

Abstract
The ability of a tumor cell population to grow exponentially represents an imbalance between cellular proliferation and cellular attrition. There is an overwhelming body of evidence suggesting the ability of tumor cells to avoid programmed cellular attrition, or apoptosis, is a major molecular force driving the progression of human tumors. Apoptotic evasion represents one of the true hallmarks of cancer and appears to be a vital component in the immunogenic, chemotherapeutic, and radiotherapeutic resistance that characterizes the most aggressive of human cancers [Hanahan and Weinberg, 2000]. The challenges in the development of effective treatment modalities for advanced prostate cancer represent a classic paradigm of the functional significance of anti-apoptotic pathways in the development of therapeutic resistance.
Keywords: prostate growth, apoptosis, survival signaling, Bcl-2, cancer therapy, molecular targets
Prostate cancer is the most commonly diagnosed cancer in American males and is second only to lung cancer in cancer-related mortality within this patient population [Jemal et al., 2005]. Most prostate tumors are initially responsive of androgen ablation therapy, which acts to devoid these tumor cells of their primary growth stimulus [Arnold and Isaacs, 2002]. Unfortunately, prostate cancer, under the physiologic stress of hormone ablation, ultimately progresses to androgen-independent disease that has proven resistant to both hormone ablation, as well as other systemic chemotherapies [Debes and Tindall, 2004]. Apoptosis appears to be the predominant form of tumor cell demise caused by both androgen ablation and chemotherapeutic agents and plays a role in prostate cancer radiosensitivity [Arnold and Isaacs, 2002; Debes and Tindall, 2004]. It is the acquisition of anti-apoptotic signal transduction that ultimately leads to the characteristic treatment resistance that typifies advanced prostate cancer.
Execution of apoptosis can occur via two distinct signaling pathways. The intrinsic and extrinsic apoptotic pathways leading to cellular death are summarized in Figure 1. The extrinsic pathway is initiated by the binding of apoptosis-inducing ligands to cell surface death receptors associated with Fas-associated death domain (FADD) [Debatin and Krammer, 2004]. Activated FADD then interacts with initiator enzymes of the caspase cascade, notably caspase 8 and 10, resulting in further downstream effector caspase activation (caspases 3 and 7) and ultimately programmed cellular destruction through proteolytic cleavage of caspase substrates [Debatin and Krammer, 2004]. The intrinsic pathway is initiated by intercellular stress, lack of growth factors, or overwhelming DNA injury and subsequently targets the mitochondrial membrane [Okada and Mak, 2004]. Loss of mitochondrial membrane potential and increased membrane permeability leads to the release of cytochrome c [Newmeyer and Miller, 2003]. Release of this protein into the cytosol results in activation of apoptotic protease activating factor-1 (APAF-1) and caspase 9 recruitment [Okada and Mak, 2004]. These proteins form a functional apoptosome that activates the effector caspase cascade again resulting in programmed cellular destruction [Debatin and Krammer, 2004]. While these two pathways are often described as separate pathways, significant cross-talk is known to exist, most notably through caspase-8 directed Bid activation leading to cytochrome c release [Kulik et al., 2001a]. Prostate cancer cells have shown the ability to acquire both intracellular survival pathways and alterations in chemokine and growth factor signal transduction that allow them to circumvent either apoptotic pathway and ultimately contribute to the androgen-independent and classically aggressive phenotype that is resistant to any form of conventional chemotherapy.
Fig. 1.
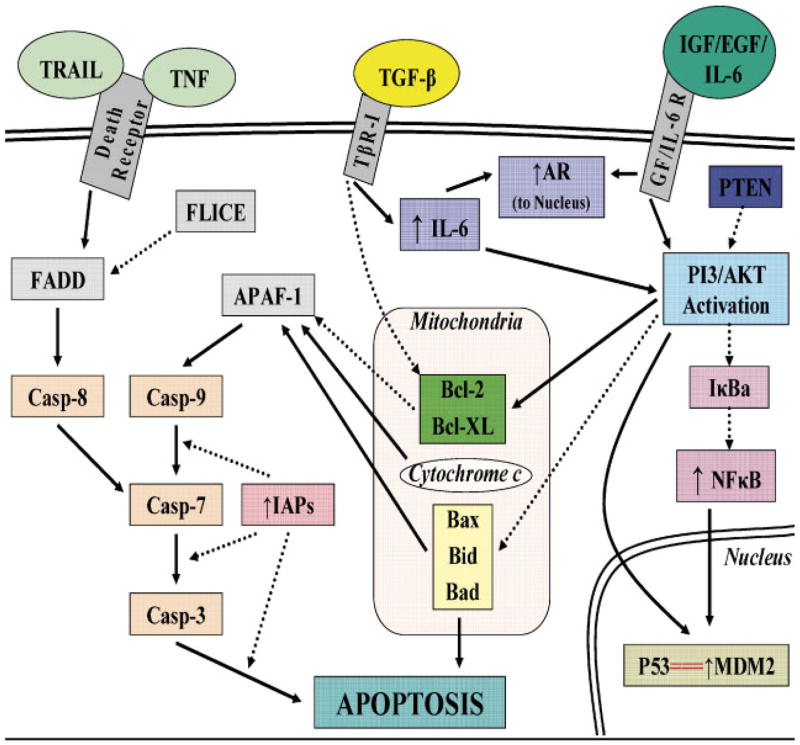
Signaling cross-talk between survival and apoptosis pathways in prostate cancer cells. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
THE APOPTOTIC PLAYERS
The Bcl-2 Family of Proteins
The Bcl-2 family proteins include a heterogeneous group of both pro-apoptotic and anti-apoptotic molecules that exert their effect on mitochondrial function [DiPaola et al., 2001]. Many Bcl-2 family members contain a hydrophobic stretch of amino acids near their carboxyl-terminus that anchors them in the outer mitochondrial membrane. In contrast, other Bcl-2 family members, such as Bid, Bim, and Bad, lack these membrane anchoring-domains, but target mitochondria. Anti-apoptotic members, most notably Bcl-2 and Bcl-xL, inhibit the release of cytochrome c from the mitochondria, consequently inhibiting mitochondrial-induced apoptosis. Their action is antagonized by proapototic members of the Bcl-2 family such as Bax, Bad, and Bid, allowing for mitochondrial cytochrome c release and caspase cascade activation [DiPaola et al., 2001]. It is the ratio of pro-apoptotic to anti-apoptotic family members that ultimately determines the survival of tumor cells.
Both in vitro and in vivo studies have established that Bcl-2 and other anti-apoptotic members of its family are significantly upregulated in aggressive prostate cancer phenotypes [Kajiwara et al., 1999; McCarty, 2004]. Over-expression of Bcl-2 and Bcl-xL has been shown in prostate cancer and other malignancies to confer resistance to both chemotherapy and radiation therapy [McCarty, 2004]. Furthermore, the Bcl-2 family plays a critical role in the androgen-signaling axis operating in prostate cancer cells. In androgen-responsive prostate cancer cells, androgens downregulate expression of pro-apoptotic Bcl-2 family members such as Bax [Coffey et al., 2002]. Increased Bcl-2 and Bcl-xL expression in androgen-independent tumors [Furuya et al., 1996] is directly linked to the ability of prostate cancer cells to survive in androgen-free environments [Kajiwara et al., 1999], evidence highlighting the functional as well as predictive significance of Bcl-2/Bcl-xL over-expression as one of the key regulators allowing for selection of androgen-independent recurrences in prolonged androgen ablation therapy [McCarty, 2004]. While recognizing that expression changes in the Bcl-2 family can contribute the emergence of therapeutic resistance, the dynamic cross-talk between this “powerful” family and other anti-apoptotic pathways influenced by exogenous ligand-receptor signaling must be acknowledged and will be discussed.
The NF-κB Intracellular Connection
The NF-κB/Rel protein family are transcription factors that regulate a multitude of immunologic and inflammatory responses as well as individual cell growth, differentiation, and apoptosis [Suh and Rabson, 2004]. While the oncogenic properties of NF-κB include augmenting angiogenesis, invasion, and metastasis formation, the most important mechanism driving the carcinogenic effect of this transcription factor is its antiapoptotic pathway [Chen, 2004; Suh and Rabson, 2004]. In the majority of cell types, NF-κB is kept inactive through compartmentalization to the cytosol via binding with inhibitors of κBs (IκBs). In response to the appropriate stimuli, IκB is phosphorylated through interaction with the IκB kinase complex (IKK), allowing nuclear translocation of NF-κB and subsequent NF-κB driven signal transduction (Fig. 1) [Chen, 2004; Suh and Rabson, 2004]. The antiapoptotic effect of NF-κB has been attributed to its ability to directly upregulate Bcl-2 and Bcl-xL expression in prostate cancer cells leading to inhibition of mitochondrial apoptosis [Shukla and Gupta, 2004]. However, dissection of the prostate apoptosis response-4 protein (PAR-4) pathway has shown that NF-κB inhibition is required for the proapoptotic effect of PAR-4 on the FAS ligand extrinsic apoptotic pathway to occur [Chakraborty et al., 2001]. Furthermore, NF-κB has been shown to upregulate FLICE inhibitory protein (c-FLIP-L) expression in prostate cancer cells; c-FLIP-L upregulation directly interferes with recruitment of caspase-8 to FADD [Zhang et al., 2004]. Downregulation of c-FLIP-L appears to restore sensitivity to Fas-mediated apoptosis in aggressive prostate cancer cells [Hyer et al., 2002]. Downstream effector caspase activation is not immune to the inhibitory effects of NK-κB either, as it has been shown to upregulate several members of the inhibitors of apoptosis proteins (IAPs) which directly inhibit caspases -3, -7, and/or -9 [McEleny et al., 2001].
