

SUMMARY
Here we show that a lupus-suppressing locus is caused by a nonsense mutation of the filamentous actin-inhibiting Coronin-1A gene. This mutation is associated with developmental and functional alterations in T cells, reduced T-dependent humoral responses, and no detectable intrinsic B cell defects. By transfer of T cells it was shown that suppression of autoimmunity could be accounted for by the presence of the Coro1aLmb3 mutation in T cells. Our results demonstrate that Coronin-1A is required for the development of systemic lupus and identify actin-cytoskeleton regulatory proteins as potential targets for modulating autoimmune diseases.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a heterogeneous multi-organ autoimmune disease associated with autoantibodies to nuclear components. Susceptibility is influenced by genetic, environmental, hormonal, and stochastic factors, among which genetic predisposition appears to be the major contributor. Consequently, there is considerable interest in defining the genetics of SLE, not only for gaining fundamental insights into its etiopathogenesis, but also for the potential of applying this information toward prevention and therapy.
Substantial progress has been made in identifying SLE loci and candidate genes (Gregersen and Behrens, 2006; Harley et al., 2006; Wong and Tsao, 2006). However, their significance for the most part remains uncertain due to a number of factors such as polygenic inheritance, extensive genetic heterogeneity, the small effect-size of most genes, and relatively low frequency of SLE, which make it difficult to firmly establish the role of specific polymorphisms. Similar efforts in mice have identified predisposing loci of varying significance in spontaneous and induced lupus-prone strains and in some instances in non-autoimmune mice (Kono and Theofilopoulos, 2006). Considerable genetic heterogeneity, instances of epistasis, different sets of loci for different autoimmune traits, and the finding that some loci are composed of several subloci, have been documented. Identifying specific susceptibility-associated polymorphisms in these strains, however, has proven difficult and only a few are known with certainty. Among these are the apoptosis-inhibiting Faslpr and related Faslgld, several H2 complex haplotypes, and Tlr7 (Kono and Theofilopoulos, 2006; Lynch et al., 1994; Pisitkun et al., 2006; Subramanian et al., 2006; Suda et al., 1993; Watanabe-Fukunaga et al., 1992). In addition, several other loci-associated candidate genes have been identified including the SLAM family and Ly108 (Sle1b locus), Cr2 (Sle1c), Ifi202 (Nba2), Fcgr2b (Nba2, Lbw7), Cd72 (Arvm1), P2rx7 (Lbw3), and CD22 (Sle5) (Boackle et al., 2001; Elliott et al., 2005; Jiang et al., 2000; Jiang et al., 1999; Kumar et al., 2006; Mary et al., 2000; Pritchard et al., 2000; Qu et al., 2000; Rozzo et al., 2001; Wandstrat et al., 2004).
In mapping studies of F2 intercross progeny of the highly lupus-prone MRL-Faslpr and much less susceptible C57BL/6(B6)-Faslpr strains, we previously identified four quantitative trait loci (QTL), named Lmb1-4, on chromosomes 4, 5, 7, and 10, that predisposed to lymphoproliferation, anti-dsDNA antibody production, or both (Vidal et al., 1998). Although it was anticipated that all lupus-predisposing Lmb alleles would come from the MRL, one QTL, Lmb1, linked susceptibility to the C57BL/6 (B6) strain. Nonetheless, analysis of combinations of Lmb QTL suggested that each locus, regardless of background origin, could contribute additively to overall severity. Subsequent generation and analysis of reciprocal interval-specific Lmb congenic mice (Santiago-Raber et al., 2007), however, revealed that only one QTL, Lmb3 on chromosome 7, strongly altered lymphoproliferation and to a lesser extent anti-chromatin antibody levels in CFA-enhanced lupus, the model originally used to map the Lmb loci (Vidal et al., 1998). Importantly, Lmb3 had a significant impact on the severity of spontaneous lupus in both MRL-Faslpr and B6-Faslpr backgrounds. Thus, interval-specific MRL.B6-Lmb3 Faslpr congenic mice (MRL-Faslpr mice containing an introgressed fragment of chromosome 7 from the B6-Faslpr/Scr) had reduced lymphoproliferation, polyclonal and autoantibody IgG levels, glomerulonephritis, and early mortality compared with the parental MRL-Faslpr, while the reciprocal B6.MRL-Lmb3 Faslpr congenic had markedly increased lymphoproliferation and autoantibody levels compared with B6-Faslpr. These findings indicated that Lmb3 plays a major role in modulating autoimmunity in Faslpr mice although the mechanism was not known.
In this study, we report the cloning of a gene alteration associated with Lmb3, which is a nonsense mutation in the Coronin-1A (Coro1a) gene, and show it is a disease-suppressing change derived from the less-susceptible B6-Faslpr strain. Further studies document that the Coro1aLmb3 mutation results in multiple T cell alterations that impair T-dependent humoral and germinal center (GC) responses. Finally, using T cell transfers, we show that the Lmb3 autoimmune suppressing phenotype can be transmitted solely by Coro1aLmb3 T cells.
RESULTS
Identification of Coro1a and Rabep2 as Candidate Genes for Lmb3
Lmb3 was mapped to a 0.9 Mb interval of chromosome 7 with ∼2500 meioses using a combination of MRL.B6-Lmb3 Faslpr subcongenic mice and later, once it was established that transmission of the susceptible Lmb3 trait followed dominant Mendelian inheritance, with (MRL-Faslpr × MRL.B6-Lmb3 Faslpr) backcrosses and intercrosses (Figure 1A). This region was densely packed with genes and contained 190 possible transcripts of which 90 were known or suspected genes.
Figure 1. Identification and Confirmation of Coro1a as the Lmb3 Gene.

(A) Fine mapping of Lmb3. The interval (indicated by shaded markers) was reduced to 0.9 Mb between D7Tsri317 and D7Tsri88 using ∼2500 meioses (see Methods). Only crossovers between D7Tsri309 and T7Tsri49 are shown. MRL and B6 phenotypes were determined by numbers of CD4+ and DN LN T cells 5 wk after subcutaneous CFA injection.
(B) B6-Faslpr/Scr.B6-Coro1awt mice express the Lmb3 phenotype. Wet weights of spleen and LN from 11-12 month-old wild-type or congenic B6-Faslpr/Scr mice that have either the original Lmb3 interval (Lmb3Scr), the Coro1awt from the B6/J, or the MRL Lmb3 interval (Lmb3MRL).
(C) Coro1aLmb3 Q262X allele. A nonsense C→T transition at residue 784 changes a glutamine (CAG) to a termination codon (TAG) within the 5th WD40 domain (5D region of the β-propeller domain). Resulting polypeptide lacks a significant portion of the β-propeller, as well as the entire distal constant (Con), unique (U) and coiled coil (CC) regions.
(D) Coro1aLmb3 protein fails to localize to the plasma membrane and immune synapse. Jurkat cells were transfected with plasmids encoding C-terminal-fluorescent protein-labeled actin, Coro1awt or Coro1aLmb3 and mixed with SED-pulsed Cy-5-labeled Raji antigen presenting cells (100X). Immune synapse (white arrows) and plasma membrane Coro1a (green arrows) are indicated. Data representative of two transfection experiments.
