

Abstract
New 4-anilidopiperidine analogues in which the phenethyl group of fentanyl was replaced by several aromatic ring-contained amino acids (or acids) were synthesized to study the biological effect of the substituents on μ and δ opioid receptor interactions. These analogues showed broad (47 nM–76 μM) but selective (up to 17-fold) binding affinities at the μ opioid receptor over the δ opioid receptor, as predicted from the message-address concept.
Keywords: 4-Anilidopiperidine analogues, Fentanyl, Dmt-Tic, Opioid receptors, Analgesic effects
Since the discovery of fentanyl1 which was shown to have potent agonist activity at the μ opioid receptor with unique biological properties, many anilidopiperidine analogues have been synthesized and evaluated for structure–activity relationships (SAR) to provide insight into the key structural features for high biological activity for the μ opioid receptor.2–4 In general, the minimal structural requirements necessary for a ligand to interact with the μ opioid receptor are an appropriate spatially oriented basic nitrogen and a hydroxylated phenyl ring.5 According to the concept of message-address domains, the tertiary nitrogen and the phenolic hydroxy group in nonpeptide opioid ligands, and the free amine and the hydroxyl groups of tyrosine residue in peptide ligands constitute a part of the message domain to anchor the ligands to opioid receptors.6
Recently our group has been developing several piperidine derivatives which can be ligated with various kinds of amino acids to create a new series of 4-anilidopiperidine analogues.7 Here new analogues in which the phenethyl group of fentanyl was replaced by several aromatic ring-contained amino acids (or acids), N-(1-substituted piperidin-4-yl)-N-phenyl-propionamide, were designed and synthesized to study the biological effect of these substituents on the δ and μ opioid receptors. The design of these analogues was based on the message-address concept. In Figure 1, the upper part of fentanyl moiety (left), N-phenyl-N-piperidin-4-yl-propionamide, was considered as an address region, and the lower part of Dmt-Tic (right), namely the 2,6-dimethyltyrosine (Dmt) moiety, a message region. By combination of the two parts shown (center) it was expected that the analogues would show a broad range of biological activities, since the modification on the C-terminus of Dmt-Tic was recognized to result in analogues that display a variety of μ/δ selectivities and agonist/antagonist combinations.8 Considering the correlation between the structures of fentanyl and Dmt-Tic, and their bioactivities, we decided to examine thoroughly other modifications with the aim of evaluating the structural requirements for binding to the opioid receptors, and identifying the variations in the receptor binding pocket. In this study, eighteen 4-anilidopiperidine analogues 1–18 in which the piperidine nitrogen was coupled with different kinds of aromatic ring-contained amino acids (or acids) were prepared and screened for their binding affinities and functional activities at the δ and μ opioid receptors.
Figure 1.
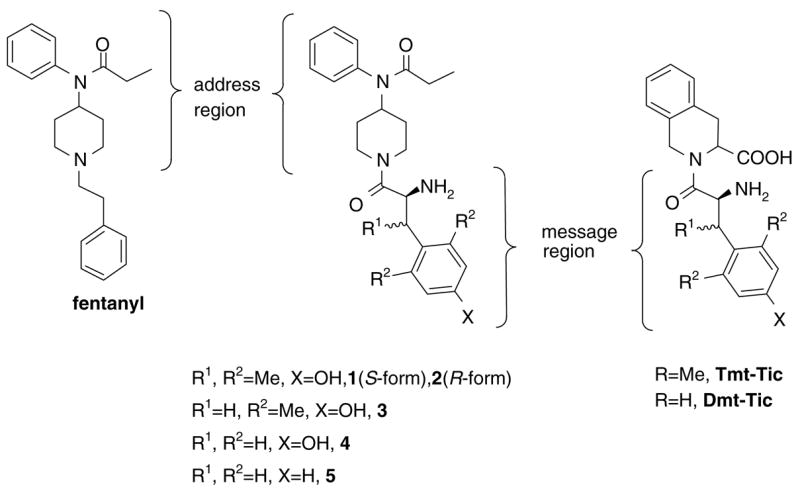
Opioid ligands.
