

Abstract
The overexpression or mutation of tyrosine kinases (TKs), such as the epidermal growth factor receptor (EGFR), can lead to the development of cancer. The most common mutation of the EGFR in glioblastomas is the deletion of exons 2–7 known as the EGFRvIII. This mutant receptor cannot bind EGF but, instead, is constitutively active. The Cbl family of ubiquitin ligases (Cbl, Cbl-b, and Cbl-c) targets the activated EGFR for degradation. As the EGFRvIII is transforming, we investigated whether it could be downregulated by the Cbl proteins. The over-expression of all three Cbl proteins resulted in the ubiquitination and degradation of the EGFRvIII. As with the wild-type EGFR, the TK-binding domain and the RING finger of Cbl-b are sufficient for the downregulation of the EGFRvIII. Also, we found that Cbl-b is recruited to the EGFRvIII and inhibits the transformation of NIH 3T3 cells by the EGFRvIII. Mutation of the Cbl-binding site (Y1045F) in the EGFRvIII inhibits its ubiquitination and downregulation by Cbl-b and enhances its ability to transform. Furthermore, the EGFR TK inhibitor, AG 1478, prevents the downregulation of the EGFRvIII by the Cbl proteins and antagonizes the ability of an immunotoxin directed against the EGFRvIII to kill cells expressing this receptor. In conclusion, the EGFR-vIII does not transform by escaping regulation by Cbl proteins and this activation-induced downregulation of the EGFRvIII has an important role in mediating the toxicity of anti-EGFRvIII immunotoxins.
Keywords: EGFRvIII, Cbl proteins, ubiquitin, cancer
Introduction
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein that plays an important role in the growth and differentiation of normal cells. The inappropriate activity of the EGFR tyrosine kinase (TK) is associated with the development of a wide range of cancers (Blume-Jensen and Hunter, 2001). Amplification of the EGFR gene occurs in approximately 50% of glioblastoma multiformes (reviewed in Kuan et al., 2001). In many cases, the amplification of the EGFR gene is associated with its genomic rearrangement. The most common of these rearrangements is known as the EGFR variant III (EGFRvIII, de2–7 EGFR, or ΔEGFR) and is characterized by the genomic deletion of exons 2–7. This deletion results in the loss of the amino-terminal amino-acid residues 6–273 in the extracellular domain of the EGFR. The expression of the EGFRvIII in glioblastoma multiforme is a negative prognostic indicator (Shinojima et al., 2003; Heimberger et al., 2005). However, it has been reported recently that the co-expression of the EGFRvIII and the tumor-suppressor protein PTEN predicts response of patients with glioblastoma to the EGFR TK inhibitors (Mellinghoff et al., 2005). In addition, the expression of the EGFRvIII has been reported in breast, non-small-cell lung, ovarian, and prostate carcinomas, but not in normal tissues (Garcia de Palazzo et al., 1993; Moscatello et al., 1995; Wikstrand et al., 1995). Therefore, the EGFRvIII is a tumor-specific target for cancer therapy (reviewed in Kuan et al., 2001; Pedersen et al., 2001).
The ectopic expression of the EGFRvIII in a glioblastoma cell line increased the speed with which these cells formed tumors in nude mice (Nishikawa et al., 1994; Nagane et al., 1996). The EGFRvIII is also capable of significantly enhancing the tumorgenicity of immortalized murine fibroblasts (Batra et al., 1995; Moscatello et al., 1996) and the breast cancer cell line MCF-7 (Tang et al., 2000). Also, the EGFRvIII increases the in vitro invasiveness of a small-cell lung cancer cell line (Damstrup et al., 2002). Using the murine hematopoietic 32D cell line, Tang et al. (2000) demonstrated that the EGFRvIII is capable of directly transforming a non-tumorigenic cell line. Unlike the wild-type (WT) EGFR, the EGFRvIII is unable to bind to EGF or transforming growth factor-α (Ekstrand et al., 1994; Nishikawa et al., 1994; Moscatello et al., 1996) but, instead, it can dimerize spontaneously (Moscatello et al., 1996; Fernandes et al., 2001; Montgomery, 2002). The spontaneous dimerization and ensuing TK activation of the EGFRvIII is necessary to transform cells (Han et al., 1996; Nagane et al., 1996; Huang et al., 1997; Antonyak et al., 1998; O’Rourke et al., 1998; Montgomery, 2002; Johns et al., 2003; Abulrob et al., 2004; Luwor et al., 2004; Pedersen et al., 2004).
The Cbl proteins (Cbl, Cbl-b, and Cbl-c) are negative regulators of WT EGFR signaling (Ettenberg et al., 1999a, b, 2001; Keane et al., 1999; Levkowitz et al., 1999; Waterman et al., 1999a; Yokouchi et al., 1999; Duan et al., 2003). The Cbl proteins all contain an amino-terminal TK-binding (TKB) domain, a RING finger domain, and a region of proline-rich sequences in their carboxy-terminus (Nau and Lipkowitz, 2003). They are recruited to the activated EGFR either by the direct binding of their TKB domain to the phosphotyrosine residue at position 1045 in the EGFR (Levkowitz et al., 1999) or by an indirect mechanism mediated by Grb2 – the SH3 domains of Grb2 bind the proline-rich region of the Cbl proteins and the SH2 domain of Grb2 binds the phosphorylated EGFR (Waterman et al., 2002; Jiang et al., 2003). The RING finger domain of the Cbl proteins allows them to function as ubiquitin ligases (E3s) and so to target the EGFR signaling complex for internalization and subsequent degradation in the lysosome (Ettenberg et al., 1999b, 2001; Levkowitz et al., 1999; Waterman et al., 1999a; Yokouchi et al., 1999; Duan et al., 2003). Thus, the Cbl proteins mediate the downregulation of the EGFR following stimulation with EGF.
It has been hypothesized that a failure to downregulate the EGFRvIII once it becomes activated contributes to its capacity to transform (Huang et al., 1997). Supporting this, a recent study reported that the EGFRvIII does not interact with either Cbl or Cbl-b and is not downregulated (Schmidt et al., 2003). Mutations in the Cbl-binding site of several receptor tyrosine kinases (RTKs) result in transforming forms of these RTKs (Peschard et al., 2001; Mancini et al., 2002; Peschard and Park, 2003). However, the intracellular domain of the EGFRvIII is not mutated and thus the Cbl protein-binding sites are intact. The Cbl proteins bind the phosphorylated EGFR (Levkowitz et al., 1999) and the phosphorylation pattern of active EGFRvIII is similar to that of the activated WT EGFR (Fernandes et al., 2001). The inability of the Cbl proteins to interact with or downregulate the EGFRvIII suggested a novel mechanism regulating the interaction between the EGFRvIII and the Cbl proteins. Therefore, we investigated the interaction between the Cbl proteins and the EGFRvIII further. In contrast to the published data, we found that the overexpression of all three Cbl proteins caused the ubiquitination and downregulation of the EGFRvIII. Also, we demonstrated that Cbl-b binds to the EGFRvIII, that the requirements for Cbl-b-mediated degradation of the EGFRvIII are identical to that of the WT EGFR, and that only active EGFRvIII is downregulated. Consequently, Cbl-b inhibits the transformation of NIH 3T3 fibroblasts by the EGFRvIII and the mutation of the Cbl-binding site in the EGFRvIII (Y1045F) enhances the ability of the EGFRvIII to transform. Finally, we demonstrated that the inhibition of the EGFRvIII TK by AG 1478, which abrogates Cbl-mediated downregulation of the EGFR-vIII, antagonizes the ability of an immunotoxin directed against the EGFRvIII to kill cells expressing this receptor. Thus, the EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins.
Results
Cbl proteins ubiquitinate and downregulate the constitutively active EGFRvIII
The overexpression of Cbl proteins enhances EGF-induced ubiquitination and downregulation of the WT EGFR (Ettenberg et al., 1999b, 2001; Levkowitz et al., 1999; Waterman et al., 1999b; Yokouchi et al., 1999). Therefore, we investigated whether the Cbl proteins also regulate the constitutively active mutant EGFRvIII in a cell line Chinese hamster ovary (CHO) that does not express the WT EGFR. The co-transfection of CHO cells with the EGFRvIII and either Cbl, Cbl-b, or Cbl-c resulted in a decrease in EGFRvIII protein levels (Figure 1a–c; compare lanes 2 and 3). Also, the co-transfection of Cbl, Cbl-b, or Cbl-c increased the amount of ubiquitinated proteins seen in immunoprecipitates of the EGFRvIII (Figure 1d–f; compare lanes 2 and 3). These ubiquitinated species represent ubiquitinated forms of the EGFRvIII suggesting that, like the active WT EGFR, Cbl proteins are capable of ubiquitinating and downregulating the EGFRvIII.
