

Abstract
Recent advances toward understanding the molecular mechanisms regulating cancer initiation and progression provide new insights into the therapeutic value of targeting tumor vascularity by interfering with angiogenic signaling pathways. The functional contribution of key angiogenic factors toward increased vascularity characterizing metastatic tumors and their therapeutic exploitation is considered in three major urologic malignancies, renal, bladder, and prostate cancer. With the realization that the success of the therapeutic efficacy of the various anti-angiogenic approaches for the treatment of urologic tumors has yet to be proven clinically, the challenge remains to select critical angiogenesis pathways that can be targeted for an individual tumor. Here we discuss the major mechanisms that support formation of vasculature in renal, bladder, and prostate tumors and the current results of targeting of specific molecules/regulators for therapeutic intervention against metastastic disease.
Keywords: vascularity, tumor growth, apoptosis, VEGF, bladder cancer, renal cancer, prostate cancer
In 2007, there will be an estimated 346,440 new cases diagnosed with urologic cancer in the United States and 54,360 Americans will die from a urologic malignancy (SEER Cancer Statistics Review, http://cancernet.nci.nih.gov/statistics). This mortality rate is alarmingly high as it translates to one individual dying every 9 min in the US due to a urologic tumor and thus a significant health issue.
Angiogenesis is an essential process in normal physiological functions such as ovarian cycle in female reproductive system [Kaczmarek et al., 2005] and a contributing factor in disease states such as chronic inflammation, arthritis, cancer, and macular degeneration [Folkman, 1995]. During the development of the embryo, mesoderm differentiates into angioblasts; these endothelial cells, not yet organized into a lumen, form primitive vessels toward development of blood vessel network, via vasculogenesis. In the adult, new blood vessels form from pre-existing vasculature, via angiogenesis [Risau, 1997], while malignant conditions induce a hypercoagulable state in their hosts [Nash et al., 2001]. By early 1960s it was evident that tumors could elaborate diffusible substances that induce angiogenesis from the host vasculature [Algira et al., 1945; Greenblatt and Shubick, 1968]. The increased tumor vascularity was originally believed to be vasodilation of the host endothelium in response to metabolic waste products from within the tumor [Folkman, 1995]. A decade later Dr. Folkman’s pioneering work identified angiogenesis as a required phenomenon for tumor growth and metastasis, first defining the potential therapeutic value of agents targeting this process [Folkman, 1995; Folkman, 1971]. Tumor blood vessels exhibit characteristic markers which are not present in normal angiogenic tissues [Ruoslahti, 2002]. After enduring the circulation “journey,” metastatic cancer cells can escape out of the endothelial vasculature and in the target tissue via extravasation. How do the metastastic cells signal activating changes in the vascular permeability of blood vessels in target organs? Vascular endothelial growth factor (VEGF) initially identified as potent vascular permeability factor is the lead candidate. Activation of Src family kinases in endothelial cells exposed to VEGF induces disruptions in endothelial cell junctions, facilitating metastatic extravasation. Hypoxia within the tumor mass applies selective pressure promoting the outgrowth of malignant cells, with diminished apoptotic ability. The cellular response to low oxygen tension involves stabilization of a hypoxia-inducible factor-1 (HIF-1) transcriptional complex genes involved in cell survival and invasion.
In this review we discuss the current knowledge on angiogenesis as a contributor to cancer progression, and the clinical exploitation of this knowledge towards molecular targeting of tumor vascularity for the treatment of urologic malignancies.
REGULATION OF ANGIOGENESIS IN TUMOR PROGRESSION
Angiogenic stimuli produced due to metabolic demands of host tissues initiate the angiogenic response [Risau, 1997]. Upon binding to membrane receptors in vascular endothelial cells, a five-step process is triggered: initially the vascular endothelial basement membrane of the parent vessel breaks down, allowing a route for the development of a new capillary sprout, this is followed by migration of endothelial cells through the basement membrane toward the angiogenic stimulus; this leading front of migrating cells is driven by enhanced proliferation of endothelial cells, followed by formation of capillary tubes via organization of the endothelial cells, and a recruitment of periendothelial cells (pericytes) and vascular smooth muscle cells for new capillary stabilization [Cotran et al., 1999; Van Moorselaar and Voest, 2002].
In normal conditions angiogenesis is maintained by an intricate balance between endogenous stimulators of angiogenesis and endogenous inhibitors of angiogenesis (Table I). Additional mechanisms include inhibition of angiogenesis via sequestration of stimulators of angiogenesis in the extracellular matrix (ECM) and changes in the endothelial cell shape, reducing their susceptibility to stimulators [Folkman, 1995]. During tumorigenesis, the angiogenic switch is activated directly via induction of angiogenic growth factors, or indirectly, by recruiting host immune cells that release mediators of angiogenesis [Folkman, 1993]. Circulating endothelial precursor cells (CEP) from the bone marrow also contribute to tumor neovascalurization [Lyden et al., 2001], while tumor cells can recruit new blood vessels due to a network from adjacent endothelial cells [Dameron et al., 1994].
TABLE I.
Endogenous Regulator of Angiogenesis
| Classification | Angiogengesis stimulators | Angiogensis inhibitors |
|---|---|---|
| Growth factor | VEGF | |
| PDGF | ||
| aFGF and bFGF | ||
| TGF-α and β | TGF-β | |
| HGF | ||
| Angiopoetins-1 | Angiopoietin-2 | |
| GCSF | ||
| Necrosis factor | TNF-arufa | |
| Chemokine | Platelet factor 4 (PF4) | |
| Membrane protein | Integrin | |
| Hormone | Proliferin | 2-Methoxyestradiol |
| Transcription factor | HIF-1, HIF-2 | |
| Cytokine | IL-8 | IL-12 |
| IFN-α and β | ||
| Protease | MMPs | |
| Protease inhibitor | TIMP-1, TIMP-2 | |
| Maspin | ||
| PEX | ||
| Plasminogen | Angiostatin | |
| Plasminogen Kringle 5 | ||
| Collagen | Endostatin | |
| Fibronectin fragment | ||
| Others | Endothelins | Antithrombin III |
| PEDF | ||
| Troponin I | ||
| TSP 1 |
TSP-1; thrombospondin-1, PEDF; pigment epithelial-derived factor, TIMP; tissue inhibitor of metal-loproteinase INF; interferon, IL; interleukin, HIF; hypoxia-inducible factor, MMPs; matrix metal-loproteinases VEGF; vascular endothelial growth factor, TGF; transforming growth factor, PDGF; platelet-derived growth factor HGF; hepatocyte growth factor, FGF; fibroblast growth factors: acidic (aFGF) and basic (bFGF) GCSF; Granulocyte colony-stimulating factor.
The Major Players: Endogenous Angiogenesis Promoters
Vascular endothelial growth factor
VEGF is the most prominent regulator of physiological angiogenesis [Lonser et al., 2003]. Genetic knockout studies revealed that loss of a single VEGF allele results in embryonic lethality [Ferrara et al., 1996], pointing to a dose-dependent requirement of VEGF for normal vasculature during development [Van Moorselaar and Voest, 2002]. VEGF-A is the protagonist member of the VEGF family that includes VEGF-B, VEGF-C, VEGF-D, and placenta growth factor [Ferrara et al., 2003]. It is a secreted heparin binding protein of four isoforms produced by alternative exon splicing [Orlandi et al., 1996; Jackson et al., 1997] and can be induced by other signaling effectors such as TGF-β, TGF-α, and PDGF [Lara et al., 2004]. VEGF functions are mediated through two tyrosinekinase receptors, VEGF-R1 (Flt-1) [deVries et al., 1992] and VEGF-R2 (Flk-1 or KDR) [Millauer et al., 1993], in vascular endothelial cells [Hanahan, 1997]. VEGF initially interacts with VEGF-R2 to promote endothelial cell proliferation, migration and vascular permeability, and subsequently with VEGF-R1 to assist the organization of new capillary tubes. Loss of VEGF-R1 impairs the ability of angioblasts to be organized into mature capillaries in vivo [Fong et al., 1995] and VEGF-R2 is of responsible for recruiting cells in the developing vasculature [Barleon, 1996]; induction of matrix metalloproteinases (MMPs) [Hiratsuka et al., 2002] and secretion of additional growth factors from the developing endothelium (Fig. 1) [LeCouter et al., 2003].
Fig. 1.
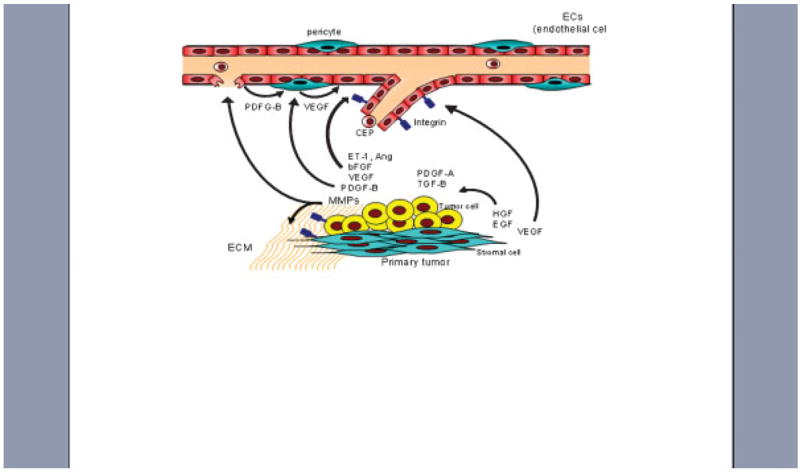
Mechanistic interaction between VEGF and FGF signaling in the tumor microenvironment. Tumor epithelial cells produce angiogenic factors such as VEGF, bFGF, ETs, and angiopoietins; BEGF is also secreted by stromal cells. Integrin (a(v)b3) is upregulated in growth-factor activated endothelial cells. MMPs mediate degradation of the ECM and cooperate to enhance angiogenesis and vascular remodeling. Tumor epithelial cells communicate with the microenvironment via growth factor signaling interactions: PDGF and TGF-β by tumor cells and EGF and HGF by stromal cells. PDGF-B produced by both endothelial and tumor epithelial cells, stimulates VEGF expression by pericytes to enhance endothelial cell survival. CEP cells from the bone marrow also contribute to new blood vessel formation.
