

Abstract
Maspin is a mammary serine protease inhibitor or serpin with tumor suppressive and antiangiogenic activity that inhibits tumor motility, invasion and metastasis, at least by its actions on cell membrane and extracellular matrix (ECM) proteins. Previous studies documented that the quinazoline-derived α1-adrenoceptor antagonist doxazosin affects the attachment and migration of prostate cancer cells. In this study, we investigated the effect of maspin overexpression on the apoptotic/antiadhesion response of prostate cancer cells to doxazosin. The response of maspin-overexpressing clones of human prostate cancer cells DU-145 to doxazosin was evaluated by determining cell viability, apoptosis and cell proliferation on the basis of the trypan blue exclusion assay/methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay, Hoechst staining and caspase-3 activation, and [3H]thymidine incorporation assay. Vascular endothelial growth factor (VEGF), transforming growth factor βRII (TGFβRII), Smad4 (a TGFβ intracellular effector) and bax expression was evaluated at the mRNA and protein level using reverse transcriptase–polymerase chain reaction and Western blotting, respectively. The effect of doxazosin on cell attachment of maspin-expressing prostate cancer cells was evaluated on collagen- and fibronectin-coated plates. Cell migration was assessed using the wounding assay. In response to tumor necrosis factor-related apoptosis-inducing ligand, DU-145-maspin expressing cells undergo apoptosis, via poly(ADP-ribose) polymerasecleavage and caspase-3 activation. DU-145-maspin cells exhibited higher sensitivity to doxazosin and an earlier temporal activation of caspase-3. The number of apoptotic cells detected in response to doxazosin was significantly higher compared to the neo control (P<0.0001). Doxazosin resulted in dramatic downregulation of the 189 isoform of VEGF in maspin transfectants, while a fivefold induction of Smad4 mRNA expression was detected in those cells after 24 h of treatment. Maspin overexpression in prostate cancer cells resulted in an increased ability to attach to ECM-coated plates, and doxazosin treatment considerably antagonized this effect by decreasing the attachment potential to collagen and fibronectin. The present study supports the ability of maspin to enhance the apoptotic threshold of prostate cancer cells to the quinazoline-based α1-adrenoceptor antagonist doxazosin. These findings may have therapeutic significance in the development of antiangiogenic targeting by doxazosin and derivative agents for advanced prostate cancer.
Keywords: prostate cancer, α-blockers, apoptosis
Introduction
Prostate cancer accounted for 230 110 new cancer cases in American men in 2004, being a leading cause of cancer death, since 29 900 patients died of the disease on that year (Jemal et al., 2004). Radical prostatectomy, open or laparoscopic, androgen ablation therapy and radiotherapy, in the form of conformal radiation or brachytherapy, are all established treatment modalities, which usually result in prolonged survival and in some patients cure (Raghavan et al., 1997; Bare and Torti, 1998). However, tumor relapse and metastasis occur frequently and the majority of prostate cancer patients become unresponsive to hormonal therapy due to the emergence of androgen-independent cancer cells (Bruckheimer and Kyprianou, 2000). The limited success associated with the use of chemotherapeutic drug protocols for hormone refractory prostate cancer (Sternberg, 2003) calls for the development of new apoptosis-based therapeutic modalities for advanced disease (Bruckheimer and Kyprianou, 2000).
Doxazosin and terazosin are α1-adrenoreceptor antagonists widely used in clinical practice for the treatment of lower urinary tract symptoms due to benign prostatic hyperplasia (BPH) (Oades et al., 2000). Recently, the quinazoline-based antagonists have been shown to exert potent apoptotic action against benign and malignant prostate cells mediated via transforming growth factor β (TGFβ) signaling and activation of NF-κβ pathway (Kyprianou and Benning, 2000; Benning and Kyprianou, 2002; Partin et al., 2003). In in vivo experimental models of both prostate cancer and BPH xenografts, doxazosin-induced apoptosis led to suppression of prostate growth without affecting their proliferative ability (Yang et al., 1997; Kyprianou and Benning, 2000). In a clinical study, terazosin resulted in an increased apoptotic index and reduced vascularity in human prostate tumor samples (Keledjian et al., 2001).
Maspin (mammary serine protease inhibitor or serpin), also known as protease inhibitor 5, was recently discovered in normal mammary epithelium (Zou et al., 1994). This 42 kDa protein, encoded by a gene located on chromosome 18q21.3, is localized to the cytoplasm, the cell membrane and the extracellular matrix (ECM) (Schaefer and Zhang, 2003). Maspin is classified as a class II tumor suppressor, since it is downregulated or silenced by hypermethylation in cancer cells (Domann et al., 2000), and suppresses tumor growth and metastasis in vivo and inhibits tumor invasion in vitro (Zou et al., 1994). Maspin also exerts a potent inhibitory action against osteolysis in a model of prostate cancer bone metastasis (Cher et al., 2003) and has been functionally linked to apoptosis of human breast and prostate cancer cells (Jiang et al., 2002). More recent evidence points to a role of maspin in angiogenesis regulation, as it reduces tumor microvessel density and endothelial cell migration in a prostate cancer xenograft model (Zhang et al., 2000). An interaction between cell surface maspin and various components of the ECM (such as collagen type I and III) might be potentially driving its ability to enhance cell adhesion and block cell migration and metastasis (Blacque and Worrall, 2002). Inhibition of pericellular proteolysis via an action on the urokinase-type plasminogen activator (uPA) might be responsible for maspin’s inhibitory effect on tumor invasion and motility (Biliran and Sheng, 2001). Recently, the proapoptotic protein bax was identified as a key mediator of the maspin apoptotic effect in DU-145 prostate cancer cells overexpressing maspin (Liu et al., 2004).
