

Abstract
Alterations in the inflammatory process, neuronal death, and glia response have been observed under manipulation of the interleukin-1 (IL-1) cytokine and subsequent signaling through the type 1 IL-1 receptor (IL-1R1). To investigate the influence of IL-1R1 activation in the pathophysiology of a chemical-induced injury to the murine hippocampus, we examined the level and pattern of neuronal death and neuroinflammation in 25-day-old male mice exposed to trimethyltin hydroxide (2.0 mg/kg, i.p.). In IL-1R1 null (IL-1R1−/−) mice, the pattern and severity of dentate granule cell death was similar as compared to wild type mice. In both groups of mice, mRNA levels for TNFα and MIP-1α were elevated and the early activation of microglia, including their ability to progress to a phagocytic phenotype, was maintained. Compared to WT mice, IL-1R1−/− mice displayed a limited glial fibrillary acidic protein (GFAP) astrocytic response, as well as a preferential induction in mRNA levels of Fas signaling components. Cumulatively, these results indicate that IL-1R1 activation is not necessary for TMT-induced death of dentate granule neurons or local activation of microglia; however, IL-1R1 signaling is involved in mediating the structural response of astrocytes to injury and may also regulate apoptotic mechanisms by influencing Fas signaling components.
Keywords: Disorders of the Nervous System, neurotoxicity, inflammation, IL-1, cytokine, trimethyltin, hippocampus
1. Introduction
Neuronal death following injury activates many cellular changes in the damaged tissue area. In addition to the activation of local glia and often the recruitment of blood-borne leukocytes, local synthesis of inflammation-related cytokines, such as interleukin-1 (IL-1) and tumor necrosis factor alpha (TNFα), exerts a wide range of effects. These effects are mediated primarily by IL-1 type 1 receptor (IL-1R1) and TNF receptor 1 (TNFp55R), respectively. While a pathophysiological role for IL-1 as part of the inflammatory process in the brain has been established (Rothwell and Luheshi, 2000; Allan and Rothwell, 2001), exactly how the signaling process may contribute to neuronal death and protection is still unclear. However, data clearly support its role as a key mediator of inflammation and neuronal death in acute brain injuries, such as stroke and trauma (Allan et al., 2005).
IL-1α and IL-1β are the more characterized forms of IL-1 (Rothwell and Luheshi, 2000). An increase in IL-1β expression has been demonstrated under various neurodegenerative conditions, as well as experimental models of brain injury (Griffin et al., 1989; Mogi et al., 1994; Zhao & Schwartz, 1998; Rothwell, 2003). The earliest source of IL-1 after injury appears to be microglia, possibly due to the expression of the converting enzyme caspase 1 (Eriksson et al., 1999). This would allow for a rapid production of the active form of IL-1β upon cleavage of pro-IL-1β. Upon stimulation, IL-1 can induce release of growth factors, decrease glutamate release, enhance the effects of GABA, increase the activation of inducible nitric oxide synthase, and increase the production of inflammatory mediators, thus establishing a cycle of inflammation (Rothwell and Luheshi, 2000; Boutin et al., 2001; Griffin, 2006).
Many of the detrimental effects of IL-1 are thought to occur through the activation of IL-1R1 (Dinarello, 1996). IL-1R1 binding initiates a sequence of protein-protein interactions forming a complex to recruit the IL-1 receptor accessory protein (IL-1RAcP) and induce cell signaling (Greenfeder et al., 1995). Subsequent binding of the IL-1β/IL-1R1 complex with the adaptor protein, MyD88, facilitates the incorporation and phosphorylation of interleukin-1 receptor associated kinase IRAK (Wesche et al., 1997). The phosphorylation of IRAK can lead to the activation of the mitogen-activated protein kinase, c-Jun N-terminal kinase, JNK (O’Neill and Green, 1998). The induction of JNK via IL-1β signaling has been reported in various cell types and within the hippocampus (Uciechowski et al., 1996; Vereker et al., 2000).
The involvement of IL-1 in excitotoxicity, seizure activity, memory, and depression suggests a susceptibility to IL-1 effects in the hippocampus. This has been supported by the differential effect of IL-1β on excitotoxicity, with the increased death of primary hippocampal neurons (Viviani et al., 2003), yet a reduction in death of cultured cortical neurons (Strijbos and Rothwell, 1995). IL-1R1 is expressed by granule cells of the dentate gyrus and by CA1 and CA3 pyramidal neurons (Cunningham et al., 1992; Ericsson et al, 1995). In addition, neutralization of IL-1 actions on IL-1R1 by the IL-1 receptor antagonist (IL-1ra) offers protection to neurons against ischemia, trauma, excitotoxic damages, and chemical induced seizures (Touzani et al., 1999; Toulmond and Rothwell, 1995). A transient decrease in receptor densities in the dentate gyrus of the hippocampus has been reported within 24 hrs following focal trauma, coinciding with an increase in IL-1ra (Gabellec et al., 1999). In the cortex, the role for IL-1R1 has been characterized and demonstrated to be associated with the progressive neuronal degeneration that follows a hypoxic-ischemic insult (Basu et al., 2002; 2005). Basu et al., (2005) found that while mice lacking IL-1R1 showed a limited protection from an the acute injury response, they did show neuroprotection with regards to the second wave of neuronal death and the progressive enlargement of infarct area suggested to occur as result of neuroinflammation.
Dentate granule neurons are not often considered to be a vulnerable population; however, damage to this cell population has been reported in experimental models of ischemia, hypoxia, and seizures (Harry and Lefebvre d’Hellencourt, 2003 review). While IL-1 receptor signaling has been implicated in cortical and hippocampal pyramidal cell death following trauma, ischemia, and excitotoxicity, the generalized nature of these effects and influence on the dentate granule cell population has not been examined. To investigate whether IL-1 signaling through IL-1R1 mediates damage to the dentate granule neurons, we employed a chemical injury that, in mice, initiates neuronal death within 24 hrs. Previous reports using the trimethyltin (TMT) murine model of brain damage indicated an associated neuroinflammatory response, including an increase in IL-1 (Bruccoleri et al., 1998; Fiedorowicz et al., 2001) and the induction of a microglia response during an active phase of neuronal death. In the current study, we evaluated the hippocampus at a timepoint representative of the initial stage of neuronal damage induced by TMT. Wildtype mice and mice deficient for IL-1R1 (IL-1R1−/−) were examined for changes in the severity of neuronal damage and the associated glia response as a framework to identify IL-1 involvement in the initiation of cell death within the dentate granule region. We report that the absence of IL-1R1 did not provide neuroprotection nor alter the associated microglia response. Consistent with the literature, IL-1R1−/− mice displayed an attenuated GFAP astrocyte response and we now report a significant induction of components of the Fas signaling pathway in receptor-null mice.
2. Results
Elevated inflammatory signals in wildtype mice
Previous studies have demonstrated elevations in TNFα and IL-1α both in vitro (Harry et al., 2002; Pompili et al., 2006) and in vivo following TMT (Bruccoleri et al., 1998; Jahnke et al., 2001). As measured by quantitative real-time PCR (qRT-PCR) a statistically significant increase was seen in TNFα and IL-1ra at 24 hrs post-TMT, as compared to mice injected with saline (Fig. 1). mRNA levels for IL-1α and IL-1β were slightly elevated by approximately 1.5-fold at 24 hrs but did not reach statistical significance (Fig. 1). mRNA levels for MyD88, an adaptor protein that is recruited to the IL-1/IL-1R1 complex, were not significantly elevated at 24 hrs post-TMT (Fig. 1). To determine if an elevation in mRNA levels for the associated cytokine receptors would occur at a later time point, hippocampal samples collected between 1 to 14 days post-TMT were examined by RNase Protection Assay (RPA). The overall pattern suggested an increase in transcript levels at 48 hrs post-TMT (Fig. 2). mRNA levels for TNF receptors were elevated at 48 hrs, with a statistically significant 2.5-fold increase in TNFp55R and a 1.6-fold increase in TNFp75R that failed to reach statistical significance (Fig. 2). Slight elevations were seen at 48 hr for IL-1R1, IL-6Rα, and gp130 that failed to reach statistical significance (Fig. 2). Any elevation in receptor transcripts was transient and returned to control levels by 14 days post-TMT. TGFβR1 and TGFβR2 mRNA levels were not increased by TMT during the 14-day period.