Constituitive activation of NF-κB is widely in many human malignancies, and while NF-κB activity occurs in low levels in androgen-dependent prostate tumors, it appears in high levels in androgen independent tumor cell lines and in highly aggressive prostate tumors and metastatic lesions [Ismail et al., 2004; Ross et al., 2004]. In a recent immunohistochemical evaluation of prostatectomy specimens, elevated NF-κB immunoreactivity correlated directly with advanced tumor stage, tumor grade, and tumor recurrence [Ross et al., 2004]. In lymph node metastasis, nuclear localization of NF-κB was significantly greater in lymph node containing tumor cells when compared to local tumor controls [Ismail et al., 2004] Interestingly, elevated NF-κB activation was also seen in tumor cell surrounding lymphocytes [Ismail et al., 2004] suggesting possible cross-talk between prostate cancer cells and surrounding lymphocytes, leading to oncogenically favorable release of paracrine prostate cancer mediators such as Il-6 and TNF-a. This apparent upregulation of NF-κB seen in aggressive prostate cancers has been correlated with resistance to both chemotherapy and radiation therapy [Suh and Rabson, 2004]. While there appears to be a direct correlation between increased NF-κB activity and androgen independence, the interaction between the NF-κB pathway and the androgen receptor (AR) pathway appears to be pleomorphic [Coffey et al., 2002; Suh and Rabson, 2004] and requires further elucidation.
p53: Guardian of Genomic Integrity
The p53 tumor suppressor gene regulates both cell cycle and apoptosis in response to numerous cellular stresses such as DNA injury, hypoxia, free radical injury, and damage to the mitotic machinery [Hernandez et al., 2003]. It is believed that the oncolytic responsibility of the p53 gene product is to invoke cell-cycle arrest and stimulate apoptosis in cells that have acquired overwhelming genetic aberrations to avoid mutation propagation [Hernandez et al., 2003]. p53 action is inhibited by MDM2, through binding to the p53 gene product and relegating it to ubiquinylation and proteasomal degradation [McCarty, 2004]. Mechanistically the ability of p53 to induce apoptosis in tumor cells results from its upregulation of BAX, leading to mitochondrial driven apoptosis [Hernandez et al., 2003]. Furthermore, recent analysis of specific p53 mutations p53 revealed that altered p53 expression can also adversely affect Fas-mediated apoptosis [Gurova et al., 2003].
Loss or mutation of p53 has been identified in over 10,000 different types of tumors analyzed and mutations of this tumor suppressor gene are found in 45%–50% of all human cancers [Soussi et al., 2000]. In prostate cancer, while mutations in p53 are uncommon in early, well-differentiated disease, mutations become abundant in both metastatic disease as well as hormone-independent tumors [Navone et al., 1993]. Moreover, upregulation of MDM2 expression has been found in up to 40% of prostatectomy specimens and correlated with advanced disease [Leite et al., 2001]. In hormone-responsive prostate cancer cells, loss of wild-type p53 leads to development of a hormone-resistant phenotype, thus implicating altered p53 function in the development of hormone refractory disease [Scherr et al., 1999; Burchardt et al., 2001]. Significantly enough altered p53 has been shown to influence chemotherapeutic response, with most mutations leading to resistance, while certain select mutations lead to increased sensitivity to specific agents such as paclitaxel [DiPaola et al., 2001]. Growing evidence indicates a direct correlation between altered p53 expression with prostate cancer resistance to radiotherapy [Pisters et al., 2004], while resolution of functional p53 status with specific p53 mutants restores the apoptotic signaling and ultimately therapeutic sensitivity in experimental models of prostate cancer [Hernandez et al., 2003; Pisters et al., 2004].
PTEN/P13K/AKT: The Downstream Intracellular Players
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a highly conserved tumor supressor gene that induces cellular apoptosis through its modulation of the P13K/AKT signal transduction pathway. Specifically, PTEN inhibits phosphorylation of AKT, which is necessary for its activation and targeting of its many downstream effectors [Wang et al., 2003]. Loss of PTEN, a common event in treatment resistant and poorly differentiated prostate cancers, leads to constituative activation of the P13K/AKT pathway and subsequent apoptotic resistance [Davies et al., 1999]. Restoration of PTEN activity in PTEN deficient prostate cancer cell lines has been shown to increase sensitivity to FADD mediated caspase-8 driven apoptosis as well as to facilitate BIDD cleavage allowing for cytochrome c release and subsequent mitochondrial driven apoptosis [Yuan and Whang, 2002]. AKTs are activated by second messengers via phosphatidylinositol 3′-kinases (P13Ks). This phosphorylation is counterbalanced by the activity of PTEN phosphatases [Stern, 2004]. In prostate cancer, AKT phosphorylation can occur constitutively through loss of PTEN activity, or be stimulated and upregulated in PTEN positive tumors through autocrine and paracrine cell membrane receptor-ligand interactions [Pfeil et al., 2004]. The ability of phosphorylated AKT to inhibit prostate cancer cellular apoptosis appears to be the result of the powerful crosstalk that exists between this effector and multiple other anti-apoptotic pathways (Fig. 1). Activated AKT has been shown to activate MDM2 leading to proteolysis of p53 and subsequent inhibition of p53 mediated apoptosis with stimulation of cell-cycle progression [Gao et al., 2003; Stern, 2004]. Activated AKT also inactivates Bad and caspase-9, allowing for Bcl-2 release and inhibition of mitochondrial apoptosis [Ghosh et al., 2003; Wang et al., 2003; Stern, 2004]. Furthermore, upregulation of P13K/AKT activity leads to phosphorylation of IκB, allowing for nuclear translocation of NF-κB and subsequent NF-κB driven suppression of apoptosis [Wang et al., 2003; Stern, 2004]. Phosphorylated AKT can induce phosphorylation of the AR, as well as upregulate AR expression, which can lead to inhibition of androgen-deprivation-induced apoptosis [Lin et al., 2001; Ghosh et al., 2003].
An expanding body of recent evidence indicates that both PTEN inactivation and AKT phosphorylation are hallmarks of aggressive prostate cancer. While PTEN inactivation is present in only 10%–15% of primary prostate cancers, PTEN loss is detected in 30%–50% of hormone-refractory tumors, as well as 60% of xenograft models derived from metastatic prostate cancer cell lines [Wang et al., 2003; Pfeil et al., 2004]. Furthermore, loss of PTEN has correlated with aggressive local disease (T3b-T4 tumors) and Gleason score >6 [McMenamin et al., 1999]. AKT phosphorylation has been shown to be a marker for aggressive disease, but also an independent prognostic indicator. High levels of AKT phosphorylation are exclusive associated with prostatic adenocarcinoma, compared to benign tissues [Ayala et al., 2004]. AKT phosphorylation has been shown to correlate with Gleason score [Liao et al., 2003], and, in poorly differentiated tumors (Gleason 8–10), strong presence of phosphorylated AKT is observed in over 90% of specimens examined [Malik et al., 2002]. In prostate cancer specimens with Gleason scores of 5–6, a notoriously difficult patient population to predict prognosis, elevated AKT phosphorylation proved to be an indicator for recurrence [Liao et al., 2003]. Furthermore, it was recently established that phosphorylation of AKT was a more effective prognostic indicator of recurrence than both mitotic index and Gleason score [Kreisberg et al., 2004]. Not surprisingly, loss of PTEN and phosphorylation of AKT are associated with resistance to chemotherapy and been implicated in the progression of refractory prostate cancer after long term androgen ablation therapy [Yuan and Whang, 2002; Ghosh et al., 2003]. Novel targeting strategies inhibiting AKT phosphorylation or restoring PTEN activity appear to cause profound apoptosis and restore sensitivity to chemotherapy in vitro and in xenograft models [Yuan and Whang, 2002; Shaw et al., 2004].
The Antagonists: Inhibitors of Apoptosis Proteins (IAPs)
Recently, a new family of apoptosis inhibitors has been identified and appears to have a role in prostate cancer treatment resistance. The IAPs are a group of caspase inhibitors that directly inhibit the effector caspases 3, 7, and 9 resulting in decreased cellular apoptosis [Schimmer, 2004]. Currently, eight human IAPs have been identified with the most studied being X-linked inhibitor of apoptosis protein (XIAP), inhibitor of apoptosis protein 1 (IAP1), inhibitor of apoptosis protein 2 (IAP2), and survivin [Krajewska et al., 2003]. While all appear capable of inhibiting effector caspases, IAP1 and IAP2 can upregulate NF-κB expression pointing to a possible positive feedback loop between these two pathways (Fig. 1) [McEleny et al., 2001]. Elevated expression of these four IAPs has been shown in both animal models of prostate cancer and prostatectomy specimens from cancer patients, and this elevation appears to be present early in prostate cancer development [Krajewska et al., 2003]. The ability of IAPs to inhibit apoptosis in response to multiple chemotherapeutic agents has been established in several tumor models [Debatin and Krammer, 2004], although the significance of IAPs in prostate cancer therapeutic resistance is an area of recently “active” investigations. Indeed recent evidence suggests that XIAP inhibition enhances chemotherapy sensitivity in otherwise resistant prostate cancer cell lines [Amantana et al., 2004]. Another small study of 23 patients revealed that IAP1 and IAP2 expression were dramatically upregulated in patients receiving neoadjuvant androgen ablation suggesting a potential role of IAPs in androgen independence [McEleny et al., 2001]. As the role of IAPs in prostate cancer treatment resistance continues to be discerned, manipulation of IAP pathways may become a valuable way to circumvent the apoptotic resistance present in the upstream intracellular apoptotic escape mechanisms such as AKT and Bcl-2.