Figure 1.
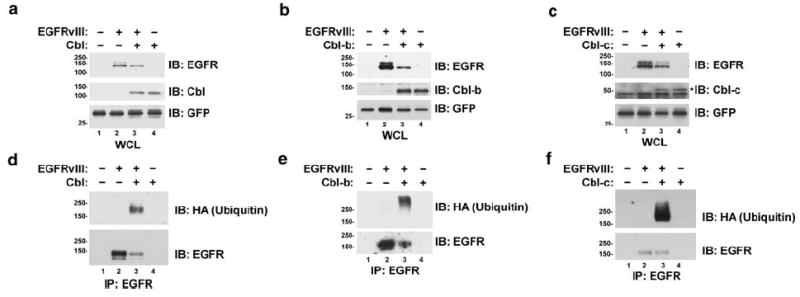
The constitutively active mutant EGFRvIII is ubiquitinated and downregulated by Cbl proteins. CHO cells were transfected with the EGFRvIII, HA-epitope-tagged ubiquitin, and either Cbl, Cbl-b, or Cbl-c as indicated. All transfections were balanced with empty vector controls; GFP was used as a transfection control. Cells were serum starved overnight and protein lysates were prepared. (a–c) Whole-cell lysates (WCL) or (d–f) EGFR immunoprecipitates (IP) were immunoblotted (IB) for the EGFR, Cbl, Cbl-b, Cbl-c, ubiquitin (with anti-HA), or GFP as indicated to the right of the blots. An asterisk indicates the band corresponding to Cbl-c. Immunoblots of the WCL were used to assess the levels of the transfected proteins. Immunoblots of the precipitated EGFRvIII were used to assess ubiquitination. Molecular weight standards (kDa) appear to the left of the panels.
As all three Cbl proteins caused the degradation of the EGFRvIII, we chose to use Cbl-b to investigate the mechanism by which they regulate this oncogenic EGFR mutant. Given that the TK activity and autophosphorylation of the WT EGFR are necessary for its ubiquitination and degradation by the Cbl proteins (Levkowitz et al., 1999), we examined whether this is also the case with the EGFRvIII. Although the WT EGFR is regulated by ligand binding, the EGFR-vIII is spontaneously active. Therefore, we used the EGFR TK inhibitor AG 1478 to inhibit the activity of the EGFRvIII. Treatment of CHO cells overexpressing the EGFRvIII with AG 1478 prevented tyrosine autophosphorylation of the EGFRvIII (Figure 2b, middle panel; compare lanes 2 and 3 to lanes 6 and 7). Inactivation of the EGFRvIII TK by AG 1478 attenuated its downregulation by Cbl-b (Figure 2a, top panel; compare lanes 2 and 3 to lanes 6 and 7). Co-expression of Cbl-b resulted in downregulation of the EGFRvIII by 73% in the absence of AG 1478 (Figure 2a, bar graph). In the presense of AG 1478, the level of the EGFRvIII was higher and co-expression of Cbl-b only resulted in 5% downregulation (Figure 2a, bar graph). AG 1478 completely abolished ubiquitination of EGFRvIII by Cbl-b (Figure 2b, top panel; compare lanes 3 and 7). Also, AG 1478 treatment inhibited the ubiquitination and downregulation of the EGFRvIII by Cbl (data not shown). Therefore, the TK activity of the EGFRvIII is necessary for its downregulation by the Cbl proteins. As AG 1478 inhibits the activation-induced downregulation of the EGFRvIII by the Cbl proteins, we examined the effects of AG 1478 upon the subcellular localization of the EGFRvIII in the murine fibroblast cell line NR-6m (Figure 2c). The NR-6m cell line is a variant of Swiss 3T3 cells which has been stably transfected with the EGFRvIII, resulting in transformation of the cells (Batra et al., 1995). It was chosen for the localization studies because the cell line does not express endogenous WT EGFR, thus allowing the use of anti-EGFR and anti-phospho-EGFR antibodies. In these cells, the EGFRvIII was localized in both the plasma membrane and intracellular vesicles. The majority of active EGFRvIII, as detected by EGFR phosphotyrosine 1173 staining, appears to be localized in intracellular vesicles. Inhibition of the TK activity of the EGFRvIII by AG 1478 treatment abolished phosphotyrosine 1173 staining and resulted in a reduction of the amount of EGFRvIII in intracellular vesicles and an increase in the proportion of the EGFRvIII located at the plasma membrane compared to intracellular vesicles. This is consistent with AG 1478 treatment preventing activation-induced internalization and downregulation of the EGFRvIII from the plasma membrane.
Figure 2.
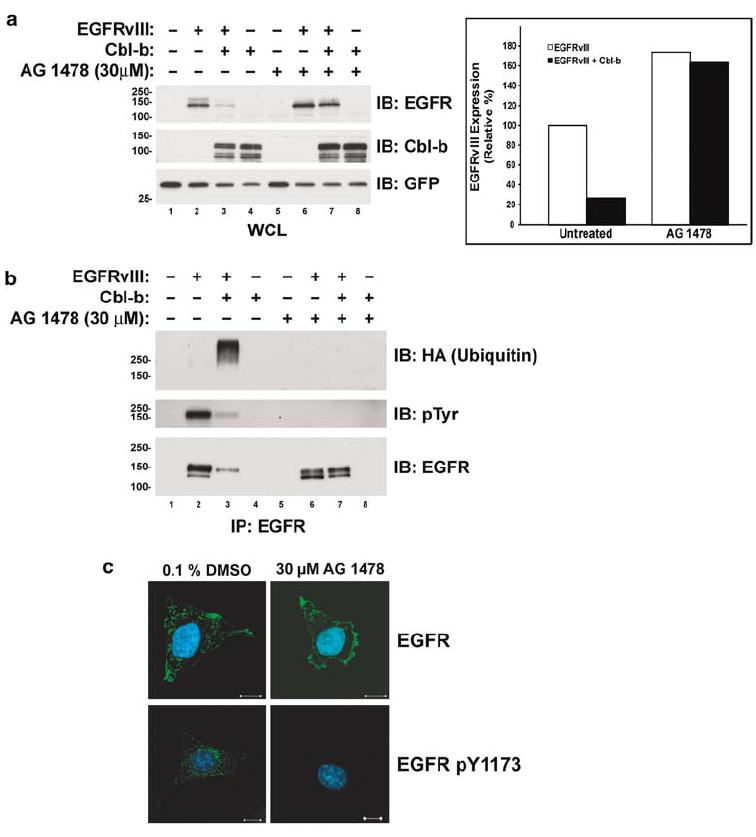
Inhibition of the TK activity of the EGFRvIII prevents its downregulation and ubiquitination by Cbl-b. CHO cells were transfected with the EGFRvIII, HA-epitope-tagged ubiquitin, and Cbl-b as indicated. All transfections were balanced with empty vector controls; GFP was used as a transfection control. Following transfection, cells were grown to 70% confluence and incubated overnight with the vehicle (0.1% DMSO) or 30 μm AG 1478. (a) Whole-cell lysates (WCL) or (b) EGFR immunoprecipitates (IP) were immunoblotted (IB) for the EGFR, Cbl-b, GFP, ubiquitin (with anti-HA), or phosphotyrosine (pTyr) as indicated to the right of the blots. The bar graph shows the EGFRvIII protein levels in the WCL blots in panel a quantified by densitometry. All values, expressed as the percentage of the amount of EGFRvIII protein in the control transfection, are adjusted for GFP levels. Molecular weight standards (kDa) appear to the left of the panels. (c) EGFRvIII-expressing NR-6m cells were incubated with 100 μg/ml cycloheximide (to inhibit new protein synthesis) and either 0.1% DMSO or 30 μm AG 1478, fixed, and stained for the EGFR or activated (pY1173) EGFR as indicated (green). Cells were counterstained with DAPI (blue). Each panel is a representative mid-level confocal slice. Bar=10 μm.