HIF-α(Hypoxic-inducible factor-α)
Tissue hypoxia is associated with rapid tumor growth and VEGF upregulation through HIF-1, which increases transcription and the stability of VEGF mRNA [Shweiki et al., 1992; Semenza, 1996]. HIF-1 is a ubiquitous bHLH/PAS (basic helix-loop-helix/Per-Arnt-Sim homology) transcription factor that is composed of the two basic helix-loop-helix PAS proteins, HIF-1α and HIF-1β [Wang et al., 1995]. Three α subunits, such as HIF-1 α, HIF-2 α, and HIF-3 α, have been identified. Under normoxia, prolyl hydroxylation of the HIF-α subunit is enhanced, and mediates interaction with von Hippel-Lindau protein (pVHL), E3 ubiqui-tin ligase that then undergoes proteosomal degradation. At low oxygen levels, prolyl hydroxylation of HIF is inhibited, escaping from degradation through pVHL. HIF-α will translocate into the nucleus to form a heterodimeric complex with HIF-β and subsequently stimulate gene transcription [Schofield and Ratcliffe, 2004]. As illustrated in Figure 2, HIF induces transcriptional activation of genes regulating key processes in tumor progression, such as angiogenesis (Ang-2), glucose metabolism, (glucose transporter 1), adhesion (E-cadherin, Vimentin), migration (TGF-α, c-Met), proteolysis (Cathepsin D, uPAR, MMP2), and pH (CA IX; carbon anhydorase 9) [Pouyssegur et al., 2006]. Tumors that exhibit abundant HIF-1 stabilization have a greater likelihood of developing metastatic relapse and shorter survival with a subset of HIF target genes acting as mediators of metastastic progression [Semenza, 2000]. HIF-1 induces the chemokine receptor CXCR4 in renal cell carcinoma cells, which promotes organ-specific metastatic dissemination [Staller et al., 2003]. Moreover a mutation in von Hippel-Lindau (VHL) suppressor gene results in VHL hereditary cancer syndrome leading to sporadic clear cell renal cell cancers (CCRCC) [Kaelin, 2002].
Fig. 2.
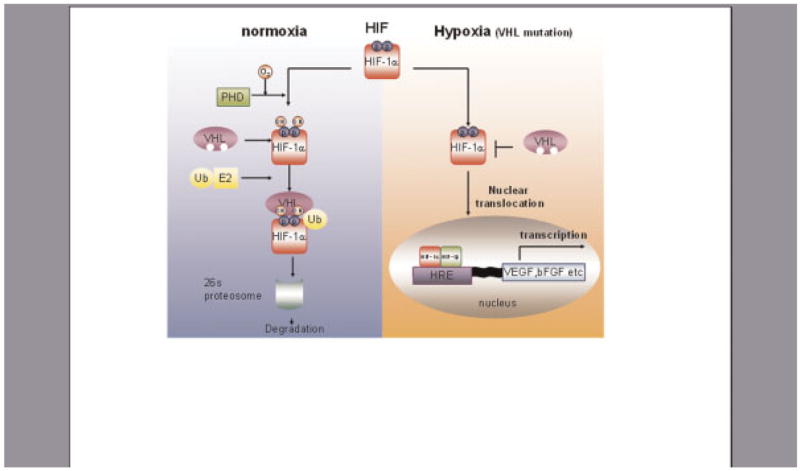
Hypoxia takes control of tumor angiogenic responses. HIF is a heterodimeric transcription factor consisting of α and β subunits. Under normoixic conditions, prolylhydroxylase domain (PHD) protein hydroxylates the two proline residues (Pro402 and Pro564) in the oxygen-dependent degradation domain (ODDD) of HIF-1 α. Hydroxylation of these proline residues allows HIF-1α to interact with von Hippel Lindau tumor suppressor protein (VHL), leading to rapid ubiquitilation and degradation of HIF-1α. HIF1-α escape from the VHL mediated degradation allows binding with HIF-1 β at the nuclear response element, thus activating transcription of target genes including VEGF and bFGF. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Angiopoietins
Angiopoietins are ligands for TIE-2 receptors located on endothelial cells. Angiopoietin-1 (Ang1) interacts with the TIE-2 receptor to recruit periendothelial cells, such as pericytes and vascular smooth muscle cells, for stabilization of the new vasculature [Luo et al., 1997]. Ang2 interacts with TIE-2 to increase vascular permeability, enhancing susceptibility to angiogenesis stimulators in the presence of VEGF, while in its absence sensitizing the endothelium to endogenous inhibitors [Davis et al., 1996]. The pattern of VEGF-dependent signaling of TIE-2 receptors dictates angiogenesis [Puri et al., 1995; Sato et al., 1995]; as suggested by embryonic lethality of TIE-2 knockout mice due to lack of brain capillary sprouting [Puri et al., 1995].
Fibroblast growth factors
Basic fibroblast growth factor (bFGF) was the first molecule to be identified as a proangiogenic agent [Shing et al., 1984]. bFGF binds tightly to heparin sulfate proteoglycans (HSPGs), on the cell surface and ECM [Vlodavsky et al., 1991], and this complex subsequently stabilizes bFGF from heat and proteolysis [Flaumenhaft et al., 1990]. FGFs interact with FGF-receptors (FGFR-1 or FGFR-2), leading to endothelial cell proliferation [Cross and Claesson-Welsh, 2001], degradation of the, stimulation of chemokines toward endothelial cell migration, regulation of integrin and cadherin expression, and modulation of cell–cell interactions [Presta et al., 2005]. Engaged in a dynamic cross-talk, FGF, can directly upregulate VEGF expression in endothelial cells [Tille et al., 2001]. A synergistic action between VEGF and FGF generates a significant angiogenic response in target cells [Xue and Greisler, 2002], and consequently VEGF targeting antibodies and VEGFR-2 antagonists inhibit angiogenesis mediated by both VEGF and FGF [Cross and Claesson-Welsh, 2001] (Fig. 1).
Matrix metalloproteinases
MMPs is a family consisting of 16 members of zinc-dependent proteases [Kleiner and Stetler-Stevenson, 1999] that mediate ECM degradation. The four major subgroups of this protease family are gelatinases, collagenases, stromelysins, and membrane associated proteases [Jiang and Muschel, 2002]. Most MMPs released in their inactive state are cleaved to activation by other MMPs or other serine proteases such as plasmin and urokinase-type plasminogen activator [Birkedal-Hansen et al., 1993; Goetzl et al., 1996]. Tissue inhibitors of matrix metalloproteinases (TIMPs) provide an additional level of regulation for MMP activation [Birkedal-Hansen et al., 1993]. ECM undergoes constant remodeling in normal homeostasis and MMPs function to remove proteins from the basement membrane during this remodeling [Kleiner and Stetler-Stevenson, 1993]. Their cooperation targets degradation of basement membrane of the vascular endothelium and ECM, thus creating a passageway in these physical barriers toward new capillary formation [Kleiner and Stetler-Stevenson, 1999].
Integrins
Integrins are transmembrane proteins that serve a role as primary mediators of cell-ECM interactions that are functionally involved in determining tumor angiogenic response during cancer progression to meta-static disease. Integrins (heterodimers containing two distinct chains, α and β subunits), recognize the major adhesive ECM components, fibronectin and laminin, toward regulation of cell proliferation, cell survival, anoikis, and migration [Giancotti and Ruoslahti, 1999; Goel and Languino, 2004]. Angiogenic growth factors such as VEGF and pFGF can exert a profound positive effect on the activity and expression of several integrins, such as αvβ3, αvβ5, αvβ1, α3β1, α3β1, α6β1, α6β4 [Klein et al., 1993]. Additional angigogenic factors, such as class 3 semaphorins (SEMA3 proteins), control vascular remodeling via targeting integrin [Serini et al., 2003]. Increased expression of integrin αvβ3 is detected in growth factor-activated endothelial cells in tumor blood vessels and granulation tissue [Enenstein et al., 1992; Brooks et al., 1994], while very low expression was detected in resting blood vessels [Arap et al., 1998]. Expression of the αvβ3 in tumor endothelium correlated with the aggressive phenotype in neuroblastoma [Erdreich-Epstein et al., 2000]. Moreover, blocking integrin αvβ3 with monoclonal antibody (Mab) mediated endothelial cell apoptosis and inhibited blood vessel formation [Enenstein et al., 1992], implicating its functional significance in angiogenesis. Consequently, αvβ3 integrin represents an attractive tumor vasculature target [Hood et al., 2002; Ruoslahti, 2002]. Nanoparticles containing agents linked with an anti-αvβ3 antibody [Sipkins et al., 1998] or bacteriophage with αvβ3 binding RGD peptide [Arap et al., 1998] effectively targeted tumor vasculature. Integrins also contribute to signal transduction from the extracelluar environment to the intracellular network mediated by integrin-activated signaling molecules, such as focal adhesion keinase (FAK), phosphatidylinositol 3-kinase (PI 3-kinase), and members of the extracellular signal-regulated kinase 1 and 2/mitogen activated protein (ERK1 and 2/MAP) kinase family to regulate cell proliferation, migration, and apoptosis of tumor and endothelial cells [Fornaro et al., 2001; Nikolopoulos et al., 2004].
Endothelins
Endothelins, a class of proteins involved in vasoconstriction, have also been linked to angiogenesis. Epithelial cells produce three types of endothelins, ET-1, 2, and 3 [Kopetz et al., 2002] that interact with two receptors, ET-A and ET-B. ET-3 stimulates ET-B receptor to induce endothelial cell growth and blood vessel formation [Lara et al., 2004]. A dynamic cross-talk between ET-1 and ET-A receptor promotes VEGF release [Goligorsky et al., 1999] leading to a strong angiogenic response [Salani et al., 2000].
TARGETING TUMOR VASCULARITY: THERAPEUTIC SIGNIFICANCE
Several angiogenesis-targeting approaches are currently evaluated in clinical trials for their therapeutic efficacy and long-term clinical benefits, while others are mechanistically exploited toward the development of novel therapeutic modalities for advanced urologic malignancies. The molecular platforms for such approaches are considered in Figure 3.
Fig. 3.