Figure 1.
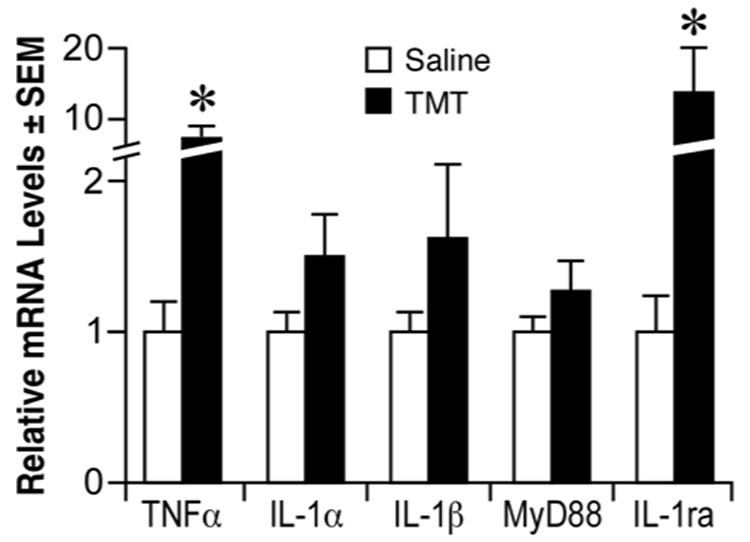
Quantitative real-time PCR for mRNA levels of TNFα, IL-1α, IL-1β, MyD88, and IL-1ra in the hippocampus 24 hours post-TMT (2.0 mg/kg, i.p.). Data are presented as fold change (mean +/− SEM) over the average control for each transcript. *Significant difference between saline controls (n=5) and TMT dosed (n=7) mice (Mann-Whitney U test, p<0.05)
Figure 2.

Time course of changes in hippocampal mRNA levels for IL-1R1, IL-1R2, TNFp55R, TNFp75R, IL-6Rα, gp130, TGFβR1, and TGFβR2 from saline control to 1, 2, 3, 7, and 14 days following TMT (2.0 mg/kg, i.p.) as determined by RNase Protection Assay (RPA). On graphs, x-axis represents day(s) post-TMT, and y-axis represents mean fragment volume over L32 volume. *Significant difference between saline controls (n=6) and TMT dosed (n=6) mice (ANOVA; independent group mean analysis - Fisher’s LSD test, p<0.05).
Lack of neuroprotection in IL-1R1−/− mice
While we failed to identify an elevation in the IL-1 receptor or ligand at the 24 hr period, previous studies report an elevation slightly later (Bruccoleri et al., 1998). Although this temporal pattern suggests that involvement of IL-1R1 may be after the time of initial cell death, an alternative explanation for the lack of significant elevation involves a dilution effect. An analysis of the entire hippocampus may dilute the ability to detect changes localized to a sub-population of cells. Thus, to examine the influence of IL-1R1, TMT-induced neuronal death and associated glia response was examined in IL-1R1−/− mice and matched wildtype (WT) controls. At 24 hrs after exposure, TMT produced localized death of dentate granule neurons characterized by nuclear pyknosis and karyolysis (Fig. 3C, D). The absence of IL-1R1 did not alter either the pattern or severity of neuronal death in the hippocampus, as demonstrated in representative images (Fig. 3D). Scoring the severity based on number and location of eosin-positive cells confirmed comparable levels of damage in WT (2.25 ± 0.46) and IL-1R1−/− (2.0 ± 0.58) mice. When mice from the same dosing cohort were examined at 72 hrs post-TMT, the severity of damage to the dentate neurons and cellular loss were similar in both groups (Fig. 3E, F); thus, the absence of the receptor did not influence the progressive nature of the insult and both groups of mice displayed an injury response consistent with a severity score of 4, inclusive of neuronal loss. Consistent with previous reports on TMT histopathology in mice, pyramidal neurons of the CA layers showed no indication of damage in either group and the average severity score remained approximately 0.5.
Figure 3.
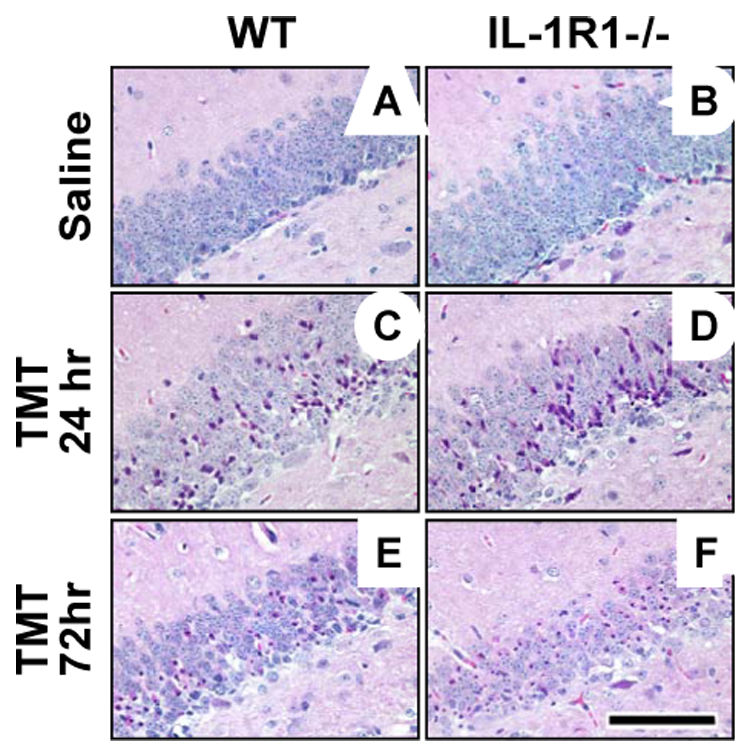
Representative hematoxylin and eosin (H&E) staining in the suprapyramidal blade of the dentate gyrus in wildtype (WT) and IL-1R1−/− mice under control conditions (A, B) and following TMT treatment (2.0 mg/kg, i.p., C–F). No differences were observed between the groups at basal level (A, B), or 24 hrs following TMT (C, D). Severity scoring of the insult based on number and location of eosin positive cells verified comparable levels of damage in WT (C, 2.25 ± 0.46) and IL-1R1−/− (D, 2.0 ± 0.58) mice at 24 hrs. Examining WT and IL-1R1−/− mice at 72 hrs post-TMT (E, F) resulted in each group reaching a severity score of 4, indicative of granule cell loss. Scale bar = 100µm.
Altered Astrocyte Response in IL-1R1−/− mice
In the normal WT mice, GFAP+ astrocytes displayed long fibrous processes projecting thorough the blades of the dentate gyrus (Fig. 4A). A progression of astrocyte response was evident following TMT. At 24 hrs, the GFAP+ astrocytes displayed increased staining density, a loss of processes extending throughout the dentate blades, and an increase in cells displaying shorten processes and slight evidence of hypertrophy (Fig. 4C). By 72 hrs, a dramatic increase in astrocyte hypertrophy was evident throughout the blades of the dentate gyrus (Fig. 4E). While CA pyramidal neurons were not damaged, the astrocyte response was evident throughout the hippocampus, including the pyramidal cell layer (Fig. 5).