Requirements for Cbl-b-mediated downregulation of the EGFRvIII
We mapped the regions of Cbl-b necessary for the downregulation of the EGFRvIII by transfecting CHO cells with the EGFRvIII and various constructs of Cbl-b (Figure 3a). As described above (Figure 1), WT Cbl-b downregulates the EGFRvIII (Figure 3b, lane 3). The deletion of the proline-rich, carboxy-terminal half of Cbl-b did not inhibit its ability to downregulate the EGFRvIII (N1/2 Cbl-b; Figure 3b, lane 4). In contrast, the deletion of the TKB domain containing the aminoterminus of Cbl-b (C2/3 Cbl-b) prevented the downregulation of the EGFRvIII by Cbl-b (Figure 3b, lane 5). Finally, a RING finger mutant of Cbl-b (C373A) that has been shown to lack E3 activity (Ettenberg et al., 2001) was unable to downregulate the EGFRvIII (Figure 3b, lane 6). Quantification of the downregulation of the EGFRvIII by the various constructs of Cbl-b revealed that N1/2 and WT Cbl-b downregulate the EGFRvIII to a similar extent, that the overexpression of C2/3 Cbl-b did not affect EGFRvIII levels, and that the RING finger mutant of Cbl-b tended to increase the amount of the EGFRvIII protein (Figure 3c). Therefore, like the WT EGFR (Ettenberg et al., 2001), the TKB and RING finger domains of Cbl-b are sufficient for the downregulation of the EGFRvIII. Also, the E3 activity of Cbl-b is necessary for the downregulation of the EGFRvIII by Cbl-b.
Figure 3.
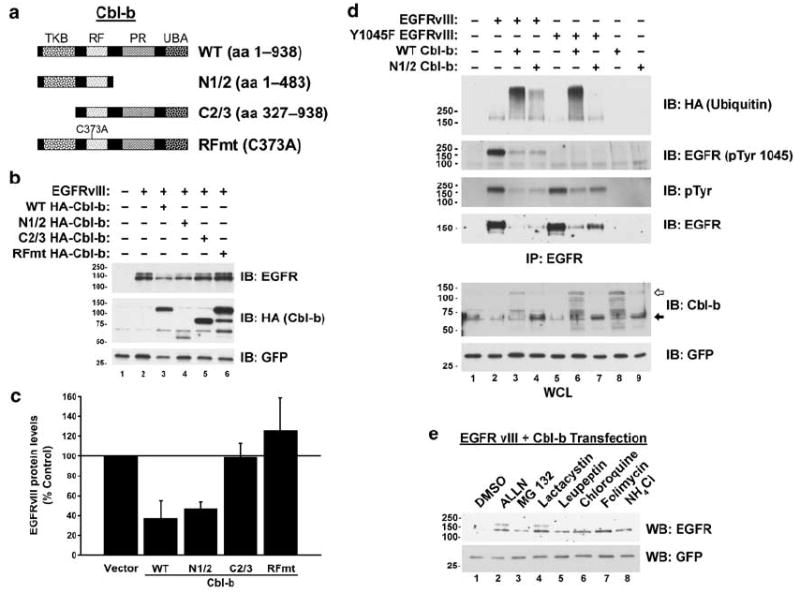
The RING finger of Cbl-b is necessary for the downregulation of the EGFRvIII. (a) WT Cbl-b (aa 1–938), N1/2 Cbl-b (aa 1–483), C2/3 Cbl-b (aa 327–938), or Cbl-b RING finger mutant (RFmt Cbl-b; C373A) expression constructs were used to map the regions of Cbl-b necessary for the downregulation of the EGFRvIII. (b) CHO cells were transfected with the EGFRvIII alone or in combination with various Cbl-b constructs as indicated. Whole-cell lysates (WCL) were immunoblotted (IB) for the EGFR, Cbl-b, or GFP as specified to the right of the blots. (c) EGFRvIII protein levels were then quantified by densitometry. All values, expressed as the percentage of the amount of EGFRvIII protein in the control transfection, are adjusted for GFP levels and are the mean and s.e.m. of n=3 experiments. (d) Mutation of Y1045 in the EGFRvIII abrogates its downregulation by Cbl-b. CHO cells were transfected with the EGFRvIII, the Y1045F EGFRvIII, HA-epitope-tagged ubiquitin, WT Cbl-b, and N1/2 Cbl-b (aa 1–483) as indicated. WCL or EGFR immunoprecipitates (IP) were immunoblotted for the EGFR, Cbl-b, or ubiquitin (with anti-HA) as indicated. The positions of WT Cbl-b (white arrow) and N1/2 Cbl-b (black arrow) are indicated along the right-hand side of the blot. Note that a non-specific artefact band migrates at the size of N1/2 Cbl-b in all lanes, a darker band corresponding to N1/2 Cbl-b can be seen in the relevant lanes. (e) Proteasomal and lysosomal inhibitors prevent Cbl-b-mediated degradation of the EGFRvIII. CHO cells were transfected with the EGFRvIII and Cbl-b. Cells were serum starved overnight and then incubated with 0.1% DMSO, 20 μm ALLN, 20 μm MG 132, 20 μm Lactacystin, 200 μm Leupeptin, 200 μm Chloroquine, 200 μm Folimycin, or 20 mM ammonium chloride (NH4Cl) for 8 h. WCL were immunoblotted for the EGFR or GFP as indicated. All transfections were balanced with empty vector controls; GFP was used as a transfection control. Molecular weight standards (kDa) appear to the left of the panels.
The TKB domain of the Cbl proteins has been shown to mediate a specific binding to a phosphotyrosine residue (Y1045) in the activated WT EGFR (Levkowitz et al., 1999; Waterman et al., 2002). The mutation of this residue attenuates the downregulation of the EGFR. We tested the ability of the equivalent mutation (Y1045F) in the EGFRvIII to affect its regulation by Cbl-b (Figure 3d). Using an antibody against phosphotyrosine 1045 EGFR, we detected phosphorylation of the EGFRvIII at this residue that was abolished by its mutation to phenylalanine (Figure 3d, second panel; compare lanes 2 and 5). As in the WT EGFR, Y1045 appears to be a minor phosphotyrosine residue (Levkowitz et al., 1999), as the loss of Y1045 phosphorylation (Figure 3d, second panel) by mutation of this residue does not decrease significantly the content of EGFRvIII phosphotyrosine (Figure 3d, third panel; compare lanes 2 and 5). As described above (Figure 3b), the EGFRvIII is ubiquitinated and downregulated by both WT and N1/2 Cbl-b (Figure 3d; lanes 3 and 4). In contrast, the Y1045F mutation in the EGFRvIII abolishes the ability of N1/2, but not WT Cbl-b to ubiquitinate the EGFRvIII (Figure 3d, top panel; lanes 6 and 7). This mutation also attenuates the downregulation of the EGFRvIII by N1/2 to a greater extent than WT Cbl-b (Figure 3d, fourth panel; compare lanes 3 and 6 and 4 and 7). Whereas N1/2 Cbl-b only contains the RING finger and TKB domains, full-length WT Cbl-b contains an extensive proline-rich region that binds Grb2. Grb2 is capable of mediating the indirect binding of the Cbl proteins to the WT EGFR (Waterman et al., 2002; Jiang et al., 2003). The ubiquitination of the Y1045F mutant EGFRvIII by WT Cbl-b, but not N1/2 Cbl-b (Figure 3d), suggests that, like the WT EGFR, the EGFRvIII can indirectly interact with the Cbl proteins.
As described above, the requirements for the downregulation of the EGFRvIII by Cbl-b appear identical to that of the WT EGFR. The targeted degradation of the active WT EGFR by Cbl-b can be blocked by both lysosomal and proteasomal inhibitors (Ettenberg et al., 2001). We investigated whether this was also the case for the degradation of the EGFRvIII by Cbl-b. EGFRvIII protein levels were stabilized by both proteasomal (ALLN, MG 132, and lactacystin) and lysosomal (leupeptin, chloroquine, folimycin, and ammonium chloride) inhibitors in CHO cells co-transfected with the EGFRvIII and Cbl-b (Figure 3e; compare lane 1 to lanes 2–8). Therefore, it appears that the degradation of the WT EGFR and the EGFRvIII by Cbl-b share a similar mechanism.
The EGFRvIII and Cbl-b associate
The ligand-induced downregulation of the WT EGFR by the Cbl proteins requires their binding to the receptor. We examined the ability of Cbl-b to bind to the EGFRvIII. In contrast to the WT EGFR following EGF stimulation, only a small proportion of the EGFRvIII is active at any given time (Huang et al., 1997; Fernandes et al., 2001). As Cbl-b targets this active pool of the EGFRvIII for degradation, the EGFRvIII bound to Cbl-b would be predicted to be a very small fraction of total EGFRvIII protein. Unlike WT Cbl-b, Cbl-b with a mutation in its RING finger (C373A) does not downregulate the EGFRvIII (see Figure 3b), thereby increasing the likelihood of observing an interaction between the EGFRvIII and Cbl-b. Indeed, when CHO cells were transfected with a combination of the EGFRvIII and a RING finger mutant of Cbl-b, we observed an association between the EGFRvIII and Cbl-b when either Cbl-b (Figure 4a, lane 3) or the EGFRvIII (Figure 4b, lane 3) were precipitated. We were also able to co-precipitate WT Cbl-b along with the EGFRvIII (data not shown).