Targeting tumor vascularity: VEGF inhibitors take central stage. VEGF binding to its receptor mediates dimerization of VEGFR and induces autophosphorylation of RTKs, which initiate activation of several downstream pathways, regulating endothelial cell proliferation (via PKC-RAF-MEK-ERK), survival (via PI3K-AKT/PKB), migration, (via p38-MAPK) and vasculature permeability (via PI3K-AKT/PKB-eNOS). Bevasizumab, a neutralizaing antibody against VEGF directly targets VEGF; Thalidomide inhibits VEGF transcription; Suramin is a non-specific inhibitor of multiple growth factor signalig pathways; Chimeric soluble receptor (VEGF-Trap) interferes with VEGF binding to its receptor; AE-941 and DC101 are anti-VEGFR-2 antibody abrogating VEGF binding to its receptor. SU011248, Bay 43-9006, PTK787 inhibit phosphorylation of VEGF receptor. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Kidney Cancer
The clinical problem
Renal cell carcinoma accounts for more than 30,000 new cases of cancer and 12,000 deaths in the USA annually. The incidence of RCC has increased by >30% in the last decade [Motzer et al., 1996] and the annual mortality-to-incidence ratio for RCC is significantly higher than for other urological tumors. In spite of recent improvement in imaging for early diagnosis, 25–30% of patients have metastasis at presentation; the 5 year survival rate for patients with metastasis is less than 10% and the overall 5-year survival rate is 60% [Kirkali et al., 2001; Schrader et al., 2006]. The current therapeutic responses in metastatic RCC patients are poor. Indeed combination regimes of recombinant cytokines with interferon or interleukin-2, currently presumed to be the most effective therapy, exhibit modest response rate of 10–20% [Schrader et al., 2006]. The options for those patients failing cytokine-based therapy are limited, hence the emerging clinical challenge. VHL syndrome is an autosomal dominant neoplasia syndrome that results from a germline mutation in the VHLgene [Crossey et al., 1994]. This gene mutation is found in 34–57% of clear cell RCC tumors [Rini, 2005; Rini and Small, 2005]; moreover, VHL gene inactivation through methylation of VHL gene is reported in 5–19% of RCC tumors [Rini and Small, 2005]. VHL inactivation in the low-grade and smaller (pT1) tumors, suggests that VHL inactivation may be involved in tumor initiation [Schrader et al., 2006]. Since VHL specifically targets the hydroxylated HIF-1 α subunit and mediates proteasomal degradation, mutations or inactivation of VHL genes that inhibit HIF-1α degradation and enhance transcription of hypoxia-related genes including VEGF [Rini, 2005].
More than 90% of hypervascular renal cell tumors express elevated VEGF mRNA compared to normal kidney [Hemmerlein et al., 2001]. Strong correlations have also been documented between VEGF up-regulation in RCC tumors with nuclear grade and TNM stage [Paradis et al., 2000; Lee et al., 2001] as well as a poor prognostic outcome [Paradis et al., 2000]. The majority of RCC tumors harboring a VHL mutation express HIF that significantly correlates with elevated VEGF [Na et al., 2003]. This evidence easily supports the concept that VHL inactivation in RCC is causally involved in promoting tumor angiogenesis via VEGF upregulation and consequently directly blocking VEGF, becomes an attractive therapeutic strategy for managing advanced RCC.
Specific single inhibitors such as TNP-470 (anti-VEGF) [Stadler et al., 1999] and SU5416 (Tyrorsine kinase inhibitor) [Kuenen et al., 2003] have failed to provide a therapeutic benefit; however, targeting multiple signaling pathways with combination srtategies bevasizumab+ erlotinib(PR:40%) [Hainsworth et al., 2004] or with the new tyrosine kinase inhibitor SU011248(PR:40%) [Mendel et al., 2003] have successfully delivered significant therapeutic responses. The leading anti-angiogenic molecules that had been investigated in clinical trials for RCC treatment are discussed.
Targeting Angiogenesis in RCC: Therapeutic Outcomes and Clinical Promise
Thalidomide therapy
Thalidomide is an anti-inflammatory and immunosuppressive agent that blocks angiogenesis through inhibiting growth factor, such as bFGF and VEGF, down regulate cytokines, such as tumor necrosis factor-α (TNF-α), modifies cell adhesion molecule expression, and promotes natural killer and T-cell activity [Rini and Small, 2005]. Clinical trials assessing the therapeutic efficacy of thalidomide as a single-agent therapy, revealed an overall response rate of 5.2% (0–16.7%) [Rini and Small, 2005]. Phase II studies with combination regimes of interferon-α or interferon-α plus capecitabine with thalidomide, yielded a distinct objective response of 20% in both trials [Hernberg et al., 2003; Amato and Rawat, 2006]. Furthermore, combination therapy of interleukin-2 plus thalidomide (in a phase II study) resulted in 41% response, with further modification enhancement by macrophage colony-simulating hormone [Amato et al., 2006]. Combination of bevasizumab with thalidomide failed to yield any therapeutic benefit over monotherapy [Elaraj et al., 2004].
Suramin
Suramin is a polysulphonate naphthylurea which has a potential to inhibit binding of a number of growth factors to their receptors, which act not only for tumor epithelial cells but also endothelial cells [Stein, 1993]. A phase II trial of suramin was conducted in 26 patients with advanced renal cell carcinoma, but the therapeutic responses were minimal (minor response observed for five patients for >3 months), while the toxic side effects were serious including an immune-mediated thrombocytopenia and Staphylococcus sepsis without neutropenia [Motzer et al., 1992]. Another suramin-based phase II clinical trial in 22 patients with advanced renal cell carcinoma revealed no objective response [Schroder et al., 2001], challenging its therapeutic value in RCC.
Bevacizumab
Bevacizumab is a recombinant human Mab which binds and neutilizes all the active isoform of the VEGF. In a preclinical model, Bevasizumab inhibits VEGF-induced proliferation of endotherial cells, leading to inhibition of tumor growth in a number of primary xenograft and metastatic models [Presta et al., 1997; Mordenti et al., 1999; Hu et al., 2002]. A Phase II clinical trial to evaluate the therapeutic efficacy of bevasizumab in RCC, resulted in a partial response rate (10% in high dose group) and prolonged time to disease progression [Yang et al., 2003]. An equally partial response was achieved by the combination of bevacizumab with the EGF-R antagonist (erlotinib), in 57 patients with metastatic RCC [Hainsworth et al., 2004].
SU011248
SU011248 is a multi-targeted receptor tyrosine kinase inhibitor which has reported to potently inhibit PDGF receptors α and β, VEGFR-1 and -2, KIT, and FLT3 (fms-related tyrosine kinase/Flk2/Stk-2) [Motzer et al., 2004, 2006]. A recently conducted A phase II trial using SU011248 in advanced 63 RCC patients (failing to respond initial cytokine treatment), demonstrated a partial response achieved in 25 patients (40%), with major toxicity including lymphopenia, elevated lipase, and fatigue/asthenia [Mendel et al., 2003].
PTK 787
PTK 787 is also a tyrosin kinase inhibitor of VEGFR-1, VEGFR-2, and PDGFR-b. In vivo experiment using murine renal carcinoma model demonstrated that oral administration of PTK787 (50 mg/kg) to mice significantly inhibit the growth of the primary tumor and metastasis compare to control group [Wood et al., 2000]. Recently George et al. reported a phase I/II clinical trial in patients with metastatic RCC. The results indicate a 5% of partial response rate, and 15% of minor response (25–50% shrinkage) rate with significant differences in vascular permeability and reduction in blood flow [George et al., 2003].
Bay 43-9006
Bay 43-9006 is an orally bio-available bi-aryl urea Raf kinase inhibitor that directly inhibits VEGFR-2, VEGFR-3, and PDGFR-b in a Ras-dependent human tumor xenograft model [Lyons et al., 2001; Wilhelm et al., 2003]. In the clinical setting, a phase II randomized study of Bay 43-9006, in refractory solid tumors including 202 metastatic RCC patients showed a positive therapeutic response [Ratain et al., 2006]. With a large number of patients (42%) achieving an initial response (tumor shrinkage), and a significantly longer median progression-free survival (PFS) in response to sorafenib treatment compared to the placebo [Ratain et al., 2006]. An interim phase III analysis indicated that Sorafenib monotherapy results in a significant increase in progression-free survival in patients with advanced RCC, delivering promising therapeutic value.
SU5416
SU5416 is a small organic molecule that blocks VEGF-mediated signaling by interfering Flk-1, a transmembrane tyrosine kinase. In preclinical studies, SU5416 inhibited endothelial cell proliferation and neovasculization [Fong et al., 1999]. In a phase II trial of SU5416 conducted in 29 patients with advanced or metastatic RCC, no objective therapeutic response was observed [Kuenen et al., 2003]. Furthermore, the combination approach of SU5416 with interferon-α, yielded no objective response, although half of the patient population demonstrated stable disease [Lara et al., 2003].
VEGF-trap
VEGF trap is a decoy receptor created from the combination of VEGFR-1 immunoglobulin (Ig) domain2 and VEGFR-2 Ig domain 3 attached to the human IgG1. This artificial protein binds VEGF with 100-fold increase affinity compare to bevasizumab [Rini, 2005]. In vitro study demonstrated that VEGF-trap efficiently inhibited phosphorylation of VEGFR-2 and VEGF-induced endothelial cell proliferation. In vivo mouse model, VEGF-trap significantly inhibits the growth and vascularity of various tumors [Holash et al., 2002]. A phase-I clinical trial using VEGF-trap involving 33 patients with refractory solid tumors, including nine metastatic RCC patients (escalating dose cohorts, 0.025–0.8 mg/kg), revealed no positive response [Dupont et al., 2004].
AE-941
The identification of anti-angiogenic and anti-tumor growth properties associated with the shark cartilage led to the synthesis of several novel compounds. AE-941 (Neovastat) is a pure shark cartilage compound that selectively inhibits −2, −9, and −12 and competes for the binding of VEGF to its receptor (VEGFR-2), resulting in endothelial cell apoptosis [Batist et al., 2002]. A phase-I trial of AE-941 was performed in 144 patients with refractory solid tumors, including patients with metastatic RCC. As shown on Table II, treatment with AE-941 (either an escalated or high single dose), resulted in an objective response in a mere 14% of patients [Batist et al., 2002].
TABLE II.