Nineteen genes or transcripts were then selected for screening because of known immunological relevance or primary expression in immune cells (Table 1). Many were attractive candidates, including Lat, Nfatc2ip, Il27, and Erk1, however, coding sequence polymorphisms were identified in only two genes, Rabep2 (RAB GTPase binding effector protein 2) and Coro1a. MRL mice had two changes in Rabep2; a synonymous C>T transition at residue 324 (T108) and a non-synonymous G>A transition at residue 764 that changed an arginine to a glutamine (R255Q). Rabep2, expressed primarily in B cells, belongs to a family of proteins that interact with intracellular membrane-associated Rab GTPases that regulate membrane traffic pathways, although the specific function of Rabep2 is not known (Kawasaki et al., 2005). The other gene, Coro1a, had a single nonsense C>T transition at residue 784 in MRL.B6-Lmb3 Faslpr mice that converted a glutamine to a stop codon in the middle of the protein (Q262X). Coro1a mRNA was expressed at higher levels in T cells from MRL.B6-Lmb3 Faslpr compared with MRL-Faslpr mice (ΔCT of 2.2±0.04 vs. 4.3±0.09, p<0.0001). However, the Lmb3-associated Coronin 1A protein, as expected because of the truncation, was not detected by Western blot (Figure S1). These findings strongly suggest that the Coro1aLmb3 contains a function-altering change. Furthermore, the fact that Coro1a is important for T cell survival and migration (Foger et al., 2006) was consistent with our previous congenic studies suggesting Lmb3 might affect T cells (Santiago-Raber et al., 2007). The unexpected finding that this mutation was derived from the less-susceptible B6-Faslpr rather than the highly-prone MRL-Faslpr strain, however, indicated that, if Coro1aLmb3 was the responsible mutation, it did not promote autoimmunity, but instead suppressed disease. Further analysis revealed that Coro1aLmb3 was present in every B6-Faslpr/Scr breeder in our colony, but not in other strains, including the B6 (Scripps and JAX colonies), B6-Faslpr/J (JAX colony), CAST, SPRET, MOLF, C3H, LG, AKR, 129, A, CBA, PL, SB, SWR, SM, NZB, NZW, or BXSB, indicating it had arisen spontaneously. Among the other Lmb3 candidates examined, no other differences between the B6 sequence in the Ensembl database and that of the B6-Faslpr/Scr were detected. Thus, within the Lmb3 interval two possible candidate genes were identified, each from a different background.
Table 1.
Candidate genes within the Lbm3 interval.
| Mb* | Gene Symbol | Gene Name; Synonym | Strain: Polymorphism ** |
|---|---|---|---|
| 125.712 | D7Tsri317 | ||
| 125.893 | Xpo6 | exportin 6 | |
| 126.064 | Sbk1 | SH3-binding kinase 1 | |
| 126.155 | Lat | linker for activation of T cells | |
| 126.161 | 2210013K02Rik | 2210013K02Rik | |
| 126.176 | Nfatc2ip | NFAT, cytoplasmic, calcineurin- dependent 2 interaction protein |
|
| 126.220 | Rabep2 | rabaptin | MRL: C324→T (T108), G764→A (R255Q) |
| 126.258 | Sh2b1 | SH2B adaptor protein 1 | |
| 126.376 | Apob48r | apolipoprotein B48 receptor | |
| 126.380 | Il27 | IL27 p28 subunit | |
| 126.414 | Nupr1 | nuclear protein 1; p8 | |
| 126.441 | Ccdc101 | coiled-coil domain containing 101 | |
| 126.487 | Bola2 | bolA-like 2 | |
| 126.491 | Coro1a | coronin, actin binding protein 1A | B6-Faslpr/Scr: C784→T (Q262Term) |
| 126.551 | Mapk3 | mitogen activated protein kinase 3; Erk1 |
|
| 126.568 | Ypel3 | yippee-like 3 | |
| 126.573 | Tbx6 | T-box 6 | |
| 126.577 | Ppp4c | protein phosphatase 4, catalytic subunit |
|
| 126.625 | D7Tsri88 |
Mb locations based on Ensembl NCBI m36 assembly. Two transcripts, D930031A20Rik and 1500016O10Rik, originally placed in this interval, but subsequently deleted from the current Ensembl assembly, were also sequenced and found not to be polymorphic.
Coding region polymorphisms that differ from the Ensembl genome sequence are shown.
Reversal of the Lmb3 Disease-suppressing Phenotype in B6-Faslpr/Scr Mice by the Wild-Type Coro1a Gene
To help determine which of these genes was responsible for the Lmb3 phenotype (increased spleen and LN weights), we backcrossed the wild-type (wt) Coro1a gene from the B6/J mice onto the B6-Faslpr/Scr strain since this approach would discriminate between the Coro1aLmb3 mutation or a gene derived from the MRL, such as Rabep2. Interestingly, the resulting B6-Faslpr/Scr.B6-Coro1awt mice had enhanced lymphoproliferation, similar to the B6.MRL-Lmb3 Faslpr congenic, which indicated that the Coro1awt gene alone can complement the B6-Faslpr/Scr Lmb3 locus and excluded any requirement for the MRL genome (Figure 1B). Thus, barring the unlikely possibility of an additional disease-suppressing B6-Faslpr/Scr mutation within the narrowed Lmb3 interval, it appeared that Coro1a was the Lmb3 gene and that the specific Lmb3 mutation suppressed autoimmunity.
Coro1aLmb3 Mutant Protein Fails to Localize to the Cell Membrane and Immune Synapse
Previous studies have suggested that the C-terminal coiled-coiled domain, missing in the Coro1aLmb3 mutant, is essential for trimerization and Coronin function (Appleton et al., 2006; Rybakin and Clemen, 2005) (Figure 1C). Furthermore, crystallographic analysis of Coronin-1A suggests that the Lmb3 mutation would also disrupt the structure of the actin-binding β-propeller because of the loss of 3 blades and the lack of the constant region thought to play a role in maintaining the propeller structure (Uetrecht and Bear, 2006). Therefore, it appeared likely that the truncated Coro1aLmb3-encoded protein was functionally impaired. Since antibodies to the mutant Coronin 1A was not available, to determine the effect of this mutation on cell localization, Jurkat cells were transfected with plasmid vectors expressing fluorescent-labeled wild-type or Lmb3 Coro1a cDNAs (Figure 1D). As expected, the wild-type Coro1a localized to the cell membrane (small green arrows) and, following activation of the transfected Jurkat cells with Staphylococcal enterotoxin D (SED)-pulsed Raji cells, became concentrated in the immune synapse (large white arrow) (Appleton et al., 2006; Rybakin and Clemen, 2005). In contrast, in none of the Coro1aLmb3-expressing mutant cells was there localization of labeled protein adjacent to the plasma membrane, or after activation, in the immune synapse. Since appropriate localization is considered important for Coronin function (Appleton et al., 2006; Rybakin and Clemen, 2005), this finding is consistent with reduced function.
Alterations in Thymic and Peripheral T Cell Populations
The finding of a range of T cell defects further supported impaired function of the Coro1aLmb3 mutant. In the thymus, DN and double positive (DP, CD4+CD8+) populations were unaffected. However, there was a variable reduction in the number of single positive (SP) CD4+ and CD8+ thymocytes that was also observed although to a lesser extent in Coro1a−/− mice (Foger et al., 2006) (Figure 2A-B). This reduction generally became apparent around 4-7 weeks, was exacerbated with age, and affected to a greater extent the later stages of SP cell development (CD69lo CD62Lhi CD24lo subset, Figure 2C and Figure S2). This was associated with increased spontaneous ex vivo apoptosis of SP, but not DP thymocytes (Figure 2D).
Figure 2. Thymic and peripheral T cell populations are altered in MRL.B6-Lmb3 Faslpr mice.

(A) Reduction in percentages of thymic CD4+ and CD8+ single positive (SP) T cells. Mean±SEM of 4-7 wk.-old mice, n=15 (MRL-Faslpr), 11 (M.B-Lmb3 Faslpr). A representative profile is shown.
(B) Thymic SP T cell numbers are reduced in MRL.B6-Lmb3 Faslpr mice. n=11-12/group.
(C) Reduction in mature (CD24lo, 62Lhi) subset of CD4+ SP thymocytes. Representative data from 4-5 mice/group (see Figure S1 for CD8+ SP subset and single markers).
(D) Increased spontaneous cell death in SP thymocytes. n=3-4/group.
(E) Reduced numbers of CD4+ and CD8+ LN T cells. n=8-15/group.
(F) LN CD44 and CD62L-defined CD4+ T cell subsets.
(G) Reduced numbers of all CD4+ T cell subsets defined in panel F.
(H) Reduced percentages of the CD44lo CD62Lhi (naïve) subset defined in panel F. Data for (FH) from 3-4/group. (see Figure S2 for CD4+ T cell subsets in the spleen and CD8+ T cell subsets in the LN and spleen).
In peripheral lymphoid organs, CD4+ and CD8+ T cell numbers were markedly decreased in the LNs and there was a moderate reduction of CD4+ T cells in the spleen (Figure 2E and Figure S3A). The decreases in CD4+ and CD8+ T cells involved the naïve and activated subsets in both the LN and spleen (Figures 2E-2G and Figures S3B-S3I). However, based on the relative percentages of cells in each subset, this reduction was most evident in the naïve T cell population (Figure 2H and Figures S3D, S3G, S3J).