This study investigated doxazosin-mediated apoptosis on maspin-transfected DU-145 prostate cancer cells. Our findings indicate that maspin sensitizes prostate cancer cells to the apoptotic action of doxazosin by interfering with their attachment to ECM.
Results
Western blot analysis revealed the maspin expression in DU-145 transfectants. As shown in Figure 1a, the DU-145-maspin cells, clone M7 (which was used for the subsequent experiments in this study), exhibited high expression of maspin protein in contrast to DU-145-neo control transfectants that totally lacked maspin. Bax expression was slightly increased in M7 cells as compared to the neo control transfectants. In response to treatment with tumor necrosis factor-related apoptosis- inducing ligand (TRAIL) (50 ng/ml; 12h), DU-145-maspin cells exhibit increased sensitivity to apoptosis, as determined by poly(ADP-ribose) polymerase (PARP) cleavage and caspase-3 activation (Figure 1b).
Figure 1.

Characterization of apoptotic response of maspin-expressing DU-145 transfectants to TRAIL. (a) Western blot analysis revealed that DU-145-maspin cells exhibit strong maspin expression (M7). (b) DU-145-maspin expressing cells undergo apoptosis in response to TRAIL, as indicated by increased PARP cleavage and caspase-3 activation after treatment with TRAIL (50 ng/ml for 12h). Western blot analysis was performed as described in ‘Materials and methods’ using the respective antibodies
A significant decrease in cell viability was observed for the DU-145-neo transfectants cells at a concentration of 15 μm doxazosin (82% viability, P<0.005) with a continuous decrease at higher doxazosin concentrations (P<0.0001). The DU-145-maspin cells exhibited a significant reduction of cell viability at a concentration of 25 μm (39.8%).
On the basis of the methylthiazolyldiphenyl-tetrazolium bromide (MTT) cell viability assay, there was a higher sensitivity for the DU-145-maspin cells to increasing doses of doxazosin compared to the DU-145-neo control cells (Figure 2a). At both 25 and 50 μm of doxazosin, significant differences were detected between the two cell lines (67.2±8.6 versus 103.2±3.7% and 3.8±0.5 versus 51.5±11.4%, P<0.005).
Figure 2.
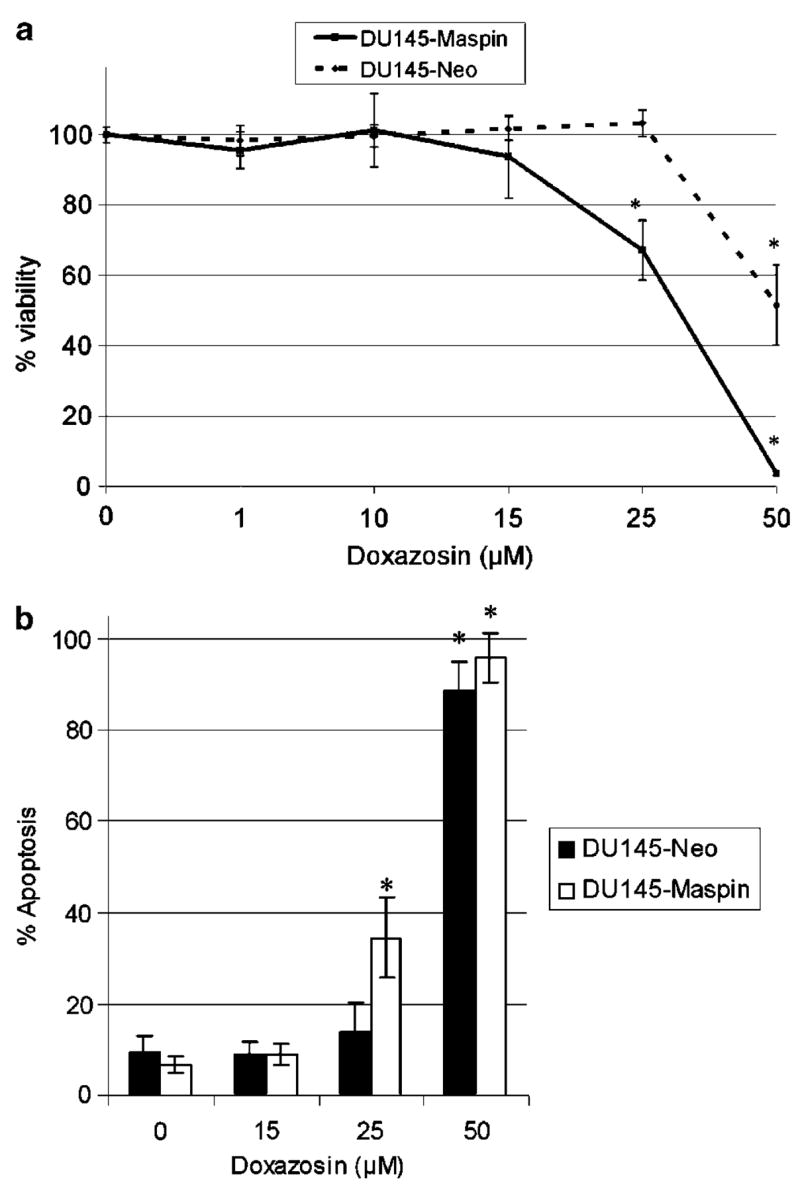
(a) Effect of doxazosin on cell viability of prostate cancer cells (MTT assay). DU-145-maspin cells were treated with increasing concentrations of doxazosin as shown. Error bars represent average±s.e. (b) Doxazosin-mediated apoptosis in maspin-expressing DU-145 cells. Apoptotic cells were detected using the Hoechst staining as described in ‘Materials and methods’. Maspin sensitizes DU-145 prostate cancer cells to the apoptotic effects of doxazosin (25 μm) after treatment for 48 h. Error bars represent average±s.d. *P<0.005
As shown in Figure 2b, a dose-dependent increase in the number of apoptotic cells was observed after treatment with doxazosin as determined by Hoechst staining. At the concentration of 25 μm, a significantly higher number of DU-145-maspin cells underwent apoptosis compared to neo controls (34.5±9 versus 13.8±6.6%, respectively, P<0.0001).