Summary of Clinical Trials
| Drug | Cancer | Objective Response | References |
|---|---|---|---|
| Anti VEGF | |||
| Suramin | RCC | 0–4% | Motzer et al. [1992, 1996, 2004, 2006] |
| Schroder et al. [2001] | |||
| HRPC | 7–15% | Small et al. [1999, 2000, 2002] | |
| Plus hydrocortisone | HRPC | 33%(PSA) | Small et al. [1999, 2000, 2002] |
| TNP-470 | RCC | 0% | Stadler et al. [1999, 2004] |
| HRPC | 0% | Logothetis et al. [2001] | |
| Thalidomide | RCC | 0–16.7% | Rini et al. [2005] |
| HRPC | 37.5%(PSA) | Drake et al. [2003] | |
| Plus Interferon-α | RCC | 20% | Hernberg et al. [2003] |
| Interferon-α+capecitabine | RCC | 20% | Amato [2006] |
| IL-2 | RCC | CR 7%, PR 33% | Amato et al. [2006] |
| Paclitaxel/doxorubicin | HRPC | 82%(PSA) | Amato et al. [2006] |
| Docetaxel | HRPC | 50%(PSA) | Leonard et al. [2003] |
| GM-CSF | HRPC | 23%(PSA) | Dreicer et al. [2005] |
| Bevacizumab | RCC | 0% | Elaraj et al. [2004] |
| 2-methoxyestradiol | HRPC | 0% | Sweeney et al. [2005] |
| Neutralizing Ab | |||
| Bevacizumab | RCC | 10% | Yang et al. [2003] |
| Plus Docetaxel+estramustine | HRPC | 77%(PSA) | Picus et al. [2003] |
| Erlotinib | RCC | 25% | Hainsworth et al. [2004] |
| Cisplatin+gemcitabin | BT | on going | Sternberg et al. [1988] |
| Paclitaxel | BT | on going | Sternberg et al. [1988] |
| Docetaxel+prednisone+thalidomide | HRPC | on going | Schofield and Ratdiffe [2004] |
| Docetaxel+prednisone | HRPC | on going | Schofield and Ratcliffe [2004] |
| TK inhibitor | |||
| SU5416 | RCC | 0% | Kauenen et al. [2003] |
| HRPC | 0% | Stadler et al. [1999, 2004] | |
| PTK 787 | RCC | 5.0% | George et al. [2003] |
| SU011248 | RCC | 40% | Mendel et al. [2003] |
| Novel approach | |||
| AE-941 | RCC | 14%(high dose) | Batist et al. [2002] |
| VEGF Trap | RCC | 0% | Dupont et al. [2004] |
RCC, renal cell carcinoma; HRPC, hormone-refractory prostate cancer; BT, bladder tumor (PSA), More than 50% reduction of serum prostate-specific antigen (PSA) is achieved after treatment.
Prostate Cancer: The Leading Tumor in Males
Prostate cancer is the most common malignancy and is the second leading cause of cancer death in males [Weir et al., 2003]. The initial standard therapy for locally advanced or meta-static disease is androgen deprivation therapy via surgical or medical castration. The initial therapeutic response to the antiandrogen-therapy, is only brief (8 month-3 years), and prostate cancer patients become refractory to additional treatment, as tumors eventually relapse to an androgen independent state [Daneshgari and Crawford, 1993] and development of hormone refractory prostate cancer (HRPC). Docetaxel was shown to prolong survival in patients with HRPC [Petrylak et al., 2004; Pronzato and Rondini, 2005], but chemotherapeutic strategies for effective disease control are still required. The significance of angiogenesis in human prostate cancer progression has been firmly established [Borre et al., 1998; Bono et al., 2002]. Several independent studies documented a significant correlation between microvessel density (MVD) with Gleason score, pathological stage, and patient survival [Borre et al., 1998; Bono et al., 2002]. Furthermore VEGF levels are significantly increased in prostate tumors (relative to normal tissue), an upregulation that directly correlates with tumor stage, differentiation and disease specific survival. A strong expression of VEGF is detected in neuroendocrine-differentiated (NE) tumor cells [Borre et al., 2000]. Serum VEGF levels are significantly higher in metastatic prostate cancer patients compared to localized disease [Duque et al., 1999]. HIF, a key mediator of VEGF expression, is highly expressed in prostate cancer, compared to normal and benign prostate tissue [Du et al., 2003]. HIF-1α mutations (homozygous P582S) in the oxygen-dependent degradation domain of HIF-1α, which mediates continuous activation of HIF, are significantly associated with increased risk for prostate cancer [Orr-Urtreger et al., 2007], indicating the importance of HIF-1α mediated VEGF regulation in prostate tumorigenesis (Fig. 2).
Targeting prostate tumor vascularity: Is there a therapeutic value?
Several clinical trials using potent anti-angiogenic drugs, TNP-470 [Logothetis et al., 2001], 2-methox-yestradiol [Sweeney et al., 2005] and SU5416 [Stadler et al., 2004] failed to deliver any positive therapeutic efficacy in prostate cancer patients with hormone refractory disease. Disappointing as these outcomes might be, a series of new angiogenesis targeted approaches have emerged from the laboratory to the clinic, promising considerable clinical benefit.
Selective vasculature targeting
Recent work by Ruoslahti et al. identified the peptides that specifically recognize the vasculature in the prostate through screening phage-displayed peptide libraries. Intravenous injection of chimeric peptide consisting of the SMSIARL homing peptide, linked to a proapoptotic peptide, induced prostate tumor destruction and delayed cancer development in TRAMP mice [Arap et al., 2002].
Doxazosin
Experimental evidence indicates that the quinazoline-based compounds, such as doxazosin and terazosin (clinically used for treatment of hypertension and BPH), may also function in inducing prostate smooth muscle cell death via apoptotic pathways involving activation of TGF-β1-mediated apoptotic signaling and inhibition of Akt [Garrison and Kyprianou, 2006]. Growing evidence also implicates anoikis (ECM detachment-induced apoptosis) in prostate tumor epithelial and endothelial cells in response to quinazolines [Rennebeck et al., 2005], potentially via targeting VEGF-mediated angiogenic response [Garrison and Kyprianou, 2006].
Suramin
The therapeutic efficacy of suramin was analyzed in a large population of patients (390) with advanced prostate cancer with a randomized dosing. The objective response rate was 9–15%, while the PSA response rates were 24–34% [Small et al., 2002]. In a randomized phase-III trial of suramin in combination with hydrocortisone conducted in 460 hormone-refractory prostate cancer patients, patients receiving suramin had higher PSA response rate, compared to the hydro-cortisone arm [Small et al., 2000] (Table II); but still, serious limitations are associated with the clinical efficacy of this agent.
Thalidomide
Preclinical experiments revealed that treatment with N-substituted thalidomide, a potent angiogenesis inhibitor, analog CPS11, (targeting platelet-derived growth factor alpha) led to 90% inhibition of tumor growth and 64% reduction in tumor vascularity of PC-3 human prostate xenografts [Ng et al., 2004]. An open trial of the efficacy and safety of thalidomide in 20 patients with androgen-independent prostate cancer resulted in significantly reduction in PSA levels in 35% of patients [Drake et al., 2003]. Prostate cancer patients with hormone refractory-disease exhibited a relatively good response to a combination treatment of docetaxel and thalidomide, by achieving a significantly improved median overall survival (28.9 months) compared to docetaxelalone (14.7 months) [Leonard et al., 2003]. In a triple-combination strategy of paclitaxel and doxorubicin with thalidomide, 9 out of 12 patients showed a 50% decrease in PSA [Amato and Sarao, 2006]. Combinational approaches using granular-macrophage colony-stimulating factor (GM-CSF) have been reported with some therapeutic promise. GM-CSF regulates the dendritic cell and tumor-specific cytokine T-cell mediated response [Small et al., 1999], and clinically GM-CSF in combination with thalidomide results in a significant PSA response (23%) [Dreicer et al., 2005].
Bevasizumab
A phase-II Cancer and Leukemia Group B (CALGB) 90006 trial of bevasizumab in combination of docetaxel, and estramustine with a premedication with decadron in chemotherapynaïve HRPC delivered promise (with the majority of patients achieving >50% PSA reduction) [Picus et al., 2003] and led to two clinical trials. The ongoing clinical trials are an NCI-phase-II study of a four-drug combination strategy of docetaxel, prednisone, thalidomide, and bevacizumab in men with chemotherapy-naïve progressive hormone-refractory prostate cancer (ClinicalTrials.gov, http://clinicaltrials.gov/ct/show); and a CALGB phase III, double-blind, placebo-controlled trial of docetaxel plus prednisone with or without bevacizumab (ClinicalTrials.gov, http://clinicaltrials.gov/ct/show).
SU5416
The initial in vivo anti-tumor effect of SU5416, a small molecule VEGFR2 inhibitor, was determined in the transgenic adenocarcinoma of the mouse prostate (TRAMP) mouse model. Administration of SU5416 to mice (16–22 weeks of age; highly expressing VEGFR-2), resulted in a dramatic decrease in tumor-associated mean vessel density and increased apoptosis [Huss et al., 2003]. Further preclinical studies using PC-3 human prostate cancer xenografts, demonstrated that a combination regime of the VEGFR2 inhibitor, SU5416 with another potent anti-angiogenesis agent, endostatin, led to a significant delay in the onset of tumor progression [Abdollahi et al., 2003].
Bladder Cancer: The Clinical Problem
Bladder cancer is the second most common malignancy in the genitourinary tract and fifth most common solid cancer in the United States and the incidence continuously increasing in recent years. Fifty-four thoudand patients are diagnosed and 12,000 patients die of bladder cancer annually [Greenlee et al., 2000]. The majority of bladder transitional cell carcinoma (TCC) of the bladder cases is of the superficial type (70%), while the rest are invasive and highly metastatic tumors [Beecken et al., 2005]. The standard treatment for operable invasive TCC is radical cystectomy and bladder tumors are initially sensitive to conventional chemotherapy approaches; the median survival in response to methotrexate, vinblastine, adriamycin, and cisplatin regimen however, is only 13–15 months due to emergence of chemoresistance [Sternberg et al., 1988]. Distinct angiogenic pathways differentiate superficial bladder tumors from invasive tumors. Superficial bladder tumor consists of a papillary morphology with an integrated branching vascular tree, expressing VEGF. In contrast, invasive solid bladder tumors contain a disorganized vascular network with necrotic regions, primarily expressing bFGF [O’Brien et al., 1997]. Elevated VEGF levels correlate with early recurrence and progression of superficial bladder tumors [O’Brien et al., 1995; Crew et al., 1996, 1997], as well as resistance to chemotherapy in patients with advanced disease [Slaton et al., 2004]. Pre-treatment of TCC cells with anti-sense VEGF increased cell sensitivity to conventional chemotherapeutic agents such as Mitomycin C, providing a promising possibility for adjuvant therapy [Krause et al., 2005]. Since bFGF also contributes to bladder cancer progression via its ECM involvement, this player becomes a prime candidate for therapeutic targeting [O’Brien et al., 1997; Guo et al., 2003].