Increased Spontaneous Apoptosis and Accumulation of Filamentous (F)-actin in T Cells
Similar to SP thymocytes, naïve T cells from Coro1aLmb3 mice displayed increased spontaneous apoptosis, which was associated with the loss of mitochondrial membrane potential and the activation of Caspases 3 and 8 (Figure S4A). CD4+ and CD8+ T cells from mutant mice also had increased amounts of cellular F-actin (Figure S4B), an alteration previously postulated to play a role in inducing spontaneous apoptosis of naïve Coro1a-deficient T cells by altering mitochondrial membrane potential (Dustin, 2006; Foger et al., 2006). In mutant mice, however, similar increases in cellular F-actin were observed in naïve and activated memory/phenotype T cells, and in B cells, which do not exhibit increased apoptosis (Figures S4B-S4C).
Impaired Lymphopenia-induced Homeostatic Proliferation in Coro1aLmb3 Mutant Mice
Another possible reason for the reduced numbers of peripheral T cells is defective lymphopenia-induced homeostatic proliferation, which is dependent on both T cell receptor (TCR) engagement with self-peptide/MHC and cytokine stimulation (Surh and Sprent, 2005). Since the required allotypic T cell markers were not available on the MRL background, experiments were conducted using B6 (Thy1.2) donors with and without the Coro1aLmb3 mutation transferred into sublethally-irradiated B6.PL (Thy1.1) recipient mice. Strikingly, CD4+ T cells from donors with the Lmb3 mutation (B6.B6-Faslpr/Scr-Coro1aLmb3 mice) had markedly fewer cell divisions compared to T cells from wild-type mice 5 days after transfer (Figure 3A).
Figure 3. Coro1aLmb3 Mutation Is Associated with Defective T Cell Activation.
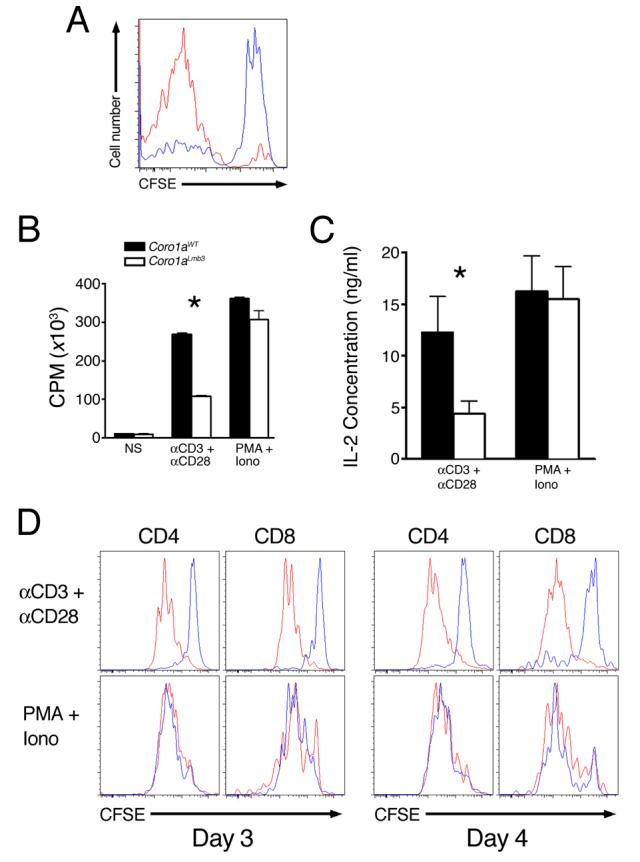
(A) Reduction in lymphopenia-induced homeostatic proliferation. CFSE-labeled B6 (red line) or B6-Coro1aLmb3 (blue line) Thy1.2 T cells were transferred into sublethally irradiated Thy1.1-recipient B6.PL mice and CFSE-staining of Thy1.1-negative LN CD4+ T cells was analyzed by flow cytometry 5 d later. n=3 mice/group.
(B) Reduced MRL.B6-Lmb3 Faslpr T cell proliferation to anti-CD3/anti-CD28 (P<0.05), but not PMA/Ionomycin.
(C) IL-2 production is lower in MRL-Faslpr T cells with Coro1aLmb3 (p<0.05). Purified T cells from MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice were activated with anti-CD3/CD28 or PMA/Ionomycin for 48 hrs and supernatants analyzed for IL-2 production.
(D) Reduced division of Coro1aLmb3 mutant T cells after stimulation with anti-CD3/anti-CD28, but not PMA/Ionomycin. Coro1awt (red lines), Coro1aLmb3 (blue lines). Data are representative of 3 independent experiments.
IL-7R signaling was examined since it is required for both survival and lymphopenia-induced homeostatic proliferation of naïve CD4+ T cells (Surh and Sprent, 2005). Surface expression of IL-7R (CD127) in naïve (CD44lo) and activated/memory (CD44hi) CD4+ and CD8+ T cells from Coro1aLmb3 and Coro1awtmice were similar, as was STAT5 phosphorylation of T cells stimulated ex vivo with exogenous IL-7 (data not shown). Levels of CD24, a surface molecule recently shown to be required for lymphopenia-induced proliferation (Li et al., 2004), was also not reduced (data not shown). Thus, reductions in IL-7 signaling or CD24 expression do not account for the reduced lymphopenia-induced proliferation, suggesting that defective TCR-mediated activation was the most likely cause of the impaired expansion of mutant T cells following lymphopenia.
Reduced Activation of Coro1aLmb3 T cells
Indeed, purified T cells from MRL.B6-Lmb3 Faslpr mice exhibited significantly reduced proliferation to anti-CD3/anti-CD28 by thymidine incorporation (Figure 3B). Since Coro1a-deficient T cells were reported to have normal activation (Foger et al., 2006), this reduction was further confirmed by measurement of IL-2 secretion, which revealed significantly less production of IL-2 (Figure 3C) and CFSE cell division analysis, which showed reduced number of cycling cells (Figure 3D). Furthermore, transfection of primary B6-Coro1aLmb3 CD4+ T cells with a plasmid expressing the wild-type Coro1a, but not a plasmid expressing the mutant Coro1aLmb3 resulted in enhanced proliferation following anti-CD3/anti-CD28-mediated activation (Figure S5A-B). In contrast, proliferation of isolated T cells to PMA/Ionomycin was not reduced by the Coro1aLmb3 mutation (Figure 3B-D), indicating that mutant cells have the capacity to proliferate, the reduced proliferation is not secondary to enhanced apoptosis, and the defect occurs proximal to PKC activation and calcium release. Activation of B cells was not altered by the Coro1aLmb3 mutation following stimulation with either LPS or anti-IgM/anti-CD40 (Figure S5C).
Reduced T Cell Proliferation to Anti-CD3/anti-CD28 Is Associated with Defective Ca2+ Flux
Ca2+ flux studies were performed to further dissect the reduced proliferation. A marked reduction in the initial Ca2+ peak in both CD4+ and CD8+ T cells from MRL.B6-Lmb3 Faslpr mice was observed following anti-CD3/anti-CD28 cross-linking (Figures 4A-4B). This was evident in all CD44 subsets except for the CD44hi CD4+ population, which as previously reported (Khaidukov and Litvinov, 2005) already has attenuation of the initial peak in Ca2+ flux. However, after exposure to Ionomycin at a concentration sufficient to permit entry of extracellular Ca2+ (1.67 μM) nearly equivalent levels of Ca2+ flux were observed (Figure 4D). Importantly, in the absence of extracellular Ca2+, engagement of anti-CD3/anti-CD28 or Ionomycin resulted in the release of similar or slightly higher levels of stored Ca2+ (Figures 5C and 5E), indicating that the TCR/CD28-mediated signaling events prior to store-operated Ca2+ release are largely unaffected. Taken together, these findings localize the Ca2+ flux defect to the interval between stored Ca2+ release and the activation of the calcium release activated calcium (CRAC) channel.
Figure 4. Coro1aLmb3 Mutation Leads to Defective CRAC Channel Activity Following TCR Engagement.
