

Abstract
Freeze-fracture electron microscopy is a technique for examining the ultrastructure of rapidly frozen biological samples by transmission electron microscopy. Of a range of approaches to freeze-fracture cytochemistry that have been developed and tried, the most successful is the technique termed freeze-fracture replica immunogold labeling (FRIL). In this technique, samples are frozen, fractured and replicated with platinum-carbon as in standard freeze fracture, and then carefully treated with sodium dodecylsulphate to remove all the biological material except a fine layer of molecules attached to the replica itself. Immunogold labeling of these molecules permits their distribution to be seen superimposed upon high resolution planar views of membrane structure. Examples of how this technique has contributed to our understanding of lipid droplet biogenesis and function are discussed.
Keywords: Freeze Fracturing; Microscopy, Immunoelectron; Immunogold Techniques
Introduction
Freeze-fracture electron microscopy is a technique in transmission electron microscopy that revolutionised our understanding of membrane structure from the 1970s onwards (1). When biological specimens are in the frozen state, membranes have a plane of weakness in their hydrophobic interior so that if the sample is broken or fractured, the fracture plane will often split the membrane into half-membrane leaflets, each corresponding to a phospholipid monolayer with associated proteins. The net result is a three-dimensional perspective of the membranous organization of the cell, with en face views of the membrane interior. These details are made visible in the electron microscope by making a very fine platinum-carbon replica of the fracture plane. The platinum is evaporated onto the specimen at an angle, so that it is deposited in varying thicknesses according to the topography of the fractured surface. In this way, high resolution details of membrane structure are revealed that cannot be seen by other techniques. Through freeze fracture, the distribution and organization of integral membrane proteins (seen as intramembrane particles), and other specialized features, are rendered visible in the membrane plane.
There are four key steps in making a standard freeze-fracture replica: i) rapid freezing of the specimen, ii) fracturing it at low temperature (-100°C or lower), iii) making the replica of the newly exposed frozen surface by vacuum-deposition of platinum and carbon; and iv) cleaning the replica using bleach or acids to remove the biological material. The replica, with no biological material remaining, is finally mounted on a grid for examination in the electron microscope. For a detailed protocol on how to carry out standard freeze fracture, see Severs (1).
The contribution of freeze fracture and associated techniques to our knowledge of membrane structure was unrivalled, but one limitation remained - the need to identify the chemical nature of the structural components visualized. Without this knowledge, the functions of newly discovered structural features remained speculative. Thus, the combination of cytochemistry with freeze fracture was a widely recognized goal which attracted numerous efforts over three decades, efforts that met with varying degrees of success (2). With the introduction of the freeze-fracture replica immunolabeling technique (FRIL) developed by Fujimoto (3,4), the technical challenges can now be said to have been effectively overcome.
A major conceptual obstacle in the development of freeze-fracture cytochemistry was the assumption that removal of all the biological material (e.g., with sodium hypochlorite or chromic acid) was essential to obtain good replicas. Remnants of biological material in imperfectly cleaned replicas were all too often a source of sub-optimal results, obstructing the electron beam and thus interfering with visualization of structural detail in the replica. Such a cleaning process inevitably leads to loss of any label that might be attached to the biological material before or after freeze-fracture replication.
An important landmark in the progress to develop an effective technique in freeze-fracture cytochemistry was the introduction of colloidal gold cytochemistry to electron microscopy in the early 1980s. Gold particles, because of their high electron density and small size, were quickly recognized to be ideal markers to use in conjunction with replicas, and this stimulated experimentation with new ideas for retaining label in such a way that it could be viewed superimposed on replicated structural detail. Strategies for integrating gold labeling were experimented with at each of the steps in the freeze-fracture procedure, i.e., before freezing, after fracturing and after replication. For a detailed discussion of the full range of techniques, the reader is referred to an earlier comprehensive review (2). For the purposes of the present article, a key foundation to the development of FRIL was the recognition, through the pioneering studies of Pinto da Silva, that half-membrane leaflets could be retained attached to the platinum/carbon replica without significantly interfering with visibility of structure, and these could provide a source of epitopes for immunogold labeling, whether conducted before freezing or after replication (5-9).