All analogues were prepared by coupling various kinds of aromatic ring-contained amino acids (or acids) to 19 (see Note 9 for the synthesis of 19) using BOP/HOBt/NMM in solution (Scheme 1). After deprotection of Nα-Boc group with TFA, crude compounds were purified by preparative RP-HPLC to give >95% pure analogues in high yields (see Table 1 for analytical data). Their opioid binding affinities at the human δ opioid receptor (hDOR) and the rat μ opioid receptor (rMOR) were determined by competition analyses against [3H]DPDPE (δ) and [3H]DAMGO (μ) using membrane preparations from transfected HN9.10 cells that constitutively express the respective receptors. Functional activities for δ and μ opioid receptors were evaluated in stimulated isolated mouse vas deferens (MVD, δ) and guinea pig isolated ileum (GPI, μ) bioassays, respectively, as previously published.10
Table 1.
Analytical data of the 4-anilidopiperidine analogues
| Compound | Molecular formula | LowMSa |
HRMSb |
HPLCc (tR, min) | |
|---|---|---|---|---|---|
| Observed | Observed | Calcd | |||
| 1 | C26H35N3O3 | 438.3 | 438.2741 | 438.2757 | 18.5 |
| 2 | C26H35N3O3 | 438.4 | 438.2763 | 438.2757 | 16.0 |
| 3 | C25H33N3O3 | 424.5 | 424.2595 | 424.2600 | 13.6 |
| 4 | C23H29N3O3 | 396.2 | 396.2296 | 396.2287 | 18.5 |
| 5 | C23H29N3O2 | 380.5 | 380.2342 | 380.2338 | 19.1 |
| 6 | C23H28ClN3O2 | 414.1 | 414.1949 | 414.1948 | 17.3 |
| 7 | C20H26N4O2S | 387.4 | 387.1851 | 387.1855 | 12.3 |
| 8 | C26H29N3O2S | 436.5 | 436.2047 | 436.2059 | 17.1 |
| 9 | C27H31N3O2 | 430.5 | 430.2489 | 430.2495 | 16.4 |
| 10 | C29H33N3O2 | 456.6 | 456.2671 | 456.2651 | 19.0 |
| 11 | C23H28ClN3O2 | 414.4 | 414.1967 | 414.1948 | 17.6 |
| 12 | C24H31N3O2 | 394.5 | 394.2489 | 394.2495 | 18.7 |
| 13 | C24H31N3O3 | 410.5 | 410.2437 | 410.2444 | 18.0 |
| 14 | C23H27BrN2O2 | 443.3d | 443.1339d | 443.1334d | 22.0 |
| 15 | C24H29ClN2O3 | 429.4d | 429.1939d | 429.1945d | 24.5 |
| 16 | C22H28N3O2 | 366.2 | 366.2185 | 366.2182 | 14.6 |
| 17 | C22H28N3O2 | 366.2 | 366.2179 | 366.2182 | 16.6 |
| 18 | C25H31N3O2 | 406.5 | 406.2501 | 406.2495 | 20.3 |
(M−TFA+H)+, ESI method [Finnigan, Thermoelectron, LCQ classic].
(M−TFA + H)+, FAB-MS (JEOL HX110 sector instrument) or MALDI-TOF.
Performed on a Hewlett Packard 1100 [C-18, Vydac, 4.6 mm × 250 mm, 5 μm, 10–90% of acetonitrile containing 0.1% TFA within 40 min and up to 100% within additional 5 min, 1 mL/min].
(M+H)+.
In Table 2, the synthesized analogues showed a broad range (47 nM to 76 μM) but selective (up to 17-fold) binding affinities for the μ opioid receptor over the δ opioid receptor. Their selectivities for the μ opioid receptor were predicted from the address region of the fentanyl structure (Fig. 1). Opioid agonist activities in the MVD and the GPI assays were very low and did not correlate with the binding affinities at both receptors. One of the analogues, 3, in which Dmt was coupled to the piperidine ring showed high binding affinities at both δ and μ opioid receptors (Ki = 50 nM and 47 nM, respectively) with very low agonist activities in the MVD and the GPI assays. Interestingly, this compound showed some μ opioid antagonist activity by its ability to shift the dose response of PL-017 twofold in the GPI assay. Compound 2 which had a β–methyl-2′,6′-dimethyltyrosine (Tmt)11 residue rather than Dmt showed the highest agonist activity in the GPI assay with no antagonist function, but low agonist activity in the MVD with some antagonist function (5-fold [3H]DPDPE shift at 1 μM), but showed low binding affinities at both δ and μ opioid receptors in the 300 nM range. Although there is no evidence that compounds 2 and 3 have potent antagonist activity, it is suggested that their partial antagonist activity could be responsible for its reduced agonist activity in the same assay. A stereo difference was found for the opioid activities of 1 [R1 = (S)-NH2, R2 = (S)-Me] and 2 [R1 = (S)-NH2, R2 = (R)-Me]. Compound 2 showed higher binding affinities for both opioid receptors and was more biologically active for the μ opioid receptor. Our results are consistent with the known fact that the preferred stereo configuration of the Tmt residue for the δ opioid receptor interaction is 2S and 3R,11 even though significant δ opioid agonist activity was not shown due to its potential interruption by some antagonist activity. It was also shown that R-configuration of R1 substituents was not tolerated in the structure 6 for the δ opioid receptor (Fig. 2).