Figure 4.
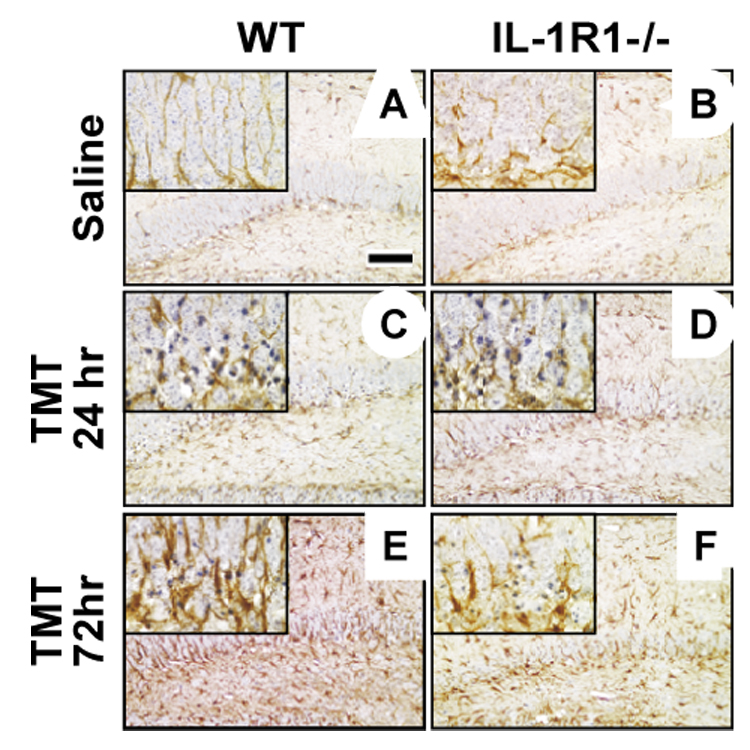
Representative images with high magnification inserts of glial fibrillary acidic protein (GFAP) staining for astrocytes in the suprapyramidal blade of the dentate gyrus in wildtype (WT) and IL-1R1−/− mice. (A) WT mice displayed astrocytes with long processes through the dentate blade under control conditions, (C) dense cells with truncated processes at 24 hrs, and (E) astrocyte hypertrophy at 72 hrs post-TMT (2.0 mg/kg i.p.). (B) IL-1R1−/− mice displayed astrocytes with fewer projections through the dentate blade under control conditions, (E) increased GFAP staining and elongated processes at 24 hrs, and (F) dense cells with truncated processes and less GFAP+ immunoreactivity compared to WT matches at 72 hrs post-TMT. Hematoxylin counterstain. Scale bar = 100µm.
Figure 5.
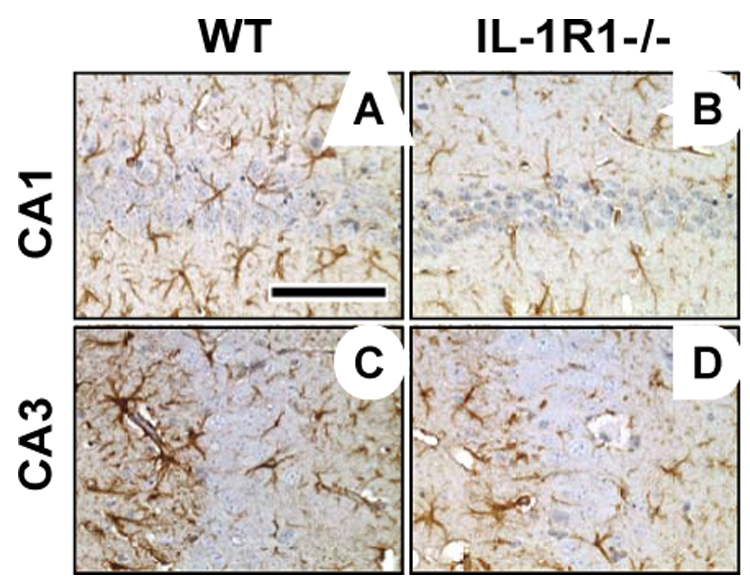
Representative glial fibrillary acidic protein (GFAP) staining for astrocytes in the uninjured CA1 (A, B) and CA3 (C, D) in wildtype (WT) and IL-1R1 knockout (IL-1R1−/−) mice at 72 hrs post-TMT (2.0 mg/kg i.p.). Treated IL-1R1−/− mice (B, D) displayed an attenuated astrocyte response in each region as compared to WT mice (A, C). Hematoxylin counterstain. Scale bar = 100µm.
In IL-1R1−/− mice, the basal expression of GFAP+ astrocytes was similar to that seen in the wildtype mice; however, a decrease was seen in astrocyte processes projecting through the blades of the dentate gyrus, as compared to the WT control (Fig. 4B). The astrocyte response to TMT was characterized at 24 hrs by an increase in immunostaining for GFAP and slightly thickened processes projecting through the blades of the dentate gyrus (Fig. 4D). At 72 hrs, astrocytes displayed shortened dense processes, yet were deficient within the blades of the dentate (Fig. 4F). This attenuation of the GFAP+ astrocyte response was evident throughout the hippocampus, including in the uninjured pyramidal cell layer (Fig. 4, 5), suggesting that the altered response of astrocytes in the IL-1R1−/− mouse represented a general response to multiple signals in addition to those from injured neurons.
Microglia Response
In the normal WT mice, lectin staining for microglia showed a minimal number of cells with very thin processes in the dentate granule cell region (Fig. 6A). By 24 hrs post-TMT, a greater number of lectin+ microglia could be detected within the dentate granule cell layer. These cells primarily displayed a ramified morphology characteristic of reactive non-phagocytic microglia (Fig. 6C). By 72 hrs, ramified lectin+ microglia were still present in the dentate gyrus; however, the prominent morphology was amoeboid and characteristic of activated microglia with a phagocytic phenotype (Fig. 6E). Consistent with the lack of neuronal damage and findings from previous studies, no amoeboid microglia response was seen in the CA pyramidal cell layer (data not shown).
Figure 6.

Representative images with high magnification inserts of Griffonia simplicifolia isolectin B4 staining for microglia activity in the suprapyramidal blade of the dentate gyrus of wildtype (WT) and IL-1R1 knockout (IL-1R1−/−) mice. In saline controls, minimal staining of microglia with thin ramified processes was seen in both the WT (A) and IL-1R1−/− (B) mice. At 24 hrs post-TMT (2.0 mg/kg i.p.), increased staining of lectin+ microglia displayed ramified microglia with shortened, thick processes in both WT (C) and IL-1R1−/− (D) mice. By 72 hrs post-TMT, reactive and activated microglia with an amoeboid phagocytic morphology were evident in both WT (E) and IL-1R1−/− (F) mice. Scale bar = 100µm.
In IL-1R1−/− mice, lectin staining in the hippocampus under control conditions showed a similar pattern to that seen in the WT mice (Fig. 6B). Twenty-four hrs following TMT, lectin staining for microglia was increased and the cells displayed a ramified morphology (Fig. 6D), and by 72 hrs, the microglia response progressed to an activated phagocytic phenotype (Fig. 6F) similar to that seen in the in the WT mice at each time point (Fig. 6C, E). The pattern of response to TMT throughout the hippocampus of IL-1R1−/− mice was comparable to that seen in WT mice.