Materials and Methods
The freeze-fracture immunolabeling technique
The principle of the FRIL technique is explained in Fig. 1. The specimen is rapidly frozen, freeze fractured and platinum/carbon replicas made following the standard protocol used for freeze-fracture electron microscopy. However, instead of removing the biological material from the replicas with bleach or acids as in the conventional technique, the replica is treated with sodium dodecylsulphate (SDS) (3,4). The aim of the SDS treatment is to dissociate most of the biological material from the replica so that structure is visible by electron microscopy, leaving just sufficient material adherent to the replica (ideally a single molecular layer) to retain epitopes for labeling. By then applying immunogold techniques, the spatial distribution of the targeted epitopes can be viewed superimposed upon standard planar freeze-fracture views of the membrane interior. Both membrane leaflets of the plasma membrane and those of intracellular membranes, as well as the variety of fracture planes through lipid droplets, are accessible to labeling.
Fig. 1.
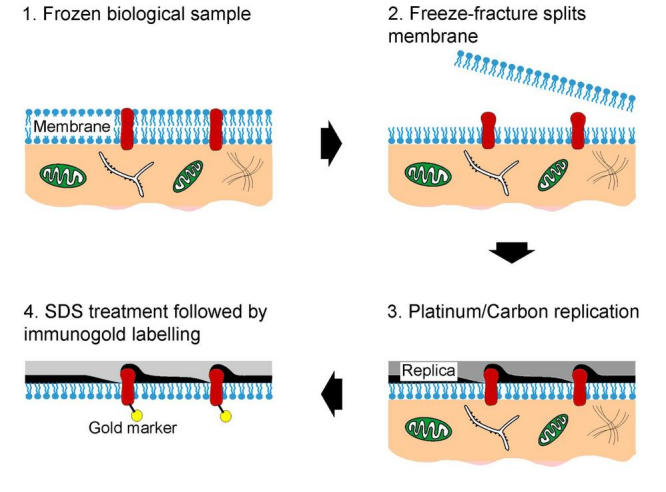
The key steps in FRIL. Tissue or cell samples are rapidly frozen and fractured (1 and 2). The freeze-fracture process splits the lipid bilayer (2) exposing the fracture face. A platinum-carbon replica is made of the fractured specimen (3). The replica is treated with SDS to remove the cellular components apart from those attached to the replica (4). Proteins still attached to the replica are then immunogold labeled. On examination in the electron microscope, the electron-dense gold label is clearly visible against the replica, marking the target molecule in the membrane plane.
To permit adequate removal of the biological material by SDS and subsequent labeling, glutaraldehyde pre-fixation (which masks epitopes) has to be avoided. Thus, in the original technique, samples were prepared by ultrarapid freezing against a helium-cooled copper block (3,4). While in principle this may be considered an ideal approach, it can make the technique time consuming and - for the novice - challenging to carry through successfully to completion. A useful compromise to avoid the need for specialized ultrarapid freezing equipment is to give a brief treatment in glycerol (without pre-fixation in glutaraldehyde) and then use standard immersion freezing. When applying this approach, it is critical to keep the period of glycerol treatment to an absolute minimum to reduce the risk of glycerol-induced artefacts. A useful alternative - to minimize such risks - is to give a brief fixation in formaldehyde prior to glycerol treatment (10-12), though this is not always compatible with subsequent primary antibody binding.
Of all the techniques in freeze-fracture cytochemistry attempted and tested over the last three decades, FRIL has proven to be the most successful, and is currently having a substantial impact in solving questions in cell biology that have hitherto been impossible to address with other ultrastructural, cell biological or molecular approaches (10-26). For the illustrative results described in this article focusing on the lipid droplet, THP-1 macrophages, 3T3-L1 fibroblasts (adipocytes) and human milk samples were used (21-26).