Table 2.
Bioactivities of 4-anilidopiperidine analogues at the δ and μ opioid receptors10
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | n | R1 | R2 | Ar | Binding Kia (μM)
|
Bioassay IC50b (μM)
|
||
| hDOR (δ) | rMOR(μ) | MVD(δ) | GPI(μ) | |||||
| 1 | 1 | (S) NH2 | (S) Me | 2,6-Dimethyl-4-hydroxyphenyl | 1.7 | 0.50 | 39% at 1 μMc | 3% at 1 μMc |
| 2 | 1 | (S) NH2 | (R) Me | 2,6-Dimethyl-4-hydroxyphenyl | 0.26 | 0.30 | 13% at 1 μMc | 3.3 |
| 3 | 1 | (S) NH2 | H | 2,6-Dimethyl-4-hydroxyphenyl | 0.050 | 0.047 | 2% at 1 μMc | 45% at 30 μMc |
| 4 | 1 | (S) NH2 | H | 4-Hydroxy phenyl | 16 | 7.9 | 18% at 1 μMc | 8% at 1 μMc |
| 5 | 1 | (S) NH2 | H | Ph | 6.9 | 3.9 | 0% at 1 μMc | 2% at 1 μMc |
| 6 | 1 | (R) NH2 | H | 4-Chlorophenyl | nc | 6.9 | 28% at 1 μMc | 0% at 1 μMc |
| 7 | 1 | (S) NH2 | H | 4-Thiazoyl | 76 | 9.7 | 8% at 1 μMc | 3% at 1 μMc |
| 8 | 1 | (S) NH2 | H | 3-Benzothiopheneyl | 15 | 2.6 | 0% at 10 μMc | 28% at 10 μMc |
| 9 | 1 | (S) NH2 | H | 2-Naphthyl | 3.3 | 1.1 | 26% at 1 μMc | 8% at 1 μMc |
| 10 | 1 | (S) NH2 | Ph | Ph | 6.5 | 0.40 | 48% at 10 μMc | 73% at 10 μMc |
| 11 | 1 | H | NH2 | 4-Chlorophenyl | 2.4 | 0.99 | 12% at 1 μMc | 5% at 1 μMc |
| 12 | 1 | H | NH2 | 4-Methylphenyl | 2.9 | 3.0 | 27% at 10 μMc | 24% at 10 μMc |
| 13 | 1 | H | NH2 | 4-Methoxyphenyl | 9.0 | 9.7 | 19% at 1 μMc | 3% at 1 μMc |
| 14 | 1 | H | H | 3-Bromophenyl | 12 | 1.9 | 0% at 1 μMc | 18% at 1 μMc |
| 15 | 1 | H | H | 3-Chloro-4-methoxyphenyl | 6.3 | 3.8 | 0% at 1 μMc | 33% at 10 μMc |
| 16 | 0 | (S) NH2 | Ph | nc | 6.4 | 7% at 1 μMc | 3% at 1 μMc | |
| 17 | 0 | (R) NH2 | Ph | nc | 5.1 | 12% at 1 μMc | 1% at 1 μMc | |
| 18 | 0 | (S) NH2 | 2-Indanyl | 15 | 2.6 | 19% at 10 μMc | 59% at 10 μMc | |
| (2S,3R)-Tmt-Tic11 | 0.0093 | 35 | 547-fold shift at 1 μM | 30% at 30 μMc | ||||
| Fentanyl12 | 0.57 | 0.0059 | — | 5.2 | ||||
| Dmt-Tic13 | 0.00097 | 0.0046 | 38.9ED50 | 500ED50 | ||||
Competition analyses against radiolabeled ligand ([3H]DPDPE for δ, [3H]DAMGO for μ) were carried out using membrane preparations from transfected HN9.10 cells that constitutively express the respective receptor types.