RPA for Pro-inflammatory Associated Genes
Under normal control conditions, basal mRNA levels for IL-1α, MIP-1α, TNFα, and TNFβ were similar in the IL-1R1−/− mice as compared to the WT control. A significantly lower basal level of TGFβ1 was observed in mice deficient in IL-1R1 (Fig. 7). At 24 hrs post-TMT, mRNA levels for MIP-1α were significantly elevated in WT mice. TNFα mRNA levels showed a slight elevation that was confirmed by qRT-PCR to be significantly elevated (11-fold, data not shown) similar to levels represented in Figure 1. No changes were seen in mRNA levels for IL-1α, TNFβ, and TGFβ1 (Fig. 7). In IL-1R1−/− mice, a statistically significant elevation was seen for MIP-1α and TGFβ1 mRNA levels, with no changes seen for IL-1α or TNFβ (Fig. 7). Elevations in TNFα mRNA levels were similar to those seen in WT mice as detected with either the RPA or qRT-PCR.
Figure 7.
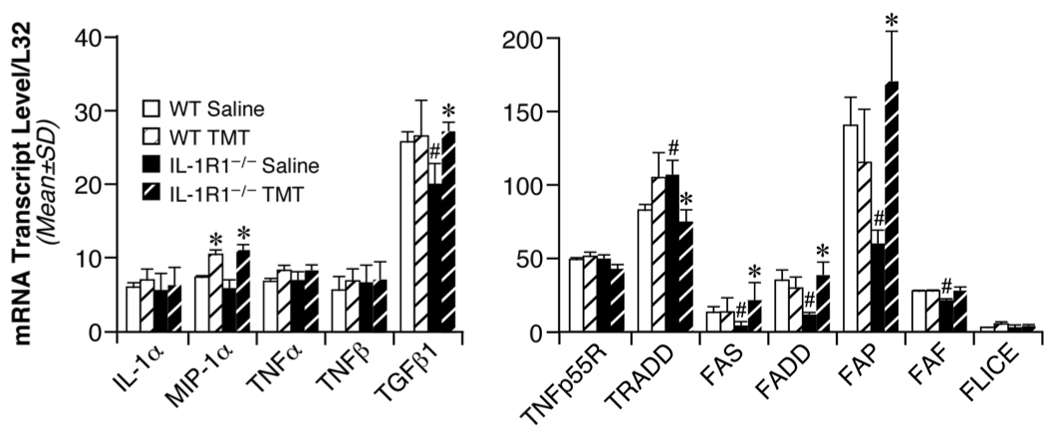
(Left) mRNA levels for inflammatory cytokines IL-1α, MIP-1α, TNFα, TNFβ, and TGFβ1 in the hippocampus of wildtype (WT) and IL-1R1 knockout (IL-1R1−/−) mice as determined by RNase Protection Assay (RPA) 24 hr post-TMT (2.0 mg/kg, i.p.). Data represent mean transcript level/L32 (+/− SD). (Right) mRNA levels for apoptotic genes TNFp55R, TRADD, Fas, FADD, FAP, FAF, and FLICE in the hippocampus of wildtype (WT) and IL-1R1 knockout (IL-1R1−/−) mice as determined by RNase Protection Assay (RPA) at 24 hrs post-TMT (2.0 mg/kg, i.p.). Data represent mean transcript level/L32 (+/− SD). #Significant difference between saline dosed WT and IL-1R1−/− mice. *Significant difference between saline and TMT-dosed mice within either the WT or IL-1R1−/− group (ANOVA; independent group mean analysis - Fisher’s LSD test, p<0.05; n=6).
RPA for Apoptosis Related Genes
Under normal control conditions, basal mRNA levels for tumor necrosis factor receptor type 1 (TNFp55R) and Fas-linked ICE-like protease (FLICE, caspase-8) were similar between the two groups of mice (Fig. 7). In the IL-1R1−/− mice, basal levels for TNF receptor-associated death domain (TRADD) were significantly elevated over WT mice; while, basal levels for Fas (TNFR superfamily member, CD95), Fas-associated death domain (FADD), Fas-associated phosphatase (FAP), and Fas-associated factor (FAF) were significantly lower than levels seen in the wildtype mice. Upon administration of TMT, no changes were seen in any of these transcripts in the wildtype mice; however, in IL-1R1−/− mice, TMT administration resulted in a significant decrease in mRNA levels for TRADD and an increase in transcripts associated with the Fas signaling pathway (Fig. 7). Significant elevations in Fas/CD95 (4.5-fold), FADD (3.2-fold), FAP (2.8-fold), and FAF (1.3-fold) mRNA levels were detected in the IL-1R1−/− mice administered TMT as compared to matched saline control.
3. Discussion
The principle findings of this study are that signaling via the IL-1R1 is not critical for the death of dentate granule neurons or the activation of microglia following chemical-induced injury. The data support previous observations of an attenuated astrocytic response to insult in mice deficient for IL-1R1 (Lin et al., 2006). In addition, the study offers new data regarding the induction of components of the Fas/FasL (CD95/CD95L) signaling pathway upon injury in IL-1R1−/− mice.
A role of IL-1 in mediating brain injury has been previously suggested. For example, diminishing actions of IL-1 by either IL-1ra or deletion of IL-1 confers a level of protection in distinct models of brain injury (Garcia et al., 1995; Loddick et al., 1997; Boutin et al., 2001). The recent work of Pinteaux et al., (2006) suggests that the neuroprotection offered by IL-1ra is due to its production and release by microglial cells (Eriksson et al., 2000). In the current study, the elevation in TNFα, TNFR1, and IL-1ra at 24 hrs suggests the induction of a neuroinflammatory response with this chemical induced hippocampal injury. While not elevated at 24 hrs, both IL-1α and IL-1β mRNA and protein levels are elevated later in the injury response corresponding to the prominent microglia phagocytosis (Bruccoleri et al., 1998; Fiedorowicz et al., 2001). An elevation of IL-1ra in the absence of an increase in IL-1 is consistent with the findings of Gabellec et al. (1999), which demonstrated a transient down regulation of receptor density and an elevation in IL-1ra in the early stages of traumatic injury. The limited induction of IL-1α and IL-1β mRNA, coupled with a lack of increase in IL-1R1 or MyD88 mRNA, at 24 hrs, along with the absence of any attenuation of the damage in IL-1R1−/− mice at 24 or 72 hrs, supports the minimal involvement of the receptor in regulating damage in the dentate granule neurons as induced by TMT.
While IL-1 shows neurotoxic actions, studies suggest that the protein does not directly initiate brain damage but rather exacerbates induced damage (Davies et al., 1999). Consistent with the neuronal data from the current study, Touzani et al. (2002) showed that, while the lack of both IL-1α and IL-1β provided neuroprotection (Boutin et al., 2001), the absence of IL-1R1 was not sufficient to significantly inhibit the extent of injury from middle cerebral artery occlusion in mice. However, in both wildtype and IL-1R1−/− mice, the addition of IL-1 exacerbated the response. This exacerbation could be inhibited with the addition of IL-1ra in the wildtype mouse with no attenuation of the injury in the knockout mice. This data suggests that IL-1 could be acting through alternate receptor(s) in the absence of the type I receptor (Touzani, 2002). A second receptor for IL-1 does exist; however, IL-1RII is considered a decoy receptor that binds IL-1 but fails to initiate signal transduction. In serving as a decoy, it can limit the biological activity of IL-1 (Colotta et al., 1994). The previously reported localized up-regulation of IL-1R1 restricted to hippocampal regions of originating seizures suggests that a strong focal neuronal activation is required to upregulate this receptor in neurons (Ravizza and Vezzani, 2006). However, this study also reported that TUNEL+ neurons were not the population of neurons expressing IL-1R1 following seizure. This would suggest a lack of association between neuron specific receptor activation and neuronal death under these conditions.