Therapeutic efficacy of anti-angiogenic approaches in bladder cancer
TNP-470
TNP-470 is a synthetic analog of fumagillin which blocks the growth of new blood vessels by inhibiting methionine amino peptidase, an enzyme critically important for endothelial cell proliferation [Ingber et al., 1990]. The therapeutic efficacy of TNP-470 in invasive bladder cancer has been investigated in mouse models using KK-47 and MGH-U1 TCC cancer cell xenografts. Treatment with TNP-470 significantly suppressed tumor growth and vascularity, with limited toxicity [Beecken et al., 2000].
Suramin
Suramin, a potent antagonist of VEGF, is a polysulphonate naphthylurea, exerts distinct anti-tumor effects in mouse models of tumorigenesis. Suramin administration in rats bearing N-methyl-N-nitrosurea (MNU)-induced bladder tumors (twice a week for 18 weeks) resulted in 0–10% of treated group developing papillary bladder tumors [Bikfalvi et al., 1991]. A dose-escalation phase-I study with suramin in a small number of patients with a history of recurrent superficial bladder cancer (n = 12), resulted in a considerable suppression of urinary VEGF [Graham et al., 1995], but failed to deliver a therapeutic effect.
VEGFR monoclonal antibody DC101
Preclinical-testing of the synergetic effect of VEGF blockade with other chemotherapeutic agents in bladder cancer promises enhanced efficacy. Combination of anti-VEGFR Mab therapy with Paclitaxel chemotherapy in TCC mouse model, resulted in a significant tumor regression and inhibition of lymph node metastasis, driven by endothelial cell apoptosis [Ord et al., 2005].
Bevacizumab
Bevacizumab is another Mab designed to target all VEGF isoforms, with an established antiproliferative profile in various cancers [Inoue et al., 2000]. Several ongoing phase-II clinical trials, including the neoajuvant Bevacizumab with cisplatin and gemcitabine followed by radical cystectomy, adjuvant bevacizumab, and paclitaxel in patients with muscle-invasive, resectable TCC, and Bevacizumab, in combination with cisplatin and gemcitabine in patients with metastatic TCC are ongoing, in anticipation of successful outcomes [de Gramont and Van Cutsem, 2005].
CONCLUSION
Recent advances toward a better understanding of the molecular mechanisms underlying urologic malignancies enables us to target angiogenic signaling pathways regulating tumor vascularity. Tumor-selectivity of angiogenic-targeting therapeutic approaches is expected to produce lower toxicity and higher efficacy than standard chemotherapeutic approaches. The functional contribution of lead angiogenic factors toward increased vascularity characterizing advanced metastatic tumors, must be considered in the context of the supporting vasculature and reactive stroma within the tumor microenvironment (Fig. 1). The fact that agents specifically inhibiting VEGF signaling have a limited efficacy in terms of tumor suppression translating into a meaningful therapeutic response, points to the realization that blocking VEGF signaling alone might not be sufficient to induce complete inhibition of tumor growth. Angiogenic factors controlling the dynamics of microenvironment with functional redundancy, are accountable for the resulting drug-resistance. Inhibition of VEGFR results in upregulation of PDGF and FGF-2 [Carmeliet, 2005; Casanovas et al., 2005]. Furthermore, one cannot underestimate targeting the supporting stroma-derived vasculature, since it may determine the meta-static potential controlled by PDGF [Blouw et al., 2003; Willett et al., 2005]. Combination modalities may provide clinical benefit by enhancing endothelial cell sensitivity to anti-angiogenic/cytotoxic agents, in addition to delaying acquisition of drug resistance [Browder et al., 2000; Dong et al., 2004].
Overexexpression of HIF correlates with the recurrence and progression of superficial urothelial bladder cancer [Klement et al., 2000], while in prostate tumors HIF expression correlated with expression of VEGF and androgen receptor levels [Theodoropoulos et al., 2005]. The recent screening of the small molecule inhibitor for HIF-1 activity [Boddy et al., 2005], may provide an effective lead in the development of novel approaches for optimizing existing angiogenesis-based therapeutic strategies in the treatment of urologic malignancies. Inorganic nanostructure “nanoparticle” technology has recently generated wide interest in the field of molecular medicine [Tan et al., 2005]. Nanoparticle has emerged as a novel intra-vascular probe for both diagnostic imaging (e.g., MRI; magnetic resonance imaging) and therapeutic intervention (drug delivery). Ruoslahti et al. recently developed a nanoparticle-tumor targeting strategy by securing delivery and stimulating its intravascular accumulation via “self-amplification,” involving coupling clot-binding peptide with an MRI enhancer [Maynard and Pui, 2007]. Subsequent efforts established a novel protease-triggered self-assembly nanoparticle, “a neutravidin-and-biotin-functionalized superparamagnetic iron oxide nanoparticles,” which amplify the assembly of nanoparticles, upon triggered by MMP-2 within the tumor site [Simberg et al., 2007]. This work provided new routes for exploiting tumor-delivered molecular therapeutics for the treatment of highly-vascular tumors without affecting normal vasculatity.
In summary, as the success of the therapeutic efficacy of the various anti-angiogenic approaches for the treatment of urologic tumors has yet to be proven in terms of long-term clinical benefit, the challenge remains to select targeting of key angiogenesis signaling pathways that can be tailor-made for and targeted to an individual tumor. The angiogenic profile of urologic tumors may vary depending on the age of individual patients and stage/grade of tumor [Harris et al., 2006]. Proteomic profiling of angiogenesis effectors dictating tumor vasculature in individual patients, will enable treatment optimization in order to bring about long-term control of urologic cancers.
Acknowledgments
These studies were supported by an NIH RO1 CA10757-03 grant (awarded to NK). A.J. Ryan was a recipient of a Markey Cancer Center Undergraduate Research Scholarship at the University of Kentucky. The authors acknowledge the expert assistance of Lorie Howard in the submission process.
Abbreviations used
- VEGF
vascular endothelial growth factor
- bFGF
basic fibroblast growth factor
- Ang
angiopoietin
- PDGF
platelet-derived growth factor
- MMPs
matrix metalloproteinases
- EGF
epidermal growth factors
- HGF
hepatocyte growth factor
- TGF-β
transforming growth factor
- ET-1
Endothelin-1
- CEP
circulating endothelial precursor cells
- E2
ubiquitine conjugated enzyme
- HIF-1 α
hypoxia-inducible factor alpha
- HIF-1β
hypoxia inducible factor beta
- HRE
hypoxia response elements
- O2
oxygen
- OH
hydroxylions
- Ub
ubiquitin
- VHL
von Hippel-Lindau protein
- RAF
Raf kinase
- MEK
mitogen and extracellular kinase
- p38MAPK
p38 mitogen-activated protein kinase
- PKC
protein kinase C
- ERK
extracellular receptor kinase
- PI3K
phosphoinositide 3-kinase
- AKT/PKB
protein kinase B
- eNOS
endothelial nitric oxide synthase
References
- Abdollahi A, Lipson KE, Sckell A, Zieher H, Klenke F, Poerschke D, Roth A, Han X, Krix M, Bischof M, Hahnfeldt P, Grone HJ, Debus J, Hlatky L, Huber PE. Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res. 2003;63:8890–8898. [PubMed] [Google Scholar]
- Algira GH, Chalkley HW, Legallaia FY, Park HD. Vascular reactions of normal and malignant tissue in vivo. I. Vascular reactions of mice to wounds and to normal and neoplastic transplants. J Natl Cancer Inst. 1945;6:73–85. [Google Scholar]
- Amato RJ, Morgan M, Rawat A. Related Articles, Links Phase I/II study of thalidomide in combination with interleukin-2 in patients with metastatic renal cell carcinoma. Cancer. 2006;106:1498–1506. doi: 10.1002/cncr.21737. [DOI] [PubMed] [Google Scholar]
- Amato RJ, Rawat A. Interferon-alpha plus capecitabine and thalidomide in patients with metastatic renal cell carcinoma: a pilot study. Invest New Drugs. 2006;24:171–175. doi: 10.1007/s10637-005-2938-5. [DOI] [PubMed] [Google Scholar]
- Amato RJ, Sarao H. A phase I study of paclitaxel/doxorubicin/thalidomide in patients with androgen-independent prostate cancer. Clin Genitourin Cancer. 2006;4:281–286. doi: 10.3816/CGC.2006.n.008. [DOI] [PubMed] [Google Scholar]
- Arap W, Pasqualini R, Ruoslahti E. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, Ellerby HM, Bredesen DE, Pasqualini R, Ruoslahti E. Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci USA. 2002;99:1527–1531. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barleon B, et al. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated by the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- Batist G, Patenaude F, Champagne P, et al. Neovastat (AE-941) in refractory renal cell carcinoma patients: Report of a phase II trial with two dose levels. Ann Oncol. 2002;13:1259–1263. doi: 10.1093/annonc/mdf195. [DOI] [PubMed] [Google Scholar]
- Beecken WD, Fernandez A, Panigrahy D, Achilles EG, Kisker O, Flynn E, Joussen AM, Folkman J, Shing Y. Efficacy of antiangiogenic therapy with TNP-470 in superficial and invasive bladder cancer models in mice. Urology. 2000;56:521–526. doi: 10.1016/s0090-4295(00)00642-7. [DOI] [PubMed] [Google Scholar]
- Beecken WD, Engl T, Hofmann J, Jonas D, Blaheta R. Clinical relevance of serum angiogenic activity in patients with transitional cell carcinoma of the bladder. J Cell Mol Med. 2005;9:655–661. doi: 10.1111/j.1582-4934.2005.tb00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikfalvi A, Sauzeau C, Moukadiri H, Maclouf J, Busso N, Bryckaert M, Plouet J, Tobelem G. Interaction of vasculotropin/vascular endothelial cell growth factor with human umbilical vein endothelial cells: Binding, internalization, degradation. and biological effects. J Cell Physiol. 1991;149:50–59. doi: 10.1002/jcp.1041490108. [DOI] [PubMed] [Google Scholar]
- Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA. Matrix metalloproteinases: A review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- Blouw B, et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Boddy JL, Fox SB, Han C, Campo L, Turley H, Kanga S, Malone PR, Harris AL. The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clin Cancer Res. 2005;11:7658–7663. doi: 10.1158/1078-0432.CCR-05-0460. [DOI] [PubMed] [Google Scholar]
- Bono AV, Celato N, Cova V, Salvadore M, Chinetti S, Novario R. Microvessel density in prostate carcinoma. Prostate Cancer Prostatic Dis. 2002;5:123–127. doi: 10.1038/sj.pcan.4500572. [DOI] [PubMed] [Google Scholar]
- Borre M, Offersen BV, Nerstrom B, Overgaard J. Microvessel density predicts survival in prostate cancer patients subjected to watchful waiting. Br J Cancer. 1998;78:940–944. doi: 10.1038/bjc.1998.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borre M, Nerstrom B, Overgaard J. Association between immunohistochemical expression of vascular endothelial growth factor (VEGF), VEGF-expressing neuroendocrine-differentiated tumor cells, and outcome in prostate cancer patients subjected to watchful waiting. Clin Cancer Res. 2000;6:1882–1890. [PubMed] [Google Scholar]
- Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin v 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, Folkman J. Anti-angiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
- Cancer Trials Support Unit. CTSU Active Protocols and Accrual Report. 2007 Available at http://www.ctsu.org/ActiveProtocolList.asp?orderby=group.