Freeze-fracture nomenclature
To apply the FRIL technique and interpret results, the practitioner has to be acquainted with the nomenclature used for describing standard freeze-fracture images (27). This nomenclature is best explained by first thinking of the membrane as consisting of two halves - a P half which lies adjacent to the protoplasm (i.e., cytoplasm or nucleoplasm), and an E half which lies adjacent to the extracellular, exoplasmic or endoplasmic space (Fig. 2). The term fracture face is applied to the interior views exposed as a result of splitting the membrane by freeze fracture. The fracture face of the P half is thus termed the P face, while that of the E half is termed the E face. The term surface is reserved for the true surface of the membrane; this view is not revealed by freeze fracture, but - when specimens are ultrarapidly frozen without glycerol treatment - may be exposed by vacuum sublimation of ice (referred to as etching). The true surfaces of the P half and the E half of the membrane are termed the P surface and the E surface, respectively.
Fig. 2.
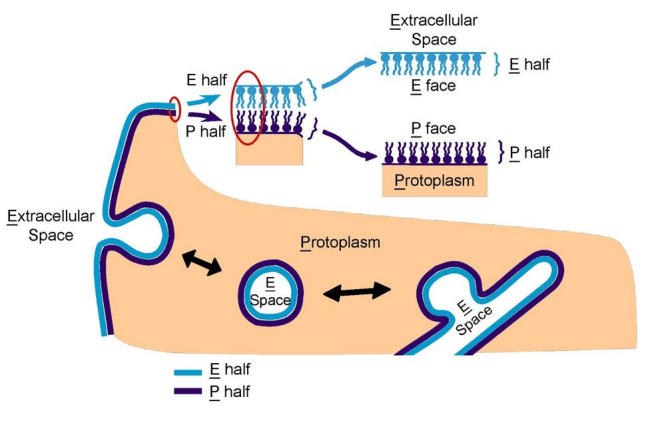
Nomenclature for describing the aspects of membranes revealed by freeze fracture. The membrane comprises a lipid bilayer with intercalated proteins. For the plasma membrane, the half-membrane leaflet adjacent to the extracellular space is termed the E half; that adjacent to the protoplasm is termed the P half. The term fracture face is applied to the interior views of membranes exposed by freeze fracturing, while the term surface is used for the true, natural surfaces of the membrane. The fracture face of the P half is thus termed the P face (or PF), while that of the E half is termed the E face (or EF). The true surfaces of the membrane are correspondingly termed the P surface and the E surface (PS and ES), respectively. The terminology is similarly applied to intracellular membranes. When membrane vesicles are produced from the plasma membrane by endocytosis, the P half of the membrane forms the outer monolayer of the vesicle membrane and the E half forms the inner monolayer. The interior of the vesicle is derived from the extracellular space. Thus, convex fractures of vesicles show the E face, and concave fractures the P face of the vesicle membrane. Corresponding spatial relationships apply to all the membrane systems of the cell. The term "E" designates the interior of all single-membrane organelles and the spaces between inner and outer membranes of all double membrane-bound organelles (nucleus, mitochondria and chloroplasts). The term "P" encompasses cytoplasm, nucleoplasm, the matrix of mitochondria and the stroma of chloroplasts.
To apply this nomenclature to the lipid droplet requires some adaptation because of the unusual structure of this organelle (Fig. 3). The lipid droplet essentially consists of a hydrophobic neutral lipid core (containing cholesterol esters and tri-acyl glycerol) surrounded by a single monolayer of phospholipids rather than the phospholipid bilayer normally found enveloping organelles. Concavely fractured droplets thus show an aspect of this monolayer that is equivalent to the P face of a normal membrane. The complementary aspect, revealed in convex fractures, would be termed an E face in the case of a normal lipid bilayer, but this designation is not appropriate since there is no E half to the enveloping structure, hence convex fractures show the outer-facing aspect of the lipid core. Some fractures through lipid droplets expose multi-layers of lipid, giving an "onion-like morphology". Lipid droplets that are cross-fractured reveal a simple homogeneous content or stacked internal fracture faces.