Concentration at 50% inhibition of muscle contraction at electrically stimulated isolated tissues.
Inhibition % of muscle contraction at electrically stimulated isolated tissues. nc: no competition at 10−5 M.
Figure 2.
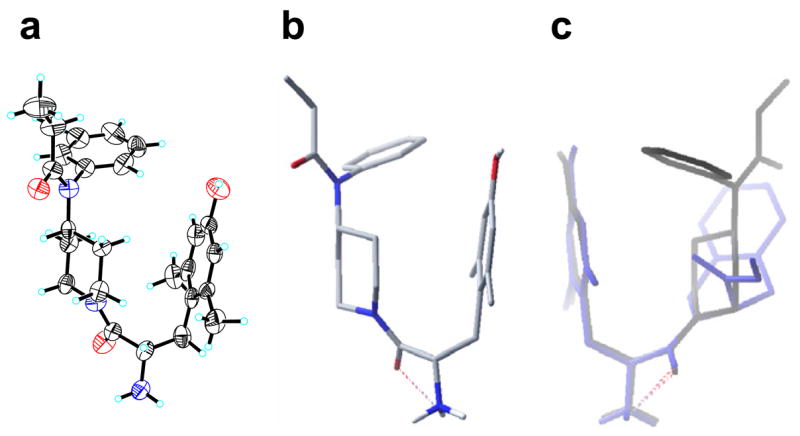
(a) X-ray structure of 3. (b) The lowest energy conformer of 3. (c) Superimposition of 3 (black) and Dmt-Tic (blue). H-bond are represented by red dashed lines. For simplicity, nonpolar hydrogens are omitted in the structures.
Among the analogues, 16 [n = 0, R1 = (S)-NH2] and 17 [n = 0, R1 = (R)-NH2] which have one less carbon between the piperidine and substituted aromatic rings than the others [n = 1] lost their binding affinities at the δ opioid receptor, while 18 [n = 0, R1 = (S)-NH2, Ar = 2-indanyl] retained its binding affinity at the same receptor. The losses of binding affinities at the δ opioid receptor could be caused by the shortened length of the molecule interrupting the ability (potential flexibility) to access the binding pocket. On the other hand, the 2-indanyl group conferred greater binding to the δ opioid receptor by extending the length of molecule through the cyclopentenyl moiety, which could play an important role in holding the piperazine ring as a rigid spacer. It is apparent that the length of molecules, especially between the piperazine and substituted aromatic rings, can be considered to be an important factor which contributes to the binding. Hydrophobic aromatic ring substituted analogues, 9 [R1 = (S)-NH2, R2 = H, Ar = naphthyl] and 10 [R1 = (S)-NH2, R2 = Ph, Ar = Ph], showed greater binding affinities at both δ and μ opioid receptors than 7 and 8. It also was observed that an additional phenyl group at the β-position of 10 increased binding affinity at the μ opioid receptor, and functional activities at the MVD and GPI assays. In comparison with analogues 11–13 [R1 = H, R2 = NH2], 14 and 15 [R1 = H, R2 = H] showed similar binding affinities at both opioid receptors. This illustrated that the position and the presence of amino group are not critical for binding to these receptors. It also was found that substitutions at the 4-position on the substituted phenyl ring did not affect the binding affinities for both opioid receptors.
In addition to the bioassays, molecular modeling experiments14 using MacroModel 8.1 were carried and yielded some insights regarding the determinants of opioid activity and selectivity. We utilized molecular modeling to probe the topographical similarities of compounds 2–5 to fentanyl and Dmt-Tic structures, and speculated that these compounds might provide an overall shape similar to the bioactive conformation of fentanyl or Dmt-Tic by controlling the orientation of the anilido moiety. The studies on 2 gave a topographically identical structure to the same part of fentanyl, which is considered as an address region, and gave a longer length between the two ends of the molecule than the others. A superimposition of the lowest energy conformers of 2 and fentanyl showed that these compounds have superimposed anilidopiperidine ring conformations, yet maintain different aromatic ring conformations. On the contrary, the lowest energy conformer of 3 looked more similar to the bioactive conformation of Dmt-Tic in the shape of the turn, even though its two aromatic rings were not oriented parallel. The lowest energy conformer fully matched the structure which was obtained from X-ray crystallography (Fig. 2).15 Superimposition of 3 and Dmt-Tic indicates that the binding pocket for the opioid receptors, preferably for the δ, and the possible π–π interactions between two aromatic rings, may be blocked by the perpendicularly oriented phenyl ring of compound 3 to result in the loss of activity.
