful reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org
This work was supported by Cancer Research (CR) U.K. and a Wellcome Trust Advanced Clinical Fellowship (to J.C.P.).
Abbreviations used in this paper: COPD, chronic obstructive pulmonary disease; TER, transepithelial electrical resistance; ZO-1, zona-occludens; LFA-1, leukocyte function associated-1; NHBE, normal human bronchial epithelial cell.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.World Health Organization . The World Health Report 2003: Shaping the Future. Geneva: World Health Organization; 2003. pp. 3–22. [Google Scholar]
- 2.Mizgerd JP. Lung infection–a public health priority. PLoS Med. 2006;3:e76. doi: 10.1371/journal.pmed.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes PJ. Chronic obstructive pulmonary disease: a growing but neglected global epidemic. PLoS Med. 2007;4:e112. doi: 10.1371/journal.pmed.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, Schmid V, Buist S. Chronic obstructive pulmonary disease: current burden and future projections. Eur. Respir. J. 2006;27:397–412. doi: 10.1183/09031936.06.00025805. [DOI] [PubMed] [Google Scholar]
- 6.Harris NL, Watt V, Ronchese F, Le Gros G. Differential T cell function and fate in lymph node and nonlymphoid tissues. J. Exp. Med. 2002;195:317–326. doi: 10.1084/jem.20011558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogan RJ, Usherwood EJ, Zhong W, Roberts AA, Dutton RW, Harmsen AG, Woodland DL. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 2001;166:1813–1822. doi: 10.4049/jimmunol.166.3.1813. [DOI] [PubMed] [Google Scholar]
- 8.De Bree GJ, van Leeuwen EM, Out TA, Jansen HM, Jonkers RE, van Lier RA. Selective accumulation of differentiated CD8+ T cells specific for respiratory viruses in the human lung. J. Exp. Med. 2005;202:1433–1442. doi: 10.1084/jem.20051365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hogan RJ, Zhong W, Usherwood EJ, Cookenham T, Roberts AD, Woodland DL. Protection from respiratory virus infections can be mediated by antigen-specific CD4+ T cells that persist in the lungs. J. Exp. Med. 2001;193:981–986. doi: 10.1084/jem.193.8.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, Kheradmand F. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodland DL, Scott I. T cell memory in the lung airways. Proc. Am. Thorac. Soc. 2005;2:126–131. doi: 10.1513/pats.200501-003AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hikono H, Kohlmeier JE, Ely KH, Scott I, Roberts AD, Blackman MA, Woodland DL. T-cell memory and recall responses to respiratory virus infections. Immunol. Rev. 2006;211:119–132. doi: 10.1111/j.0105-2896.2006.00385.x. [DOI] [PubMed] [Google Scholar]
- 13.Panina-Bordignon P, Papi A, Mariani M, Di Lucia P, Casoni G, Bellettato C, Buonsanti C, Miotto D, Mapp C, Villa A, et al. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J. Clin. Invest. 2001;107:1357–1364. doi: 10.1172/JCI12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, Bellettato CM, Papi A, Corbetta L, Zuin R, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002;165:1404–1409. doi: 10.1164/rccm.2107139. [DOI] [PubMed] [Google Scholar]
- 15.Hussell T, Snelgrove R, Humphreys IR, Williams AE. Co-stimulation: novel methods for preventing viral-induced lung inflammation. Trends Mol. Med. 2004;10:379–386. doi: 10.1016/j.molmed.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphreys IR, Walzl G, Edwards L, Rae A, Hill S, Hussell T. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J. Exp. Med. 2003;198:1237–1242. doi: 10.1084/jem.20030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guyot-Revol V, Innes JA, Hackforth S, Hinks T, Lalvani A. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. Am. J. Respir. Crit. Care Med. 2006;173:803–810. doi: 10.1164/rccm.200508-1294OC. [DOI] [PubMed] [Google Scholar]
- 18.Sheppard D. Killed by leukocytes that don't know when to leave. Nat. Immunol. 2002;3:337–338. doi: 10.1038/ni0402-337. [DOI] [PubMed] [Google Scholar]
- 19.Uller L, Persson CG, Kallstrom L, Erjefalt JS. Lung tissue eosinophils may be cleared through luminal entry rather than apoptosis: effects of steroid treatment. Am. J. Respir. Crit. Care Med. 2001;164:1948–1956. doi: 10.1164/ajrccm.164.10.2011135. [DOI] [PubMed] [Google Scholar]