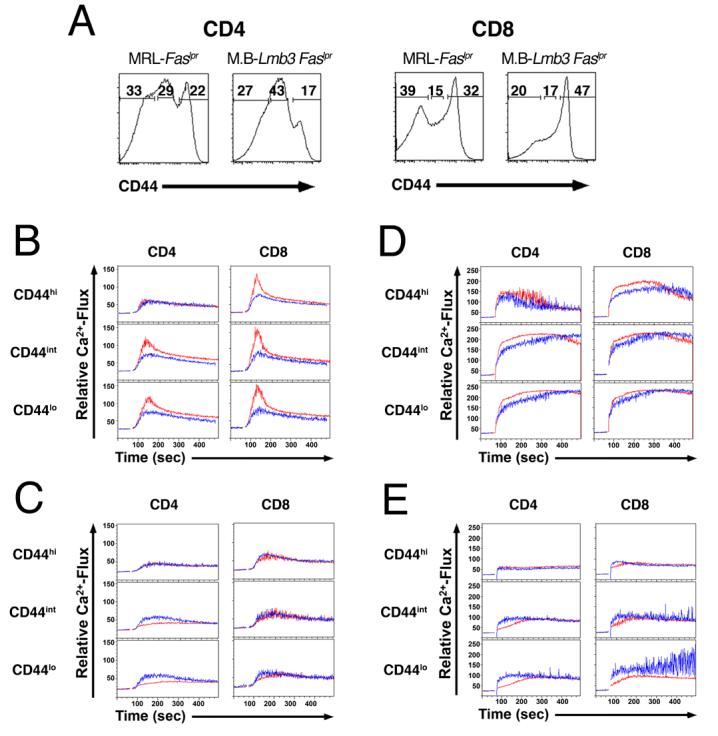
(A) CD44 low, intermediate and high subsets of CD4+ and CD8+ T from Coro1awt and Coro1aLmb3 mice.
(B) Ca2+-flux in CD44 low, intermediate and high subsets of CD4+ and CD8+ T cells in the presence of exogenous calcium following TCR engagement (anti-CD3/anti-CD38 antibodies).
(C) Ca2+-flux in CD44 low, intermediate and high subsets of CD4+ and CD8+ T cells in the absence of exogenous calcium following TCR engagement.
(D) Ca2+-flux in CD44 low, intermediate and high subsets of CD4+ and CD8+ T cells in the presence of exogenous calcium following Ionomycin stimulation.
(E) Ca2+-flux in CD44 low, intermediate and high subsets of CD4+ and CD8+ T cells in the absence of exogenous calcium following Ionomycin stimulation. Coro1awt (red lines), Coro1aLmb3 (blue lines). Data are representative of 3 independent experiments.
Figure 5. Reduced IgG Autoantibody Levels, IgG Humoral Responses, and GC Formation in MRL.B6-Lmb3 Faslpr Mice.
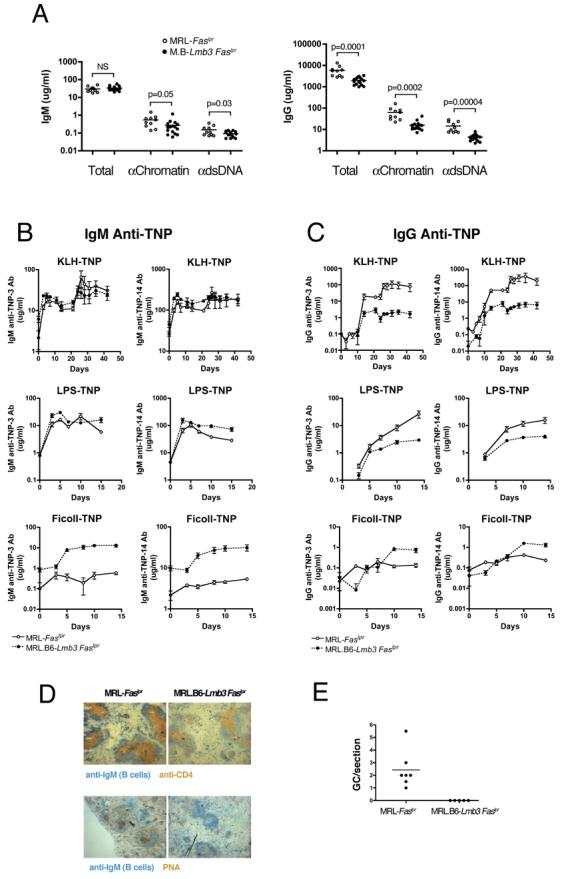
(A) IgG polyclonal (total) and anti-nuclear antibodies from 4 mo. old mice (right panel) are reduced to a greater degree than IgM antibodies (left panel).
(B-C) IgM and IgG TNP levels to TNP-14 (low and high affinity antibodies) and TNP-3 (high affinity antibodies) after immunization with T-D (TNP-KLH), T-I type 1, (TNP-LPS), or T-I type 2 (TNP-Ficoll) antigens. n=5-6 (T-D) or 9-12 (T-I). T-I groups were immunized on day 0 and T-D groups on day 0 and 21.
(D) Representative spleen sections stained for CD4+ T cells, B cells (IgM+), and GC (PNA+) 10 days after i.p. TNP-KLH in CFA.
(E) Number of GC per spleen section 10 d after i.p. TNP-KLH in CFA. n= 5 for congenic and 7 for wild-type MRL-Faslpr mice (D,E).
Reduced Migration of T cells from MRL.B6-Lmb3 Faslpr Mice
Further evidence that the Coro1aLmb3 mutant was nonfunctional came from the observation that Coro1aLmb3 mutant T cells had impaired migration to chemokines (SDF1/MIP-3b cocktail, Supplemental Video 1). In our system, Coro1awt T cells moved at an average speed of 3.0±0.66 μm/min, while Coro1aLmb3 T cells migrated at 0.32±0.14 μm/min toward the chemokine gradient (p<0.0005).
Serology of MRL.B6-Lmb3 Faslpr Mice is Consistent with Reduced T Helper Activity
Taken together, the data suggested that reduced T cell activation, survival, and migration caused by the Coro1aLmb3 mutation were responsible for the suppression of autoimmunity. This was further supported by the effects of the Coro1aLmb3 mutation on IgM and IgG antibodies (Figure 5A). In this instance, there was no reduction in total polyclonal IgM and only a modest decrease in anti-chromatin (2-fold) and anti-dsDNA (1.7-fold) IgM antibodies. In contrast, levels of polyclonal IgG (2.9-fold) and IgG anti-chromatin (4-fold) and anti-dsDNA (3.4-fold) antibodies were significantly reduced. Importantly, as shown here, all were decreased to a greater extent than their IgM counterparts.
MRL.B6-Lmb3 Faslpr Mice Have Impaired T-dependent B Cell Response
To directly examine the effect of the Coro1aLmb3 mutation on T-dependent humoral responses, we immunized wild-type and mutant mice with the T-dependent antigen TNP-KLH and compared their IgM and IgG TNP responses. T-independent responses to both type I (TNP-LPS) and type II (TNP-Ficoll) antigens were also examined. The IgM anti-TNP responses of mutant MRL.B6-Lmb3 Faslpr mice to all three antigens were normal or even slightly increased (Figure 5B). Likewise, the T-independent type I and II IgG responses in wild-type and mutant mice were similar. In contrast, the T-dependent IgG anti-TNP primary response in Coro1aLmb3 mice was substantially lower (p<0.002 at day 21) and there was no increase after recall immunization (p<0.002 for all days tested from day 24, Figure 5C). Overall, these findings suggest that B cells in Coro1aLmb3 mice can respond normally, but are unable to effectively class-switch or develop into memory B cells because of insufficient T helper activity. Indeed, this possibility was further supported by the striking absence of detectable germinal centers (GCs) in spleens 10 days after injection with TNP-KLH in CFA (MRL 2.4±0.6 GC per section, MRL.B6-Lmb3 Faslpr no GCs, p<0.008, Figures 5D-5E).