Tumor microenvironment: extracellular forces driving intracellular resistance
Recently, the focus of tumor biology has embraced a crucial paradigm shift implicating not only the intracellular pathophysiology inherent to carcinoma cells, but also the critical role that the tumor microenvironment plays in developing aggressive cancer phenotypes. It has become apparent that the cross-talk that exists between prostate epithelial tumor cells and their surrounding stromal and endothelial partners, as well as the localized inflammatory cells attracted to the neoplastic region, is a driving force towards both androgen independence and subsequent treatment resistant recurrence [Arnold and Isaacs, 2002; Chung et al., 2005]. The ability of tumor microenvironment to alter the apoptotic outcomes of prostate cancer cells is exemplified by recent advances in the understanding of tumor hypoxia. The ability of solid organ tumors, including prostate adenocarcinoma, to outgrow their own blood supply coupled with their innate propensity for disordered neovascularization creates an intratumoral environment with often severely diminished oxygen tension. Consequently, tumor hypoxia has been shown to correlate significantly with prostate cancer stage, aggressiveness, androgen independence, and treatment resistance [Movsas et al., 2000; Cvetkovic etal., 2001; Hochachka etal., 2002; Ghafar etal., 2003]. Furthermore, this hypoxia driven aggressive behavior can, at least in part, be attributed to enhanced apoptotic resistance in hypoxic prostate cancer cells due to suppression of p53 activity and upregulation of AKT [Skinner et al., 2004; Liu et al., 2005]. Along with tumor hypoxia, the paracrine and subsequent autocrine release of both growth factors and cytokines has also been shown to affect apoptotic sensitivity and, ultimately, tumor aggressiveness.
Growth factor signaling pathways
Mechanistic dissection of the pathways leading to the emergence of hormone independent-prostate cancer identified the dynamic contribution of an array of growth factors, in addition to the androgen-signaling axis. While a full understanding of the pro-survival characteristics of these growth factor pathways is still evolving, the impact that growth factors such as epidermal growth factor, insulin-like growth factor 1, and transforming growth factor-β can be appreciated by the robust development of new cancer therapies targeting their signal transduction. As the medical and scientific community enthusiastically witnessed the development of the therapeutically promising tyrosine kinase inhibitor Iressa, the role of the epidermal growth factor (EGF) system in apoptosis evasion and prostate cancer progression has been exposed. EGF can be secreted in a paracrine followed by autocrine manner in prostate tumors [Mimeault et al., 2003]. Upregulation of its membrane receptor in invasive prostate carcinoma cells has also been well characterized [Di Lorenzo et al., 2002; Hernes et al., 2004; Shuch et al., 2004]. While upregulation of this pathway has been associated with increased cellular proliferation and increased invasion [Mimeault et al., 2003], its role in prostate cancer therapeutic resistance and androgen independent status can be attributed to its ability to protect prostate cancer cells from apoptosis. Indeed, the EGF-EGFR system can activate Pi3K leading to AKT phosphorylation and subsequent inhibition of proapoptotic BAD (Fig. 1) [Mimeault et al., 2003]. Interestingly, while disruption of the EGF-EGFR pathway leads to robust prostate cancer cell apoptosis [Harper et al., 2002; Farhana et al., 2004], the enhanced apoptotic sensitivity can be only partially explained by downregulation of the PI3K/AKT pathway. Recent mechanistic analysis of this pathway has exposed the ability of the EGF-EGFR system to rescue prostate cancer cells from the proapototic effects of PI3k/AKT inhibition [Torring et al., 2003]. The ability of the EGF-EGFR system to provide apoptotic evasion in a PI3K/AKT independent manner can be attributed, at least in part, to its effects on AR signaling. While the ability of epidermal growth factor to induce AR transcriptional activity alone has been a topic of debate [Orio et al., 2002; Mellinghoff et al., 2004], the ability of this systemto, atminimum, coactivate the AR, sensitize it to the low levels of androgen characteristic of hormone ablation therapy, and synergize androgenic stimulation of AR transcriptional activity has been established [Orio et al., 2002; Bonaccorsi et al., 2004; Gregory et al., 2004]. These characteristics predict the clinical experience with EGF-EGFR signal transduction and its relationship with prostate cancer progression and treatment resistance.
Tissue analysis of tumor specimens from both mouse xenografts and human patients has shown EGFR expression to be a predictor of aggressive disease. Elevations of EGFR expression occur in prostate cancer cells and associated endothelial cells from bony metastases, as opposed to other metastatic sites in experimental xenograft models [Kim et al., 2003]. Furthermore, elevated EGFR expression in human tumor specimens has been correlated with increased stage, Gleason grade, PSA, invasiveness and metastatic disease [Di Lorenzo et al., 2002; Shuch et al., 2004]. Elevated receptor expression is also associated with the molecular switch to androgen independence [Hernes et al., 2004] and has been implicated in the racial disparities that exist in prostate cancer disease behavior and outcomes [Shuch et al., 2004]. This constellation of data provided a strong molecular basis for the development of inhibitors of the EGF-EGFR pathway. Both in vitro and in vivo experimental models firmly established that EGF-EGFR pathway disruption leads to increased apoptosis and growth inhibition in prostate cancer cells [Harper et al., 2002; Kim et al., 2003; Vicentini et al., 2003; Farhana et al., 2004]. This has led to clinical trials with compounds, such as the EGFR tyrosine kinase inhibitor IRESSA. Although early results in prostate cancer have met with a certain degree of variability, one has to recognize that disruption of this pathway emerges as an attractive target with therapeutic promise [Blackledge, 2003].
Like the EGF-EGFR pathway, the insulin-like growth factor (IGF) axis has proven to be a critical player in the progression of prostate cancer. Unlike EGF, the IGF pathway may be equally important in the development of early prostate cancer. The IGF signaling pathway is a complex balance of interactions between IGF-1 ligand, multiple IGF binding proteins (IGFBPs), IGF receptor (IGFR), and IGFBP proteases. IGF-1 is synthesized in nearly every human tissue, but in the prostate, appears to exert its action on prostate cancer cells through paracrine release from the prostate stroma [Moschos and Mantzoros, 2002]. There are six known IGFBPs described in humans, and these compounds determine both the bioavailability of IGF-1 as well as guide its effects on target tissue [Djavan et al., 2001; Moschos and Mantzoros, 2002]. Ninety-nine percent of the circulating IGF-1 is bound to IGFBPs with 75% of IGF-1 bound specifically to IGFBP-3 [Djavan et al., 2001; Moschos and Mantzoros, 2002]. IGFR is also constituitively expressed in human tissues but quantitative receptor expression can be altered and will affect tissue response to IGF-1 [Djavan et al., 2001; Moschos and Mantzoros, 2002; Krueckl et al., 2004]. IGF-1 function can be further regulated by IGFBP proteases of which PSA is included. In prostate cancer cells, binding of IGF-1 to IGFR initiates two predominant apoptotic resistance pathways: the P13K/AKT pathway and, to a lesser extent, the NF-κB pathway (Fig. 1) [Djavan et al., 2001; Moschos and Mantzoros, 2002; Bogdanos et al., 2003]. In a pattern to the EGF-EGFR signaling events, in the presence of elevated IGFR, which is common in advanced disease, IGF-1 is able to rescue prostate cancer cells from apoptosis induced by P13K/AKT pathway disruption [Miyake et al., 2000]. IGF-1 has also been shown to stimulate the AR [Moschos and Mantzoros, 2002] and has been directly implicated in the progression to androgen independence [Krueckl et al., 2004]. Furthermore, the differential expression of IGFBPs by prostate cancer cells also influences apoptotic sensitivity. While several of the IGFBPs have been implicated in prostate cancer progression, IGFBP 3 appears to be the most influential player [Djavan et al., 2001; Hong et al., 2002; Moschos and Mantzoros, 2002; Li et al., 2003]. IGFBP 3 binding to IGF-1 attenuates the upregulation of the P13K/AKT pathway leading to increased prostate cancer cell apoptosis [Djavan et al., 2001; Moschos and Mantzoros, 2002]. Recent evidence shows that IGFBP 3 is able to sensitize prostate cancer cells to apoptosis in the absence of IGF-1 binding [Hong et al., 2002]. It is not surprising that downregulation of IGFBP 3 is common in prostate cancer and is also degraded by the known IGFBP protease PSA [Djavan et al., 2001].