We subsequently investigated the effect of doxazosin on cell proliferation. Figure 3 indicates a decrease in [3H]thymidine uptake in both cell lines after doxazosin treatment that was statistically significant at 25 μm and there was a further significant decrease at 50 μm (46.1±7.7 and 43.1±5.4% for the DU-145-maspin and -neo cells, respectively). DU-145-maspin cells exhibited a higher sensitivity at 25 μm of doxazosin, showing a significant reduction in thymidine uptake compared to neo controls (52.3±5.9 versus 82.1±3.6%, P<0.005).
Figure 3.

Effect of doxazosin on DNA synthesis: [3H]thymidine uptake assay. Prostate cancer DU-145-maspin cells treated with increasing concentrations of the drug as shown are more sensitive at the concentration of 25 μm doxazosin (P<0.005). Error bars represent average±s.e. *P<0.005
Considering the ability of maspin to increase adhesion to ECM components, we subsequently examined the attachment potential of prostate cancer cells after doxazosin treatment. Overall, untreated prostate cancer maspin transfectants exhibited an enhanced ability to attach to coated plates, which was more pronounced on fibronectin-coated plates (maspin: 89.9±7.3 versus neo: 63.1±4.1 cells/100 × power field, P<0.005, Figure 4b), whereas the difference on collagen was not that prominent. There was progressive loss of cell attachment to collagen for both cell lines treated with doxazosin (Figure 4a); however, the DU-145-maspin cells exhibited a relatively higher ability to remain attached under treatment with doxazosin compared to DU-145-neo controls (at 50 μm, P<0.01). A similar significant decrease in cell attachment to fibronectin was observed after doxazosin treatment at both concentrations (Figure 4b).
Figure 4.
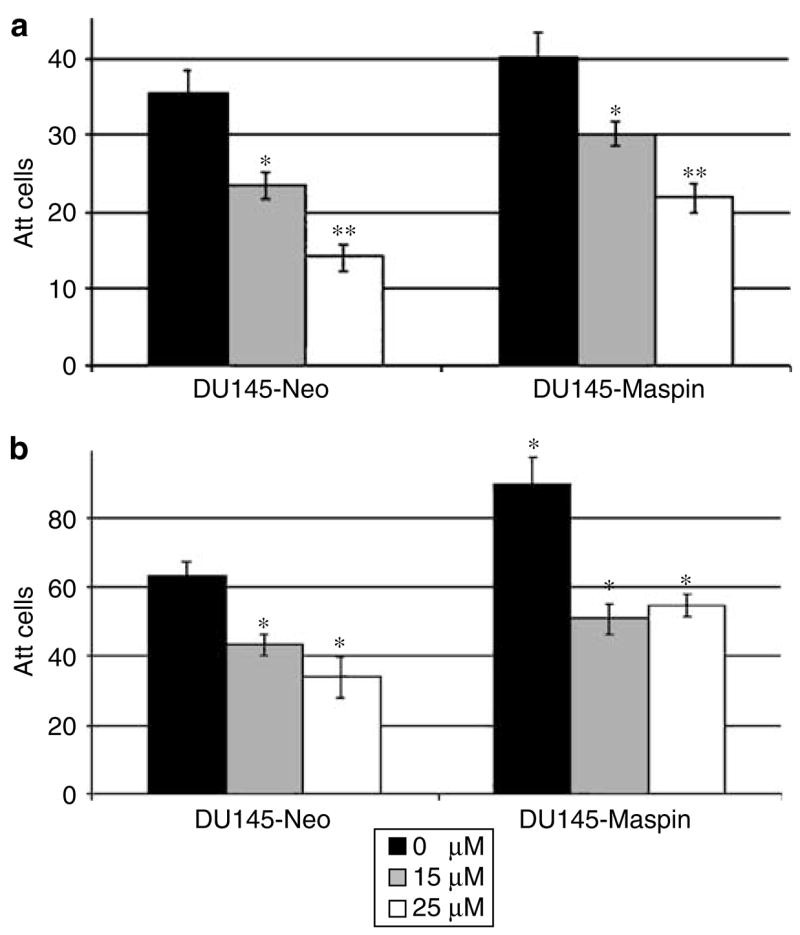
Effect of doxazosin on cell attachment potential of prostate cancer cells overexpressing maspin. Doxazosin-treated DU-145-maspin cells exhibit increased adhesion to collagen compared to neo controls (P<0.01). (Panel a) Untreated maspin transfectants, M7, show significantly increased attachment on fibronectin-coated plates (panel b), expressed as attached cells/100 × field. Error bars represent average±s.e. *P<0.01, **P<0.0001
There was a significant loss of cell migration after treatment with 15 and 25μm doxazosin in both cell lines (Figure 5a–c). At a concentration of 15 μm (Figure 5d), DU-145-maspin cells had a decreased ability to migrate compared to neo controls; however, this difference did not reach significance. The addition of vascular endothelial growth factor (VEGF) (Figure 5e) did not result in a significant increase in the migration of either cell line. Doxazosin, in the presence of VEGF (Figure 5e), could still significantly inhibit the migration of DU-145 cells, resulting in a comparable decrease to that achieved when used alone (P<0.01).