Lmb3 T Cells Transfer the Lmb3 Phenotype
To directly determine whether the presence of the Coro1aLmb3 mutation in T cells was sufficient to reduce lupus-like disease, isolated LN T cells from the MRL.B6-Lmb3 Faslpr or MRL-Faslpr mice were transferred into MRL-Faslpr Tcrb−/− recipients and the effects of having the wild-type or mutant Coro1a on the development of autoimmunity was assessed over a 22-week period (Figures 6A-6C). Recipients of mutant T cells had significantly less autoimmunity with reduced lymphoproliferation as evidenced by lower peripheral lymphoid organ weights and cell numbers, decreased percentages of CD4+ and DN T cells, lower polyclonal serum IgG, and, importantly, virtually absent IgG anti-chromatin autoantibodies. Similar to Coro1aLmb3 mutant MRL mice, the reduction in cell numbers and lymphoid organ weights was more pronounced in the LN than spleen. These findings directly demonstrate that the multiple effects of Coro1aLmb3 on T cells are sufficient to reduce autoimmunity in MRL.B6-Lmb3 Faslpr congenic mice.
Figure 6. Presence of the Coro1aLmb3 Mutation in T cells Is Sufficient to Suppress Autoimmunity.

Purified LN T cells from MRL-Faslpr or MRL.B6-Lmb3 Faslpr (M.B-Lmb3 Faslpr) were transferred into T cell receptor β-chain-deficient MRL-Faslpr mice. Lupus-related traits in panels A and B were determined 22 wk. after transfer.
(A) Body and lymphoid organ weights. Total cell numbers of lymphoid organs.
(B) Percentages of B cells and T cell subsets in spleen and LN.
(C) Total and anti-chromatin IgG serum levels.
DISCUSSION
Here, we show by mapping and complementation that the genetic alteration responsible for the lupus-modifying Lmb3 locus is most likely a function-impairing nonsense Q262X mutation of the Coro1a gene on distal chromosome 7. Furthermore, we document that this mutation is an autoimmunity suppressing allele that arose spontaneously in our B6-Faslpr/Scr colony. We further show that the Coro1aLmb3 mutation reduces humoral and cellular manifestations of lupus in Faslpr mice by significantly impairing the development, migration, survival, and activation of T cells, T-dependent immune responses, and germinal center formation. These findings define the critical role of Coronin 1A in normal immune responses and autoimmunity, and identify it as a potential therapeutic target.
Coronins are actin-associated cytoskeletal proteins present in prokaryotes to eukaryotes that share an actin-binding 7-bladed β propeller, a common C-terminal extension flanking the propeller domain, a unique region that differs among Coronin members, and, in the single β propeller Coronins, such as Coronin-1A, a heptad repeat coiled-coil domain necessary for homotrimerization (Rybakin and Clemen, 2005; Uetrecht and Bear, 2006). They act as inhibitors of F-actin by sequestering the nucleation-promoting Arp2/3 complex in its inactive open form (Rodal et al., 2005) and by serving as a cofactor for cofilin/Aip1-mediated F-actin disassembly (Brieher et al., 2006). As such, Coronins play a critical role in the assembly and turnover of actin during lamellipodia formation and cell migration (Cai et al., 2007) and are implicated in vesicular trafficking, morphogenesis, cell division, and cell survival, although some functions are species-specific. Of the seven Coronin family members found in mammals, Coronin-1A alone is expressed primarily in hematopoietic cells and has been associated in neutrophils with the NADPH oxidase complex subunit p40phox (Grogan et al., 1997), in macrophages with phagocytosis and lamellipodia formation (Ferrari et al., 1999; Yan et al., 2005), and in T cells with high expression throughout ontogeny and with localization to actin-rich regions of the immune synapse (Nal et al., 2004). Recent characterization of Coro1a-deficient mice, however, revealed alterations limited essentially to T cells, with T lymphopenia from reduced migration and increased apoptosis, but normal T cell development and activation (Foger et al., 2006). More recent characterization of macrophages from Coro1a−/− mice showed no defects in phagocytosis, motility, or membrane ruffling (Jayachandran et al., 2007).
Consistent with the Coro1aLmb3 mutant encoding a nonfunctional product, several T cell alterations reported with Coro1a deficiency (Foger et al., 2006) were also observed in MRL.B6-Lmb3 Faslpr mice. These include defective chemokine-mediated T cell migration, enhanced spontaneous apoptosis of naïve T cells, reduced numbers of peripheral T cells, and the accumulation of F-actin in cells. Also similar to the findings in Coro1a−/− mice, we observed fewer SP thymocytes in Coro1aLmb3 mice that was associated with increased apoptosis of SP, but not DP thymocytes. As both SP thymocytes and naïve peripheral Coro1aLmb3 T cells exhibit increased spontaneous apoptosis, the same pathologic process likely affects both populations. In this regard, the apoptosis in Coronin-1A deficiency was previously suggested to be secondary to a decrease in mitochondrial membrane potential caused by the increase in F-actin levels (Foger et al., 2006). Although how this occurs is not yet defined, it was postulated that the accumulated F-actin might either sequester factors that modulate mitochondrial voltage-dependent anion channels or else boost the delivery of proapoptotic molecules to the mitochondria (Dustin, 2006; Foger et al., 2006). Our finding of significant loss of mitochondrial membrane potential in Coro1aLmb3 T cells is certainly consistent with these possibilities. By contrast, the finding that B cells, which do not display increased apoptosis, also have increased cellular F-actin, suggests that either other factors are involved or that there is an alternative mechanism. One such possibility is that the apparent spontaneous apoptosis might involve T cells that are already committed to die because of inadequate survival signals in vivo from defective migration and/or reduced TCR-mediated signaling. Indeed, chemokine-mediated migration (Schaerli and Moser, 2005) and engagement of TCR by self-peptide/MHC complexes (Eck et al., 2006; Surh et al., 2006) have been shown to be required for retaining a normal T cell homeostasis.
In our study, Coro1aLmb3 T cells also displayed impaired activation as shown ex vivo by thymidine incorporation, cell division analysis, and IL-2 production, as well as in vivo by the degree of lymphopenia-induced homeostatic proliferation. Furthermore, the observation that transfection of Coro1aLmb3 CD4+ T cells with wild-type Coronin-expressing plasmids enhanced proliferation clearly implicates Coro1a. Additional studies demonstrated that proliferation to PMA/Ionomycin by both thymidine incorporation and cell division analysis was not reduced. This showed that Coro1aLmb3 mutant T cells had the capacity to proliferate and suggested that the impairment involved a proximal signaling event somewhere up to and including PKC activation and Ca2+ entry, which we identified as a reduction in peak Ca2+ flux. That Coronins might play a role in Ca2+ mobiliztion is supported by a recent report in macrophages implicating Coro1a in the activation of calcineurin, a signaling protein downstream of Ca2+ entry (Jayachandran et al., 2007). Why Coro1aLmb3, but not Coro1a-deficient (Foger et al., 2006) T cells exhibit reduced activation is not readily apparent, but there are possible explanations. One possibility is that background strain differences between the Coro1a−/− mice (mixed 129;B6 background) and the MRL background (this study) may be responsible. Indeed, MRL-Faslpr CD4+ T cells have an intrinsic threshold defect in activation associated with TCR-mediated hyperproliferation (Zielinski et al., 2005). In this case, the intrinsic hyperproliferation may be more sensitive to changes in Ca2+ flux. Another possibility is that the difference may be related to the fact that only transgenic T cells expressing a single antigen receptor were analyzed in the Coro1a knockout study whereas we studied polyclonal populations.
Our findings also suggest that Coro1aLmb3 T cells have a defect in Ca2+ flux between store-operated Ca2+ release and opening of the CRAC channels. Recent studies (Lewis, 2007) have shown that stored Ca2+ release induces STIM1, a membrane Ca2+ sensor located throughout the ER, to migrate to the junctional ER, the part of the ER located parallel and adjacent to the plasma membrane. This process is also associated with a modest increase in the amount of junctional ER. The resulting close juxtaposition of STIM1 in the ER membrane to the CRAC channels (Orai or CRACM1) in the plasma membrane leads to Ca2+ entry at those locations by a mechanism that has yet to be defined. Thus, one possible explanation for the reduced peak Ca2+ flux is that Coronin-1A, through its effects on actin filament mobilization, may be required for migration of STIM1 and/or formation of junctional ER. Another possibility is that the interaction of STIM1 and CRAC channels might be blocked by the Coro1aLmb3-associated accumulation of F-actin adjacent to the plasma membrane. A third possibility is that Coronin-1A might play a direct role in the formation of functional CRAC channel complexes. Interestingly, mice deficient for WAVE2, another actin cytoskeletal-regulatory protein, also exhibit a similar T cell defect in CRAC channel-mediated Ca2+ entry (Nolz et al., 2006), and mice deficient for HS1, another actin-regulatory protein that modulates F-actin at the immune synapse, also have a T cell Ca2+ flux defect that occurs after anti-CD3-mediated crosslinking (Gomez et al., 2006). Our findings further demonstrate the importance of actin-regulatory proteins in controlling Ca2+ mobilization in T cells.