The clinical impact of the IGF/IGFB/IGFR/PSA axis cannot be overemphasized. While there is some debate whether this axis is more important during the development of prostate cancer or during the progression to metastatic, treatment refractory disease, it is clear that IGF signal transduction is crucial to the pathogenesis of prostate cancer from initiation through to metastatic formation. Increased IGFR expression is common in androgen-independent metastatic lesions and increased IGFBP 2 and 5 levels in prostate cancer specimens correlate with increased Gleason grade [Djavan et al., 2001]. Furthermore, elevated serum IGF-1 levels as well as an elevated IGF/IGFBP 3 ratio has been found in multiple clinical studies to be an independent predictor of prostate cancer risk and to also improve the diagnostic yield of PSA screening [Chan et al., 1998; Li et al., 2003; Stattin et al., 2004]. Recent insight into the bone microenvironment has implicated the IGF1 axis as the predominant survival factor pathway responsible for the androgen ablation and chemotherapy refractoriness seen in prostate cancer bony metastasis [Bogdanos et al., 2003]. As metastatic prostate cancer cells release urokinase-type plasminogen activator (uPA), hydrolosis of IGFBPs occurs, resulting in a local increase in IGF-1 bioavailability and subsequent apoptotic resistance and osteoblastic reaction [Bogdanos et al., 2003]. As both uPA and IGF-1 have promoter region binding sites for the glucocorticoid receptor, glucocorticoid therapy has become a component of rescue therapy in the case of skeletal metastatic disease [Bogdanos et al., 2003]. Several similar treatments aimed at disrupting IGF signal transduction including GNRH antagonism, somatostatin analogs, and IGFBP protease inhibition, are currently under active investigation [Djavan et al., 2001]. To date, while quality of life measurements with such therapeutic strategies are encouraging, no significant changes in survival have been appreciated [Bogdanos et al., 2003].
The role of transforming growth factor-β1 (TGF-β1) in prostate cancer pathogenesis represents a classic ability of cancer cells to alter signal transduction in the presence of an abundant apoptosis-inducing ligand in order to evade cell death and promote disease progression. TGF-β1 signaling in normal prostate epithelium and in early prostate cancer can be characterized by proliferation inhibition and tumor suppression [Bello-DeOcampo and Tindall, 2003]. The ability of TGF-β1 to suppress early prostate cancer tumorigenesis requires intact signal transduction via interaction with the TGF-β1 receptors TGFβR-I and TGFβR-II and subsequent downstream targeting through regulation of the SMAD family of protein effectors [Bello-DeOcampo and Tindall, 2003]. Upregulation of this pathway from TGF-β1 and receptor binding leads to caspase-1 activation, upregulation of BAX, and downregulation of Bcl-2, ultimately resulting in tumor cell apoptosis [Guo and Kyprianou, 1999; Kyprianou, 1999]. Furthermore, the enhanced expression of TGF-β1 and its receptors that occurs after medical or surgical castration has been implicated as the main driving force for the pronounced prostate cancer cell apoptosis seen with such therapy [Wikstrom et al., 1999]. Unfortunately, TGF-β1’s ability to induce prostate cancer apoptosis eventually gives way to disease promotion and metastatic formation.
Increased TGF-β1 ligand expression directly correlates with prostate cancer progression, while there is loss expression of its receptors [Wikstrom et al., 1999]. This disruption of normal TGF-β1 signaling tips the axis in favor of enhanced angiogenesis, extracellular matrix remodeling favorable for invasion and, most importantly, immununosuppression [Matthews et al., 2000; Tuxhorn et al., 2002; Bello-DeOcampo and Tindall, 2003]. TGF-β1 overexpression, common in advanced prostate cancer, has been shown to directly inhibit the ability of tumor specific cytotoxic T lymphocytes (CTLs) and NK cells to induce prostate cancer cell apoptosis; downregulation of TGF-β1 can restore immunogenicity of prostate cancer cells and suppress metastasis formation [Matthews et al., 2000; Teicher, 2001; Shah et al., 2002], potentially via activation of Il-6 expression (Fig. 1), a powerful inhibitor of prostate cancer cell apoptosis and metastasis promoter [Park et al., 2003]. Bcl-2 overexpression, another common finding in prostate cancer, is also able to inhibit TGF-β1 induced apoptosis [Bruckheimer and Kyprianou, 2002]. Moreover TGF-β1 can synergize with AR transactivation in response to androgen and upregulate downstream targets, such as PSA, which have been implicated in apoptotic evasion [Kang et al., 2001].
TGF-β1 ligand overexpression coupled with the downregulation of TGβR-I and TGFβ R-II are hallmarks of advanced prostate cancer. Numerous clinical studies of both prostatectomy specimens as well as serum analysis of patients both before and after prostatectomy have revealed that upregulation of TGF-β1 along with downregulation of TGFβR-I and TGFβR-II is associated with invasive disease, increased Gleason grade, and treatment refractory disease [Shariat et al., 2004a,b; Zeng et al., 2004]. The prognostic power of TGF-β1 is exemplified by its inclusion in current pre-operative nomograms that have proven more effective at predicting recurrent disease than standard parameters used today such as pre-operative PSA or Gleason grade [Kattan et al., 2003]. Attempts to target TGF-β1 signaling have included quinazoline-based α1-adrenoceptor antagonists, restoration of TGF-β receptor expression through gene delivery, and anti-sense inhibition of TGF-β1 expression [Guo and Kyprianou, 1999; Matthews et al., 2000; Partin et al., 2003]. The encouraging results in the laboratory have yet to be translated to the clinical setting.
The role of cytokines and inflammatory response in prostate tumor progression
Our current understanding of the contribution of inflammation to the tumorigenic process points to an enticing question: could it be possible that the immune response generated to combat cancer initiation and progression, provides yet another opportunistic interaction within the tumor microenvironment? While a connection between inflammation and the development and progression of cancer has been suspected for some time, new insights into the importance of this relationship are emerging. Two recent experimental models have convincingly implicated the inflammatory response, occurring both in intestinal colitis and chronic hepatitis, as a key mediator not only in the development of solid tumors but also in tumor progression [Balkwill and Coussens, 2004]. In both the colitis and hepatitis model, the NF-κB system appears to be the intracellular pathway link that allows the inflammatory response to be a potential co-conspirator in tumor progression [Viatour et al., 2005]. Further dissection by Greten and colleagues revealed the importance of secretion inflammatory mediators such as TNF-a, IL-1, IL-6, and IL-8 in driving the NF-κB pathway towards apoptotic resistance and tumor progression [Greten et al., 2004]. While the NF-κB link between cancer and inflammation has been proposed in other tumor systems [Viatour et al., 2005], it has yet to be validated in prostate cancer. Considering the evidence linking prostate cancer development to chronic inflammation [Konig et al., 2004; Nelson et al., 2004], one must recognize the immediate need for molecular exploration of this relationship. Perhaps the most important lesson learned so far from the experimental and clinical studies on prostate cancer, is the vital role of cytokines in the host inflammatory response during tumor progression. The two cytokines most often implicated in this dual capacity, TNF-a and IL-6, will be examined.
Tumor necrosis factor-α
TNF-a is a pleiomorphic cytokine involved in both inflammation and cancer biology. The cellular response to TNF-α ligand-receptor binding can invoke either the apoptotic cascade or promote tumor cell survival. There are two well-described TNF-α receptors, TNFRI and TNFRII. Ligand binding to TNFRI usually results in FADD recruitment and subsequent caspase 8 activation, which ultimately results in apoptosis [Guseva et al., 2004]. Ligand binding to TNFRII leads to activation in the MAPK and NF-κB pathways resulting in proliferation and apoptotic resistence [Guseva et al., 2004]. However, receptor expression alone does not dictate the tumor cell’s fate, as TNFRI binding can also stimulate NF-κB activation through TRAF2 activation of IKK mediated IκB-α phosphorylation [Chopra et al., 2004; Guseva et al., 2004]. In prostate cancer cells, the response to TNF-α appears to be linked to androgen responsiveness. Data from both in vitro and in vivo model experimental studies confirm that androgen responsive prostate cancer cell lines are sensitive to TNF-α induced apoptosis via both p53 accumulation, as well as BID cleavage and subsequent caspase cascade initiation leading to cytochrome c release [Rokhlin et al., 2000; Kulik et al., 2001b]. Prolonged exposure of androgen sensitive prostate cancer cells to TNF-α leads to increased AR activity and hypersensitivity to low-androgen levels [Harada et al., 2001]. Furthermore, in metastatic androgen-insensitive prostate cancer cells, exposure to TNF-α can actually promote apoptotic resistance rather than sensitivity. This axial shift towards tumor promotion rather than apoptotic sensitivity has been attributed to the high levels of constitutive NF-κB expression in androgen insensitive prostate cancer cells, as well as TNF-α mediated upregulation of IKK activity and subsequent NF-κB activation through PI3K-AKT dependent and independent pathways (Fig. 1) [Sumitomo et al., 1999; Gustin et al., 2001; Dhanalakshmi et al., 2002; Chopra et al., 2004]. Moreover, fibronectin can protect aggressive prostate cancer cells from TNF-α induced apoptosis via the Akt/survivin pathway, with surviving maintaining a critical anti-apoptotic threshold [Fornaro et al., 2003].
Examination of prostate cancer specimens reveals increased TNF-α expression in both epithelial tumor cells and tumor-associated macrophages [Muenchen et al., 2000; Michalaki et al., 2004]. Furthermore, serum levels of TNF-α taken from patients prostate cancer correlated with both bulky local disease and metastatic progression, and elevated levels were shown to be an independent prognostic indicator for survival [Michalaki et al., 2004]. Due to the pleiomorphic response to TNF-α in prostate cancer cell lines it has been widely used as a therapeutic target via signal transduction inhibition. The majority of the therapeutic exploration has centered around the radiation sensitizing effects of TNF-α on multiple tumor systems including prostate cancer [Chung et al., 1998; Kimura et al., 1999]. However, recent studies have targeted inhibition of the NF-κB pathway, driving the response to TNF-α downstream in the apoptotic pathway [Dhanalakshmi et al., 2002; Papandreou and Logothetis, 2004]. A recent weapon in the cancer armamentarium, proteosome inhibition, has been shown to target numerous signaling pathways, most notably NF-κB activation, with promising results in patients with hormone refractory disease [Papandreou and Logothetis, 2004].