A decrease in neocortical microglia activation has been reported in IL-1R1−/− mice after hypoxia/ischemia (Basu et al., 2005); however, whether this is due to a direct effect on the microglia or secondary to the diminished neuronal death is unknown. Given that the response of microglia cells is often linked to the level of cell damage within any specific brain region, evaluation and conclusions drawn regarding microglia actions can be confounded if neuronal/tissue damage is not equivalent for comparison. In the current study, a focal injury to dentate granule neurons was not altered with the absence of IL-1R1 and the response of microglia was equivalent to that seen in the wildtype mouse. In addition, the temporal and spatial progression of the microglia response followed a similar pattern for both groups. Within 24 hrs, pyknotic dentate granule cell neurons were present and this was accompanied by a response of microglia with a reactive phenotype. This response progressed to an activated phenotype and the phagocytosis of neuronal debris. Within each section and across groups, the relationship between the severity of neuronal death and microglia response was maintained, suggesting that the timing of reaction and functional role of the microglia is not altered in the absence of IL-1R1. This data suggests that IL-1R1 signaling is either not critical for this specific model of damage or more general, for dentate granule neurons. It also demonstrates that the microglia response is not regulated by IL-1R1 signaling given equivalent levels of neuronal damage.
While both the neuronal and microglia response were similar, the astrocyte response was very distinct between the two groups. Parker et al., (2002) suggested that astrocytes are the primary target for IL-1 actions with IL-1R1-dependent activity demonstrated in reactive astrocytes. The expression of IL-1R1 has been linked to reactive astrocytes in a transient manner over 18–48 hrs post-seizure (Ravizza and Vezzanini, 2006) and at 24 hrs following a puncture wound (Friedman, 2001). Mice deficient for IL-1R1 displayed limited astrocytic processes within the dentate blades and a diminished GFAP induction as compared to wildtype mice in response to the injury process. Corresponding with the time at which IL-1 appears to be most influential in the TMT injury (Bruccoleri et al., 1998; Fiedorowicz et al., 2001), the most distinct differences in GFAP immunoreactivity between the groups occurred at 72 hrs. The diminished structural and GFAP response of astrocytes seen in the current study is similar to observations previously reported using a penetrating brain wound (Lin et al., 2006). It is also consistent with observations that hippocampal astrocytes respond to IL-1 (Friedman et al., 2001) and that astrocyte over-expression of IL-1R1 contributes to reactive astrogliosis (Giulian et al., 1988). The specific elevation of TGFβ1 seen in IL-1R1−/− mice following TMT may contribute to the attenuated astrocytes response given its actions on astrocytes and role in scar formation (Lindholm et al., 1992; Laping et al., 1994). These immunosuppressive and wound healing actions of TGFβ1 are thought to control inflammation and to limit the extent of neuronal injury (Sei et al., 1995).
While IL-1R1 mediates IL-1β inhibition of astrocytic glutamate re-uptake (Ye and Sontheimer, 1996; Hu et al., 2000), which could lead to a higher glutamate environment and subsequent neuronal death, the structural differences in astrocytes seen in the IL-1R1−/− mice has not necessarily been correlated with functional changes in glutamate uptake (Lin et al., 2006). Stimulation of IL-1R1 can also induce the production of soluble factors such as, cytokines and growth factors (Croll et al., 2004) and the activation of NFκB in astrocytes (Krohn et al., 1999). These effects can be either beneficial or detrimental to neurons. A recent study by Thornton et al. (2006) used primary cortical cell cultures to examine the influence of astrocytes in the IL-1β toxicity to neurons. The authors concluded that the activation of IL-1R1 in astrocytes and the release of free radicals induce a caspase-dependent death of neurons. This was supported by the in vivo study of mild hypoxia/ischemia showing an attenuation of iNOS-mediated free radical damage in IL-1R1−/− mice (Lazovic et al., 2005). In an acute brain injury, where the intervention of astrocytes to either provide growth factor support or to provide a physical glia barrier, a diminished astrocyte response may indeed be a critical factor in determining the severity of neuronal damage. However, in brain injury that targets a neuronal population directly, such as occurs in the TMT model and in targeted neuronal degenerative diseases, the involvement of astrocytes in the acute phase of neuronal death may not be as critical. Thus, the diminished response of astrocytes in the IL-1R1−/− mice would have little direct consequences on the severity of dentate granule cell death, yet would have a significant impact on a severe neuronal injury involving glia scarring.
The neurotoxicity of IL-1β has been linked to the upregulation of FasL in astrocytes (Ghorpade et al., 2003) and the induction of neuronal apoptosis (Allan and Rothwell, 2001; Deshpande et al., 2005). While we found no protection from the hippocampal injury, the distinct changes seen in the IL-1R1−/− mice suggests an active role for the intracellular Fas signaling pathway that could contribute to the neuroprotection often seen in other models of brain injury. The lack of detectable FasL transcript levels would be consistent with the lack of a prominent astrocyte response at 24 hrs. Both FADD and FAP-1 are Fas receptor associated proteins and, as such, changes in gene expression may reflect the levels and activity of Fas. In the IL-1R1−/− mice, this pattern seems to hold with the lower basal transcript levels and the increase following TMT for all three transcripts. FADD is an adaptor protein for the extrinsic apoptotic receptor Fas, and is essential for multiple signaling events downstream of Fas (Juo et al., 1999). FAP, however, is an anti-apoptotic tyrosine phosphatase that is located in the cytosol and serves as an intracellular regulator of Fas translocation to block the transduction of the death signal (Sato et al., 1995). There is little data regarding the apoptotic signaling pathways recruited via IL-1 or in the absence of IL-1R1 activation; however, it is interesting to note that the changes in Fas-associated genes following TMT occurred exclusively in IL-1R1−/− animals. Although there are numerous mechanisms of signal transduction that are not reflected in transcript levels, such as protein phosphorylation, the fact that message levels for none of the components are modulated in the wildtype animals may suggest that this is not a primary mechanism of cell death in the TMT model. In addition, any activation of the Fas signaling pathway may not automatically be associated with the neuronal damage observed in this model. Up-regulation of FasL/CD95L has been attributed to astrocytic-induced elimination of activated T cells (Bechmann et al., 2002) and apoptosis in neurons (Deshpande et al., 2005), but the mechanism may not necessarily be applied to certain neuronal populations. Indeed, we have investigated the contribution of Fas-specific death within the TMT injury model through immunohistochemical detection of the Fas/CD95 receptor in the dentate gyrus, and its involvement appears to be minimal (data not shown). In injury models where Fas/FasL activation is a significant contributor to neuronal apoptosis, the upregulation of the intracellular phosphatase FAP may provide a mechanism for neuronal survival and contribute to the neuroprotection often seen with the absence of IL-1R1 stimulation. Finally, the possibility that distinct apoptotic mechanisms are contributing to the neuronal damage seen in each group cannot be excluded. It is possible that in the absence of IL-1R1, Fas signaling may be a significant contributor to the observed cell death, but is not as active in wildtype mice. Whether the observed changes in mRNA translate into downstream signaling of Fas/FasL would require further investigation.
Given the similar levels of damage, the TMT injury offers a model to identify differential responses in IL-1R1−/− mice that may contribute to the neuroprotection often seen in other models of brain injury. Inhibition of IL-1 activity on IL-1R1 through various interventions has been proposed as a possible therapeutic intervention for brain injury in the human population. However, data from the current study suggests that the neuroprotective effects of IL-1 inhibition may be injury specific and may not be a process that can be generalized. While IL-1β has been implicated in neurodegeneration, there are also observations indicating that, under certain conditions, IL-1 can contribute beneficial effects (Brenneman et al., 1992), including regulation of ischemic tolerance in CA1 hippocampal neurons (Ohtsuki et al., 1996), anti-convulsive properties such as, slowing the rate of amygdala kindling (Sayyah et al., 2005), and the induction of various neurotrophic factors. In addition, previous evidence reveals that IL-1 demonstrates properties signifying its role as a modulator of memory functions, including effects on long term potentiation and synaptic transmission (Bellinger et al., 1993), as well as, regulating N-methyl-D-aspartate (NMDA) receptors (Ma et al., 2002) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Lai et al., 2006). These properties of IL-1 suggest additional intracellular interactions that have yet to be identified. Thus, future efforts to target IL-1 signaling for therapeutic intervention require a greater level of understanding of the cell biology associated with IL-1 and its pleitrophic actions in the brain.