The Coro1aLmb3 mutation is thus associated with three major intrinsic T cell alterations involving migration, cell survival, and activation. These contribute to the reduced numbers of SP thymocytes and peripheral T cells, the inability to form GC, and ineffective T-D antibody responses, which ultimately suppress the development of humoral and cellular manifestations of lupus. This is consistent with our T cell transfer study, which demonstrated that the Coro1aLmb3 mutation in T cells is sufficient for suppression of autoimmunity and with the lack of obvious alterations in other hematopoietic lineages in this study and in Coro1a knockout mice (Foger et al., 2006). Nevertheless, to assess the possible role of non-T cell lineages on lupus susceptibility, we are currently backcrossing the Coro1aLmb3 mutation onto the MRL-Faslpr Tcrb−/− background to test, in T cell transfer experiments, the effect of having wild-type Coro1a expressed in T cells alone.
That Lmb3 is caused by a lupus-suppressing mutation was certainly unexpected and illustrates the fact that genetic mapping studies do not distinguish between predisposing or suppressive alleles. Thus, it is likely that other lupus-related loci, particularly in mouse studies that involve homogeneous backgrounds, might in some instances be similarly associated with suppressive alleles. Also highlighted is the fact that, in addition to traditional predisposing genes, both disease-suppressing genes and spontaneous mutations, as was the case for CoroLmb3, are likely to be significant contributors to the repertoire of genetic variations that modulate induction, severity, and phenotypic heterogeneity of disease in individuals with lupus. Furthermore, different mutations within a single gene could in some instances lead to either increased susceptibility or disease suppression. These types of variants will complicate the identification of susceptibility genes and reduce the clinical utility of genetic associations. Nevertheless, in the case of disease-suppressing genes, such as Coro1a, their identification can provide important clues to pathogenesis and possibly therapy.
The actin cytoskeleton mobilizes molecular scaffolding essential for many crucial cellular functions and involves complex regulatory mechanisms that control and compartmentalize these activities in specific cell types. Our findings demonstrate the critical importance of actin regulation in lupus pathogenesis and further document that alteration of an actin-regulatory protein can have limited, but significant effects, on several specific functions of the immune system despite being highly expressed in all hematopoietic cell lineages. This provides strong impetus, from both the basic and clinical perspectives, to examine the precise roles of other actin cytoskeleton regulatory proteins in normal and self-reactive immune responses.
EXPERIMENTAL PROCEDURES
Mice
MRL-Faslpr/MpScr (MRL-lpr), C57BL/6- Faslpr/Scr (B6-lpr), and derivative mice were bred and maintained at TSRI animal facility. MRL.B6-Lmb3 Faslpr were generated as previously noted (Santiago-Raber et al., 2007). B6-Faslpr/Scr mice congenic for the normal Coro1a gene (termed B6-Faslpr/Scr.B6-Coro1awt) were generated by backcrossing the B6 Coro1awt gene onto the B6-Faslpr/Scr background. MRL-Faslpr Tcrb−/− (T cell receptor β-chain-deficient) mice were a kind gift from Dr. J. Craft (Yale University) and B6.PL (Thy1.1) mice were kindly provided by Drs. R. Baccala and C. Surh (TSRI). Mice were followed for spontaneous or CFA-accelerated lupus as described (Santiago-Raber et al., 2007; Vidal et al., 1998). DNA from various strains was from the Jackson Laboratories, Bar Harbor, ME. Fas genotyping was preformed as described (Feeney et al., 2001). All mouse studies were approved by TSRI Institutional Animal Care and Use Committee.
Mapping
Localization of Lmb3 was first performed using MRL.B6-Lmb3 Faslpr subcongenic mice, and later with (MRL.B6-Lmb3 FaslprxMRL-Faslpr)F1xMRL.B6-Lmb3 backcrosses or (MRL.B6-Lmb3 FaslprxMRL-Faslpr)F2 intercrosses for dominant transmission. Since mapping required B6 and F1 genotypes, in some instances, intercross offspring with intervals involving MRL and F1 genotypes were backcrossed to MRL.B6-Lmb3 Faslpr mice. Assignment of F1 or B6 Lmb3 phenotypes was based on LN weight and percentages of CD4+ and DN T cells. Polymorphic microsatellite markers were obtained by screening MRL-Faslpr and B6-Faslpr DNA with PCR primers flanking dinucleotide repeats located within the desired interval (Table S1). Paired PCR primers were generated using Primer3 (http://frodo.wi.mit.edu/cgibin/primer3/primer3_www.cgi) and sequentially numbered as D7Tsri DNA segments. Placement of markers and genes was based on the Ensembl mouse genome map.
Quantitative PCR
Primers spanning exons 9-11 of Coro1a were generated using the Primer3 program (forward 5'-AGTGCCTAGAAAGTCGGACCTGTT; reverse 5'-CTTGACACGGTATCCGAGCTG). HGPRT primers were kindly provided by Drs. D. Cauvi and K. M. Pollard (forward 5'-GCTCGAGATGTCATGAAGGAGATGGG; reverse 5'-CCCATCTCCTTCATGACATCTCGAGC. Real-time PCR was performed in two steps. First, first strand cDNA was synthesized (SuperScript III, Invitrogen) from DNase-treated RNA (PureLink, Invitrogen) isolated from negatively selected purified pooled spleen and LN T cells (Miltenyi Biotec). Next, real time PCR using SYBR Green was performed using the ABI 7900HT platform (ABI, Foster City, CA) using 0.4 μM primer concentrations and PCR conditions of 95°C for 10 min then 40 cycles of 96°C for 15 sec/60°C for 1 min. Relative quantitation was calculated using ABI 7900HT software.
Immunopathology
Serum was obtained and autopsies performed at indicated times as previously described (Kono et al., 1994).
Cell Transfection
Coro1aWT or Coro1aLmb3 cDNA were cloned into pECFP, pGFP, or pEYFP plasmids to generate C-terminal fusion proteins. pEYFP-actin and pECFP-actin were kindly provided by Dr. D. Soong (King's College London, UK). To analyze immune synapse formation, Jurkat cells were transfected with individual constructs (Nucleofector kit, Amaxa, Gaithersburg, MD) then mixed with SED-pulsed Cy-5-labelled Raji B cells at 37°C for 30 min. Cells were then gently resuspended before addition to slide chambers and fixation. Conjugates were visualized using a 200M microscope (Carl Zeiss, Jena, Germany).
Flow Cytometry
Cells were stained with combinations of fluorescent dye-labeled antibodies to B220, CD3, CD4, CD8, CD19, CD24, CD25, CD44, CD62L, and CD69, as well as fluorescent dye-labeled annexin V and Phalloidin, and 7AAD (BD Biosciences, San Diego, CA). Data were acquired on a LSR II (BD Biosciences) and analyzed by FlowJo (Tree Star, Ashland, OR).
Spontaneous T Cell Apoptosis
Splenocytes from wild-type MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice (n=3 mice/group) were placed in culture for spontaneous apoptosis assessment. Five hours later, cells were harvested and stained with antibodies to CD4, CD8, CD44, Annexin V (BD PharMingen), and levels of active Caspase 3, active Caspase 8 and mitochondrial membrane potential (ΔΨM) were analyzed in T cell subsets (Cell Technology, Mountain View, CA).
T Cell Homeostatic Proliferation
Purified T cells were negatively sorted and pooled from wild-type B6 or B6-Coro1aLmb3 mice (Thy1.2), stained with CFSE, and injected i.v. into B6.PL (Thy1.1) recipient mice (n=3 mice/time point) at 2 × 106 T cells/mouse. Recipient mice were sacrificed on day 5, LN CD4+ and CD8+ T cells were stained with appropriately labeled fluorescent-conjugated antibodies, and cells analyzed for division by flow cytometry as described (Lawson et al., 2001).