Fig. 3.
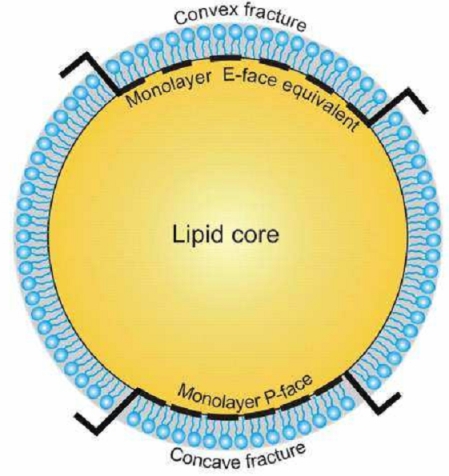
Nomenclature for describing freeze-fracture images of lipid droplets. The diagram shows the phospholipid monolayer surrounding the hydrophobic lipid core. Concavely fractured droplets show an aspect of the monolayer that is equivalent to the P face of a normal membrane. The complementary aspect, revealed in convex fractures, would be termed an E face in a normal lipid bilayer. However, as there is no E half, the "E-face equivalent" seen in convex fractures is best envisaged as the outer-facing aspect of the surface of the lipid core.
Results and Discussion
To illustrate the value of FRIL in addressing topical questions in cell biology, we will here summarize some of its current contributions to our understanding of the function of the lipid droplet. Largely ignored for many years, recent research on these structures and their associated proteins has led to a new appreciation of their importance. The assembly, fusion and degradation of lipid droplets, resulting in storage and release of their lipid components, is controlled by a series of proteins, in particular lipid transport proteins, acyl-CoA synthetases, caveolins and PAT family proteins (the collective term for perilipin, adipophilin and TIP47). As a storage depot, lipid droplets not only serve as a source of cellular fuel and constituents for membrane construction, but also provide precursors for lipid signaling molecules and hormones. They are thus intimately involved in the cellular influx and efflux of lipids and in the signaling and transcriptional networks central to lipid homeostasis in health and disease. Lipoatrophy (lack of mature lipid-droplet containing adipocytes) leads to diabetes and fatty liver pathology, while lipodystrophy (abnormal fat distribution), resulting from sedentary lifestyle and excess food intake, is associated with obesity and diabetes. Moreover, lipid accumulation is a critical step in the pathogenesis of atherosclerosis which, by causing coronary heart disease, is a major cause of death and disability throughout the world. Understanding how lipid droplets form in the cell is thus fundamental to our knowledge of the pathogenesis of these disease conditions.
It is widely held that the formation of lipid droplets involves accumulation of neutral lipids within the lipid bilayer of the endoplasmic reticulum membrane followed by budding-off into the cytoplasm. It is through this budding off process that the lipid droplet is presumed to acquire its phospholipid monolayer and associated proteins from the P half of the ER membrane.
Results from FRIL refute this model on several counts (21). First, freeze fracture, by permitting unique three-dimensional views of the spatial relationships of membranes and organelles, demonstrates that at sites of close association, the lipid droplet is not situated within the ER membrane, but adjacent to it (Figs. 4-6). Both ER membranes clearly lie external to and follow the contour of the lipid droplet (Figs. 4, 5), enclosing it in a manner akin to an egg-cup (the ER) holding an egg (the lipid droplet) (Fig. 6A-C). Application of FRIL demonstrates that the PAT family protein, adipophilin, is concentrated in prominent clusters in the P half of the ER membrane at the site of the closely apposed lipid droplet (Fig. 6A, B), as well as in the lipid droplet surface apposed to the ER (Fig. 6D, E). Adipophilin is thus positioned to play a role in lipid droplet growth by facilitating lipid transfer from the ER to the droplet. FRIL demonstrates unequivocally that lipid droplets develop adjacent and external to specialized domains of the ER membrane enriched in adipophilin, not within the bilayer of the ER as previously supposed.