4. Experimental Procedure
Murine model of dentate granule cell damage
Pathogen free, 30-day-old male C57BL/6J mice (Charles River; Raleigh, NC) were randomly assigned to experimental groups and administered a single intraperitoneal (i.p.) injection of 2.0 mg/kg trimethyltin hydroxide (TMT, originally obtained from Alpha Products, Danvers, MA) or saline, in a dosing volume of 2 mL/kg body weight. Tremor and seizure activity was evident at 24 hrs with no acute morbidity or death. Animals were individually housed in a dual corridor, semi-barrier animal facility (21° + 2°C; 50% + 5% humidity; 12-hr light/dark cycle). Food (autoclaved NIH 31 rodent chow) and deionized, reverse osmotic-treated water were available ad libitum. Sentinel animals recorded negative for pathogenic bacteria, mycoplasma, viruses, ectoparasites, endoparasites. All procedures were conducted in compliance with NIEHS/NIH Animal Care and Use Committee approved animal protocol.
mRNA levels of IL-1, IL-1ra, MyD88, and pro-inflammatory cytokine receptors
C57BL/6J mice were anesthetized with CO2 and decapitated, brains were removed from the cranium, and the hippocampus quickly dissected and immediately frozen on dry ice. Total RNA was isolated with TRIzol® Reagent (Invitrogen™; Carlsbad, CA). Reverse transcription was performed with 2.5 µg total RNA, using a SuperScript™ II Reverse Transcriptase (Invitrogen™; Carlsbad, CA). Real time PCR was carried out on a Perkin Elmer ABI Prism™ 7700 Sequence Detector using 2.5 µL cDNA as template, combined with reaction mixture to give final concentrations of 1X Power SYBR® Green Master Mix (Applied Biosystems; Foster City, CA), and optimized concentrations of forward and reverse primers. 25 µl reaction mixtures were held at 50°C for 2 min, 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 1 min at 60°C. Specificity of primers was verified through analysis of dissociation curves. Amplification curves from individual real-time PCR reactions were generated within Sequence Detection System (SDS) 1.9.1 software program. From amplification plots, threshold cycle values were determined, and mean fold changes were calculated for each transcript induced by TMT over the average saline-treated control using the comparative CT method (Livak and Schmittgen, 2001). GAPDH was used as a housekeeping gene for normalization to an internal control for each sample. Data are presented as a mean fold-change over the average saline-treated control for each mRNA transcript. Sequences and concentrations for primers used in PCR reactions are given in Table 1.
Table 1.
Q-RT-PCR primer sequences and concentrations
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| TNFα | TGGCCTCCCTCTCATCAGTT | GCTTGTCACTCGAAATTTGAGAAG |
| (300nM) | (900nM) | |
| IL-1α | TCGGGAGGAGACGACTCTAA | GGCAACTCCTTCAGCAACAC |
| (600nM) | (600nM) | |
| IL-1β | TGGTGTGTGACGTTCCCATT | CAGCACGAGGCTTTTTTGTTG |
| (300nM) | (300nM) | |
| IL-1ra | CTCCTTCTCATCCTTCTGTTTCATT | TGCATCTTGCAGGGTCTTTTC |
| (300nM) | (300nM) | |
| MyD88 | ACTGGCCTGAGCAACTAGGA | TGTCCCAAAGGAAACACACA |
| (300nM) | (300nM) | |
| GAPDH | GGGAAGCTCACTGGCATGG | CTTCTTGATGTCATCATACTTGGCAG |
| (200nM) | (200nM) |
To determine the elevation and temporal pattern of pro-inflammatory cytokine receptor genes, hippocampal tissue was collected at various time points following TMT (1–14 days). RNase protection assays (RPA) were conducted using the mCR4 multi-probe template set (BD Pharmingen; San Diego, CA) containing probes for IL1-R1, TNF receptor 2 (TNFR2; p75), TNF receptor 1(TNFR1; p55; TNFRSF1A), IL-6Rα, GP130, transforming growth factor beta receptor 1 (TGFβR1), and TGFβR2. A one µl aliquot of an equimolar pool of plasmid templates was used for the synthesis of a 32[P]-labeled cRNA probe set using T7 RNA polymerase (Promega; Madison, WI, USA) and UTP α-32[P] (New England Nuclear; Wilmington, DE). Briefly, a 20 µg aliquot of total RNA was hybridized with 4.4 × 105 cpm of 32[P]-labeled probe overnight at 56°C. Samples were digested in a 100 µl solution of RNase A and RNase T1 cocktail (1:400, Ambion, Austin, TX), treated with 0.5 mg/ml proteinase K (Boeringher Mannheim, Indianapolis, IN), and individually protected fragments separated by 5% acrylamide/8M urea sequencing gel electrophoresis. Radioactivity in each protected fragment was visualized by autoradiography (Kodak XAR film) and phosphor-imaging (Storm Imager, Molecular Dynamics, Sunnyvale, CA). The relative volumes of fragments were determined using Imagequant (Molecular Dynamics). Testing for homogeneity of variance determined that L32 mRNA levels remained relatively constant over time. Therefore, all data was normalized relative to corresponding L32 volume.
Hippocampal Response in IL-1R1ko mice
IL-1R1ko (B6;129S1-IL1r1<tm1Rom1>) and matched wildtype B6:129 controls (Jackson Labs, Bar Harbor, ME) were used for experiments examining differences between groups in response to TMT. Pathogen free, 25-day-old male mice were randomly assigned to experimental groups and administered a single intraperitoneal (i.p.) injection of trimethyltin hydroxide (TMT; 2.0 – 2.4 mg/kg body weight) or saline in a dosing volume of 2 mL/kg body weight. Hippocampal tissue was collected for mRNA levels and histopathology at 24 and 72 hrs following TMT injections.
Based upon the time course for neuronal death and glial response (Bruccoleri et al., 1998), morphological changes in the hippocampus were examined at 24 and 72 hrs post-TMT. Mice were anesthetized with CO2 and decapitated. Brains were removed from the cranium, bisected in the midsagittal plane, and one hemisphere immersion-fixed in 4% paraformaldehyde/0.1M phosphate buffer (PB; pH 7.2; RNase/DNase free) overnight at room temperature (RT). The hippocampus from the contralateral hemisphere was frozen for RNA analysis to maintain individual comparisons. Brains were rinsed with PB, dehydrated in ethanol, embedded in paraffin, and 8-µm sections cut and collected from DEPC water on ProbeOn+™ slides (Fisher Scientific, Norcross, GA). Sections were deparaffinized and ethanol rehydrated and stained with hematoxylin and eosin (H&E) to determine general cellularity and cell death via positive eosin staining. Severity (scale of 0–4) of dentate granule cell death was based upon number and location of eosin+ cells, and structural characteristics of dense and collapsed chromatin.
In staining for microglia and astrocytes, rehydrated 8-µm paraffin sections were transferred to 0.01 M citrate buffer (pH 6.0) and subjected to heat-induced epitope retrieval (HIER) using a decloaking chamber (Biocare Medical, Walnut Creek, CA). Sections were blocked with 10% normal goat serum in PBS containing 1% BSA for 30 min prior to incubation with rabbit polyclonal anti-glial fibrillary acidic protein (GFAP; 1:2000, Dako Corp, Carpinteria, CA; 1 hr) and visualized by diaminobenzidine (DAB) solution. Microglia were identified by lectin binding including pre-incubation for 30 min in 1X Automation Buffer (Biomedia Corp, Foster City, CA) containing 0.1 M CaCl2, MgCl2, MnCl2, and 0.1% Triton X-100, followed by incubation in isolectin B4 from Griffonia simplicifolia (1:75, Sigma, St. Louis, MO; 4°C overnight) and visualized by DAB.