Figure 5.
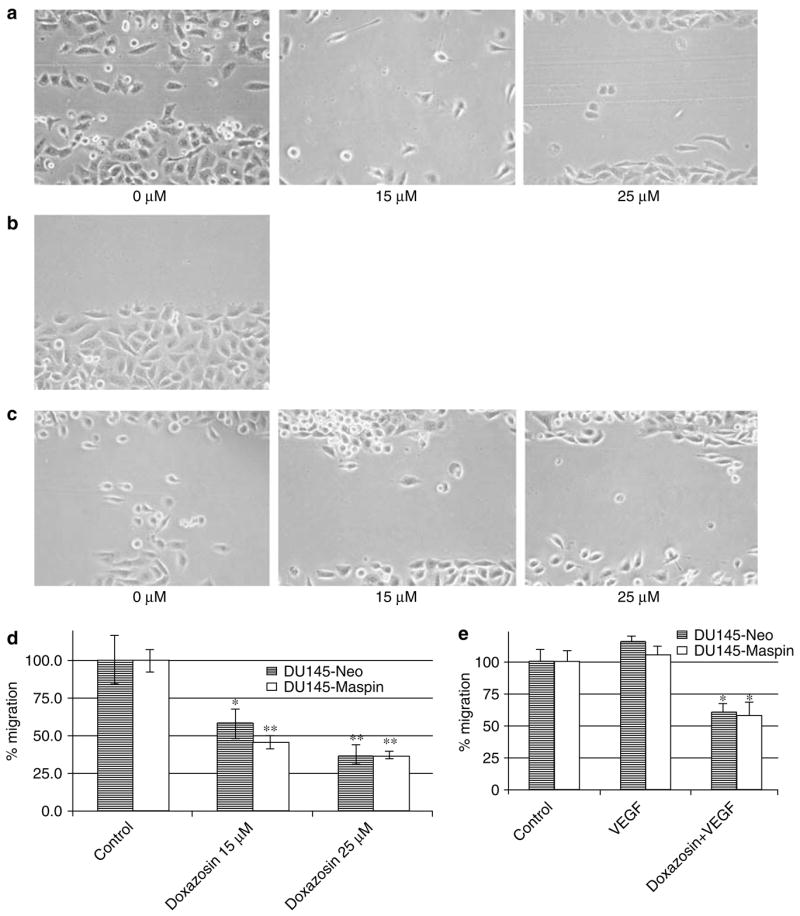
Effect of doxazosin on cell migration: impact of maspin. (a, c) Reduction of cell migration in DU-145-maspin (a) and DU- 145-neo cells (c) with 15 and 25 μm doxazosin. (b) Example of a wounded area. (d) Quantitative analysis of the results. (e) VEGF does not result in a significant increase in the migration of DU-145 cells. Error bars represent average±s.e. *P<0.05, **P<0.005
To characterize these effects at the molecular level, we performed reverse transcriptase–polymerase chain reaction (RT–PCR) analysis for a number of key apoptosis and angiogenesis molecules. Figure 6 indicates a dramatic induction of Smad4 mRNA after 24 h in DU-145-maspin cells (Figure 6b, 47.6% increase versus 7.8% increase for controls). RT–PCR analysis of VEGF expression (Figure 7a) indicated a similar progressive decrease after doxazosin treatment for the different isoforms of VEGF in both DU-145-maspin and neo controls. As shown in Figure 7b, isoform 189 of VEGF was dramatically downregulated in M7 (by 84%) within the first 6 h of doxazosin treatment compared with a 34% reduction in VEGF mRNA in neo controls. Doxazosin downregulated all other VEGF isoforms in a similar manner in the two groups. A similar downregulation by doxazosin was detected for transforming growth factor βRII (TGFβRII) mRNA (data not shown).
Figure 6.

(a) RT–PCR of Smad4 expression in DU-145 cells after treatment with doxazosin. mRNA analysis was performed as described in the ‘Materials and methods’ section. (b) Quantitative analysis of Smad4 expression in DU-145 cells
Figure 7.

(a) RT–PCR of VEGF expression in DU-145 cells after treatment with doxazosin, showing the different isoforms of VEGF. (b) Quantitative analysis of VEGF expression (189 isoform) in DU-145 cells indicates differential downregulation in DU-145-maspin cells
The apoptotic nature of the response to doxazosin treatment was confirmed with caspase-3 activation documented by Western blotting (Figure 8). The caspase-3 activity assay failed, however, to show any significant caspase-3 activation after doxazosin treatment in any cell line (data not shown). Doxazosin- treated DU-145-maspin cells exhibited cleavage of caspase-3, within the first 12h of exposure to doxazosin, compared to neo controls (after 24 h). No significant differences in caspase-8 cleavage were noted in either cell line after doxazosin treatment (data not shown). A significant induction of bax, however, was noted in both cell lines after treatment for 24 h, and was stronger in the maspin-transfected cells (30.5% versus 175% for neo controls, Figure 9a and b). Western blotting revealed downregulation of VEGF for both cell lines in response to doxazosin (Figure 10a and b), while there was a parallel increase in Smad4 protein levels (Figure 11a and b). Phosphorylated VEGF receptor 2 expression was not detected in either cell line (data not shown).
Figure 8.

Caspase-3 activation by doxazosin in prostate cancer cells. DU-145 cells were treated with 15 μm doxazosin for 6, 12 and 24 h, and cell lysates were subjected to Western blotting for caspase-3. A temporally accelerated apoptosis induction and caspase-3 cleavage was detected in DU-145-maspin expressing cells (after 12h of treatment with doxazosin) compared to 24 h for DU-145-neo controls
Figure 9.