Figure 4.
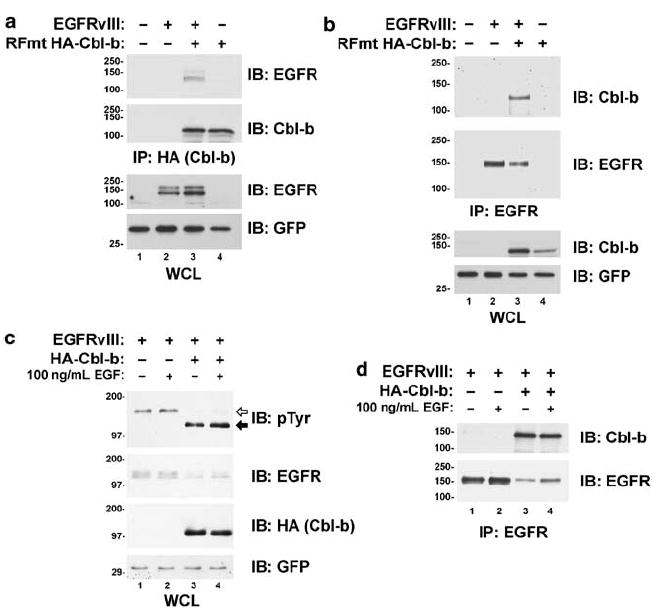
Cbl-b associates with the EGFRvIII. CHO cells were transfected with the EGFRvIII and HA-epitope-tagged RING finger mutant (RFmt; C373A) Cbl-b. Cells were serum starved overnight, lysed, and either (a) Cbl-b (with anti-HA) or (b) the EGFRvIII was immunoprecipitated. Whole-cell lysates (WCL) or the immunoprecipitates (IP) were immunoblotted (IB) for the EGFR, Cbl-b, or GFP as indicated to the right-hand of the blots. (c,d) Cbl-b associates with and downregulates the EGFRvIII in HEK 293T cells. HEK 293T cells were transfected with the EGFRvIII and HA-epitope-tagged Cbl-b as indicated. The cells were serum starved overnight and incubated with or without 100 ng/ml EGF for 30 min. (c) WCL or (d) EGFR IP were immunoblotted for the EGFR, Cbl-b (with anti-HA), phosphotyrosine (pTyr), or GFP as indicated to the right-hand side of the blots. The positions of the tyrosine-phosphorylated EGFRvIII (white arrow) and HA-Cbl-b (black arrow) proteins are indicated along the right-hand side of the blot. All transfections were balanced with empty vector controls; GFP was used as a transfection control. Molecular weight standards (kDa) appear to the left of the panels.
As in CHO cells (Figures 2 and 3d), the co-transfection of the EGFRvIII and Cbl-b into human embryonic kidney (HEK) 293T cells decreased EGFR-vIII protein levels and tyrosine phosphorylation (Figure 4c). In addition, we were also able to co-precipitate the EGFRvIII and WT Cbl-b from the lysates of HEK 293T cells transfected with these proteins (Figure 4d). Activation of the endogenous EGFR by EGF (100 ng/ml; 30 min) did not affect significantly the downregulation of the EGFRvIII by Cbl-b, nor did it affect the association between these proteins. Similarly, the co-expression of the WT EGFR with the EGFRvIII in CHO cells did not appear to affect the regulation of EGFRvIII by Cbl-b (data not shown).
Cbl-b prevents the ability of the EGFRvIII to induce transformation of NIH 3T3 fibroblasts
The EGFRvIII has been shown to mediate cell transformation as a consequence of its constitutively active TK (Nagane et al., 1996; Huang et al., 1997). As Cbl-b downregulates active EGFRvIII, we tested the ability of Cbl-b to inhibit EGFRvIII-induced transformation using a cell focus forming assay. Immortalized NIH 3T3 cells were transfected with either the EGFRvIII, Cbl-b, RING finger mutant Cbl-b, or a combination of the EGFRvIII and Cbl-b or RING finger mutant Cbl-b. All transfections were balanced with empty control vectors. Stable Zeocin- and G-418-resistant clones were pooled and a focus-forming assay was performed. We found that cells ectopically expressing the EGFRvIII gave rise to foci 10–14 days after inoculation (Figure 5a, plate 2). The overexpression of Cbl-b alone did not induce foci formation (Figure 5a, plate 3), instead it inhibited the formation of foci by the EGFRvIII (Figure 5a, plate 4). Western blotting of the pooled Zeocin- and G-418-resistant clones indicated that Cbl-b downregulates the EGFRvIII in NIH 3T3 cells (data not shown). In contrast, a RING finger mutant of Cbl-b (C373A) failed to suppress the induction of foci by the EGFRvIII (Figure 5a, plate 6). Therefore, Cbl-b inhibits the ability of the EGFRvIII to transform and this inhibition is dependent upon the E3 activity of Cbl-b.
Figure 5.
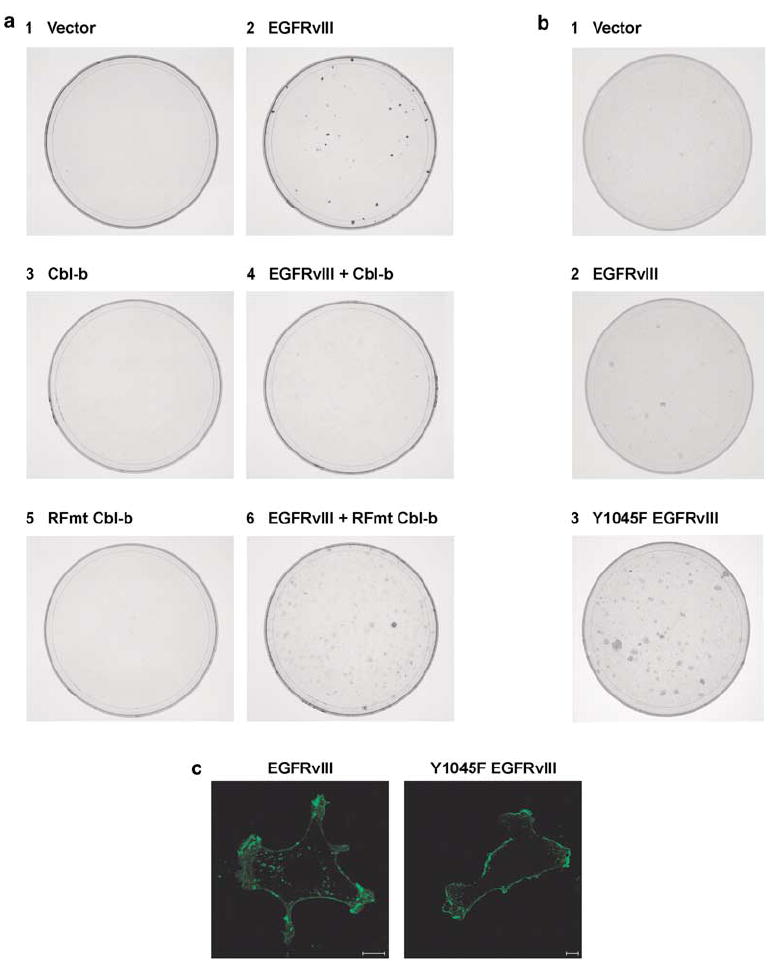
The transformation of NIH 3T3 cells by the EGFRvIII is regulated by Cbl proteins. (a) NIH 3T3 cells were transfected with 2 μg of the EGFRvIII, HA-epitope-tagged WT Cbl-b, HA-epitope-tagged RING finger mutant (RFmt; C373A) Cbl-b, or empty vector controls as indicated before selection in medium containing 600 μg/ml Zeocin and 600 μg/ml G-418. At passage 3, foci assays were performed. (b) NIH 3T3 cells were transfected with 1 μg of the EGFRvIII, EGFRvIII Y1045F, or empty vector before selection in medium containing 600 μg/ml Zeocin. At passage 3, foci assays were performed. (c) EGFRvIII- or Y1045 EGFRvIII-expressing NIH 3T3 cells were fixed and stained for the EGFR (green). Each panel is a representative mid-level confocal slice. Bar=10 μm.