Light microscopy was performed with a Leica DMRBE microscope (Wetzlar, Germany). Digital images were acquired using a SpotRT™ cooled, charged-couple device (CCD) camera (Diagnostic Instruments, Sterling Heights, MI) under the control of Metamorph™ software (Universal Imaging Co., Downingtown, PA).
RPA for inflammatory and apoptotic signals in IL-1R1 knockout mice
From the contralateral brain hemisphere to that collected for histological analysis, the hippocampus was dissected quickly and frozen immediately on dry ice. Total RNA was isolated, samples prepared, and RPAs conducted as previously described in Methods section. The probe set for inflammatory signals (Pharmingen; San Diego, CA) contained probes for TNFα, TNFβ, IL-1α, Mip-1α, TGFβ1, GAPDH, and L32. The mAPO-3 multi-probe template set (Pharmingen, San Diego, CA) contained probes related to both the TNFR1 and Fas pathways, including Fas associated death domain (FADD), FADD-like ICE (FLICE; Caspase 8), FasL, Fas, Fas associated phosphatase (FAP), Fas associated factor (FAF), TNF-related apoptosis-inducing ligand (TRAIL), TNF Receptor 1 associated death domain (TRADD), TNFR1 (p55; TNFRSF1A), receptor interacting protein (RIP), L32, and GAPDH.
Statistical analysis
Statistical significance of group differences between saline controls and TMT exposed wildtype mice for RPA data was conducted by ANOVA and subsequent independent group comparisons for each transcript were conducted using a Fisher’s Least Significant Difference (LSD) post hoc analysis. Group differences between wildtype and knockout mice with and without TMT were assessed using a 2×2 multi-factorial ANOVA and subsequent independent group comparisons were conducted using a Fisher's Least Significant Difference (LSD) post hoc analysis. A one-way ANOVA was used for individual Q-RT-PCR data found to meet criteria of homogeneity of variance; while, a Mann-Whitney U test was used for non-parametric data. All data is presented as mean +/− SEM. All statistical significance levels were set at p < 0.05.
Acknowledgements
This work was supported by the division of intramural research of the National Institute of Environmental Health Science, National Institutes of Health, Department of Health and Human Services. The authors wish to thank Mr. Christopher McPherson, Sue Edelstein, and Drs Michelle Block and Ramen Saha for their review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Basu A, Lazovic J, Krady JK, Mauger DT, Rothstein RP, Smith MB, Levison SW. Interleukin-1 and the interleukin-1 type 1 receptor are essential for the progressive neurodegeneration that ensues subsequent to a mild hypoxic/ischemic injury. J Cereb Blood Flow Metab. 2005;25:17–29. doi: 10.1038/sj.jcbfm.9600002. [DOI] [PubMed] [Google Scholar]
- Basu A, Krady JK, O'Malley M, Styren SD, DeKosky ST, Levison SW. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J Neurosci. 2002;22:6071–6082. doi: 10.1523/JNEUROSCI.22-14-06071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechmann I, Steiner S, Gimsa U, Mor G, Wolf S, Beyer M, Nitsch R, Zipp F. Astrocyte-induced T cell elimination is CD95 ligand dependent. J Neuroimmunol. 2002;132:60–65. doi: 10.1016/s0165-5728(02)00311-9. [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21:5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Schultzberg M, Bartfai T, Gozes I. Cytokine regulation of neuronal survival. J Neurochem. 1992;58:454–460. doi: 10.1111/j.1471-4159.1992.tb09743.x. [DOI] [PubMed] [Google Scholar]
- Bruccoleri A, Brown H, Harry GJ. Cellular localization and temporal elevation of tumor necrosis factor-alpha, interleukin-1 alpha, and transforming growth factor-beta 1 mRNA in hippocampal injury response induced by trimethyltin. J Neurochem. 1998;71:1577–1587. doi: 10.1046/j.1471-4159.1998.71041577.x. [DOI] [PubMed] [Google Scholar]
- Colotta F, Dower SK, Sims JE, Mantovani A. The type II 'decoy' receptor: a novel regulatory pathway for interleukin 1. Immunol Today. 1994;15:562–566. doi: 10.1016/0167-5699(94)90217-8. [DOI] [PubMed] [Google Scholar]
- Croll SD, Goodman JH, Scharfman HE. Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword. Adv Exp Med Biol. 2004;548:57–68. doi: 10.1007/978-1-4757-6376-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham ET, Jr, Wada E, Carter DB, Tracey DE, Battey JF, De Souza EB. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci. 1992;12:1101–1114. doi: 10.1523/JNEUROSCI.12-03-01101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1999;19:87–98. doi: 10.1097/00004647-199901000-00010. [DOI] [PubMed] [Google Scholar]
- Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, Ghorpade A. Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res. 2005;7:183–192. doi: 10.1007/BF03036448. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol. 1995;361:681–698. doi: 10.1002/cne.903610410. [DOI] [PubMed] [Google Scholar]
- Eriksson C, Nobel S, Winblad B, Schultzberg M. Expression of interleukin 1 alpha and beta, and interleukin 1 receptor antagonist mRNA in the rat central nervous system after peripheral administration of lipopolysaccharides. Cytokine. 2000;12:423–431. doi: 10.1006/cyto.1999.0582. [DOI] [PubMed] [Google Scholar]
- Eriksson C, Van Dam AM, Lucassen PJ, Bol JG, Winblad B, Schultzberg M. Immunohistochemical localization of interleukin-1beta, interleukin-1 receptor antagonist and interleukin-1beta converting enzyme/caspase-1 in the rat brain after peripheral administration of kainic acid. Neuroscience. 1999;93:915–930. doi: 10.1016/s0306-4522(99)00178-5. [DOI] [PubMed] [Google Scholar]
- Fiedorowicz A, Figiel I, Kaminska B, Zaremba M, Wilk S, Oderfeld-Nowak B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001;912:116–127. doi: 10.1016/s0006-8993(01)02675-0. [DOI] [PubMed] [Google Scholar]
- Friedman WJ. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp Neurol. 2001;168:23–31. doi: 10.1006/exnr.2000.7595. [DOI] [PubMed] [Google Scholar]
- Gabellec MM, Crumeyrolle-Arias M, Le Saux F, Auriou N, Jacque C, Haour F. Expression of interleukin-1 genes and interleukin-1 receptors in the mouse brain after hippocampal injury. Neurosci Res. 1999;33:251–260. doi: 10.1016/s0168-0102(99)00014-0. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons inrats with middle cerebral artery occlusion. Am J Pathol. 1995;147:1477–1486. [PMC free article] [PubMed] [Google Scholar]
- Ghorpade A, Holter S, Borgmann K, Persidsky R, Wu L. HIV-1 and IL-1 beta regulate Fas ligand expression in human astrocytes through the NF-kappa B pathway. J Neuroimmunol. 2003;141:141–149. doi: 10.1016/s0165-5728(03)00222-4. [DOI] [PubMed] [Google Scholar]