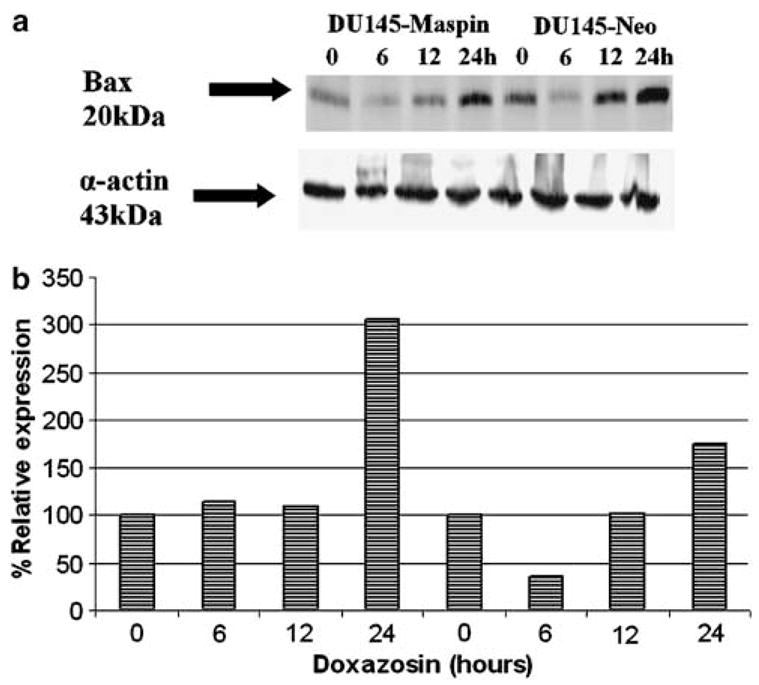
(a) Bax protein expression after treatment with doxazosin in DU-145 cells. (b) Quantitative analysis of bax expression normalized to actin expression. Maspin transfectants exhibit stronger activation of bax after doxazosin treatment
Figure 10.
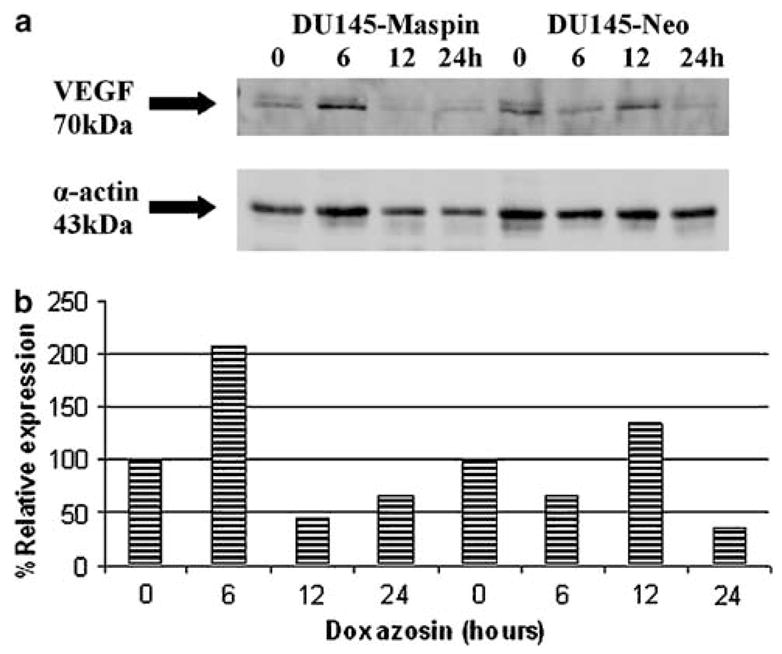
(a) Western blot analysis of VEGF expression in response doxazosin in DU-145 cells. (b) Quantitative analysis of VEGF expression normalized to actin expression. Downregulation of VEGF after a transient peak in both cell lines
Figure 11.

(a) Smad4 protein expression after treatment with doxazosin in DU-145 cells. (b) Quantitative analysis of Smad4 expression normalized to actin expression. Earlier induction of Smad4 at 6 h was observed for the maspin transfectants, M7
Discussion
The identification of maspin in breast epithelial cells provided a new insight on a possible mechanism of tumor suppression through inhibition of angiogenesis (Zhang et al., 2000), invasion of basement membranes (Sheng et al., 1994) and metastasis (Shi et al., 2001), and increased susceptibility of cancer cells to induced apoptosis (Jiang et al., 2002). The present study provides proof-of-principle that maspin alters the sensitivity of prostate cancer cells to doxazosin. In a similar way, it was demonstrated that it could modify the ability of mammary MDA-MB-435 carcinoma cells to undergo apoptosis in response to staurosporine (Jiang et al., 2002). Our findings demonstrate an increased sensitivity of DU-145-maspin cells to the apoptotic actions of doxazosin, as evidenced by an increased apoptotic index and earlier caspase-3 cleavage. Also, stronger bax activation was evident after 24 h of treatment for maspin cells (M7). Our observations are similar to the previous study (Jiang et al., 2002) that reported increased activation of caspase-3 and increased fragmentation of DNA as determined by the TUNEL assay in staurosporine- treated mammary carcinoma cells. Also, a recent study demonstrated that the upregulation of bax in maspin cells may be a possible mechanism for the sensitization of prostate cancer cells to apoptosis by tipping the balance of pro- versus antiapoptotic apoptosis regulators in response to a variety of drugs including TRAIL, staurosporine and brefeldin (Liu et al., 2004).