An analysis of the binding modes indicated the most potent fentanyl derivatives possess an extended conformation for the phenethyl group with θ1 = trans (C–C bond of phenethyl group) and θ2 = gauche (C–Npiperidine bond).3 This also suggested that minor differences in the conformations of substituted fentanyls may have a significant impact on ligand binding. In comparing fentanyl with the classic μ opiate morphine, certain similarities are apparent. Both compounds possess a nitrogen which can be easily protonated and aromatic groups that are commonly thought to mimic the N-terminal tyrosine moiety of opioid peptides. However, SAR studies of N-phenolic derivatives of fentanyl have shown that hydroxyl substitution into the aromatic ring does not influence the morphinomimetic potency of these compounds. In our study, it was also shown that the phenolic hydroxyl group is not a necessary part for the opioid receptor interactions. Analogues 4 and 5 had no significant differences in their biological activities and both showed completely superimposed lowest energy conformations with the two aromatic rings faced perpendicularly in computer modeling experiments.
In conclusion, we have examined the pharmacological effects of novel 4-anilidopiperidine analogues on the μ and δ opioid receptors. In the structural modifications described here, all analogues bind more effectively to the μ opioid receptor than to the δ opioid receptor. It is likely that the selective binding to the μ opioid receptor is due to the N-phenyl-N-piperidin-4-yl-propionamide moiety. All analogues showed a wide range of binding affinities at the δ and μ opioid receptors depending on their respective structures. From the SAR studies of these analogues, it is proposed that the constraints caused by the methyl groups on the substituted aromatic ring or the β-carbon serve as the most critical factor in determining molecular conformation along with the length between the piperazine and the aromatic rings. Overall, neither of the amino groups led to an increase in binding. The substituents at the 3-, or 4-positions also did not play a role in the binding affinities.
Scheme 1.
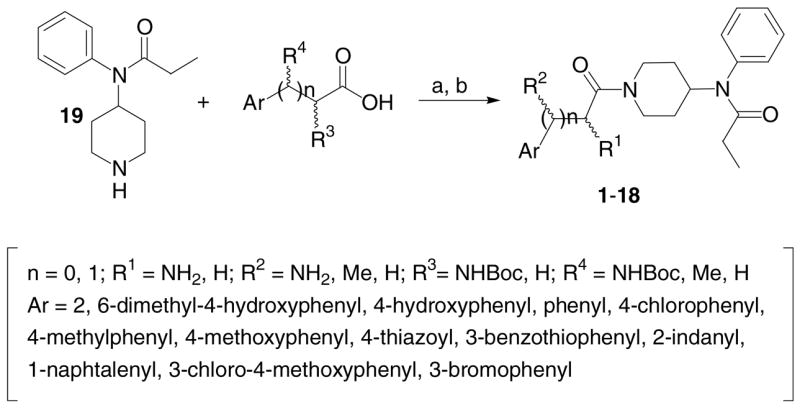
Synthesis of 4-anilidopiperidine analogues. Reagents and conditions: (a) 1.1 equiv BOP, 1.1 equiv HOBt, 2.2 equiv NMM, rt, 2–4 h, 85–95%; (b) TFA, 0 °C, 20 min, quantitative.
Acknowledgments
The work was supported by grants from the USDHS, National Institute on Drug Abuse (DA-12394 and DA-06284). We thank Margie Colie for assistance with the manuscript.
References and notes
- 1.Janssen PAJ. Br J Anaesth. 1962;34:260. doi: 10.1093/bja/34.4.260. [DOI] [PubMed] [Google Scholar]
- 2.Casy AF, Huckstep MR. J Pharm Pharm. 1988;40:605. doi: 10.1111/j.2042-7158.1988.tb05318.x. [DOI] [PubMed] [Google Scholar]
- 3.Subramanian G, Paterlini MG, Portoghese PS, Ferguson DM. J Med Chem. 2000;43:381. doi: 10.1021/jm9903702. [DOI] [PubMed] [Google Scholar]
- 4.Zaveri N, Polgar WE, Olsen CM, Kelson AB, Grundt P, Lewis JW, Toll L. Eur J Pharm. 2001;428:29. doi: 10.1016/s0014-2999(01)01282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hruby VJ, Gehrigh CA. Med Res Rev. 1989;9:343. doi: 10.1002/med.2610090306. [DOI] [PubMed] [Google Scholar]
- 6.Portoghese PS, Sultana M, Takemori AE. J Med Chem. 1990;33:1714. doi: 10.1021/jm00168a028. [DOI] [PubMed] [Google Scholar]
- 7.Petrov RR, Vardanyan RS, Lee YS, Ma SW, Davis P, Begay LJ, Lai J, Porreca F, Hruby VJ. Bioorg Med Chem Lett. 2006:4946. doi: 10.1016/j.bmcl.2006.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Biopolymers. 2003;71:86. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
-
9.