- 20.Erjefalt JS, Uller L, Malm-Erjefalt M, Persson CG. Rapid and efficient clearance of airway tissue granulocytes through transepithelial migration. Thorax. 2004;59:136–143. doi: 10.1136/thorax.2003.004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corry DB, Rishi K, Kanellis J, Kiss A, Song Lz LZ, Xu J, Feng L, Werb Z, Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat. Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:735–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 24.Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, Butcher EC. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 2005;6:895–901. doi: 10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 26.Cole AM, Ganz T, Liese AM, Burdick MD, Liu L, Strieter RM. Cutting edge: IFN-inducible ELR- CXC chemokines display defensin-like antimicrobial activity. J. Immunol. 2001;167:623–627. doi: 10.4049/jimmunol.167.2.623. [DOI] [PubMed] [Google Scholar]
- 27.Sauty A, Dziejman M, Taha RA, Iarossi AS, Neote K, Garcia-Zepeda EA, Hamid Q, Luster AD. The T cell-specific CXC chemokines IP-10, Mig, and I-TAC are expressed by activated human bronchial epithelial cells. J. Immunol. 1999;162:3549–3558. [PubMed] [Google Scholar]
- 28.Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. 2005;289:L85–L95. doi: 10.1152/ajplung.00397.2004. [DOI] [PubMed] [Google Scholar]
- 29.Jiang D, Liang J, Hodge J, Lu B, Zhu Z, Yu S, Fan J, Gao Y, Yin Z, Homer R, et al. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J. Clin. Invest. 2004;114:291–299. doi: 10.1172/JCI16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taguchi M, Sampath D, Koga T, Castro M, Look DC, Nakajima S, Holtzman MJ. Patterns for RANTES secretion and intercellular adhesion molecule 1 expression mediate transepithelial T cell traffic based on analyses in vitro and in vivo. J. Exp. Med. 1998;187:1927–1940. doi: 10.1084/jem.187.12.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller LA, Butcher EC. Human airway epithelial monolayers promote selective transmigration of memory T cells: a transepithelial model of lymphocyte migration into the airways. Am. J. Respir. Cell Mol. Biol. 1998;19:892–900. doi: 10.1165/ajrcmb.19.6.3245mrev. [DOI] [PubMed] [Google Scholar]
- 32.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol. Biol. Cell. 2002;13:3218–3234. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Signoret N, Pelchen-Matthews A, Mack M, Proudfoot AE, Marsh M. Endocytosis and recycling of the HIV coreceptor CCR5. J. Cell Biol. 2000;151:1281–1294. doi: 10.1083/jcb.151.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dransfield I, Cabanas C, Craig A, Hogg N. Divalent cation regulation of the function of the leukocyte integrin LFA-1. J. Cell Biol. 1992;116:219–226. doi: 10.1083/jcb.116.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehmann C, Wilkening A, Leiber D, Markus A, Krug N, Pabst R, Tschernig T. Lymphocytes in the bronchoalveolar space reenter the lung tissue by means of the alveolar epithelium, migrate to regional lymph nodes, and subsequently rejoin the systemic immune system. Anat. Rec. 2001;264:229–236. doi: 10.1002/ar.1163. [DOI] [PubMed] [Google Scholar]
- 36.Pabst R, Binns RM. Lymphocytes migrate from the bronchoalveolar space to regional bronchial lymph nodes. Am. J. Respir. Crit. Care Med. 1995;151:495–499. doi: 10.1164/ajrccm.151.2.7842212. [DOI] [PubMed] [Google Scholar]
- 37.Thomas SY, Banerji A, Medoff BD, Lilly CM, Luster AD. Multiple chemokine receptors, including CCR7 and CXCR3, regulate antigen-induced T cell homing to the human asthmatic airway. J. Immunol. 2007;179:1901–1912. doi: 10.4049/jimmunol.179.3.1901. [DOI] [PubMed] [Google Scholar]
- 38.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 39.Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 40.Dan N, Cutler DF. Transcytosis and processing of intrinsic factor-cobalamin in Caco-2 cells. J. Biol. Chem. 1994;269:18849–18855. [PubMed] [Google Scholar]