Maspin is further known to exert its tumor suppressor action by increasing adhesion to ECM components (Abraham et al., 2003). This evidence is in accordance with the present findings that maspin-transfected cells have a higher attachment potential to ECM-coated surface compared to the neo transfectants. Maspin-overexpressing prostate cancer cells have a greater ability to remain attached to collagen-coated plates when treated with doxazosin, a potential anoikis-inducing agent (Keledjian and Kyprianou, 2003). These results may be related to the ability of maspin to interact with collagen I and III, an interaction that may contribute to increased adhesion, thus preventing cell migration (Blacque and Worrall, 2002), whereas interactions with fibronectin and laminin have also been reported (Abraham et al., 2003).
We recently reported that the key identified molecular players of doxazosin action were members of the TGFβ pathway such as Smad4 and TGFβ1-inducible early gene (TIEG1) (Partin et al., 2003). In particular, increased expression of Smad4 was reported at the protein level 6 h after doxazosin treatment. Our results show an earlier increase in Smad4 protein expression in DU-145 maspin cells (6 versus 12h), and at the mRNA level, there was a clear induction of Smad4 even after 24 h of treatment. The earlier Smad4 induction could be linked to the earlier activation of caspase-3 detected in maspin transfectants.
Maspin is a potent inhibitor of motility and invasion, as shown in modified Boyden chamber assays both with tumor transfectants and with the addition of exogenous recombinant maspin (Sheng et al., 1996; Abraham et al., 2003). A direct link between maspin and angiogenesis was recently suggested by evidence that maspin exerts a role as an angiogenesis inhibitor (Zhang et al., 2000). In the xenograft model of LNCaP human prostate cancer, maspin resulted in a 50% reduction of microvessel density of prostate tumors (28.6±3.6 versus 15.3±1.8 vessels). It was documented in that study, however, that this ability was associated with a direct action on angiogenic factors such as VEGF. On the other hand, doxazosin has been shown to result in a downregulation of VEGF at the protein and mRNA levels (Keledjian and Kyprianou, 2003). Our results show a differential downregulation of the 189 isoform of VEGF in maspin transfectants under doxazosin treatment, as well as earlier downregulation at the protein level. Exciting as these findings might be, further mechanistic analysis is required to dissect the contribution of the crosstalk between maspin and VEGF toward prostate cancer development and progression.
In conclusion, this is the first evidence that maspin can augment the apoptotic action of doxazosin in prostate cancer cells. We found that doxazosin can significantly suppress the migratory ability of prostate cancer cells, even in the presence of VEGF, although there were no significant differences between the two cell lines. Ongoing studies focus on determining whether maspin can inhibit prostate cancer metastasis in vivo, in a manner similar to mammary epithelial cells (Zou et al., 1994) and to further dissect the mechanism underlying this (anoikis-driven) effect.
Materials and methods
Cell culture
Human prostate cancer DU-145-mock-transfectant cells (neo-mycin) and the stable DU-145-maspin transfectants (M7) were cultured in G418 (Geneticin, Gibco Brl, Grand Island, NY, USA) containing RPMI1640 medium (Gibco Brl, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Gibco Brl, Grand Island, NY, USA) and penicillin/strepto-mycin mixture with L-glutamine (BioWhittaker, Walkersville, MD, USA). Doxazosin (doxazosin mesylate, Pfizer Pharmaceuticals, New York, NY, USA) was generously provided by the manufacturer.
Apoptosis evaluation
Cell viability assay
Cells were seeded on six-well plates (105 cells/well) and treated with increasing concentrations of doxazosin (1, 10, 15, 25 and 50 μm) for 48 h. Cell viability was determined and expressed as the number of viable cells divided by the number of total cells × 100%. Each experiment was performed twice in duplicate (duplicate wells for each treatment).
MTT assay
Cells were seeded in 24-well plates at a concentration of 5 × 104 cells/well and treated with increasing concentrations of doxazosin (1, 10, 15, 25 and 50 μm) for 48 h. After treatment, the medium was replaced with 250 μl sterile MTT solution (Sigma, St Louis, MO, USA; 1 mg/ml in PBS) per well and cells were incubated for 2 h in a cell culture incubator at 37°C. The MTT crystals were solubilized in DMSO (Sigma, St Louis, MO, USA) (overnight at 37°C), and duplicates of 100 μl each from each well were subjected to optical density analysis. Samples were evaluated in a μQuant Universal Microplate Spectrophotometer (Biotek Instruments, Winooski, VT, USA) at a wavelength of 570 nm with back-ground subtraction at 630 nm.
Hoechst staining
Cells were treated with different concentrations of doxazosin (15, 25 and 50 μm) for 48 h. After treatment, cells were fixed in 4% paraformaldehyde for 10 min. After washing with PBS, Hoechst dye was added to a final concentration of 10 μg/ml with Triton X-100 to permeabilize the cells, and plates were incubated overnight at 4°C. Apoptotic cells were counted in three random fields/well and percent apoptosis was determined based on the total number of cells. Each experiment was performed twice in duplicate.
Caspase-3 activity assay
Cells were treated with 15 μm of doxazosin in six-well plates for 0, 12, 24 and 48 h, and the Caspase-3 colorimetric activity assay kit (Chemicon, Temecula, CA, USA) was used to assess caspase-3 activity according to the manufacturer’s instructions. Briefly, cells were lysed, centrifuged (5 min) in a microcentrifuge and the supernatant was collected and 150 μg of protein/reaction was incubated with the substrate Ac-DEVD-pNA for 2 h at 37°C. Samples were read at 405 nm on a μQuant microtiter plate reader (Biotek, Vinooski, VT, USA).
Cell proliferation ([3H]thymidine uptake assay)
Cells were seeded in 24-well plates at 5 × 104/well. After 24 h, doxazosin was added at increasing concentrations (1, 15, 25 and 50 μm). The [3H]thymidine uptake assay was performed as described previously (Guo and Kyprianou, 1998). Each experiment was performed twice in triplicate.