- Giulian D, Woodward J, Young DG, Krebs JF, Lachman LB. Interleukin-1 injected into mammalian brain stimulates astrogliosis and neovascularization. J Neurosci. 1988;8:2485–2490. doi: 10.1523/JNEUROSCI.08-07-02485.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J Biol Chem. 1995;270:13757–13765. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- Griffin WS. Inflammation and neurodegenerative diseases. Am J Clin Nutr. 2006;83:470S–474S. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry GJ, Lefebvre d'Hellencourt C. Dentate gyrus: alterations that occur with hippocampal injury. Neurotoxicology. 2003;24:343–356. doi: 10.1016/S0161-813X(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Tyler K, d'Hellencourt CL, Tilson HA, Maier WE. Morphological alterations and elevations in tumor necrosis factor-alpha, interleukin (IL)-1alpha, and IL-6 in mixed glia cultures following exposure to trimethyltin: modulation by proinflammatory cytokine recombinant proteins and neutralizing antibodies. Toxicol Appl Pharmacol. 2002;180:205–218. doi: 10.1006/taap.2002.9390. [DOI] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7:153–159. doi: 10.1159/000026433. [DOI] [PubMed] [Google Scholar]
- Jahnke GD, Brunssen S, Maier WE, Harry GJ. Neurotoxicant-induced elevation of adrenomedullin expression in hippocampus and glia cultures. J Neurosci Res. 2001;66:464–474. doi: 10.1002/jnr.1237. [DOI] [PubMed] [Google Scholar]
- Juo P, Woo MS, Kuo CJ, Signorelli P, Biemann HP, Hannun YA, Blenis J. FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ. 1999;10:797–804. [PubMed] [Google Scholar]
- Krohn K, Rozovsky I, Wals P, Teter B, Anderson CP, Finch CE. Glial fibrillary acidic protein transcription responses to transforming growth factor-beta 1 and interleukin-1 beta are mediated by a nuclear factor-1-like site in the near-upstream promoter. J Neurochem. 1999;72:1353–1361. doi: 10.1046/j.1471-4159.1999.721353.x. [DOI] [PubMed] [Google Scholar]
- Lai AY, Swayze RD, El-Husseini A, Song C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. 2006;175:97–106. doi: 10.1016/j.jneuroim.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Laping NJ, Morgan TE, Nichols NR, Rozovsky I, Young-Chan CS, Zarow C, Finch CE. Transforming growth factor-beta 1 induces neuronal and astrocyte genes: tubulin alpha 1, glial fibrillary acidic protein and clusterin. Neuroscience. 1994;58:563–572. doi: 10.1016/0306-4522(94)90081-7. [DOI] [PubMed] [Google Scholar]
- Lazovic J, Basu A, Lin HW, Rothstein RP, Krady JK, Smith MB, Levison SW. Neuroinflammation and both cytotoxic and vasogenic edema are reduced in interleukin-1 type 1 receptor-deficient mice conferring neuroprotection. Stroke. 2005;36:2226–2231. doi: 10.1161/01.STR.0000182255.08162.6a. [DOI] [PubMed] [Google Scholar]
- Lin HW, Basu A, Druckman C, Cicchese M, Krady JK, Levison SW. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J Neuroinflammation. 2006;3:15. doi: 10.1186/1742-2094-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression using real-time quantitative PCR and the 2(−δδC(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Wong ML, Bongiorno PB, Gold PW, Licinio J, Rothwell NJ. Endogenous interleukin-1 receptor antagonist is neuroprotective. Biochem Biophys Res Commun. 1997;234:211–215. doi: 10.1006/bbrc.1997.6436. [DOI] [PubMed] [Google Scholar]
- Ma XC, Gottschall PE, Chen LT, Wiranowska M, Phelps CP. Role and mechanisms of interleukin-1 in the modulation of neurotoxicity. Neuroimmunomodulation. 2002;10:199–207. doi: 10.1159/000068322. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Greene C. Signal transduction pathways activated by the IL-1 receptor family: ancient signaling machinery in mammals, insects, and plants. J Leukoc Biol. 1998;63:650–657. [PubMed] [Google Scholar]
- Ohtsuki T, Ruetzler CA, Tasaki K, Hallenbeck JM. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J Cereb Blood Flow Metab. 1996;16:1137–1142. doi: 10.1097/00004647-199611000-00007. [DOI] [PubMed] [Google Scholar]
- Parker LC, Luheshi GN, Rothwell NJ, Pinteaux E. IL-1 beta signalling in glial cells in wildtype and IL-1RI deficient mice. Br J Pharmacol. 2002;136:312–320. doi: 10.1038/sj.bjp.0704715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinteaux E, Rothwell NJ, Boutin H. Neuroprotective actions of endogenous interleukin-1 receptor antagonist (IL-1ra) are mediated by glia. Glia. 2006;53:551–556. doi: 10.1002/glia.20308. [DOI] [PubMed] [Google Scholar]
- Pompili E, Fabrizi C, Fumagalli L. PAR-1 upregulation by trimethyltin and lipopolysaccharide in cultured rat astrocytes. Int J Mol Med. 2006;18:33–39. [PubMed] [Google Scholar]
- Ravizza T, Vezzani A. Status epilepticus induces time-dependent neuronal and astrocytic expression of interleukin-1 receptor type I in the rat limbic system. Neuroscience. 2006;137:301–308. doi: 10.1016/j.neuroscience.2005.07.063. [DOI] [PubMed] [Google Scholar]
- Rothwell N. Interleukin-1 and neuronal injury: mechanisms, modification, and therapeutic potential. Brain Behav Immun. 2003;17:152–157. doi: 10.1016/s0889-1591(02)00098-3. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 2000;23:618–625. doi: 10.1016/s0166-2236(00)01661-1. [DOI] [PubMed] [Google Scholar]
- Sato T, Irie S, Kitada S, Reed JC. FAP-1: a protein tyrosine phosphatase that associates with Fas. Science. 1995;268:411–415. doi: 10.1126/science.7536343. [DOI] [PubMed] [Google Scholar]
- Sayyah M, Beheshti S, Shokrgozar MA, Eslami-far A, Deljoo Z, Khabiri AR, Haeri Rohani A. Antiepileptogenic and anticonvulsant activity of interleukin-1 beta in amygdala-kindled rats. Exp Neurol. 2005;191:145–153. doi: 10.1016/j.expneurol.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Sei Y, Vitkovic L, Yokoyama MM. Cytokines in the central nervous system: regulatory roles in neuronal function, cell death and repair. Neuroimmunomodulation. 1995;2:121–133. doi: 10.1159/000096881. [DOI] [PubMed] [Google Scholar]
- Strijbos PJ, Rothwell NJ. Interleukin-1 beta attenuates excitatory amino acid-induced neurodegeneration in vitro: involvement of nerve growth factor. J Neurosci. 1995;15:3468–3474. doi: 10.1523/JNEUROSCI.15-05-03468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem. 2006;98:258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- Touzani O, Boutin H, LeFeuvre R, Parker L, Miller A, Luheshi G, Rothwell N. Interleukin-1 influences ischemic brain damage in the mouse independently of the interleukin-1 type I receptor. J Neurosci. 2002;22:38–43. doi: 10.1523/JNEUROSCI.22-01-00038.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzani O, Boutin H, Chuquet J, Rothwell N. Potential mechanisms of interleukin-1 involvement in cerebral ischaemia. J Neuroimmunol. 1999;100:203–215. doi: 10.1016/s0165-5728(99)00202-7. [DOI] [PubMed] [Google Scholar]
- Uciechowski P, Saklatvala J, von der Ohe J, Resch K, Szamel M, Kracht M. Interleukin 1 activates jun N-terminal kinases JNK1 and JNK2 but not extracellular regulated MAP kinase (ERK) in human glomerular mesangial cells. FEBS Lett. 1996;394:273–278. doi: 10.1016/0014-5793(96)00967-2. [DOI] [PubMed] [Google Scholar]
- Vereker E, O'Donnell E, Lynch MA. The inhibitory effect of interleukin-1beta on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J Neurosci. 2000;20:6811–6819. doi: 10.1523/JNEUROSCI.20-18-06811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport. 1996;7:2181–2185. doi: 10.1097/00001756-199609020-00025. [DOI] [PubMed] [Google Scholar]
- Zhao B, Schwartz JP. Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res. 1998;52:7–16. doi: 10.1002/(SICI)1097-4547(19980401)52:1<7::AID-JNR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]