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Clinical Trials, gov. [Accessed June 28, 2005];Docetaxel, Thalidomide, Prednisone, and Bevacizumab to Treat Metastatic Prostate Cancer. 2007 Available at http://www.clinicaltrials.gov/ct/show/NCT00089609.
- Cotran RS, Kumar V, Collins T, editors. Robbins pathologic basis of disease. 6. Philadelphia: W.B. Saunders; 1999. Tissue repair: Cellular growth, fibrosis, and wound healing; pp. 103–106. [Google Scholar]
- Crew JP, O’Brien TS, Harris AL. Bladder cancer angiogenesis, its role in recurrence, stage progression and as a therapeutic target. Cancer Metastasis Rev. 1996;15:221–230. doi: 10.1007/BF00437475. [DOI] [PubMed] [Google Scholar]
- Crew JP, O’Brien T, Bradburn M, Fuggle S, Bicknell R, Cranston D, Harris AL. Vascular endothelial growth factor is a predictor of relapse and stage progression in superficial bladder cancer. Cancer Res. 1997;57:5281–5285. [PubMed] [Google Scholar]
- Cross MJ, Claesson-Welsh L. VEGF and FGF function in angiogenesis: Signaling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201–207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- Crossey PA, Foster K, Richards FM, et al. Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: Analysis of allele loss in VHL tumours. Hum Genet. 1994;93:53–58. doi: 10.1007/BF00218913. [DOI] [PubMed] [Google Scholar]
- Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- Daneshgari F, Crawford ED. Endocrine therapy of advanced carcinoma of the prostate. Cancer. 1993;71:1089–1097. doi: 10.1002/1097-0142(19930201)71:3+<1089::aid-cncr2820711431>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–1169. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- de Gramont A, Van Cutsem E. Investigating the potential of bevacizumab in other indications: Metastatic renal cell, non-small cell lung, pancreatic and breast cancer. Oncology. 2005;3:46–56. doi: 10.1159/000088483. [DOI] [PubMed] [Google Scholar]
- deVries C, et al. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- Dong J, Grunstein J, Tejada M, Peale F, Frantz G, Liang WC, Bai W, Yu L, Kowalski J, Liang X, Fuh G, Gerber HP, Ferrara N. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibro-blast recruitment for tumorigenesis. EMBO J. 2004:2800–2810. doi: 10.1038/sj.emboj.7600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MJ, Robson W, Mehta P, Schofield I, Neal DE, Leung HY. An open-label phase II study of low-dose thalidomide in androgen-independent prostate cancer. Br J Cancer. 2003;88:822–827. doi: 10.1038/sj.bjc.6600817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreicer R, Klein EA, Elson P, Peereboom D, Byzova T, Plow EF. Phase II trial of GM-CSF + thalidomide in patients with androgen-independent metastatic prostate cancer. Urol Oncol. 2005;23:82–86. doi: 10.1016/j.urolonc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Du ZQ, Du TX, Wang ZD, Li GL. Expression of hypoxia-inducible factor 1alpha in human normal, benign, and malignant prostate tissue. Chin Med J (Engl) 2003;116:1936–1939. [PubMed] [Google Scholar]
- Dupont J, Schwartz L, Koutcher J, et al. Phase I and pharmacokinetic study of VEGF trap administered subcutaneously (sc) to patients (pts) with advanced solid malignancies. Proc Am Soc Clin Oncol. 2004;23:197. [Google Scholar]
- Duque JL, Loughlin KR, Adam RM, Kantoff PW, Zurakowski D, Freeman MR. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology. 1999;54:523–527. doi: 10.1016/s0090-4295(99)00167-3. [DOI] [PubMed] [Google Scholar]
- Elaraj DM, White DE, Steinberg SM, Haworth L, Rosen-berg SA, Yang JC. A pilot study of antiangiogenic therapy with bevacizumab and thalidomide in patients with metastatic renal cell carcinoma. J Immunother. 2004;27:259–264. doi: 10.1097/00002371-200407000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enenstein J, Waleh NK, Kramer RH. Basic FGF and TGF-beta differentially modulate integrin expression of human microvascular endothelial cells. Exp Cell Res. 1992;203:499–503. doi: 10.1016/0014-4827(92)90028-7. [DOI] [PubMed] [Google Scholar]
- Erdreich-Epstein A, Shimada H, Groshen S, Liu M, Metelitsa LS, Kim KS, Stins MF, Seeger RC, Durden DL. Integrins alpha(v)beta3 and alpha(v)beta5 are expressed by endothelium of high-risk neuroblastoma and their inhibition is associated with increased endogenous ceramide. Cancer Res. 2000;60:712–721. [PubMed] [Google Scholar]
- Ferrara N, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R, Moscatelli D, Rifkin DB. Heparin and heparan sulfate increase the radius of diffusion and action of basic fibroblast growth factor. J Cell Biol. 1990;111:1651–1659. doi: 10.1083/jcb.111.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: Therapeutic implications. N Eng J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis. In: Holland JF, Frei E III, Bast RC Jr, Kufe DW, Morton DL, Weichselbaum RR, editors. Cancer medicine. 3. Vol. 1. Philadelphia: Lea & Febiger; 1993. pp. 153–170. [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Folkman J. Clinical applications of research on angiogenesis. New Eng J Med. 1995;333:1757–1763. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis. In: Mendelson J, Howley PM, Israel MA, Liotta LA, editors. The molecular basis of cancer. Philadelphia: W. B. Saunders; 1995. pp. 206–232. [Google Scholar]
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;576:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- Fong TA, Shawver LK, Sun L, Tang C, App H, Powell TJ, Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth KP, McMahon G. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59:99–106. [PubMed] [Google Scholar]
- Fornaro M, Manes T, Laugunino LR. Integrin and prostate cancer metastases. Cancer Metastasis Rev. 2001;20:321–331. doi: 10.1023/a:1015547830323. [DOI] [PubMed] [Google Scholar]
- Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Cancer Res. 2006;66:464–472. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George D, Michaelson D, Oh WK, Reitsma D, Laurent D, Mietlowski W, Wang Y, Dugan M, Kaelin WG, Kantoff P. Phase I study of PTK787/ZK222584 (PTK/ZK) in metastaic renal cell carcinoma [abstract 1548] Proc Am Soc Clin Oncol. 2003;22:385. [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Goel HL, Languino LR. Integrin signaling in cancer. Cancer Treat Res. 2004;119:15–31. doi: 10.1007/1-4020-7847-1_2. [DOI] [PubMed] [Google Scholar]
- Goetzl EJ, Banda MJ, Leppert D. Matrix metal-loproteinases in immunity. J Immunol. 1996;156:1–4. [PubMed] [Google Scholar]
- Goligorsky MS, Budzikowski AS, Tsukahara H, Noiri E. Co-operation between endothelin and nitric oxide in promoting endothelial cell migration and angiogene-sis. Clin Exper Pharm Phys. 1999;26:269–271. doi: 10.1046/j.1440-1681.1999.03029.x. [DOI] [PubMed] [Google Scholar]
- Graham SD, Jr, Napalkov P, Oladele A, Keane TE, Petros JA, Clarke HS, Kassabian VS, Dillehay DL. Intravesical suramin in the prevention of transitional cell carcinoma. Urology. 1995;45:59–63. doi: 10.1016/s0090-4295(95)96720-6. [DOI] [PubMed] [Google Scholar]
- Greenblatt M, Shubick P. Tumor angiogenesis: Transfilter diffusion studies in the hamster by the transparent chamber technique. J Natl Cancer Inst. 1968;41:111–124. [PubMed] [Google Scholar]
- Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics. Ca Cancer J clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- Guo ZH, Mei H, Huang J, Li SY. Vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) expression in superficial transitional cell bladder carcinoma. Ai Zheng. 2003;22:307–309. [PubMed] [Google Scholar]
- Hainsworth J, Sosman J, Spigel DR, et al. Phase II trial of bevacizumab and erlotinib in patients with metastatic renal carcinoma (RCC) [abstract 4502] Proc Am Assoc Cancer Res. 2004;23:381. [Google Scholar]
- Hanahan D. Signaling vascular morphogenesis and maintenance. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- Harris TJ, von Maltzahn G, Derfus AM, Ruoslahti E, Bhatia SN. Proteolytic actuation of nanoparticle self-assembly. Angew Chem Int Ed Engl. 2006;45:3161–3165. doi: 10.1002/anie.200600259. [DOI] [PubMed] [Google Scholar]
- Hemmerlein B, Kugler A, Ozisik R, et al. Vascular endothelial growth factor expression, angiogenesis, and necrosis in renal cell carcinomas. Virchows Arch. 2001;439:645–652. doi: 10.1007/s004280100464. [DOI] [PubMed] [Google Scholar]