Interleukin-6
Similar to TNF-α, IL-6, while traditionally described as a key mediator in the inflammatory response, has proven to be an integral part of prostate cancer biology. The ability of IL-6 to affect the intracellular apoptotic machinery can be attributed to its effects on both the PI3K/AKT pathway and the AR pathway (Fig. 1). Elevations in IL-6 lead to activation in the PI3K/AKT pathway with subsequent increases in Bcl-xl [Pu et al., 2004; Xie et al., 2004] and resistence to standard chemotherapy-induced apoptosis. This increase in AKT activation has also been associated with neuroendocrine differentiation, a common phenotype of treatment resistant prostate cancer cells [Xie et al., 2004]. Additional evidence demonstrated that IL-6 stimulates AR activity in the absence of androgen via the STAT3 pathway [Lee et al., 2004]. This ability to bypass the receptor ligand interaction of androgen and its receptor allows IL-6 to protect hormone sensitive prostate cancer cells from apoptosis induced by androgen deprivation [Lee et al., 2004]. Interestingly, there is an inversely proportional correlation between IL-6 expression and androgen expression, in aging, healthy male subjects, and the effects of IL-6 on hormone responsive cell lines can be blunted with the addition of androgen [Kim et al., 2004; Xie et al., 2004]. IL-6 overexpression in the prostate cancer microenvironment is due to both autocrine and paracrine feedback loops. The constitutive overexpression of IL-6 in hormone resistant prostate cancer cell lines has been attributed to an autocrine loop governed by the NF-κB activity [Zerbini et al., 2003]. Within the bone microenvironment IL-6 plays a critical role in osteoblast paracrine interactions with metastatic prostate cancer cells [Garcia-Moreno et al., 2002; Lu et al., 2004].
The clinical significance of IL-6 is exemplified in its prognostic capabilities. Studies including serum measurements of IL-6 with or without the addition of its soluble receptor have shown both to be powerful predictors of PSA failure, disease progression, and mortality [Shariat et al., 2001; George et al., 2005]. Furthermore, preoperative serum IL-6 measurements appear to be an effective screening tool for occult metastatic disease at the time of resection [Shariat et al., 2001] and may prove valuable in the development of adjuvant therapy protocols. Attempts to directly target IL-6 expression with monoclonal antibody therapy have had moderate success in animal models [Smith and Keller, 2001]. One could easily argue that as autocrine IL-6 production is apparently NF-κB driven, NF-κB targeting and proteosome inhibition may ultimately inhibit this cytokine’s prosurvival activity as well.
CONCLUSION
As one dissects the mechanisms underlying prostate cancer progression to hormone independence and treatment resistance during the clinical course of the disease, the role of apoptotic evasion takes center stage. The ability of prostate cancer cells to activate intracellular survival pathways coupled with the critically dynamic intracellular cross-talk and anti-apoptotic pathway redundancy leaves a formidable opponent for the most powerful of cytotoxic therapies, much less hormone withdrawal. Furthermore, the ability of these cells to adapt to their extracellular microenvironment by alterations in: (1) epithelial-stromal interactions; (2) pathophysiologic cellular stress responses; (3) growth factor-receptor pathways; or (4) the inflammatory response; allows the most hostile of tumor microenvironments to promote rather than inhibit cancer cell survival and, ultimately, encourage aggressive phenotypes. Whether it is the upregulation of intracellular survival pathways or the extracellular influence that upregulates intracellular anti-apototic signal transduction that allows for such aggressive adaptation remains a subject of debate. It is becoming increasingly apparent however that our ability to positively improve the therapeutic response and survival of patients with hormone-refractory metastatic prostate cancer will ultimately require a therapeutic arsenal that targets multiple and often functionally overlapping signal transduction pathways rather than the current, frequently ineffective attempts at monotherapy for advanced disease.
Acknowledgments
The authors thank Ryan Reynolds for his expert assistance with the preparation of the figure, and Lorie Howard for help with the manuscript submission.
Grant sponsor: NIH/NIDDK; Grant number: R01 DK53525-06.
References
- Amantana A, London CA, Iversen PL, Devi GR. X-linked inhibitor of apoptosis protein inhibition induces apoptosis and enhances chemotherapy sensitivity in human prostate cancer cells. Mol Cancer Ther. 2004;3(6):699–707. [PubMed] [Google Scholar]
- Arnold JT, Isaacs JT. Mechanisms involved in the progression of androgen-independent prostate cancers: It is not only the cancer cell’s fault. Endocr Relat Cancer. 2002;9:61–73. doi: 10.1677/erc.0.0090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala G, Thompson T, Yang G, Frolov A, Li R, Scardino P, Ohori M, Wheeler T, Harper W. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin Cancer Res. 2004;10(19):6572–6578. doi: 10.1158/1078-0432.CCR-04-0477. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Coussens LM. An inflammatory link. Nature. 2004;431:405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- Bello-DeOcampo D, Tindall DJ. TGF-β/Smad signaling in prostate cancer. Curr Drug Targets. 2003;4(3):197–207. doi: 10.2174/1389450033491118. [DOI] [PubMed] [Google Scholar]
- Blackledge G. Growth factor receptor tyrosine kinase inhibitors; clinical development and potential for prostate cancer therapy. J Urol. 2003;170:S77–S83. doi: 10.1097/01.ju.0000095022.80033.d3. [DOI] [PubMed] [Google Scholar]
- Bogdanos J, Karamanolakis D, Tenta R, Tsintavis A, Milathianakis C, Mitsiades C, Koutsilieris M. Endocrine/paracrine/autocrine survival factor activity of bone microenvironment participates in the development of androgen ablation and chemotherapy refractoriness of prostate cancer metastasis in skeleton. Endocr Relat Cancer. 2003;10(2):279–289. doi: 10.1677/erc.0.0100279. [DOI] [PubMed] [Google Scholar]
- Bonaccorsi L, Carloni V, Muratori M, Formigli L, Zecchi S, Forti G, Baldi E. EGF receptor (EGFR) signaling promoting invasion is disrupted in androgen-sensitive prostate cancer cells by an interaction between EGFR and androgen receptor (AR) Int J Cancer. 2004;112(1):78–86. doi: 10.1002/ijc.20362. [DOI] [PubMed] [Google Scholar]
- Bruckheimer EM, Kyprianou N. Bcl-2 antagonizes the combined apoptotic effect of transforming growth factor-β and dihydrotestosterone in prostate cancer cells. Prostate. 2002;53(2):133–142. doi: 10.1002/pros.10143. [DOI] [PubMed] [Google Scholar]
- Burchardt M, Burchardt T, Shabsigh A, Ghafar M, Chen MW, Anastasiadis A, de la Taille A, Kiss A, Buttyan R. Reduction of wild type p53 function confers a hormone resistant phenotype on LNCaP prostate cancer cells. Prostate. 2001;48(4):225–230. doi: 10.1002/pros.1101. [DOI] [PubMed] [Google Scholar]
- Chakraborty M, Qiu SG, Vasudevan KM, Rangnekar VM. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Res. 2001;61(19):7255–7263. [PubMed] [Google Scholar]
- Chan JM, Stampfer MJ, Giovannucci E, et al. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science. 1998;279:563–566. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- Chen F. Endogenous inhibitors of nuclear factor-κB, an opportunity for cancer control. Cancer Res. 2004;64:8135–8138. doi: 10.1158/0008-5472.CAN-04-2096. [DOI] [PubMed] [Google Scholar]
- Chopra DP, Menard RE, Januszewski J, Mattingly RR. TNF-alpha-mediated apoptosis in normal human prostate epithelial cells and tumor cell lines. Cancer Lett. 2004;203(2):145–154. doi: 10.1016/j.canlet.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Chung TD, Mauceri HJ, Hallahan DE, Yu JJ, Chung S, Grdina WL, Yajnik S, Kufe DW, Weichselbaum RR. Tumor necrosis factor-alpha-based gene therapy enhances radiation cytotoxicity in human prostate cancer. Cancer Gene Ther. 1998;5(6):344–349. [PubMed] [Google Scholar]
- Chung LWK, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: The missing link of tumor microenvironment. J Urol. 2005;173:10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- Coffey RNT, Watson WG, O’Neill AJ, McEleny K, Fitzpatrick JM. Androgen-mediated resistance to apoptosis. The Prostate. 2002;53:300–309. doi: 10.1002/pros.10159. [DOI] [PubMed] [Google Scholar]