Fig. 4.
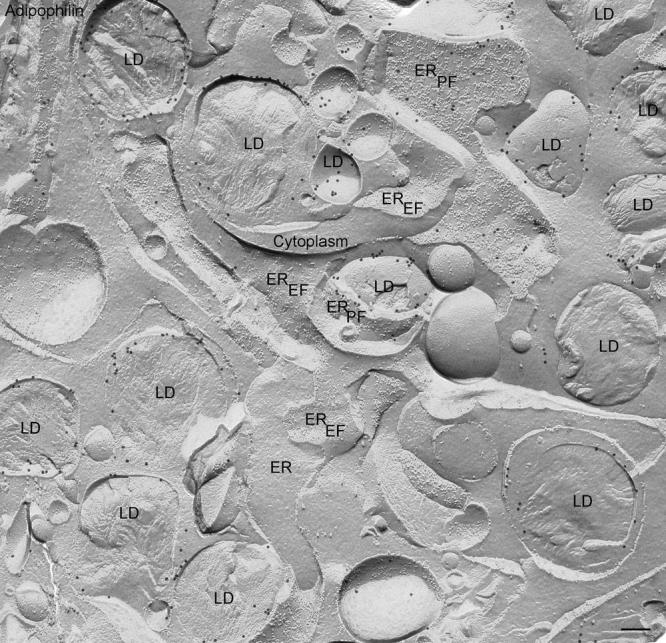
Freeze-fracture overview of lipid droplets (LD) in a lipid laden macrophage and their associations with endoplasmic reticulum (ER) membranes after labeling for adipophilin using the FRIL technique. In freeze-fracture, lipid droplets have a unique smooth appearance enabling their unambiguous discrimination from other organelles. Where the fracture follows the outermost layer of the droplet to give a concave fracture, the enveloping outer phospholipid monolayer is seen en face; convex fractures give mirror image (complementary) views. Some fractures skip along successive layers of the lipid, revealing a multilayered appearance, other cross-fracture the droplet to give what is essentially a cross-section of the interior. The gold label, seen as sharply defined black particles, can be seen in abundance on the outer phospholipid monolayer of the droplet, and some label is also present deeper in the interior. Adipophilin label is also apparent on areas of the ER membrane P face (PF) but not on the E face (EF).
Fig. 6.
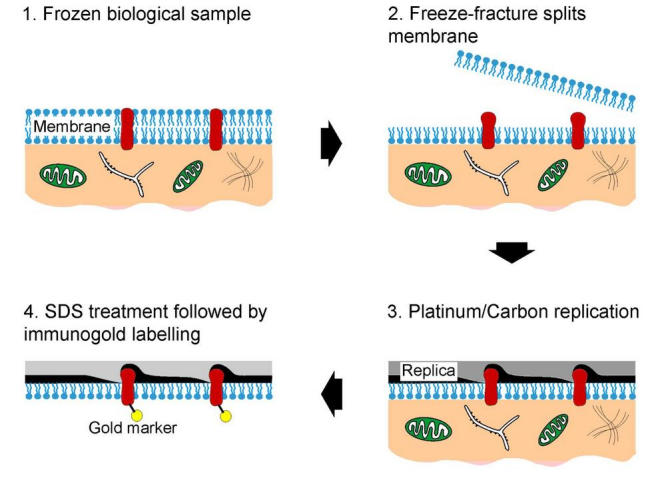
(A) Lipid droplet situated in a cup formed from ER membranes. Both ER membranes are visible (seen in P face (PF) and E face (EF)) view following the contour of the lipid droplet. The lipid droplet has been convexly fractured, and lies adjacent to and not within both ER membranes exposed. This example is from an unlabeled (standard freeze-fracture) preparation. (B, C) Similar views to (A), but with labeling for adipophilin using the FRIL technique. Abundant gold label is visible on the ER membranes (P face, PF) immediately adjacent to the lipid droplet. (D) Lipid droplet seen in cross fracture and in (E) concave fracture. FRIL demonstrates abundant labeling for adipophilin in the outer phospholipid monolayers surrounding the lipid droplets (P face, PF) exposed in these views. Bars: 0.2 µm
Fig. 5.