The mutation of the Cbl-binding site (Y1045F) in the EGFRvIII attenuates its downregulation by Cbl-b (Figure 3c). This mutation increased the number of foci formed by the EGFRvIII (Figure 5b, compare plates 2 and 3). In NIH 3T3 cells, the EGFRvIII is localized in both the plasma membrane and in intracellular vesicles (Figure 5c). However, the proportion of EGFRvIII located at the plasma membrane compared to intracellular vesicles is increased by mutation of Y1045F (Figure 5c). In cells, the only proteins known to bind Y1045 when it is phosphorylated are the Cbl proteins. As both Cbl and Cbl-b are endogenous to NIH 3T3 cells this change in localization – similar to that seen with the inhibition of the EGFRvIII TK activity (Figure 2c) – is consistent with the Y1045F EGFRvIII being defective in Cbl-mediated downregulation. Although the Y1045F mutation affected the localization of the EGFRvIII and markedly enhanced foci formation in NIH 3T3 cells, this mutation had a relatively modest effect upon the downregulation of the EGFRvIII by Cbl-b in CHO cells (Figure 3d). This is likely due to the low endogenous levels of the Cbl proteins present in the NIH 3T3 cells used in the focus-forming assay compared to the levels of Cbl-b when it is overexpressed in CHO cells. Similarly, Waterman et al. (2002) reported that mitogenic signaling from the WT EGFR was increased significantly by the Y1045F mutation in the context of endogenous Cbl proteins.
As the formation of foci is increased by the mutation of the Cbl-binding site in the EGFRvIII and decreased by the overexpression of Cbl-b (Figure 5), the ability of the EGFRvIII to transform is regulated by the Cbl proteins.
The cytotoxicity of an EGFRvIII-specific immunotoxin is antagonized by an EGFRvIII TK inhibitor
To confirm further that the EGFRvIII undergoes activation-dependent downregulation, we investigated the effects of an EGFR TK inhibitor, AG 1478, upon the activity of an anti-EGFRvIII immunotoxin (MR1-1(scFv)-PE38) (Beers et al., 2000). Immunotoxins must be internalized upon binding to their receptor in order to kill cells (Pastan et al., 1992). As we have shown above (Figure 2), AG 1478 treatment inhibits the activation-induced downregulation of the EGFRvIII by the Cbl proteins. Therefore, the inhibition of the EGFRvIII TK would be expected to decrease the efficacy of the anti-EGFRvIII immunotoxin MR1-1(scFv)-PE38. The effect of MR1-1(scFv)-PE38 treatment upon the viability of a murine fibroblast cell line (NR-6) and a subclone that stably expresses the EGFRvIII (NR-6m) (Batra et al., 1995) was measured using an MTS dye reduction assay (Figure 6a). Previously, we have shown that this indirect measurement of cytotoxicity correlates with cell death (Keane et al., 1996). A 24 h incubation with MR1-1 (scFv)-PE38 causes a concentration-dependent decrease in the viability of NR-6m cells. In contrast, the viability of the parental cell line (NR-6), which does not express the EGFRvIII, is not affected by treatment with the fusion toxin. Treatment with 30 μm AG 1478 attenuated the decrease in viability of NR-6m cells caused by MR1-1(scFv)-PE38 (Figure 6b). The concentration of MR1-1(scFv)-PE38 necessary to reduce cell viability by 50% was approximately 1000-fold higher when cells were incubated with 30 μm AG 1478 (IC50≈10 μg/ml) than when they were incubated with the vehicle (IC50≈10 ng/ml). Therefore, the TK activity of the EGFRvIII has an important role in mediating the toxicity of anti-EGFRvIII immunotoxins. In addition, this result is consistent with the EGFRvIII undergoing activation-induced downregulation.
Figure 6.

The EGFR TK inhibitor, AG 1478, prevents MR1-1(scFv)-PE38-mediated cell death of EGFRvIII-expressing cells. (a) The murine fibroblast cell lines NR-6 (●) and the EGFRvIII-expressing NR-6m (■) were incubated with increasing concentrations (0.01–10 000 ng/ml) of MR1-1(scFv)-PE38 for 24 h. The cell viability, measured by an MTS dye reduction assay, is expressed as a percentage of maximum. Values are the mean±1 s.d. of a representative experiment. (b) EGFRvIII-expressing NR-6m cells were pretreated with either the vehicle (0.1% DMSO;■) or 30 μm AG 1478 (▲) for 4 h. The cells were then incubated with a combination of 0.01–10 000 ng/ml MR1-1(scFv)-PE38 in the continued presence of either the vehicle or 30 μm AG 1478. The cell viability, measured by an MTS dye reduction assay, is expressed as a percentage of maximum. Values are the mean±s.e.m. of n=3 experiments.
Discussion
The ability of all three members of the Cbl family of E3s (Cbl, Cbl-b, and Cbl-c) to ubiquitinate and downregulate the EGFR following stimulation with EGF is well-characterized (Ettenberg et al., 1999b, 2001; Levkowitz et al., 1999; Waterman et al., 1999a; Yokouchi et al., 1999; Duan et al., 2003). In this study, we establish that the Cbl proteins can downregulate the constitutively active mutant of the EGFR known as the EGFRvIII. The overexpression of Cbl, Cbl-b, or Cbl-c caused a decrease in the level of EGFRvIII protein in CHO cells (Figure 1a–c). We observed also that the co-expression of the Cbl proteins enhanced the ubiquitination of the EGFRvIII (Figure 1d–f). This downregulation of the EGFRvIII by Cbl-b was blocked by the use of an EGFR TK inhibitor, AG 1478 (Figure 2), and by the Y1045F mutation of the EGFRvIII (Figures 3 and 5). As in the active WT EGFR, Y1045 is phosphorylated in the EGFRvIII and the Y1045F mutation prevents phosphorylation of this residue (Figure 3d). This prevents the direct binding of the Cbl proteins, the only proteins known to interact with this phosphotyrosine residue in cells. The abrogation of the interaction of the EGFRvIII with endogenous Cbl proteins by either EGFRvIII Y1045F mutation (Figure 5b and c) or TK inhibition (Figures 2 and 6b) blocks EGFRvIII downregulation. Therefore, it appears that the Cbl proteins mediate the activation-induced downregulation of the EGFRvIII.
The ligand-induced activation of the WT EGFR results in its autophosphorylation and the subsequent recruitment of Cbl-b (Ettenberg et al., 1999a). Therefore, we investigated the interaction between the EGFRvIII and Cbl-b using a cell line that expresses endogenous EGFR (HEK 293T) and a cell line that does not (CHO). We observed an association between the EGFRvIII and Cbl-b in both of these cell lines (Figure 4). The interaction between the EGFRvIII and Cbl-b in HEK 293T cells appears to be unaffected by the activation of WT EGFR by EGF. In addition, the co-transfection of the WT EGFR and the EGFRvIII into CHO cells did not appear to prevent the downregulation of either of these proteins by Cbl-b (data not shown). Therefore, it appears that the constitutive association between Cbl-b and the EGFRvIII is independent of the WT EGFR. Like the WT EGFR, we found that the recruitment of Cbl-b to the EGFRvIII involves two mechanisms: one that involves the TKB domain of Cbl-b, the other that involves the proline-rich carboxy-terminus of Cbl-b. Using the end point of receptor degradation, we found that the EGFRvIII is downregulated by both WT Cbl-b and a truncated form of Cbl-b (N1/2) that contains its TKB and RING finger domains, but not its extensive proline-rich carboxy-terminus (Figure 3). Mutation of the Cbl TKB-binding site in the WT EGFR (Y1045F) impairs the ligand-induced ubiquitination and downregulation of the EGFR (Levkowitz et al., 1999). When we mutated the equivalent residue in the EGFRvIII, we prevented the ubiquitination and downregulation of this receptor by N1/2 Cbl-b (Figure 3d). However, the mutation of this residue does not appear to have as significant an effect upon the interaction between the EGFRvIII and WT Cbl-b. As the proline-rich region of the Cbl proteins can indirectly bind to the WT EGFR via Grb2 (Waterman et al., 2002; Jiang et al., 2003), this is likely also the case with the EGFRvIII. The EGFRvIII has been shown to bind to Grb2 in NIH 3T3 fibroblasts (Moscatello et al., 1996). Interestingly, stable clones of NIH 3T3 cells expressing high levels of the EGFRvIII have decreased levels of Grb2 protein (Moscatello et al., 1996). This is consistent with the ability of the Cbl proteins to downregulate the EGFR signaling complex, including Grb2 (Ettenberg et al., 2001).