- Cvetkovic D, Movsas B, Dicker AP, Hanlon AL, Greenberg RE, Chapman JD, Hanks GE, Tricoli JV. Increased hypoxia correlates with increased expression of the angiogenesis marker vascular endothelial growth factor in human prostate cancer. Urology. 2001;57(4):821–825. doi: 10.1016/s0090-4295(00)01044-x. [DOI] [PubMed] [Google Scholar]
- Davies MA, Koul D, Dhesi H, Berman R, McDonnell TJ, McConkey D, Yung WK, Steck PA. Regulation of Akt/PKB activity, cellular growth, and apoptosis in prostate carcinoma cells by MMAC/PTEN. Cancer Res. 1999;59(11):2551–2556. [PubMed] [Google Scholar]
- Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23(16):2950–2966. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. NEJM. 2004;351(15):1488–1490. doi: 10.1056/NEJMp048178. [DOI] [PubMed] [Google Scholar]
- Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits constitutive and TNFα-induced activation of NF-κB and sensitizes human prostate carcinoma DU145 cells to TNF-α induced apoptosis. Oncogene. 2002;21(11):1759–1767. doi: 10.1038/sj.onc.1205240. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo G, Tortora G, D’Armiento FP, De Rosa G, Staibano S, Autorino R, D’Armiento M, De Laurentiis M, De Placido S, Catalano G, Bianco AR, Ciardiello F. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002;8(11):3438–3444. [PubMed] [Google Scholar]
- DiPaola RS, Patel J, Rafi MM. Targeting apoptosis in prostate cancer. Heme/onc Clin North Am. 2001;15(3):1–14. doi: 10.1016/s0889-8588(05)70229-x. [DOI] [PubMed] [Google Scholar]
- Djavan B, Waldert M, Seitz C, Marberger M. Insulin-like growth factors and prostate cancer. World J Urol. 2001;19(4):225–233. doi: 10.1007/s003450100220. [DOI] [PubMed] [Google Scholar]
- Farhana L, Dawson MI, Huang Y, Zhang Y, Rishi AK, Reddy KB, Freeman RS, Fontana JA. Apoptosis signaling by the novel compound 3-Cl-AHPC involves increased EGFR proteolysis and accompanying decreased phosphatidylinositol 3-kinase and AKT kinase activities. Oncogene. 2004;23(10):1874–1884. doi: 10.1038/sj.onc.1207311. [DOI] [PubMed] [Google Scholar]
- Fornaro M, Plescia J, Chheang S, Tallini G, Zhu Y-M, King M, Altieri D, Languino LR. Fibronectin protects prostate cancer cells from tumor necrosis factor-induced apoptosis via the AKT/survivin pathway. J Biol Chem. 2003;50:50402–50411. doi: 10.1074/jbc.M307627200. [DOI] [PubMed] [Google Scholar]
- Furuya Y, Krajewski S, Epstein JI, Reed JC, Isaacs JT. Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clin Cancer Res. 1996;2:389–398. [PubMed] [Google Scholar]
- Gao N, Zhang Z, Jiang BH, Shi X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem Biophys Res Commun. 2003;310(4):1124–1132. doi: 10.1016/j.bbrc.2003.09.132. [DOI] [PubMed] [Google Scholar]
- Garcia-Moreno C, Mendez-Davila C, de La Piedra C, Castro-Errecaborde NA, Traba ML. Human prostatic carcinoma cells produce an increase in the synthesis of interleukin-6 by human osteoblasts. Prostate. 2002;50(4):241–246. doi: 10.1002/pros.10050. [DOI] [PubMed] [Google Scholar]
- George DJ, Halabi S, Shepard TF, Sanford B, Vogelzang NJ, Small EJ, Kantoff PW. The prognostic significance of plasma interleukin-6 levels in patients with metastatic hormone-refractory prostate cancer: Results from cancer and leukemia group B 9480. Clin Cancer Res. 2005;11:1815–1820. doi: 10.1158/1078-0432.CCR-04-1560. [DOI] [PubMed] [Google Scholar]
- Ghafar MA, Anastasiadis AG, Chen MW, Burchardt M, Olsson LE, Xie H, Benson MC, Buttyan R. Acute hypoxia increases the aggressive characteristics and survival properties of prostate cancer cells. Prostate. 2003;54(1):58–67. doi: 10.1002/pros.10162. [DOI] [PubMed] [Google Scholar]
- Ghosh PM, Malik S, Bedolla R, Kreisberg JI. Akt in prostate cancer: Possible role in androgen-independence. Curr Drug Metab. 2003;4(6):487–496. doi: 10.2174/1389200033489226. [DOI] [PubMed] [Google Scholar]
- Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, Wilson EM. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279(8):7119–7130. doi: 10.1074/jbc.M307649200. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckman L, Greten TF, Park JM, Egan LJ, Karin M. IKKB links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Guo Y, Kyprianou N. Restoration of transforming growth factor beta signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer Res. 1999;59(6):1366–1371. [PubMed] [Google Scholar]
- Gurova KV, Rokhlin OW, Budanov AV, Burdelya LG, Chumakov PM, Cohen MB, Gudkov AV. Cooperation of two mutant p53 alleles contributes to Fas resistance of prostate carcinoma cells. Cancer Res. 2003;63(11):2905–2912. [PubMed] [Google Scholar]
- Guseva NV, Taghiyev AF, Rokhlin OW, Cohen MB. Death receptor-induced cell death in prostate cancer. J Cell Biochem. 2004;91(1):70–99. doi: 10.1002/jcb.10707. [DOI] [PubMed] [Google Scholar]
- Gustin JA, Maehama T, Dixon JE, Donner DB. The PTEN tumor suppressor protein inhibits tumor necrosis factor-induced nuclear factor kappa B activity. J Biol Chem. 2001;276(29):27740–27744. doi: 10.1074/jbc.M102559200. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harada S, Keller ET, Fujimoto N, Koshida K, Namiki M, Matsumoto T, Mizokami A. Long-term exposure of tumor necrosis factor alpha causes hypersensitivity to androgen and anti-androgen withdrawal phenomenon in LNCaP prostate cancer cells. Prostate. 2001;46(4):319–326. doi: 10.1002/1097-0045(20010301)46:4<319::aid-pros1039>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Harper ME, Goddard L, Glynne-Jones E, Assender J, Dutkowski CM, Barrow D, Dewhurst OL, Wakeling AE, Nicholson RI. Multiple responses to EGF receptor activation and their abrogation by a specific EGF receptor tyrosine kinase inhibitor. Prostate. 2002;52(1):59–68. doi: 10.1002/pros.10069. [DOI] [PubMed] [Google Scholar]
- Hernandez I, Maddison LA, Wei Y, DeMayo F, Petras T, Li B, Gingrich JR, Rosen JM, Greenberg NM. Prostate-specific expression of p53(R172L) differentially regulates p21, Bax, and mdm2 to inhibit prostate cancer progression and prolong survival. Mol Cancer Res. 2003;1(14):1036–1047. [PubMed] [Google Scholar]
- Hernes E, Fossa SD, Berner A, Otnes B, Nesland JM. Expression of the epidermal growth factor receptor family in prostate carcinoma before and during androgen-independence. Br J Cancer. 2004;90(2):449–454. doi: 10.1038/sj.bjc.6601536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Rupert JL, Goldenberg L, Gleave M, Kozlowski P. Going malignant: The hypoxia-cancer connection in the prostate. Bioessays. 2002;24(8):749–757. doi: 10.1002/bies.10131. [DOI] [PubMed] [Google Scholar]
- Hong J, Zhang G, Dong F, Rechler MM. Insulin-like growth factor (IGF)-binding protein-3 mutants that do not bind IGF-I or IGF-II stimulate apoptosis in human prostate cancer cells. J Biol Chem. 2002;277(12):10489–10497. doi: 10.1074/jbc.M109604200. [DOI] [PubMed] [Google Scholar]
- Hyer ML, Sudarshan S, Kim Y, Reed JC, Dong JY, Schwartz DA, Norris JS. Downregulation of c-FLIP sensitizes DU145 prostate cancer cells to Fas-mediated apoptosis. Cancer Biol Ther. 2002;1(4):401–406. doi: 10.4161/cbt.1.4.15. [DOI] [PubMed] [Google Scholar]
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Chafoor A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- Ismail HA, Lessard L, Mes-Masson AM, Saad F. Expression of NF-kappaB in prostate cancer lymph node metastases. Prostate. 2004;58(3):308–313. doi: 10.1002/pros.10335. [DOI] [PubMed] [Google Scholar]
- Kajiwara T, Takeuchi T, Ueki T, Moriyama N, et al. Effect of bcl-2 overexpression in human prostate cancer cells in vitro and in vivo. Int J Urol. 1999;6:520–525. doi: 10.1046/j.1442-2042.1999.00102.x. [DOI] [PubMed] [Google Scholar]
- Kang HY, Lin HK, Hu YC, Yeh S, Huang KE, Chang C. From transforming growth factor-β signaling to androgen action: Identification of Smad3 as an androgen receptor coregulator in prostate cancer cells. Proc Natl Acad Sci U S A. 2001;98(6):3018–3023. doi: 10.1073/pnas.061305498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattan MW, Shariat SF, Andrews B, Zhu K, Canto E, Matsumoto K, Muramoto M, Scardino PT, Ohori M, Wheeler TM, Slawin KM. The addition of inter-leukin-6 soluble receptor and transforming growth factor beta1 improves a preoperative nomogram for predicting biochemical progression in patients with clinically localized prostate cancer. J Clin Oncol. 2003;21(19):3573–3579. doi: 10.1200/JCO.2003.12.037. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Uehara H, Karashima T, Shepherd DL, Killion JJ, Fidler IJ. Blockade of epidermal growth factor receptor signaling in tumor cells and tumor-associated endothelial cells for therapy of androgen-independent human prostate cancer growing in the bone of nude mice. Clin Cancer Res. 2003;9(3):1200–1210. [PubMed] [Google Scholar]