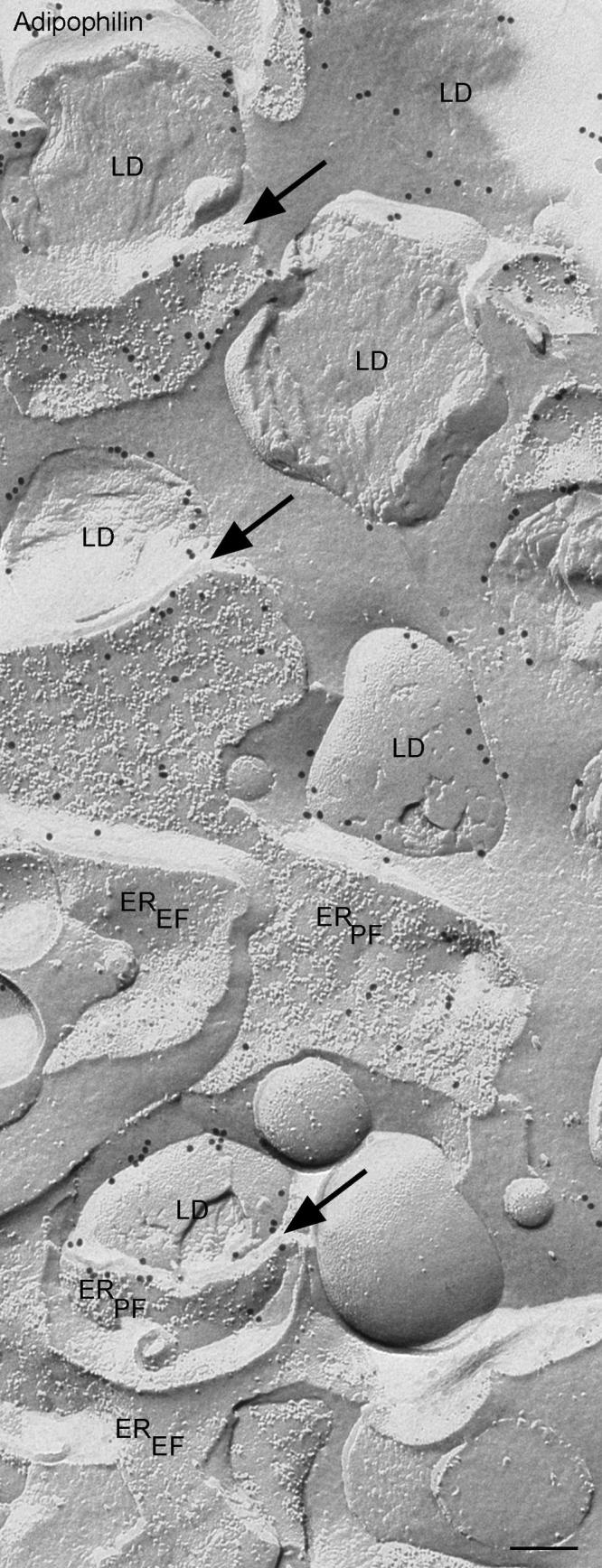
Similar view to Fig 4. at higher magnification. Gold label for adipophilin is clearly seen on the ER membrane (P Face, PF) immediately adjacent to the lipid droplets (arrows). The lower example arrowed shows a clear demonstration of how the ER membrane curves around the contour of the lipid droplet.