- 41.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 2007;179:4053–4064. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 42.Allport JR, Muller WA, Luscinskas FW. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J. Cell Biol. 2000;148:203–216. doi: 10.1083/jcb.148.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Millan J, Ridley AJ. Rho GTPases and leukocyte-induced endothelial remodelling. Biochem. J. 2005;385:329–337. doi: 10.1042/BJ20041584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am. J. Physiol. 2003;285:C343–C352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- 45.Shaw SK, Bamba PS, Perkins BN, Luscinskas FW. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J. Immunol. 2001;167:2323–2330. doi: 10.4049/jimmunol.167.4.2323. [DOI] [PubMed] [Google Scholar]
- 46.Van Buul JD, Voermans C, van den Berg V, Anthony EC, Mul FP, van Wetering S, van der Schoot CE, Hordijk PL. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J. Immunol. 2002;168:588–596. doi: 10.4049/jimmunol.168.2.588. [DOI] [PubMed] [Google Scholar]
- 47.Edens HA, Levi BP, Jaye DL, Walsh S, Reaves TA, Turner JR, Nusrat A, Parkos CA. Neutrophil transepithelial migration: evidence for sequential, contact-dependent signaling events and enhanced paracellular permeability independent of transjunctional migration. J. Immunol. 2002;169:476–486. doi: 10.4049/jimmunol.169.1.476. [DOI] [PubMed] [Google Scholar]
- 48.Choi H, Fleming NW, Serikov VB. Contact activation via ICAM-1 induces changes in airway epithelial permeability in vitro. Immunol. Invest. 2007;36:59–72. doi: 10.1080/08820130600745703. [DOI] [PubMed] [Google Scholar]
- 49.Aksoy MO, Yang Y, Ji R, Reddy PJ, Shahabuddin S, Litvin J, Rogers TJ, Kelsen SG. CXCR3 surface expression in human airway epithelial cells: cell cycle dependence and effect on cell proliferation. Am. J. Physiol. 2006;290:L909–L918. doi: 10.1152/ajplung.00430.2005. [DOI] [PubMed] [Google Scholar]
- 50.Kelsen SG, Aksoy MO, Yang Y, Shahabuddin S, Litvin J, Safadi F, Rogers TJ. The chemokine receptor CXCR3 and its splice variant are expressed in human airway epithelial cells. Am. J. Physiol. 2004;287:L584–L591. doi: 10.1152/ajplung.00453.2003. [DOI] [PubMed] [Google Scholar]
- 51.Shahabuddin S, Ji R, Wang P, Brailoiu E, Dun N, Yang Y, Aksoy MO, Kelsen SG. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am. J. Physiol. 2006;291:C34–C39. doi: 10.1152/ajpcell.00441.2005. [DOI] [PubMed] [Google Scholar]
- 52.Soejima K, Rollins BJ. A functional IFN-γ-inducible protein-10/CXCL10-specific receptor expressed by epithelial and endothelial cells that is neither CXCR3 nor glycosaminoglycan. J. Immunol. 2001;167:6576–6582. doi: 10.4049/jimmunol.167.11.6576. [DOI] [PubMed] [Google Scholar]
- 53.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hogg N, Henderson R, Leitinger B, McDowall A, Porter J, Stanley P. Mechanisms contributing to the activity of integrins on leukocytes. Immunol. Rev. 2002;186:164–171. doi: 10.1034/j.1600-065x.2002.18614.x. [DOI] [PubMed] [Google Scholar]
- 55.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat. Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- 57.Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat. Cell. Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 58.Smith A, Bracke M, Leitinger B, Porter JC, Hogg N. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J. Cell Sci. 2003;116:3123–3133. doi: 10.1242/jcs.00606. [DOI] [PubMed] [Google Scholar]
- 59.Smith A, Carrasco YR, Stanley P, Kieffer N, Batista FD, Hogg N. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J. Cell Biol. 2005;170:141–151. doi: 10.1083/jcb.200412032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J. Immunol. 2006;176:537–543. doi: 10.4049/jimmunol.176.1.537. [DOI] [PubMed] [Google Scholar]
- 61.Strieter RM, Belperio JA, Keane MP. Cytokines in innate host defense in the lung. J. Clin. Invest. 2002;109:699–705. doi: 10.1172/JCI15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mohan K, Ding Z, Hanly J, Issekutz TB. IFN-γ-inducible T cell α chemoattractant is a potent stimulator of normal human blood T lymphocyte transendothelial migration: differential regulation by IFN-γ and TNF-α. J. Immunol. 2002;168:6420–6428. doi: 10.4049/jimmunol.168.12.6420. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez-Madrid F, Gonzalez-Amaro R. Drugs, inflammation and cell adhesion receptors. Expert Opin. Pharmacother. 2001;2:3–17. doi: 10.1517/14656566.2.1.3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.