In contrast to the present study, Schmidt et al. (2003) reported that the EGFRvIII does not interact with either Cbl or Cbl-b. In their investigation, HEK 293 cells were transfected with EGFRvIII and either Cbl or Cbl-b. Then the EGFRvIII was precipitated with an anti-EGFRvIII-specific antibody. Although they observed the co-precipitation of both Cbl and Cbl-b with the EGFRvIII, the WT EGFR was also precipitated in their experiments. They concluded that the anti-EGFRvIII antibody was crossreacting with the WT receptor, so in subsequent experiments they precleared the lysate with an anti-EGFR antibody before the precipitation of the EGFRvIII. Following preclearing of the lysates, they failed to observe either Cbl or Cbl-b when the EGFRvIII was precipitated. In addition, they were also unable to observe any ubiquitination of the EGFRvIII following this preclearing. As the EGFRvIII and the WT EGFR are capable of heterodimerizing (Luwor et al., 2004), it is possible that this preclearing step removed any of the EGFRvIII that is bound to the WT EGFR. As this heterodimerized protein may be the active pool of the EGFRvIII, this could account for any differences between the two studies. Our experiments in CHO cells, which do not express the WT EGFR, allowed us to investigate the interaction between the EGFRvIII and the Cbl proteins in the absence of the WT receptor. In addition, we used a mutant (C373A) of Cbl-b deficient in E3 activity to test an interaction between the EGFRvIII and Cbl-b in CHO cells. As this mutant cannot target the complex of Cbl-b and the EGFRvIII for lysosomal degradation, the amount of active EGFRvIII bound to Cbl-b should be increased relative to cells transfected with WT Cbl-b. Therefore, any association between these proteins should be detected with a greater sensitivity than if WT Cbl-b was used. Only a small fraction (approximately 10%) of the EGFRvIII protein is active at any given time compared to the WT EGFR that has been stimulated by EGF (Huang et al., 1997; Fernandes et al., 2001). Thus, it is possible that the interaction between the EGFRvIII and the Cbl proteins was below the level of sensitivity of the immunoprecipitation and immunoblotting procedure used by Schmidt et al. (2003).
The constitutive TK activity of the EGFRvIII results in the malignant transformation of cells (Han et al., 1996; Nagane et al., 1996; Huang et al., 1997; Antonyak et al., 1998; Montgomery, 2002; Johns et al., 2003; Abulrob et al., 2004; Luwor et al., 2004; Pedersen et al., 2004). In this study, we found that the EGFRvIII is regulated by the Cbl proteins in an identical manner to the WT EGFR. This is unsurprising given that the activity and phosphorylation pattern of the dimerized EGFRvIII is similar to that of the WT EGFR following EGF stimulation (Fernandes et al., 2001). Indeed, we were able to detect phosphorylation of the Cbl TKB-binding site (Y1045) on the EGFRvIII using a specific antibody (Figure 3d). In addition, Reist et al. (1995) reported that the EGFRvIII is internalized rapidly from the surface of fibroblasts transfected with the EGFR-vIII, suggesting that it is downregulated. Conversely, in a study using glioblastoma cells transfected with either the WT EGFR or the EGFRvIII, Huang et al. (1997) reported that, while the EGF-stimulated WT EGFR is rapidly endocytosed, the EGFRvIII is internalized at a similar rate to that of the unstimulated WT EGFR. This suggests that the EGFRvIII is not downregulated. However, only a small proportion of the total EGFR-vIII protein is active when compared to the ligand bound EGFR (Huang et al., 1997; Fernandes et al., 2001). It is likely that, compared to the spontaneous endocytosis of the overexpressed WT EGFR, the enhanced internalization of the small amount of active EGFRvIII does not significantly affect the overall rate of endocytosis.
Our work indicates that active EGFRvIII is degraded by a Cbl protein-dependent mechanism. However, cancer cells with amplification of the EGFRvIII constitutively synthesize new inactive EGFRvIII protein. Experiments using the EGFR inhibitor AG 1478 demonstrate that the Cbl proteins do not mediate ubiquitination or degradation of inactive EGFRvIII (Figure 2). The amplification and overexpression of the EGFRvIII creates a large pool of inactive receptor, a small fraction of which spontaneously activates to replenish the pool of downregulated active EGFRvIII. Thus, at steady-state equilibrium, there always will be active EGFRvIII and this results in the transformation of cells. The overexpression of Cbl-b inhibits the transformation of fibroblasts by the EGFRvIII by enhancing the degradation of the active EGFRvIII. Conversely, the mutation of the Cbl-binding site in the EGFRvIII increases its capacity to transform by preventing degradation of the active EGFRvIII.
The anti-EGFRvIII immunotoxin, MR1-1(scFv)-PE38, kills glioblastoma cells that ectopically express the EGFRvIII (Beers et al., 2000). In this study, we used an MTS dye reduction assay to test the ability of this immunotoxin to kill a Swiss 3T3-derived cell line (NR-6) that does not express the WT EGFR (Pruss and Herschman, 1977). Although MR1-1(scFv)-PE38 did not effect the growth of NR-6 cells, it caused a concentration-dependent death of EGFRvIII-expressing NR-6m cells (Figure 6a). This finding confirmed the previous report (Beers et al., 2000) that MR1-1(scFv)-PE38 specifically kills EGFRvIII-expressing cells. The IC50 of MR1-1(scFv)-PE38 in this study (approximately 10 ng/ml) is similar to previously reported values (Beers et al., 2000). To function, immunotoxins must be internalized upon binding to their receptors (Pastan et al., 1992); indeed anti-EGFRvIII monoclonal antibodies — including MR1-1(scFv)-PE38 — are rapidly internalized by EGFRvIII-expressing cells (Reist et al., 1995; Kuan et al., 2000). These internalized antibodies become localized to vesicles in the perinuclear Golgi region and are rapidly catabolized, suggesting that the internalized EGFRvIII:monoclonal antibody complex is trafficked to the lysosome. The Cbl proteins are critical regulators of the trafficking of the WT EGFR to the lysosome (Duan et al., 2003) and this study has established that they regulate the constitutively active EGFRvIII. Furthermore, the inhibition of the TK activity of the EGFRvIII prevents its downregulation by the Cbl proteins and decreases the amount of EGFRvIII located in intracellular vesicles (Figure 2). Therefore, we tested whether inhibition of the EGFR-vIII TK affects the efficacy of MR1-1(scFv)-PE38. Consistent with the ability of the EGFRvIII to undergo activation-induced downregulation, we found that treatment with AG 1478 caused an approximately 1000-fold increase in the IC50 of MR1-1(scFv)-PE38 (Figure 6b). Thus, the inhibition of the TK activity of the EGFRvIII appears to antagonize MR1-1(scFv)-PE38 in vitro. Like the WT EGFR, the EGFRvIII also can be spontaneously endocytosed in an activation-independent manner. Thus, MR1-1(scFv)-PE38 is still capable of killing cells in the presence of AG1478, albeit with an IC50 1000-fold higher than untreated cells. This finding suggests that TK inhibitors and immunotoxins may be antagonistic if used together for the treatment of EGFRvIII-expressing tumors.
This study has demonstrated that the EGFRvIII undergoes activation-induced downregulation by the Cbl proteins. This suggests that the ability of the EGFRvIII to transform cells is not a consequence of unattenuated signaling from this mutant, but is due rather to the spontaneous activity of this TK. The ability of the EGFRvIII to be regulated by the Cbl proteins has implications for the treatment of malignancies. Therapies, such as immunotoxins, that exploit the down-regulation of the EGFRvIII or therapies aimed at enhancing the activation-induced degradation of this mutant offer a promising approach to the treatment of EGFRvIII-expressing tumors. However, the use of TK inhibitors in conjunction with these therapies may decrease their efficacy.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin, streptomycin sulfate, and Zeocin were obtained from Invitrogen (Carlsbad, CA, USA). Dulbecco’s phosphate-buffered saline (DPBS) and G-418 sulfate were purchased from Mediatech Inc. (Herndon, VA, USA). AG 1478, ALLN (MG 101), cycloheximide, MG 132, lactacystin, and folimycin (concanamycin A) were acquired from EMD Biosciences Inc. (San Diego, CA, USA). Leupeptin hemisulfate was bought from MP Biomedicals (Irvine, CA, USA). Chloroquine, ammonium chloride, and DMSO were obtained from Sigma-Aldrich Corp. (St Louis, MO, USA). Recombinant human EGF was purchased from BD Biosciences, Inc. (San Jose, CA, USA). A recombinant immunotoxin generated from an EGFRvIII-specific single chain Fv domain fused to domains I and II of the Pseudomonas exotoxin (MR1-1(scFv)-PE38) was provided by Dr Ira Pastan (Beers et al., 2000). Tissue culture plastic ware and other laboratory consumables were purchased from commercial sources.