- Kim O, Jiang T, Xie Y, Guo Z, Chen H, Qiu Y. Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene. 2004;23(10):1838–1844. doi: 10.1038/sj.onc.1207304. [DOI] [PubMed] [Google Scholar]
- Kimura K, Bowen C, Spiegel S, Gelmann EP. Tumor necrosis factor-alpha sensitizes prostate cancer cells to gamma-irradiation-induced apoptosis. Cancer Res. 1999;59(7):1606–1614. [PubMed] [Google Scholar]
- Konig JE, Senge T, Allhoff EP, Konig W. Analysis of the inflammatory network in benign prostate hyperplasia and prostate cancer. Prostate. 2004;58(2):121–129. doi: 10.1002/pros.10317. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Krajewski S, Banares S, Huang X, et al. Elevated expression of inhibitors of apoptosis proteins in prostate cancer. Clin Cancer Res. 2003;9:4914–4925. [PubMed] [Google Scholar]
- Kreisberg JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, Kreisberg S, Ghosh PM. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64(15):5232–5236. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- Krueckl SL, Sikes RA, Edlund NM, Bell RH, Hurtado-Coll A, Fazli L, Gleave ME, Cox ME. Increased insulin-like growth factor I receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res. 2004;64(23):8620–8629. doi: 10.1158/0008-5472.CAN-04-2446. [DOI] [PubMed] [Google Scholar]
- Kulik G, Carson JP, Vomastek T. Tumor necrosis factor alpha induces BID cleavage and bypasses anti-apoptotic signals inprostate cancer LNCaP cells. Cancer Res. 2001a;61:2713–2719. [PubMed] [Google Scholar]
- Kulik G, Carson JP, Vomastek T, Overman K, Gooch BD, Srinivasula S, Alnemri E, Nunez G, Weber MJ. Tumor necrosis factor alpha induces BID cleavage and bypasses antiapoptotic signals in prostate cancer LNCaP cells. Cancer Res. 2001b;61(6):2713–2719. [PubMed] [Google Scholar]
- Kyprianou N. Activation of TGF-beta signalling in human prostate cancer cells suppresses tumorigenicity via deregulation of cell cycle progression and induction of caspase-1 mediated apoptosis: Significance in prostate tumorigenesis. Prostate Cancer Prostatic Dis. 1999;2(S3):S18. doi: 10.1038/sj.pcan.4500344. [DOI] [PubMed] [Google Scholar]
- Lee SO, Lou W, Johnson CS, Trump DL, Gao AC. Interleukin-6 protects LNCaP cells from apoptosis induced by androgen deprivation through the Stat3 pathway. Prostate. 2004;60(3):178–186. doi: 10.1002/pros.20045. [DOI] [PubMed] [Google Scholar]
- Leite KR, Franco MF, Srougi M, Nesrallah LJ, Nesrallah A, Bevilacqua RG, Darini E, Carvalho CM, Meirelles MI, Santana I, Camara-Lopes LH. Abnormal expression of MDM2 in prostate carcinoma. Mod Pathol. 2001;14(5):428–436. doi: 10.1038/modpathol.3880330. [DOI] [PubMed] [Google Scholar]
- Li L, Yu H, Schumacher F, Casey G, Witte JS. Relation of serum insulin-like growth factor-I (IGF-I) and IGF binding protein-3 to risk of prostate cancer (United States) Cancer Causes Control. 2003;14(8):721–726. doi: 10.1023/a:1026383824791. [DOI] [PubMed] [Google Scholar]
- Liao Y, Grobholz R, Abel U, Trojan L, Michel MS, Angel P, Mayer D. Increase of AKT/PKB expression correlates with Gleason pattern in human prostate cancer. Int J Cancer. 2003;107(4):676–680. doi: 10.1002/ijc.11471. [DOI] [PubMed] [Google Scholar]
- Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci U S A. 2001;98(13):7200–7205. doi: 10.1073/pnas.121173298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Kirschenbaum A, Yu K, Yao S, Levine AC. Cyclooxygenase-2 suppresses hypoxia-induced apoptosis via a combination of direct and indirect inhibition of p53 activity in a human prostate cancer cell line. J Biol Chem. 2005;280(5):3817–3823. doi: 10.1074/jbc.M406577200. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zhang J, Dai J, Dehne LA, Mizokami A, Yao Z, Keller ET. Osteoblasts induce prostate cancer proliferation and PSA expression through interleukin-6-mediated activation of the androgen receptor. Clin Exp Metastasis. 2004;21:399–408. doi: 10.1007/s10585-005-0056-6. [DOI] [PubMed] [Google Scholar]
- Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R, Kreisberg JI. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin Cancer Res. 2002;8(4):1168–1171. [PubMed] [Google Scholar]
- Matthews E, Yang T, Janulis L, Goodwin S, Kundu SD, Karpus WJ, Lee C. Down-regulation of TGF-beta1 production restores immunogenicity in prostate cancer cells. Br J Cancer. 2000;83(4):519–525. doi: 10.1054/bjoc.2000.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty MF. Targeting multiple signaling pathways as a strategy for managing prostate cancer: Multifocal signal modulation therapy. Integr. Cancer Ther. 2004;3(4):349–380. doi: 10.1177/1534735404270757. [DOI] [PubMed] [Google Scholar]
- McEleny KR, Watson RWG, Fitzpatrick JM. Defining the role for the inhibitors of apoptosis proteins in prostate cancer. Prost Ca Prostat Dis. 2001;4:28–32. doi: 10.1038/sj.pcan.4500502. [DOI] [PubMed] [Google Scholar]
- McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers R. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–4296. [PubMed] [Google Scholar]
- Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. Her2/neu kinase dependent modulation of androgen receptor function through the effects on DNA binding and stability. Cancer Cell. 2004;6:517–527. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Michalaki V, Syrigos K, Charles P, Waxman J. Serum levels of IL-6 and TNF-a correlate with clinicopathological features and patient survival in patients with prostate cancer. Br J Cancer. 2004;90:2312–2316. doi: 10.1038/sj.bjc.6601814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimeault M, Pommery N, Henichart JP. New advances on prostate carcinogenesis and therapies: Involvement of EGF-EGFR transduction system. Growth Factors. 2003;21(1):1–14. doi: 10.1080/0897719031000094921. [DOI] [PubMed] [Google Scholar]
- Miyake H, Nelson C, Rennie PS, Gleave ME. Overexpression of insulin-like growth factor binding protein-5 helps accelerate progression to androgen-independence in the human prostate LNCaP tumor model through activation of phosphatidylinositol 3′-kinase pathway. Endocrinology. 2000;141(6):2257–2265. doi: 10.1210/endo.141.6.7520. [DOI] [PubMed] [Google Scholar]
- Moschos SJ, Mantzoros CS. The role of the IGF system in cancer: From basic to clinical studies and clinical applications. Oncology. 2002;63(4):317–332. doi: 10.1159/000066230. [DOI] [PubMed] [Google Scholar]
- Movsas B, Chapman JD, Greenberg RE, Hanlon AL, Horwitz EM, Pinover WH, Stobbe C, Hanks GE. Increasing levels of hypoxia in prostate carcinoma correlate significantly with increasing clinical stage and patient age: An Eppendorf pO(2) study. Cancer. 2000;89(9):2018–2024. doi: 10.1002/1097-0142(20001101)89:9<2018::aid-cncr19>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- Muenchen HJ, Lin DL, Walsh MA, Keller ET, Pienta KJ. Tumor necrosis factor-alpha-induced apoptosis in prostate cancer cells through inhibition of nuclear factor-κB by an IκBα “super-repressor”. Clin Cancer Res. 2000;6(5):1969–1977. [PubMed] [Google Scholar]
- Navone NM, Troncoso P, Pisters LL, Goodrow TL, Palmer JL, Nichols WW, von Eschenbach AC, Conti CJ. p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. J Natl Cancer Inst. 1993;85(20):1657–1669. doi: 10.1093/jnci/85.20.1657. Abstract. [DOI] [PubMed] [Google Scholar]
- Nelson WG, De Marzo AM, DeWeese TL, Isaacs WB. The role of inflammation in the pathogenesis of prostate cancer. J Urol. 2004;172(5Pt 2):S6–S11. doi: 10.1097/01.ju.0000142058.99614.ff. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Miller SF. Mitochondria: Releasing the power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4(8):592–603. doi: 10.1038/nrc1412. [DOI] [PubMed] [Google Scholar]
- Orio F, Jr, Terouanne B, Georget V, Lumbroso S, Avances C, Siatka C, Sultan C. Potential action of IGF-1 and EGF on androgen receptor nuclear transfer andt ransactivation in normal and cancer human prostate cell lines. Mol Cell Endocrinol. 2002;198(1–2):105–114. doi: 10.1016/s0303-7207(02)00374-x. [DOI] [PubMed] [Google Scholar]
- Papandreou CN, Logothetis CJ. Bortezomib as a potential target for prostate cancer. Cancer Res. 2004;64:5036–5043. doi: 10.1158/0008-5472.CAN-03-2707. [DOI] [PubMed] [Google Scholar]
- Park J, Lee M, Cho K, Park B, Chae K, et al. Transforming growth factor-β1 activates interleukin-6 expression in prostate cancer cells through synergistic collaboration of the smad-2, p38-NF-kB, JNK, and Ras signaling pathways. Oncogene. 2003;22:4314–4332. doi: 10.1038/sj.onc.1206478. [DOI] [PubMed] [Google Scholar]