The spatial distribution of caveolin 1, a putative mediator of intracellular lipid transport, adds to the evidence discrediting the popular model of lipid droplet biogenesis (22,23). As with other lipid droplet associated proteins, caveolin 1 was thought to become incorporated into the lipid droplet from the ER membrane by the budding off process. As it is the P half of the ER membrane that is proposed to enclose the lipid droplet during budding off, proteins such as caveolin would have to be resident in this membrane leaflet. Contrary to this prediction, FRIL demonstrates the caveolin-1 is situated in the E half of the ER membrane. As this membrane half does not, according to the existing model, participate in the lipid droplet budding process, caveolin is actually in a location in the ER membrane that would make it impossible to gain access to the forming lipid droplet.
Another common assumption is that caveolin and the PAT family proteins are confined exclusively to the droplet surface, and that PAT family proteins are specific to lipid droplets and not present in any other organelle or membrane system of the cell. FRIL, however, demonstrates that in macrophages and adipocytes 1) PAT family proteins and caveolin are distributed not only in the surface but also throughout the lipid droplet core (Fig. 7); and 2) PAT family proteins are integral components of the plasma membrane (Fig. 8) (24,25). Under normal culture conditions, these proteins are dispersed in the P half of the plasma membrane. Stimulation of lipid droplet formation by incubation of the cells with acetylated low density lipoprotein leads to clustering of the PAT family proteins in raised plasma membrane domains (Fig. 8A, B). Fractures penetrating beneath the plasma membrane demonstrate that lipid droplets are closely apposed to these domains (Fig. 8C, D). A similar distribution pattern of labeling in the form of linear aggregates within the clusters is apparent in the P half of the plasma membrane and the immediately adjacent outer monolayer of the lipid droplet (24,25). The aggregation of the PAT family proteins into such assemblies may facilitate carrier-mediated lipid influx from the extracellular environment into the lipid droplet. The findings point to a common cellular mechanism of intracellular lipid loading in the macrophage as part of the pathogenesis of atherosclerosis and in the adipocyte during development of obesity.
Fig. 7.
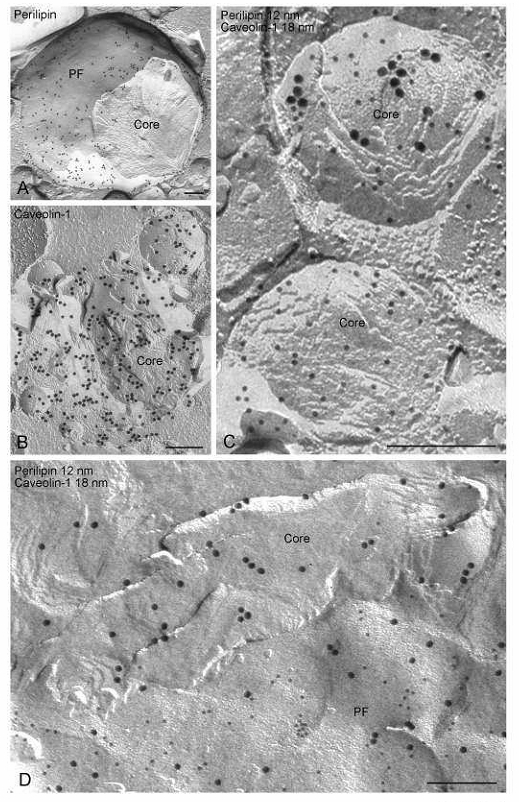
FRIL images demonstrating that PAT family and other proteins are distributed not only at the lipid droplet surface but also in the cross-fractured lipid droplet core. These examples come from adipocytes and show labeling for perilipin and caveolin-1. (A) Example in which perilipin label is seen predominantly in the outer phospholipid monolayer (P face, PF) of the lipid droplet. (B) Example of a lipid droplet in which caveolin-1 label is seen in the core. (C) Examples in which abundant perilipin label (12nm gold) and simultaneous labeling for caveolin-1 (18 nm gold) and perilipin (12 nm gold) is seen in the core. (D) Example in which abundant caveolin-1 label (18 nm gold) is seen in the core and a mixture of perilipin (12 nm gold) and caveolin-1 (18 nm gold) label is predominantly in the phospholipid monolayer P face (PF).