IL-7 Signaling Analysis by STAT5 Phosphorylation
Pooled splenocytes from wild-type MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice (n=3 mice/group) were activated with increasing concentrations (0, 0.01, 0.1, 1 and 10 ng/ml) of recombinant mouse IL-7 (BioLegend) for 10 min. Cells were fixed in 1.6% paraformaldehyde, permeabilized with ice-cold methanol, surface phenotyped with antibodies to CD4, CD8 and CD44, and levels of intracellular phosphorylated STAT5 (Cell Signaling Technology) were determined by flow cytometry.
T Cell Proliferation Assays
Purified T cells from wild-type MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice (n=3 mice/group) were either stimulated with plate-bound anti-CD3 (10 μg/ml) plus soluble anti-CD28 (5 μg/ml), PMA (50 ng/ml)/ Ionomycin (1 μg/ml) for ∼64 hrs. [3H]-TdR was added 16 h prior to harvesting and incorporation measured by liquid scintillation. Purified T cells from both genotypes were prestained with CFSE (Invitrogen), activated with plate-bound CD3 (10 μg/ml) plus CD28 (5 μg/ml) as above, then analyzed for division 3 days later by flow cytometry.
IL-2 Production Following Anti-CD3/CD28 T Cell Activation
Purified T cells were negatively sorted from wild-type MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice (n=5 mice/group) then activated with plate-bound anti-CD3 (10 μg/ml) plus soluble anti-CD28 (5 μg/ml) or PMA (50 ng/ml)/Ionomycin (1 μg/ml). Supernatants were collected 48 hrs later and analyzed by ELISA for IL-2 levels (BioLegend, San Diego, CA, USA).
T Cell Calcium Flux
Pooled purified splenic T cells from wild-type MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice (n=3 mice/group) using a negative T cell magnetic sort kit (Miltenyi Biotec, Auburn, CA, USA) were loaded with Indo-1, AM (2 μg/ml), previously dissolved in pluronic-127 and FBS, and incubated for 30 min at 37° C in the presence of probenecid (all from Invitrogen, Carlsbad, CA, USA). Simultaneously, T cells were surface stained with biotinylated anti-CD3 plus anti-CD28 antibodies (BD PharMingen). During the last 10 min of Indo-1 loading, T cells were surface-labeled with fluorescent conjugated antibodies to CD4, CD44, CD8, and Annexin (all from BD PharMingen). Cells were washed and resuspended in calcium loading buffer (PBS containing 1% FBS and 1 mM calcium) or calcium-free PBS/FBS. Samples were warmed to 37° C prior to and during analysis. Baseline fluorescence was monitored for ∼1 min before the addition of a saturating concentration of Avidin D (60 μg/sample, Vector Laboratories, Burlingame, CA) to crosslink the biotinylated anti-CD3/28 antibodies or Ionomycin (2 μg/ml). Calcium flux data was acquired for 6-8 min at 351 nm on a LSR II cytometer and analyzed with FlowJo. Relative Ca2+ flux was expressed as relative value x10−3. Activation of cells in calcium-free buffer measures the release of Ca2+ from intracellular stores. Three separate experiments with pooled cells were performed.
Serology and T-independent and –dependent Antibody Responses
Immunoglobulin and autoantibody levels were determined by ELISA as previously reported (Santiago-Raber et al., 2007). T-independent (T-I) and -dependent (T-D) B cell responses to TNP were assessed by serial IgM and IgG anti-TNP-3 (high affinity) and anti-TNP-14 (low affinity) antibody levels following immunization with 100 μg of either TNP-LPS or TNP-Ficoll in PBS, or with 100 μg TNP-KLH i.p. in CFA on day 1 and in PBS on day 21.
Immunohistochemistry
Staining was performed per manufacturer (Vector Laboratories, Burlingame, CA). Briefly, acetone-fixed 5 μ-thick sections of Tissue Tek OCT (Sakura Finetek, Torrance, CA)-embedded splenic tissue were incubated sequentially after blocking with anti-mouse IgM-alkaline phosphatase, PNA-biotin (Vector Lab) or anti-mouse CD4-biotin (Accurate, Westbury, NY), and streptavidin-HRP (Invitrogen, San Diego, CA), then developed with DAB and Vector Blue (Vector Lab). For GC analysis, widest parts of spleens were submitted for sections.
T Cell Transfer
Ten wk.-old MRL-Faslpr-Tcrb−/− mice were injected i.v. with 2.5 ×106 purified T cells from MRL-Faslpr or MRL.B6-Lmb3 Faslpr mice and followed for 22 wk. for development of systemic autoimmunity.
Statistical Analyses
Two-tailed unpaired t test or Mann-Whitney U test were used to compare groups, unless indicated. Data are expressed as mean±standard error unless otherwise indicated.
Supplementary Material
ACKNOWLEDGEMENTS
This is publication No. 18901-IMM from the Department of Immunology, The Scripps Research Institute. We greatly appreciate materials from Drs. J. Craft, R. Baccala, C. Surh, D. Soong, A. Chan, D. Cauvi, and K. M. Pollard. We thank Dr. M. Rodriguez for sequencing, Kat Occhipinti for editing, and C. Thompson and J. Lee for technical assistance. This work was supported by NIH grants AR39555, AR42242, AR31203, AI051977, and GM65230.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Appleton BA, Wu P, Wiesmann C. The crystal structure of murine coronin-1: a regulator of actin cytoskeletal dynamics in lymphocytes. Structure. 2006;14:87–96. doi: 10.1016/j.str.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Boackle SA, Holers VM, Chen X, Szakonyi G, Karp DR, Wakeland EK, Morel L. Cr2, a candidate gene in the murine Sle1c lupus susceptibility locus, encodes a dysfunctional protein. Immunity. 2001;15:775–785. doi: 10.1016/s1074-7613(01)00228-x. [DOI] [PubMed] [Google Scholar]
- Brieher WM, Kueh HY, Ballif BA, Mitchison TJ. Rapid actin monomerinsensitive depolymerization of Listeria actin comet tails by cofilin, coronin, and Aip1. J Cell Biol. 2006;175:315–324. doi: 10.1083/jcb.200603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Marshall TW, Uetrecht AC, Schafer DA, Bear JE. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128:915–929. doi: 10.1016/j.cell.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML. Immunology. When F-actin becomes too much of a good thing. Science. 2006;313:767–768. doi: 10.1126/science.1131714. [DOI] [PubMed] [Google Scholar]
- Eck SC, Zhu P, Pepper M, Bensinger SJ, Freedman BD, Laufer TM. Developmental alterations in thymocyte sensitivity are actively regulated by MHC class II expression in the thymic medulla. J Immunol. 2006;176:2229–2237. doi: 10.4049/jimmunol.176.4.2229. [DOI] [PubMed] [Google Scholar]
- Elliott JI, McVey JH, Higgins CF. The P2X7 receptor is a candidate product of murine and human lupus susceptibility loci: a hypothesis and comparison of murine allelic products. Arthritis Res Ther. 2005;7:R468–475. doi: 10.1186/ar1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney AJ, Lawson BR, Kono DH, Theofilopoulos AN. Terminal deoxynucleotidyl transferase deficiency decreases autoimmune disease in MRL-Faslpr mice. J Immunol. 2001;167:3486–3493. doi: 10.4049/jimmunol.167.6.3486. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 1999;97:435–447. doi: 10.1016/s0092-8674(00)80754-0. [DOI] [PubMed] [Google Scholar]
- Foger N, Rangell L, Danilenko DM, Chan AC. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science. 2006;313:839–842. doi: 10.1126/science.1130563. [DOI] [PubMed] [Google Scholar]
- Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, Zhu P, Freedman BD, Clark MR, Rawlings DJ, et al. HS1 functions as an essential actinregulatory adaptor protein at the immune synapse. Immunity. 2006;24:741–752. doi: 10.1016/j.immuni.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen PK, Behrens TW. Genetics of autoimmune diseases--disorders of immune homeostasis. Nat Rev Genet. 2006;7:917–928. doi: 10.1038/nrg1944. [DOI] [PubMed] [Google Scholar]