- Hernberg M, Virkkunen P, Bono P, Ahtinen H, Maenpaa H, Joensuu H. Interferon alfa-2b three times daily and thalidomide in the treatment of metastatic renal cell carcinoma. J Clin Oncol. 2003;21:3770–3776. doi: 10.1200/JCO.2003.01.536. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, Shipley JM, Senior RM, Shibuya M. MMP-9 induction by vascular endothelial growth facto receptor-1 is involved in lung specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA. Science. 2002;296:2404–2407. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- Hu L, Hofmann J, Zaloudek C, Ferrara N, Hamilton T, Jaffe RB. Vascular endothelial growth factor immunoneutralization plus Paclitaxel markedly reduces tumor burden and ascites in athymic mouse model of ovarian cancer. Am J Pathol. 2002;161:1917–1924. doi: 10.1016/S0002-9440(10)64467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss WJ, Barrios RJ, Greenberg NM. SU5416 selectively impairs angiogenesis to induce prostate cancer-specific apoptosis. Mol Cancer Ther. 2003;2:611–616. [PubMed] [Google Scholar]
- Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, Folkman J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppresses tumor growth. Nature. 1990;348:555–557. doi: 10.1038/348555a0. [DOI] [PubMed] [Google Scholar]
- Inoue K, Slaton JW, Davis DW, Hicklin DJ, McConkey DJ, Karashima T, Radinsky R, Dinney CP. Treatment of human metastatic transitional cell carcinoma of the bladder in a murine model with the anti-vascular endothelial growth factor receptor monoclonal antibody DC101 and paclitaxel. Clin Cancer Res. 2000;6:2635–2643. [PubMed] [Google Scholar]
- Jackson MW, Bentel JM, Tilley WD. Vascular endothelial growth factor (VEGF) expression in prostate cancer and benign prostatic hyperplasia. J Urology. 1997;157:2323–2328. [PubMed] [Google Scholar]
- Jiang Y, Muschel R. Regulation of matrix metal-loproteinase-9 (MMP-9) by translational efficiency in murine prostate cancer cells. Cancer Research. 2002;62:1910–1914. [PubMed] [Google Scholar]
- Kaczmarek MM, Schams D, Ziecik AJ. Role of vascular endothelial growth factor in ovarian physiology—an overview. Reprod Biol. 2005;5:111–136. [PubMed] [Google Scholar]
- Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- Kirkali Z, Tuzel E, Mungan MU. Recent advances in kidney cancer and metastatic disease. BJU Int. 2001;88:818–824. doi: 10.1046/j.1464-4096.2001.02442.x. [DOI] [PubMed] [Google Scholar]
- Klein S, Giancotti FG, Presta M, Albelda SM, Buck CA, Rifkin DB. Basic fibroblast growth factor modulates integrin expression in microvascular endothelial cells. Mol Biol Cell. 1993;4:973–982. doi: 10.1091/mbc.4.10.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Stetler-Stevenson WG. Structural biochemistry and activation of matrix metalloproteinases. Curr Opinion Cell Biol. 1993;5:891–897. doi: 10.1016/0955-0674(93)90040-w. [DOI] [PubMed] [Google Scholar]
- Kleiner D, Stetler-Stevenson W. Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol. 1999;43:S42–S51. doi: 10.1007/s002800051097. [DOI] [PubMed] [Google Scholar]
- Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105:R15–R24. doi: 10.1172/JCI8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopetz ES, Nelson JB, Carducci MA. Endothelin-1 as a target for therapeutic intervention in prostate cancer. Invest New Drugs. 2002;20:173–182. doi: 10.1023/a:1015630513908. [DOI] [PubMed] [Google Scholar]
- Krause S, Forster Y, Kraemer K, Fuessel S, Kotzsch M, Schmidt U, Wirth MP, Meye A, Schwenzer B. Vascular endothelial growth factor antisense pretreatment of bladder cancer cells significantly enhances the cytotoxicity of mitomycin C, gemcitabine and Cisplatin. J Urol. 2005;174:328–331. doi: 10.1097/01.ju.0000161588.94827.27. [DOI] [PubMed] [Google Scholar]
- Kuenen BC, Tabernero J, Baselga J, Cavalli F, Pfanner E, Conte PF, Seeber S, Madhusudan S, Deplanque G, Huisman H, Scigalla P, Hoekman K, Harris AL. Efficacy and toxicity of the angiogenesis inhibitor SU5416 as a single agent in patients with advanced renal cell carcinoma, melanoma, and soft tissue sarcoma. Clin Cancer Res. 2003;9:1648–1655. [PubMed] [Google Scholar]
- Lara PN, Jr, Quinn DI, Margolin K, Meyers FJ, Longmate J, Frankel P, Mack PC, Turrell C, Valk P, Rao J, Buckley P, Wun T, Gosselin R, Galvin I, Gumerlock PH, Lenz HJ, Doroshow JH, Gandara DR California Cancer Consortium. SU5416 plus interferon alpha in advanced renal cell carcinoma: A phase II California Cancer Consortium Study with biological and imaging correlates of angiogenesis inhibition. Clin Cancer Res. 2003;9:4772–4781. [PubMed] [Google Scholar]
- Lara PN, Jr, Przemyslaw T, Quinn DL. Angiogenesis-targeted therapies in prostate cancer. Clin Prost Cancer. 2004;3:165–173. doi: 10.3816/cgc.2004.n.027. [DOI] [PubMed] [Google Scholar]
- LeCouter J, Moritz DF, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, Ferrara N. Angiogenesis-independent endothelial protection of the liver: Role of VEGF- R1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- Lee JS, Kim HS, Jung JJ, Kim YB, Lee MC, Park CS. Expression of vascular endothelial growth factor in renal cell carcinoma and the relation to angiogenesis and p53 protein expression. J Surg Oncol. 2001;77:55–60. doi: 10.1002/jso.1066. [DOI] [PubMed] [Google Scholar]
- Leonard GD, Dahut WL, Gulley JL, Arlen PM, Figg WD. Docetaxel and thalidomide as a treatment option for androgen-independent, nonmetastatic prostate cancer. Rev Urol. 2003;3:S65–S70. [PMC free article] [PubMed] [Google Scholar]
- Logothetis CJ, Wu KK, Finn LD, Daliani D, Figg W, Ghaddar H, Gutterman JU. Phase I trial of the angiogenesis inhibitor TNP-470 for progressive androgen-independent prostate cancer. Clin Cancer Res. 2001;7:1198–1203. [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- Luo G, Gu YZ, Jain S, Chan WK, Carr KM, Hogenesch JB, Bradfield CA. Molecular characterization of the murine Hif-1 alpha locus. Gene Expr. 1997;6:287–299. [PMC free article] [PubMed] [Google Scholar]
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu ZP, Hackett NK, Crystal RG, Moore MAS, Hajjar KA, Manova K, Benezra R, Rafii S. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer. 2001;8:219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- Maynard AD, Pui DYH. Nanotechnology and occupational health: New technologies-new challenges. J Nano-particle Res. 2007;9:1–3. [Google Scholar]
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A. High affinity VEGF binding and developmental expression suggests flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- Mordenti J, Thomsen K, Licko V, Chen H, Meng YG, Ferrara N. Efficacy and concentration-response of murine anti-VEGF monoclonal antibody in tumor-bearing mice and extrapolation to humans. Toxicol Pathol. 1999;27:14–21. doi: 10.1177/019262339902700104. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Nanus DM, O’Moore P, Scher HI, Bajorin DF, Reuter V, Tong WP, Iversen J, Louison C, Albino AP, George JB. Phase II trial of suramin in patients with advanced renal cell carcinoma: Treatment results, pharmacokinetics, and tumor growth factor expression. Cancer Res. 1992;52:5775–5779. [PubMed] [Google Scholar]
- Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865–875. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI. SU011248, a novel tyrosine kinase inhibitor, shows antitumor activity in secondline therapy for patients with metastatic renal cell carcinoma. Results of a phase 2 trial. J Clin Oncol. 2004;22:4500. [Google Scholar]
- Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- Na X, Wu G, Ryan CK, Schoen SR, di’Santagnese PA, Messing EM. Overproduction of vascular endothelial growth factor related to von Hippel-Lindau tumor suppressor gene mutations and hypoxia-inducible factor-1 alpha expression in renal cell carcinomas. J Urol. 2003;170:588–592. doi: 10.1097/01.ju.0000074870.54671.98. [DOI] [PubMed] [Google Scholar]
- Nash GF, Walsh DC, Kakkar AK. The role of the coagulation system in tumour angiogenesis. Lancet Oncol. 2001;2:608–613. doi: 10.1016/s1470-2045(01)00518-6. [DOI] [PubMed] [Google Scholar]
- NCI homepage. http://www.cancer.gov/clinicaltrials/ct-types-list.
- Ng SS, MacPherson GR, Gutschow M, Eger K, Figg WD. Antitumor effects of thalidomide analogs in human prostate cancer xenografts implanted in immuno-deficient mice. Clin Cancer Res. 2004;10:4192–4197. doi: 10.1158/1078-0432.CCR-03-0700. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Giancotti FG. Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell. 2004;6:471–483. doi: 10.1016/j.ccr.2004.09.029. [DOI] [PubMed] [Google Scholar]
- O’Brien T, Cranston D, Fuggle S. Different angiogenic pathways characterize superficial and invasive bladder cancer. Cancer Res. 1995;55:510–513. [PubMed] [Google Scholar]
- O’Brien T, Cranston D, Fuggle S, Bicknell R, Harris AL. Two mechanisms of basic fibroblast growth factor-induced angiogenesis in bladder cancer. Cancer Res. 1997;57:136–140. [PubMed] [Google Scholar]
- OncoLink. [Accessed June 28, 2005];Bevacizumab (Avastin) in Advanced Prostate Cancer. Available at http://www.oncolink.com/conferences/article.cfm?c=3&s=24&ss=150&id=1003.