Expression constructs
The expression plasmids for full-length WT and HA-epitope-tagged Cbl, Cbl-b, and Cbl c along with HA-epitope-tagged full-length RING finger mutant (C373A) Cbl-b, C2/3 Cbl-b (aa 327–938), N1/2 Cbl-b (aa 1–483), and the control vector (pCEFL) have been described previously (Ettenberg et al., 1999a; Ettenberg et al., 2001). The cDNA for the EGFRvIII was a gift from Dr Gordon N Gill and was cloned into pSVZeo(−) (Invitrogen, Carlsbad, CA, USA). Site-directed mutagenesis of EGFRvIII (Y1045F) was performed using the Quick Change Kit (Stratagene, La Jolla, CA, USA). All of the constructs were confirmed by DNA sequencing. The GFP expression plasmid (pCDNA3.1/zeo) was obtained from Invitrogen (Carlsbad, CA, USA). The HA-epitope-tagged ubiquitin expression plasmid was provided by Dr Dirk Bohmann (Treier et al., 1994).
Cell culture, transfections, and foci assays
CHO, HEK 293T, and NIH 3T3 cells were maintained in culture in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. NR-6 cells were maintained in DMEM supplemented with 5% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. NR-6m cells, a subclone of NR-6 that stably expresses the EGFRvIII, were provided by Dr Darrel Bigner and were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate, and 750 μg/ml G-418. CHO cells were transfected with various constructs using FuGENE 6 (Roche Diagnostics Corp., Indianapolis, IN, USA), whereas HEK 293T cells were transfected using calcium phosphate (Profection; Promega Corp., Madison, WI, USA). Following transfection, cells were grown to 70% confluence and starved overnight in DMEM supplemented with 0.5% FBS. Then, cells were treated as described in the figure legends before the preparation of cell lysates.
NIH 3T3 cells were transfected with the EGFRvIII, Y1045F EGFRvIII, HA-Cbl-b, C373A HA-Cbl-b, or empty vector controls as indicated using Effectine (Qiagen Inc., Valencia, CA, USA). A day after the transfection, the cells were split 1:3 and grown for 14 days in selection medium containing either 600 μg/ml Zeocin alone or a combination of 600 μg/ml Zeocin and 600 μg/ml G-418. Stable clones were pooled and foci assays were performed at passage 3 by plating 1×106 cells per 100 mm tissue culture dish. Cells were incubated 1–2 weeks, fixed with 10% methanol, 10% acetic acid solution for 15 min, and stained with 20% ethanol, 0.4% crystal violet for 5 min.
Immunoblotting and immunoprecipitation
To harvest proteins, cells were washed twice in ice-cold DPBS containing 200 μm sodium orthovanadate (Fisher Chemicals, Fairlawn, NJ, USA) and then lysed in ice-cold lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10% glycerol, 100 mM iodoacetamide (Sigma-Aldrich Corp., St Louis, MO, USA), 2 mM sodium orthovanadate, and protease inhibitors (Complete tabs®, Roche Diagnostics Corp., Indianapolis, IN, USA)). The lysates were cleared of debris by centrifugation at 16 000 g for 10 min at 4°C. Supernatant protein concentrations were determined using a BioRad protein assay (BioRad, Hercules, CA, USA). For immunoblotting, lysates (2 μg protein/μl) were boiled in loading buffer (62.5 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 1 mg/ml bromphenol blue, 0.3573M β-mercaptoethanol) for 5 min. For immunoprecipitation, lysates containing 500 μg protein were incubated with either a mouse monoclonal anti-EGFR antibody (528; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and Protein A/G+ agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or HA-affinity matrix (Roche Diagnostics Corp., Indianapolis, IN, USA) overnight at 4°C with tumbling. Immune complexes were washed five times in cold lysis buffer, resuspended in 2× loading buffer and boiled for 5 min. The proteins were resolved by SDS–PAGE and transferred to PVDF membranes (Immobilon P; Millipore, Billerica, MA, USA). Membranes were probed with either rabbit polyclonal anti-EGFR (2232; Cell Signaling Technology Inc., Beverly, MA, USA), rabbit polyclonal anti-phosphotyrosine 1045 EGFR (2237; Cell Signaling Technology Inc., Beverly, MA, USA), rabbit polyclonal anti-Cbl (C15; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-Cbl-b (H121 or H454; Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat polyclonal anti-Cbl-c (C15; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-HA (12CA5; Roche Diagnostics Corp., Indianapolis, IN, USA), mouse monoclonal anti-GFP (C163; Zymed, San Francisco, CA, USA), mouse monoclonal anti-α-Tubulin (DM 1A; Sigma-Aldrich Corp., St Louis, MO, USA), or peroxidase-linked anti-phosphotyrosine (4G10; Upstate Biotechnology Inc., Waltham, MA, USA) antibodies. Horse-radish peroxidase-linked donkey anti-rabbit (GE Healthcare, Piscataway, NJ, USA), donkey anti-mouse (GE Healthcare, Piscataway, NJ, USA), or rabbit anti-goat (Santa Cruz Biotechnology, Santa Cruz, CA, USA) immunoglobulin was used with SuperSignal (Pierce Biotechnology Inc., Rockford, IL, USA) to visualize the blots. Immunoblots were quantified on a PC computer using the public domain NIH Image program (developed at the US National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Immunofluorescence and confocal microscopy
NR-6m cells or pooled clones of NIH 3T3 cells expressing the EGFRvIII or Y1045F EGFRvIII were plated at 2×104 cells/ well in four-well chambered coverslips (Nalgene Nunc International, Rochester, NY, USA) and incubated overnight. Then, the NR-6m cells were incubated for 3 h with 100 μg/ml cycloheximide and either 30 μm AG 1478 or 0.1% DMSO. Following a rinse with PBS, both NR-6m and NIH 3T3 cells were fixed with 2% paraformaldehyde in PBS for 30 min at room temperature. The chambers were rinsed three times with PBS, washed three times with PFNS buffer (PBS, containing 10% FBS, 0.02% sodium azide (Sigma-Aldrich Corp., St Louis, MO, USA), and 10% saponin (Sigma-Aldrich Corp., St Louis, MO, USA)) and blocked with PFNS-G (PFNS buffer supplemented with 2% normal goat serum) for 30 min at room temperature. Blocked chambers were then incubated overnight at 4°C with either mouse monoclonal anti-EGFR (528; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse monoclonal anti-phosphotyrosine 1173 EGFR (9H2; Upstate Biotechnology Inc., Waltham, MA, USA) antibodies diluted in PFNS-G, washed three times with PFNS, and incubated with Alexa-Fluor 488-conjugated goat anti-mouse antibody diluted in PFNS-G for 1 h at room temperature. The chambers were then washed three times with PBS containing 2% saponin, stained with 300 nM DAPI in PBS for 3 min, and rinsed three times with PBS. All images were collected using a Ziess 510 META confocal microscope with a ×63 (NA 1.4) Plan-Apochromat oil immersion objective (Carl Zeiss Microimaging Inc., Thornwood, NY, USA). Alexa-Fluor 488 staining was imaged using a 488 nm Argon Laser line in conjunction with a HFT 405/488/543/633 multiple beam splitter, NFT 545 dichroic, and a BP 505–570 emission filter. DAPI was imaged using a 405 nm laser diode line, HFT 405/ 488/543/633 multiple beam splitter, NFT 505 dichroic, and a BP 420–480 emission filter. The laser power was set to 4% transmission with the pinhole opened to 1 Airy unit. Confocal image series were recorded with a frame size of 512×512 pixels and a pixel size of 110–140 nm. Images were processed with Zeiss LSM Image Browser (version 3.5). Adobe Photoshop (version 7.0) was used to prepare composite images.
Cytotoxicity assays
NR-6 or NR-6m cells were plated at 1.5×104 cells/well in 96-well microtiter plates and incubated overnight. Then, NR-6m cells were incubated for 4 h with either 30 μm AG 1478 or 0.1% DMSO. NR-6m cells were then incubated for a further 24 h with either 30 μm AG 1478 or 0.1% DMSO alone or in combination with 0.1–10 000 ng/ml MR1-1(scFv)-PE38. NR-6 cells were incubated with MR1-1(scFv)-PE38 (0.1–10 000 ng/ml) or mock-treated for 24 h. Cell viability was assessed by the MTS dye reduction assay (Cell Titer 96 AQueous One Solution Cell Proliferation assay; Promega Corp., Madison, WI, USA). Six wells were used in each experimental group. The cell viability in each experimental group was expressed as the mean percentage of maximum viability. Values are the mean±1 s.d. of a representative experiment or the mean±s.e.m. for n=3 experiments.