Attachment assay
Cells were treated with doxazosin (15 and 25μm). After 24 h, cells were seeded in six-well plates (5 × 104 cells/well) coated with fibronectin (BD Biosciences Discovery Labware, Bedford, MA, USA; 2.8 μg/cm2) or type I collagen (1 μg/cm2; Sigma, St Louis, MO, USA). Following a 10-min attachment period, attached cells were fixed with methanol (100%) and maintained at 4°C for image analysis on a Nikon Eclipse TE2000-U microscope (Nikon, Melville, NY, USA). Cells were counted in three 100 × fields/well and average values were determined.
Cell migration assay
The wounding assay was applied in cells growing in six-well plates. Upon confluency, the cell monolayer was wounded with a toothpick, and cells were treated with doxazosin (0, 15 and 25 μm) for 24 h. Cells that migrated to the wounded areas were counted and migration was defined as the average number of cells in three random fields (200 × ) per well (Keledjian et al., 2005). The experiment was performed three times, with and without the addition of 15 ng/ml VEGF (recombinant human VEGF; R&D Systems, Minneapolis, MN, USA) in combination with doxazosin.
RT–PCR analysis
Cells were treated with doxazosin (15 μm) for 6, 24 and 48 h and total cellular RNA was isolated from untreated control and treated cells using the Trizol reagent (Gibco Brl, Grand Island, NY, USA). RNA (2 μg) was reverse transcribed into cDNA using the SuperScript II system (Gibco Brl, Grand Island, NY, USA) in a Gene Amp 2400 thermocycler (Perkin-Elmer, Wellesley, MA, USA).
The PCR for VEGF, TGFβRII and Smad4 was performed in a total volume of 25 μl containing 0.2 mm DNTPs, 1.5 mm MgCl2, 1 U Platinum Taq DNA polymerase, 0.15 m of each sense and antisense primer, and distilled water. The following cycling conditions were used for 37 cycles in a Gene Amp 2400 thermocycler (Perkin-Elmer, Wellesley, MA, USA): initial denaturation, 5 min at 94°C; denaturation 30 s at 94°C; annealing 30 s at 55°C; and elongation 30 s at 72°C. The program was followed by a final elongation step for 5 min at 72°C. The primers used for TGFβRII PCR were 5′-CGC TGG GGG CTC GGT CTA TG-3′ (sense) and 5′-CCA CTG TCT CAA ACT GCT CT-3′ (antisense) (Wang et al., 1995) and for VEGF 5′-TGC ACC CAT GGC AGA AGG AGG-3′ (sense) and 5′-TCA CCG CCT CGG CTT GTC ACA-3′ (antisense) (Burchardt et al., 1999).
Relative quantitative RT–PCR using RNA from untreated control and doxazosin-treated cells was performed with the QuantumRNA ™ 18 S Internal Standards kit (Ambion, Austin, TX, USA) according to the manufacturer’s protocol. Samples of PCR products were subjected to electrophoretic analysis on agarose gels (1.8% (w/v)) and bands were visualized with SYBR Gold nucleic acid stain (Molecular Probes, Eugene, OR, USA). Densitometric analysis was performed using an EpiChemi3 Darkroom (UVP Bioimaging System, Upland, CA, USA). Each experiment was performed three times.
Western blot analysis
Cell lysates were prepared from doxazosin-treated and -untreated cultures of DU-145-neo and DU-145-maspin cells (15 μm) after treatment for 0, 6, 12 and 24 h. Cells were harvested and lysed with lysis buffer consisting of 50 mm Tris-HCl (pH 7.4), 1% Triton X-100, 150 mm NaCl, 5 mm MgCl2 and 1 mm PMSF. Cell lysate samples (protein content 20 μg) were subjected to electrophoretic analysis through 15% (w/v) sodium dodecyl sulfate (SDS)–polyacrylamide gel and transferred to Hybond C (Amersham Biosciences, Piscataway, NJ, USA) membrane. Membranes were incubated with caspase-3 (8G10) rabbit monoclonal antibody (Cell Signaling Technology, Beverly, MA, USA) overnight at 4°C and were subsequently exposed to the secondary antibody, goat anti-rabbit IgG (Jackson Immunoresearch, West Grove, PA, USA) for 1 h at room temperature. For VEGF and Smad4 detection, the VEGF (A-20) rabbit polyclonal antibody and the Smad4 (B-8) mouse monoclonal antibody were used, respectively (Santa Cruz, Santa Cruz, CA, USA). For VEGF receptor 2, bax and caspase-8 detection, the antibodies used were, respectively, the phospho-VEGF receptor 2 (7H11) antibody, human bax antibody and caspase-8 monoclonal antibody (1C12) obtained from Cell Signaling Technology (Beverly, MA, USA). PARP cleavage (as the apoptosis end point) was detected using the antibody from Pharmacia (Piscataway, NJ, USA). Protein expression was normalized to α-actin, as determined using the actin Ab-1 mouse ascites monoclonal antibody (Oncogene Research Products, San Diego, CA, USA). For signal detection, membranes were incubated with SuperSignal West Dura Extended Duration Substrate (Pierce Biotechnology, Rockford, IL, USA) and visualized using chemiluminescent imaging on an EpiChemi3 Darkroom (UVP Bioimaging System, Upland, CA, USA).
Acknowledgments
This study was supported by an NIH Grant CA10757-01 (awarded to NK). We wish to thank Dr Shuping Yin (Department of Pathology, Wayne State University School of Medicine) for her skillful technical assistance. We thank Lorie Howard (University of Kentucky) for her expert assistance in the preparation and submission of the manuscript and James Partin (University of Kentucky) for his expertise in the preparation of the figures.