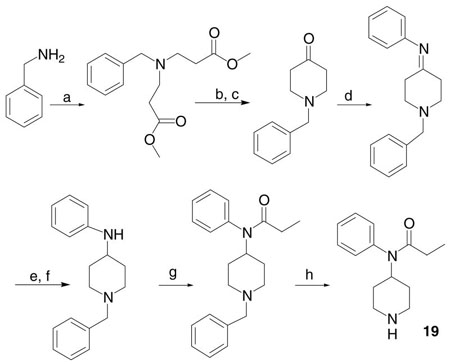 Synthesis of N-phenyl-N-piperidin-4-yl-propionamide (19). Reagents and conditions: (a) acrylic acid methyl ester, MeOH, rt, 2 h, reflux, 4 h, 95%; (b) Na/toluene, reflux, 2 h; (c) HCl at 0 °C, heat, 3–4 h, 41%; (d) aniline, acetic acid, toluene, Dean–Stark, 2 h, 73%; (e) NaBH4, MeOH, reflux, 1 h; (f) NaOH, 81%; (g) propionyl chloride, TEA/benzene, 1 day, 54%; (h) Pd/C, EtOH, rt, 2.5 days, 61%.
Synthesis of N-phenyl-N-piperidin-4-yl-propionamide (19). Reagents and conditions: (a) acrylic acid methyl ester, MeOH, rt, 2 h, reflux, 4 h, 95%; (b) Na/toluene, reflux, 2 h; (c) HCl at 0 °C, heat, 3–4 h, 41%; (d) aniline, acetic acid, toluene, Dean–Stark, 2 h, 73%; (e) NaBH4, MeOH, reflux, 1 h; (f) NaOH, 81%; (g) propionyl chloride, TEA/benzene, 1 day, 54%; (h) Pd/C, EtOH, rt, 2.5 days, 61%.
- 10.Lee YS, Agnes RS, Davis P, Ma S, Badghisi H, Lai J, Porreca F, Hruby VJ. J Med Chem. 2006;49:1773. doi: 10.1021/jm05085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao S, Lin J, Shenderovich MD, Han Y, Hasohata K, Davis P, Qui W, Porreca F, Yamamura HI, Hruby VJ. Bioorg Med Chem Lett. 1997;23:3049. [Google Scholar]
- 12.Jagerovic N, Cano C, Elguero J, Goya P, Callado LF, Meana JJ, Giron R, Abalo R, Ruiz D, Goicoechea C, Martin IM. Bioorg Med Chem. 2002;10:817. doi: 10.1016/s0968-0896(01)00345-5. [DOI] [PubMed] [Google Scholar]
- 13.Salvadori S, Attila M, Balboni G, Bianchi C, Bryant SD, Crescenzi O, Guerrini R, Picone D, Tancredi T, Temussi PA, Lazarus LH. Mol Med. 1995;1:678. [PMC free article] [PubMed] [Google Scholar]
- 14.Molecular modeling experiments were performed on MacroModel 8.1 equipped with Maestro 5.0 graphical interface installed on a Linux RedHat 8.0. Molecular structures were minimized using MM2 force field and Polak–Ribier conjugate gradient (PRCG). Optimizations were converged to a gradient RMSD less than 0.05 kJ/mol or continued until a limit of 10,000 iterations was reached.
- 15.Compound 3 was crystallized from MeOH, ether, and hexane as colorless rods (0.13 mm × 0.15 mm × 0.78 mm). X-ray crystallography on a Bruker SMART 1000 CCD detector X-ray diffractometer at 170(2) K and a power setting of 50 kV, 40 mA showed measurable diffraction to at least theta = 18.23 deg. Data were collected on the SMART1000 system using graphite monochromated MoKα radiation (λ = 0.71073 Å).