- Partin JV, Anglin IE, Kyprianou N. Quinazoline-based α1-adrenoceptor antagonists induce prostate cancer cell apoptosis via TGF-β signaling and IκBα induction. Br J Cancer. 2003;88(10):1615–1621. doi: 10.1038/sj.bjc.6600961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeil K, Eder IE, Putz T, Ramoner R, Culig Z, Ueberall F, Bartsch G, Klocker H. Long-term androgen-ablation causes increased resistance to PI3K/Akt pathway inhibition in prostate cancer cells. Prostate. 2004;58(3):259–268. doi: 10.1002/pros.10332. [DOI] [PubMed] [Google Scholar]
- Pisters LL, Pettaway CA, Troncoso P, McDonnell TJ, Stephens LC, Wood CG, Do KA, Brisbay SM, Wang X, Hossan EA, Evans RB, Soto C, Jacobson MG, Parker K, Merritt JA, Steiner MS, Logothetis CJ. Evidence that transfer of functional p53 protein results in increased apoptosis in prostate cancer. Clin Cancer Res. 2004;10(8):2587–2593. doi: 10.1158/1078-0432.ccr-03-0388. [DOI] [PubMed] [Google Scholar]
- Pu YS, Hour TC, Chuang SE, Cheng AL, Lai MK, Kuo ML. Interleukin-6 is responsible for drug resistance and anti-apoptotic effects in prostatic cancer cells. Prostate. 2004;60:120–129. doi: 10.1002/pros.20057. [DOI] [PubMed] [Google Scholar]
- Rokhlin OW, Gudkov AV, Kwek S, Glover RA, Gewies AS, Cohen MB. p53 is involved in tumor necrosis factor-alpha-induced apoptosis in the human prostatic carcinoma cell line LNCaP. Oncogene. 2000;19(15):1959–1968. doi: 10.1038/sj.onc.1203453. [DOI] [PubMed] [Google Scholar]
- Ross JS, Kallakury BV, Sheehan CE, Fisher HA, Kaufman RP, Jr, Kaur P, Gray K, Stringer B. Expression of nuclear factor-kappa B and IκBα proteins in prostatic adenocarcinomas: Correlation of nuclear factor-κB immunoreactivity with disease recurrence. Clin Cancer Res. 2004;10(7):2466–2472. doi: 10.1158/1078-0432.ccr-0543-3. [DOI] [PubMed] [Google Scholar]
- Scherr DS, Vaughan ED, Jr, Wei J, Chung M, Felsen D, Allbright R, Knudsen BS. BCL-2 and p53 expression in clinically localized prostate cancer predicts response to external beam radiotherapy. J Urol. 1999;162:12–16. doi: 10.1097/00005392-199907000-00003. Discussion 16–17. [DOI] [PubMed] [Google Scholar]
- Schimmer AD. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004;64:7183–7190. doi: 10.1158/0008-5472.CAN-04-1918. [DOI] [PubMed] [Google Scholar]
- Shah AH, Tabayoyong WB, Kundu SD, Kim SJ, Van Parijs L, Liu VC, Kwon E, Greenberg NM, Lee C. Suppression of tumor metastasis by blockade of transforming growth factor beta signaling in bone marrow cells through a retroviral-mediated gene therapy in mice. Cancer Res. 2002;62(24):7135–7138. [PubMed] [Google Scholar]
- Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58:1008–1015. doi: 10.1016/s0090-4295(01)01405-4. [DOI] [PubMed] [Google Scholar]
- Shariat SF, Menesses-Diaz A, Kim IY, Muramoto M, Wheeler TM, Slawin KM. Tissue expression of transforming growth factor-beta1 and its receptors: Correlation with pathologic features and biochemical progression in patients undergoing radical prostatectomy. Urology. 2004a;63(6):1191–1197. doi: 10.1016/j.urology.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Shariat SF, Kattan MW, Traxel E, Andrews B, Zhu K, Wheeler TM, Slawin KM. Association of pre- and postoperative plasma levels of transforming growth factor β (1) and interleukin 6 and its soluble receptor with prostate cancer progression. Clin Cancer Res. 2004b;10(6):1992–1999. doi: 10.1158/1078-0432.ccr-0768-03. [DOI] [PubMed] [Google Scholar]
- Shaw YJ, Yang YT, Garrison JB, Kyprianou N, Chen CS. Pharmacological exploitation of the alpha1-adrenoreceptor antagonist doxazosin to develop a novel class of antitumor agents that block intracellular protein kinase B/Akt activation. J Med Chem. 2004;47(18):4453–4462. doi: 10.1021/jm049752k. [DOI] [PubMed] [Google Scholar]
- Shuch B, Mikhail M, Satagopan J, Lee P, Yee H, Chang C, Cordon-Cardo C, Taneja SS, Osman I. Racial disparity of epidermal growth factor receptor expression in prostate cancer. J Clin Oncol. 2004;22(23):4725–4729. doi: 10.1200/JCO.2004.06.134. [DOI] [PubMed] [Google Scholar]
- Shukla S, Gupta S. Suppression of constitutive and tumor necrosis factor alpha- induced nuclear factor (NF)-κB activation and induction of apoptosis by apigenin in human prostate carcinoma PC-3 cells: Correlation with down-regulation of NF-κB-responsive genes. Clin Cancer Res. 2004;10(9):3169–3178. doi: 10.1158/1078-0432.ccr-03-0586. [DOI] [PubMed] [Google Scholar]
- Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279(44):45643–45651. doi: 10.1074/jbc.M404097200. [DOI] [PubMed] [Google Scholar]
- Smith PC, Keller ET. Anti-interleukin-6 monoclonal antibody induces regression of human prostate cancer xenografts in nude mice. Prostate. 2001;48:47–53. doi: 10.1002/pros.1080. [DOI] [PubMed] [Google Scholar]
- Soussi T, Dehouche K, Beroud C. p53 website and analysis of p53 gene mutations in human cancer: Forging a link between epidemiology and carcinogenesis. Hum Mutat. 2000;15:105–113. doi: 10.1002/(SICI)1098-1004(200001)15:1<105::AID-HUMU19>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Stattin P, Rinaldi S, Biessy C, Stenman UH, Hallmans G, Kaaks R. High levels of circulating insulin-like growth factor-I increase prostate cancer risk: A prospective study in a population-based nonscreened cohort. J Clin Oncol. 2004;22(15):3104–3112. doi: 10.1200/JCO.2004.10.105. [DOI] [PubMed] [Google Scholar]
- Stern DF. More than a marker … phosphorylated Akt in prostatic carcinoma. Clin Cancer Res. 2004;10:6407–6410. doi: 10.1158/1078-0432.CCR-04-1783. [DOI] [PubMed] [Google Scholar]
- Suh J, Rabson AB. NF-κB activation in human prostate cancer: Important mediator or epiphenomenon? J Cell Biochem. 2004;91:100–117. doi: 10.1002/jcb.10729. [DOI] [PubMed] [Google Scholar]
- Sumitomo M, Tachibana M, Nakashima J, Murai M, Miyajima A, Kimura F, Hayakawa M, Nakamura H. An essential role for nuclear factor kappa B in preventing TNF-alpha-induced cell death in prostate cancer cells. J Urol. 1999;161(2):674–679. [PubMed] [Google Scholar]
- Teicher BA. Malignant cells, directors of the malignant process: Role of transforming growth factor-β. Cancer Metastasis Rev. 2001;20(1–2):133–143. doi: 10.1023/a:1013177011767. [DOI] [PubMed] [Google Scholar]
- Torring N, Hansen FD, Sorensen BS, Nexo E, Hynes NE. ErbB1 and prostate cancer: ErbB1 activity is essential for androgen-induced proliferation and protection from the apoptotic effects of ly294002. The Prostate. 2003;56:142–149. doi: 10.1002/pros.10245. [DOI] [PubMed] [Google Scholar]
- Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. Inhibition of transforming growth factor-beta activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res. 2002;62(21):6021–6025. [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Vicentini C, Festuccia C, Gravina GL, Angelucci A, Marronaro A, Bologna M. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell lines and primary cultures. J Cancer Res Clin Oncol. 2003;129(3):165–174. doi: 10.1007/s00432-003-0420-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4(3):209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- Wikstrom P, Westin P, Stattin P, Damber JE, Bergh A. Early castration-induced upregulation of transforming growth factor beta1 and its receptors is associated with tumor cell apoptosis and a major decline in serum prostate-specific antigen in prostate cancer patients. Prostate. 1999;38(4):268–277. doi: 10.1002/(sici)1097-0045(19990301)38:4<268::aid-pros2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Xie S, Lin HK, Ni J, Yang L, Wang L, di Sant’Agnese PA, Chang C. Regulation of interleukin-6-mediated PI3K activation and neuroendocrine differentiation by androgen signaling in prostate cancer LNCaP cells. Prostate. 2004;60:61–67. doi: 10.1002/pros.20048. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Whang YE. PTEN sensitizes prostate cancer cells to death receptor-mediated and drug-induced apoptosis through a FADD-dependent pathway. Oncogene. 2002;21(2):319–327. doi: 10.1038/sj.onc.1205054. [DOI] [PubMed] [Google Scholar]
- Zeng L, Rowland RG, Lele SM, Kyprianou N. Apoptosis incidence and protein expression of p53, TGF-beta receptor II, p27Kip1, and Smad4 in benign, premalignant, and malignant human prostate. Hum Pathol. 2004;35(3):290–297. doi: 10.1016/j.humpath.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Wang Y, Cho JY, Libermann TA. Constitutive activation of nuclear factor kappaB p50/p65 and Fra-1 and JunD is essential for deregulated interleukin 6 expression in prostate cancer. Cancer Res. 2003;63(9):2206–2215. [PubMed] [Google Scholar]
- Zhang X, Jin TG, Yang H, DeWolf WC, Khosravi-Far R, Olumi AF. Persistent c-FLIP(L) expression is necessary and sufficient to maintain resistance to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in prostate cancer. Cancer Res. 2004;64(19):7086–7091. doi: 10.1158/0008-5472.CAN-04-1498. [DOI] [PubMed] [Google Scholar]