Abbreviations
- BPH
benign prostatic hyperplasia
- ECM
extracellular matrix
- MTT
methylthiazolyldiphenyl-tetrazolium bromide
- PARP
poly(ADP-ribose) polymerase
- TGFβ1
transforming growth factor β1
- TIEG1
TGFβ1-inducible early gene
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- uPA
urokinase-type plasminogen activator
- VEGF
vascular endothelial growth factor
References
- Abraham S, Zhang W, Greenberg N, Zhang M. J Urol. 2003;169:1157–1161. doi: 10.1097/01.ju.0000040245.70349.37. [DOI] [PubMed] [Google Scholar]
- Bare RL, Torti FM. Cancer Treat Res. 1998;94:69–87. doi: 10.1007/978-1-4615-6189-7_5. [DOI] [PubMed] [Google Scholar]
- Benning CM, Kyprianou N. Cancer Res. 2002;62:597–602. [PubMed] [Google Scholar]
- Biliran H, Jr, Sheng S. Cancer Res. 2001;61:676–682. [PubMed] [Google Scholar]
- Blacque OE, Worrall DM. J Biol Chem. 2002;277:10783–10788. doi: 10.1074/jbc.M110992200. [DOI] [PubMed] [Google Scholar]
- Bruckheimer EM, Kyprianou N. Cell Tissue Res. 2000;301:153–162. doi: 10.1007/s004410000196. [DOI] [PubMed] [Google Scholar]
- Burchardt M, Burchardt T, Chen MW, Shabsigh A, de la Taille A, Buttyan R, Shabsigh R. Biol Reprod. 1999;60:398–404. doi: 10.1095/biolreprod60.2.398. [DOI] [PubMed] [Google Scholar]
- Cher ML, Biliran HR, Jr, Bhagat S, Meng Y, Che M, Lockett J, Abrams J, Fridman R, Zachareas M, Sheng S. Proc Natl Acad Sci USA. 2003;100:7847–7852. doi: 10.1073/pnas.1331360100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domann FE, Rice JC, Hendrix MJ, Futscher BW. Int J Cancer. 2000;85:805–810. doi: 10.1002/(sici)1097-0215(20000315)85:6<805::aid-ijc12>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Guo Y, Kyprianou N. Cell Growth Differ. 1998;9:185–193. [PubMed] [Google Scholar]
- Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- Jiang N, Meng Y, Zhang S, Mensah-Osman E, Sheng S. Oncogene. 2002;21:4089–4098. doi: 10.1038/sj.onc.1205507. [DOI] [PubMed] [Google Scholar]
- Keledjian K, Borkowski A, Kim G, Isaacs JT, Jacobs SC, Kyprianou N. Prostate. 2001;48:71–78. doi: 10.1002/pros.1083. [DOI] [PubMed] [Google Scholar]
- Keledjian K, Garrison JB, Kyprianou N. J Cell Biochem. 2005;94:374–388. doi: 10.1002/jcb.20240. [DOI] [PubMed] [Google Scholar]
- Keledjian K, Kyprianou N. J Urol. 2003;169:1150–1156. doi: 10.1097/01.ju.0000042453.12079.77. [DOI] [PubMed] [Google Scholar]
- Kyprianou N, Benning CM. Cancer Res. 2000;60:4550–4555. [PubMed] [Google Scholar]
- Liu J, Yin S, Reddy N, Spencer C, Sheng S. Cancer Res. 2004;64:1703–1711. doi: 10.1158/0008-5472.can-03-2568. [DOI] [PubMed] [Google Scholar]
- Oades GM, Eaton JD, Kirby RS. Curr Urol Rep. 2000;1:97–102. doi: 10.1007/s11934-000-0043-z. [DOI] [PubMed] [Google Scholar]
- Partin JV, Anglin IE, Kyprianou N. Br J Cancer. 2003;88:1615–1621. doi: 10.1038/sj.bjc.6600961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan D, Koczwara B, Javle M. Eur J Cancer. 1997;33:566–574. doi: 10.1016/s0959-8049(96)00510-2. [DOI] [PubMed] [Google Scholar]
- Schaefer JS, Zhang M. Curr Mol Med. 2003;3:653–658. doi: 10.2174/1566524033479519. [DOI] [PubMed] [Google Scholar]
- Sheng S, Carey J, Seftor EA, Dias L, Hendrix MJ, Sager R. Proc Natl Acad Sci USA. 1996;93:11669–11674. doi: 10.1073/pnas.93.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng S, Pemberton PA, Sager R. J Biol Chem. 1994;269:30988–30993. [PubMed] [Google Scholar]
- Shi HY, Zhang W, Liang R, Abraham S, Kittrell FS, Medina D, Zhang M. Cancer Res. 2001;61:6945–6951. [PubMed] [Google Scholar]
- Sternberg CN. Eur J Cancer. 2003;39:136–146. doi: 10.1016/s0959-8049(02)00665-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Sun L, Myeroff L, Wang X, Gentry LE, Yang J, Liang J, Zborowska E, Markowitz S, Willson JK, Brattain MG. J Biol Chem. 1995;270:22044–22049. doi: 10.1074/jbc.270.37.22044. [DOI] [PubMed] [Google Scholar]
- Yang G, Timme TL, Park SH, Wu X, Wyllie MG, Thompson TC. Prostate. 1997;33:157–163. doi: 10.1002/(sici)1097-0045(19971101)33:3<157::aid-pros2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Zhang M, Volpert O, Shi YH, Bouck N. Nat Med. 2000;6:196–199. doi: 10.1038/72303. [DOI] [PubMed] [Google Scholar]
- Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]