- Grogan A, Reeves E, Keep N, Wientjes F, Totty NF, Burlingame AL, Hsuan JJ, Segal AW. Cytosolic phox proteins interact with and regulate the assembly of coronin in neutrophils. J Cell Sci. 1997;110(Pt 24):3071–3081. doi: 10.1242/jcs.110.24.3071. [DOI] [PubMed] [Google Scholar]
- Harley JB, Kelly JA, Kaufman KM. Unraveling the genetics of systemic lupus erythematosus. Springer Semin Immunopathol. 2006;28:119–130. doi: 10.1007/s00281-006-0040-5. [DOI] [PubMed] [Google Scholar]
- Jayachandran R, Sundaramurthy V, Combaluzier B, Mueller P, Korf H, Huygen K, Miyazaki T, Albrecht I, Massner J, Pieters J. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell. 2007;130:37–50. doi: 10.1016/j.cell.2007.04.043. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Hirose S, Abe M, Sanokawa-Akakura R, Ohtsuji M, Mi X, Li N, Xiu Y, Zhang D, Shirai J, et al. Polymorphisms in IgG Fc receptor IIB regulatory regions associated with autoimmune susceptibility. Immunogenetics. 2000;51:429–435. doi: 10.1007/s002510050641. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Hirose S, Sanokawa-Akakura R, Abe M, Mi X, Li N, Miura Y, Shirai J, Zhang D, Hamano Y, Shirai T. Genetically determined aberrant down-regulation of FcgammaRIIB1 in germinal center B cells associated with hyper-IgG and IgG autoantibodies in murine systemic lupus erythematosus. Int Immunol. 1999;11:1685–1691. doi: 10.1093/intimm/11.10.1685. [DOI] [PubMed] [Google Scholar]
- Kawasaki M, Nakayama K, Wakatsuki S. Membrane recruitment of effector proteins by Arf and Rab GTPases. Curr Opin Struct Biol. 2005;15:681–689. doi: 10.1016/j.sbi.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Khaidukov SV, Litvinov IS. Calcium homeostasis change in CD4+ T lymphocytes from human peripheral blood during differentiation in vivo. Biochemistry (Mosc) 2005;70:692–702. doi: 10.1007/s10541-005-0170-8. [DOI] [PubMed] [Google Scholar]
- Kono DH, Burlingame RW, Owens DG, Kuramochi A, Balderas RS, Balomenos D, Theofilopoulos AN. Lupus susceptibility loci in New Zealand mice. Proc Natl Acad Sci U S A. 1994;91:10168–10172. doi: 10.1073/pnas.91.21.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono DH, Theofilopoulos AN. Genetics of SLE in mice. Springer Semin Immunopathol. 2006;28:83–96. doi: 10.1007/s00281-006-0030-7. [DOI] [PubMed] [Google Scholar]
- Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312:1665–1669. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- Lawson BR, Koundouris S, Barnhouse M, Dummer W, Baccala R, Kono DH, Theofilopoulos AN. The role of αβ+ cells and homeostatic T cell proliferation in Yaa+-associated murine lupus. J Immunol. 2001;167:2354–2360. doi: 10.4049/jimmunol.167.4.2354. 446, 284-287. [DOI] [PubMed] [Google Scholar]
- Li O, Zheng P, Liu Y. CD24 expression on T cells is required for optimal T cell proliferation in lymphopenic host. J Exp Med. 2004 doi: 10.1084/jem.20040779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;200:1083–1089. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- Lynch DH, Watson ML, Alderson MR, Baum PR, Miller RE, Tough T, Gibson M, Davis-Smith T, Smith CA, Hunter K, et al. The mouse Fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity. 1994;1:131–136. doi: 10.1016/1074-7613(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Mary C, Laporte C, Parzy D, Santiago ML, Stefani F, Lajaunias F, Parkhouse RM, O'Keefe TL, Neuberger MS, Izui S, Reininger L. Dysregulated expression of the Cd22 gene as a result of a short interspersed nucleotide element insertion in Cd22a lupusprone mice. J Immunol. 2000;165:2987–2996. doi: 10.4049/jimmunol.165.6.2987. [DOI] [PubMed] [Google Scholar]
- Nal B, Carroll P, Mohr E, Verthuy C, Da Silva MI, Gayet O, Guo XJ, He HT, Alcover A, Ferrier P. Coronin-1 expression in T lymphocytes: insights into protein function during T cell development and activation. Int Immunol. 2004;16:231–240. doi: 10.1093/intimm/dxh022. [DOI] [PubMed] [Google Scholar]
- Nolz JC, Gomez TS, Zhu P, Li S, Medeiros RB, Shimizu Y, Burkhardt JK, Freedman BD, Billadeau DD. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol. 2006;16:24–34. doi: 10.1016/j.cub.2005.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B Cell Responses to RNA-Related Antigens Due to TLR7 Gene Duplication. Science. 2006 doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- Pritchard NR, Cutler AJ, Uribe S, Chadban SJ, Morley BJ, Smith KG. Autoimmune-prone mice share a promoter haplotype associated with reduced expression and function of the Fc receptor FcgammaRII. Curr Biol. 2000;10:227–230. doi: 10.1016/s0960-9822(00)00344-4. [DOI] [PubMed] [Google Scholar]
- Qu W, Miyazaki T, Terada M, Lu L, Nishihara M, Yamada A, Mori S, Nakamura Y, Ogasawara H, Yazawa C, et al. Genetic dissection of vasculitis in MRL/lpr lupus mice: a novel susceptibility locus involving the CD72c allele. Eur J Immunol. 2000;30:2027–2037. doi: 10.1002/1521-4141(200007)30:7<2027::AID-IMMU2027>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rodal AA, Sokolova O, Robins DB, Daugherty KM, Hippenmeyer S, Riezman H, Grigorieff N, Goode BL. Conformational changes in the Arp2/3 complex leading to actin nucleation. Nat Struct Mol Biol. 2005;12:26–31. doi: 10.1038/nsmb870. [DOI] [PubMed] [Google Scholar]
- Rozzo SJ, Allard JD, Choubey D, Vyse TJ, Izui S, Peltz G, Kotzin BL. Evidence for an interferon-inducible gene, Ifi202, in the susceptibility to systemic lupus. Immunity. 2001;15:435–443. doi: 10.1016/s1074-7613(01)00196-0. [DOI] [PubMed] [Google Scholar]
- Rybakin V, Clemen CS. Coronin proteins as multifunctional regulators of the cytoskeleton and membrane trafficking. Bioessays. 2005;27:625–632. doi: 10.1002/bies.20235. [DOI] [PubMed] [Google Scholar]
- Santiago-Raber M-L, Haraldsson MK, Theofilopoulos AN, Kono DH. Characterization of Reciprocal Lmb1-4 Interval MRL-Faslpr and B6-Faslpr Congenic Mice Reveals Significant Effects from Lmb3. J Immunol. 2007;178:8195–8202. doi: 10.4049/jimmunol.178.12.8195. [DOI] [PubMed] [Google Scholar]
- Schaerli P, Moser B. Chemokines: control of primary and memory T-cell traffic. Immunol Res. 2005;31:57–74. doi: 10.1385/IR:31:1:57. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17:183–191. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Uetrecht AC, Bear JE. Coronins: the return of the crown. Trends Cell Biol. 2006;16:421–426. doi: 10.1016/j.tcb.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Vidal S, Kono DH, Theofilopoulos AN. Loci predisposing to autoimmunity in MRL-Faslpr and C57BL/6-Faslpr mice. J Clin Invest. 1998;101:696–702. doi: 10.1172/JCI1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, Yim YS, Pertsemlidis A, Garner HR, Jr., Morel L, Wakeland EK. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–780. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferative disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- Wong M, Tsao BP. Current topics in human SLE genetics. Springer Semin Immunopathol. 2006;28:97–107. doi: 10.1007/s00281-006-0031-6. [DOI] [PubMed] [Google Scholar]
- Yan M, Collins RF, Grinstein S, Trimble WS. Coronin-1 Function Is Required for Phagosome Formation. Mol Biol Cell. 2005 doi: 10.1091/mbc.E04-11-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski CE, Jacob SN, Bouzahzah F, Ehrlich BE, Craft J. Naive CD4+ T cells from lupus-prone Fas-intact MRL mice display TCR-mediated hyperproliferation due to intrinsic threshold defects in activation. J Immunol. 2005;174:5100–5109. doi: 10.4049/jimmunol.174.8.5100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.