- Ord JJ, Streeter E, Jones A, Le Monnier K, Cranston D, Crew J, Joel SP, Rogers MA, Banks RE, Roberts IS, Harris AL. Phase I trial of intravesical Suramin in recurrent superficial transitional cell bladder carcinoma. Br J Cancer. 2005;92:2140–2147. doi: 10.1038/sj.bjc.6602650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi M, Marconcini L, Ferruzi R, Oliviero S. Identification of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Proc Natl Acad Sci USA. 1996;93:11675–11680. doi: 10.1073/pnas.93.21.11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bar-Shira A, Matzkin H, Mabjeesh NJ. The homozygous P582S mutation in the oxygen-dependent degradation domain of HIF-1alpha is associated with increased risk for prostate cancer. Prostate. 2007;67:8–13. doi: 10.1002/pros.20433. [DOI] [PubMed] [Google Scholar]
- Paradis V, Lagha NB, Zeimoura L, Blanchet P, Eschwege P, Ba N, Benoit G, Jardin A, Bedossa P. Expression of vascular endothelial growth factor in renal cell carcinomas. Virchows Arch. 2000;436:351–356. doi: 10.1007/s004280050458. [DOI] [PubMed] [Google Scholar]
- Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- Picus J, Halabi S, Rini B, Vogelzang N, Whang Y, Kaplan E, Kelly W, Small E. The use of bevacizumab (B) with docetaxel (D) and estramustine (E) in hormone refractory prostate cancer (HRPC): Initial results of CALGB 90006. Proc Am Soc Clin Oncol. 2003;22:393. [Google Scholar]
- Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnat M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Pronzato P, Rondini M. Ann Oncol. 2005;16:iv80–iv84. doi: 10.1093/annonc/mdi913. [DOI] [PubMed] [Google Scholar]
- Puri M, Rossant J, Alitolo K, Bernstein A, Partanen J. The receptor tyrosine kinase tie is required for integrity and survival of vascular endothelial cells. EMBO J. 1995;14:5884–5891. doi: 10.1002/j.1460-2075.1995.tb00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, Gore M, Desai AA, Patnaik A, Xiong HQ, Rowinsky E, Abbruzzese JL, Xia C, Simantov R, Schwartz B, O’Dwyer PJ. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24:2505–2512. doi: 10.1200/JCO.2005.03.6723. [DOI] [PubMed] [Google Scholar]
- Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment: Is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–11235. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini BI. VEGF-targeted therapy in metastatic renal cell carcinoma. Oncologist. 2005;10:191–197. doi: 10.1634/theoncologist.10-3-191. [DOI] [PubMed] [Google Scholar]
- Rini BI, Small EJ. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol. 2005;23:1028–1043. doi: 10.1200/JCO.2005.01.186. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Antiangiogenics meet nanotechnology. Cancer Cell. 2002;2:97–98. doi: 10.1016/s1535-6108(02)00100-9. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Specialization of tumour vasculature. Nat Rev Cancer. 2002;2:83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- Salani D, Garagoletti G, Rosano L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A. Endothelin-1 induces an angiogenic phenotype in cultures endothelial cells and stimulates neovascularization in vivo. Amer J Path. 2000;157:1703–1711. doi: 10.1016/S0002-9440(10)64807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinase tie-1 and tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Schrader AJ, Varga Z, Pfoertner S, Goelden U, Buer J, Hofmann R. Treatment targeted at vascular endothelial growth factor: a promising approach to managing metastatic kidney cancer. BJU Int. 2006;97:461–465. doi: 10.1111/j.1464-410X.2006.05873.x. [DOI] [PubMed] [Google Scholar]
- Schroder LE, Lew D, Flanigan RC, Eisenberger MA, Seay TE, Hammond N, Needles BM, Crawford ED. Phase II evaluation of suramin in advanced renal cell carcinoma. A Southwest Oncology Group study. Urol Oncol. 2001;6:145–148. doi: 10.1016/s1078-1439(00)00126-5. [DOI] [PubMed] [Google Scholar]
- SEER. SEER Cancer Statistics Review. National Cancer Institute Web site. Available at http://cancernet.nci.nih.gov/statistics.
- Semenza GL. Transcriptional regulation by hypoxia-inducible factor 1-molecular mechamisms of oxygen homeostasis. Trends Cardiovasc. 1996;6:151–157. doi: 10.1016/1050-1738(96)00039-4. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, et al. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 2003;424:391–397. doi: 10.1038/nature01784. [DOI] [PubMed] [Google Scholar]
- Shing Y, Folkman J, Sullivan R, Butterfield C, Murray J, Klagsbrun M. Heparin affinity: Purification of a tumor-derived capillary endothelial cell growth factor. Science. 1984;223:1296–1299. doi: 10.1126/science.6199844. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, Derfus AM, Yang M, Hoffman RM, Bhatia S, Sailor MJ, Ruoslahti E. Biomimetic amplification of nano-particle homing to tumors. Proc Natl Acad Sci USA. 2007;104:932–936. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipkins DA, Cheresh DA, Kazemi MR, Nevin LM, Bednarski MD, Li KCP. Nat Med. 1998;4:623–626. doi: 10.1038/nm0598-623. [DOI] [PubMed] [Google Scholar]
- Slaton JW, Millikan R, Inoue K, Karashima T, Czerniak B, Shen Y, Yang Y, Benedict WF, Dinney CP. Correlation of metastasis related gene expression and relapse-free survival in patients with locally advanced bladder cancer treated with cystectomy and chemotherapy. J Urol. 2004;171:570–574. doi: 10.1097/01.ju.0000108845.91485.20. [DOI] [PubMed] [Google Scholar]
- Small EJ, Reese DM, Um B, Whisenant S, Dixon SC, Figg WD. Therapy of advanced prostate cancer with granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 1999;5:1738–1744. [PubMed] [Google Scholar]
- Small EJ, Meyer M, Marshall ME, Reyno LM, Meyers FJ, Natale RB, Lenehan PF, Chen L, Slichenmyer WJ, Eisenberger M. Suramin therapy for patients with symptomatic hormone-refractory prostate cancer: Results of a randomized phase III trial comparing suramin plus hydrocortisone to placebo plus hydro-cortisone. J Clin Oncol. 2000;18:1440–1450. doi: 10.1200/JCO.2000.18.7.1440. [DOI] [PubMed] [Google Scholar]
- Small EJ, Halabi S, Ratain MJ, Rosner G, Stadler W, Palchak D, Marshall E, Rago R, Hars V, Wilding G, Petrylak D, Vogelzang NJ. Randomized study of three different doses of suramin administered with a fixed dosing schedule in patients with advanced prostate cancer: Results of Intergroup Cancer and Leukemia Group B 9480. J Clin Oncol. 2002;20:3369–3375. doi: 10.1200/JCO.2002.10.022. [DOI] [PubMed] [Google Scholar]
- Stadler WM, Kuzel T, Shapiro C, Sosman J, Clark J, Vogelzang NJ. Multi-institutional study of the angiogenesis inhibitor TNP-470 in metastatic renal carcinoma. J Clin Oncol. 1999;17:2541–2545. doi: 10.1200/JCO.1999.17.8.2541. [DOI] [PubMed] [Google Scholar]
- Stadler WM, Cao D, Vogelzang NJ, Ryan CW, Hoving K, Wright R, Karrison T, Vokes EE. A randomized Phase II trial of the antiangiogenic agent SU5416 in hormone-refractory prostate cancer. Clin Cancer Res. 2004;10:3365–3370. doi: 10.1158/1078-0432.CCR-03-0404. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Stein CA. Suramin: A novel antineoplastic agent with multiple potential mechanisms of action. Cancer Res. 1993;53:2239–2248. [PubMed] [Google Scholar]
- Sternberg CN, Yagoda A, Scher HI, Watson RC, Herr HW, Morse MJ, Sogani PC, Vaughan ED, Jr, Bander N, Weiselberg LR, et al. M-VAC (methotrexate, vinblastine, doxorubicin and cisplatin) for advanced transitional cell carcinoma of the urothelium. J Urol. 1988;139:461–469. doi: 10.1016/s0022-5347(17)42494-3. [DOI] [PubMed] [Google Scholar]
- Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, Fife K, Armstrong V, Treston A, Sidor C, Wilding G. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxy-estradiol capsules in hormone-refractory prostate cancer. Clin Cancer Res. 2005;11:6625–6633. doi: 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]
- Tan C, de Noronha RG, Roecker AJ, Pyrzynska B, Khwaja F, Zhang Z, Zhang H, Teng Q, Nicholson AC, Giannakakou P, Zhou W, Olson JJ, Pereira MM, Nicolaou KC, Van Meir EG. Identification of a novel small-molecule inhibitor of the hypoxia-inducible factor 1 pathway. Cancer Res. 2005;65:605–612. [PubMed] [Google Scholar]
- Theodoropoulos VE, Lazaris AC, Kastriotis I, Spiliadi C, Theodoropoulos GE, Tsoukala V, Patsouris E, Sofras F. Evaluation of hypoxia-inducible factor 1alpha overexpression as a predictor of tumour recurrence and progression in superficial urothelial bladder carcinoma. BJU Int. 2005;95:425–431. doi: 10.1111/j.1464-410X.2005.05314.x. [DOI] [PubMed] [Google Scholar]
- Tille JC, Wood J, Manriota SJ, Schnell C, Ferrari S, Mestan J, Zhu Z, Witte L, Pepper MS. Vascular endothelial growth factor receptor-2 antagonists inhibit VEGF and basic fibroblast growth factor-induced angiogenesis in vivo and vitro. J Pharmacol Exp Ther. 2001;299:1073–1085. [PubMed] [Google Scholar]
- Van Moorselaar RJA, Voest EE. Angiogenesis in prostate cancer: Its role in disease progression and possible therapeutic approaches. Mol Cel Endocrinol. 2002;197:239–250. doi: 10.1016/s0303-7207(02)00262-9. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Bar-Shavit R, Ishai-Michaeli R, Bashkin P, Fuks Z. Extracellular sequestration and release of fibroblast growth factor: A regulatory mechanism? Trends Biochem Sci. 1991;16:268–271. doi: 10.1016/0968-0004(91)90102-2. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, Jemal A, Ward E, Anderson RN, Edwards BK. Annual Report to the Nation on the Status of Cancer, 1975–2000, featuring the uses of surveillance data for cancer prevention and control. J Natl Cancer Inst. 2003;95:1276–1299. doi: 10.1093/jnci/djg040. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum anti-tumor activity and targets raf/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Proc Am Assoc Cancer Res. 2003;44:100. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT, Kozin SV, Petit L, Jain RK, Chung DC, Sahani DV, Kalva SP, Cohen KS, Scadden DT, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Shellito PC, Mino-Kenudson M, Lauwers GY. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: Continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136–8139. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O’Reilly T, Persohn E, Rosel J, Schnell C, Stover D, Theuer A, Towbin H, Wenger F, Woods-Cook K, Menrad A, Siemeister G, Schirner M, Thierauch KH, Schneider MR, Drevs J, Martiny-Baron G, Totzke F. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178–2189. [PubMed] [Google Scholar]
- Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]