Acknowledgments
We thank Marion M Nau for helpful discussion and critical review of the manuscript. We thank Jeremy Karlin for helpful discussion and for help with quantitation of the immunoblots. Also, we thank Valarie A Barr for help with the confocal microscopy. Philip E Ryan is a graduate student in the Genetics Program of the George Washington University Institute of Biomedical Sciences. The work presented here is in partial fulfillment for the degree of PhD. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- Abulrob A, Giuseppin S, Andrade MF, McDermid A, Moreno M, Stanimirovic D. Oncogene. 2004;23:6967–6979. doi: 10.1038/sj.onc.1207911. [DOI] [PubMed] [Google Scholar]
- Antonyak MA, Moscatello DK, Wong AJ. J Biol Chem. 1998;273:2817–2822. doi: 10.1074/jbc.273.5.2817. [DOI] [PubMed] [Google Scholar]
- Batra SK, Castelino-Prabhu S, Wikstrand CJ, Zhu X, Humphrey PA, Friedman HS, et al. Cell Growth Differ. 1995;6:1251–1259. [PubMed] [Google Scholar]
- Beers R, Chowdhury P, Bigner D, Pastan I. Clin Cancer Res. 2000;6:2835–2843. [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Damstrup L, Wandahl Pedersen M, Bastholm L, Elling F, Skovgaard Poulsen H. Int J Cancer. 2002;97:7–14. doi: 10.1002/ijc.1572. [DOI] [PubMed] [Google Scholar]
- Duan L, Miura Y, Dimri M, Majumder B, Dodge IL, Reddi AL, et al. J Biol Chem. 2003;278:28950–28960. doi: 10.1074/jbc.M304474200. [DOI] [PubMed] [Google Scholar]
- Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, Collins VP, et al. Oncogene. 1994;9:2313–2320. [PubMed] [Google Scholar]
- Ettenberg SA, Keane MM, Nau MM, Frankel M, Wang LM, Pierce JH, et al. Oncogene. 1999a;18:1855–1866. doi: 10.1038/sj.onc.1202499. [DOI] [PubMed] [Google Scholar]
- Ettenberg SA, Magnifico A, Cuello M, Nau MM, Rubinstein YR, Yarden Y, et al. J Biol Chem. 2001;276:27677–27684. doi: 10.1074/jbc.M102641200. [DOI] [PubMed] [Google Scholar]
- Ettenberg SA, Rubinstein YR, Banerjee P, Nau MM, Keane MM, Lipkowitz S. Mol Cell Biol Res Commun. 1999b;2:111–118. doi: 10.1006/mcbr.1999.0157. [DOI] [PubMed] [Google Scholar]
- Fernandes H, Cohen S, Bishayee S. J Biol Chem. 2001;276:5375–5383. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- Garcia de Palazzo IE, Adams GP, Sundareshan P, Wong AJ, Testa JR, Bigner DD, et al. Cancer Res. 1993;53:3217–3220. [PubMed] [Google Scholar]
- Han Y, Caday CG, Nanda A, Cavenee WK, Huang HJ. Cancer Res. 1996;56:3859–3861. [PubMed] [Google Scholar]
- Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Clin Cancer Res. 2005;11:1462–1466. doi: 10.1158/1078-0432.CCR-04-1737. [DOI] [PubMed] [Google Scholar]
- Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. J Biol Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- Jiang X, Huang F, Marusyk A, Sorkin A. Mol Biol Cell. 2003;14:858–870. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns TG, Luwor RB, Murone C, Walker F, Weinstock J, Vitali AA, et al. Proc Natl Acad Sci USA. 2003;100:15871–15876. doi: 10.1073/pnas.2036503100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane MM, Ettenberg SA, Lowrey GA, Russell EK, Lipkowitz S. Cancer Res. 1996;56:4791–4798. [PubMed] [Google Scholar]
- Keane MM, Ettenberg SA, Nau MM, Banerjee P, Cuello M, Penninger J, et al. Oncogene. 1999;18:3365–3375. doi: 10.1038/sj.onc.1202753. [DOI] [PubMed] [Google Scholar]
- Kuan CT, Wikstrand CJ, Archer G, Beers R, Pastan I, Zalutsky MR, et al. Int J Cancer. 2000;88:962–969. doi: 10.1002/1097-0215(20001215)88:6<962::aid-ijc20>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Kuan CT, Wikstrand CJ, Bigner DD. Endocr Relat Cancer. 2001;8:83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygan-kov AY, Alroy I, et al. Mol Cell. 1999;4:1029–1040. doi: 10.1016/s1097-2765(00)80231-2. [DOI] [PubMed] [Google Scholar]
- Luwor RB, Zhu HJ, Walker F, Vitali AA, Perera RM, Burgess AW, et al. Oncogene. 2004;23:6095–6104. doi: 10.1038/sj.onc.1207870. [DOI] [PubMed] [Google Scholar]
- Mancini A, Koch A, Wilms R, Tamura T. J Biol Chem. 2002;277:14635–14640. doi: 10.1074/jbc.M109214200. [DOI] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- Montgomery RB. Int J Cancer. 2002;101:111–117. doi: 10.1002/ijc.10560. [DOI] [PubMed] [Google Scholar]
- Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, et al. Cancer Res. 1995;55:5536–5539. [PubMed] [Google Scholar]
- Moscatello DK, Montgomery RB, Sundareshan P, McDanel H, Wong MY, Wong AJ. Oncogene. 1996;13:85–96. [PubMed] [Google Scholar]
- Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. Cancer Res. 1996;56:5079–5086. [PubMed] [Google Scholar]
- Nau MM, Lipkowitz S. Gene. 2003;308:103–113. doi: 10.1016/s0378-1119(03)00471-2. [DOI] [PubMed] [Google Scholar]
- Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. Proc Natl Acad Sci USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke DM, Nute EJ, Davis JG, Wu C, Lee A, Murali R, et al. Oncogene. 1998;16:1197–1207. doi: 10.1038/sj.onc.1201635. [DOI] [PubMed] [Google Scholar]
- Pastan I, Chaudhary V, FitzGerald DJ. Annu Rev Biochem. 1992;61:331–354. doi: 10.1146/annurev.bi.61.070192.001555. [DOI] [PubMed] [Google Scholar]
- Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. Ann Oncol. 2001;12:745–760. doi: 10.1023/a:1011177318162. [DOI] [PubMed] [Google Scholar]
- Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS. Int J Cancer. 2004;108:643–653. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, et al. Mol Cell. 2001;8:995–1004. doi: 10.1016/s1097-2765(01)00378-1. [DOI] [PubMed] [Google Scholar]
- Peschard P, Park M. Cancer Cell. 2003;3:519–523. doi: 10.1016/s1535-6108(03)00136-3. [DOI] [PubMed] [Google Scholar]
- Pruss RM, Herschman HR. Proc Natl Acad Sci USA. 1977;74:3918–3921. doi: 10.1073/pnas.74.9.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reist CJ, Archer GE, Kurpad SN, Wikstrand CJ, Vaidyanathan G, Willingham MC, et al. Cancer Res. 1995;55:4375–4382. [PubMed] [Google Scholar]
- Schmidt MH, Furnari FB, Cavenee WK, Bogler O. Proc Natl Acad Sci USA. 2003;100:6505–6510. doi: 10.1073/pnas.1031790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Cancer Res. 2000;60:3081–3087. [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Waterman H, Alroy I, Strano S, Seger R, Yarden Y. EMBO J. 1999a;18:3348–3358. doi: 10.1093/emboj/18.12.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, et al. EMBO J. 2002;21:303–313. doi: 10.1093/emboj/21.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H, Levkowitz G, Alroy I, Yarden Y. J Biol Chem. 1999b;274:22151–22154. doi: 10.1074/jbc.274.32.22151. [DOI] [PubMed] [Google Scholar]
- Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN, et al. Cancer Res. 1995;55:3140–3148. [PubMed] [Google Scholar]
- Yokouchi M, Kondo T, Houghton A, Bartkiewicz M, Horne WC, Zhang H, et al. J Biol Chem. 1999;274:31707–31712. doi: 10.1074/jbc.274.44.31707. [DOI] [PubMed] [Google Scholar]