Fig. 8.
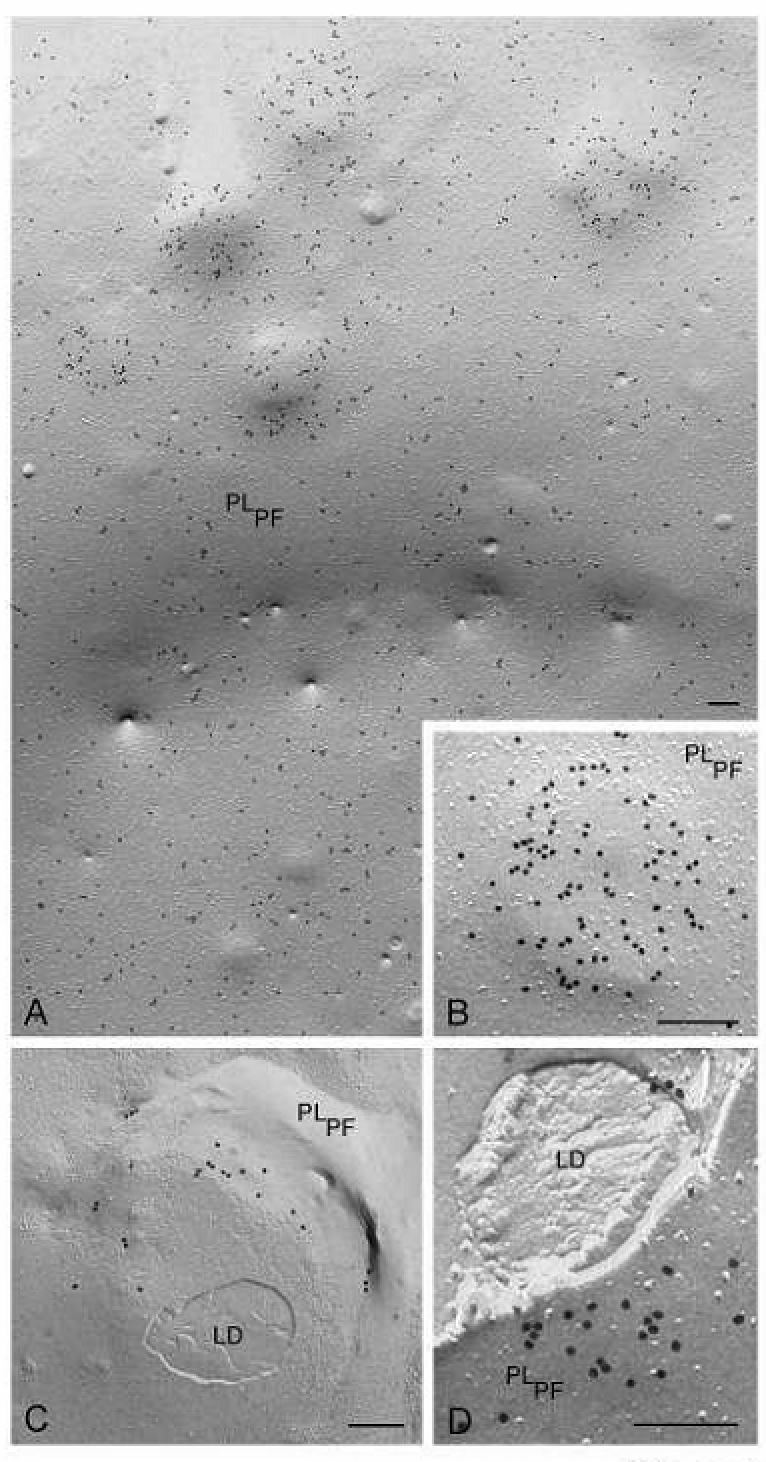
FRIL images demonstrating that PAT family proteins are present in the plasma membrane, seen in P-face view (PLPF), and undergo profound changes in distribution under conditions of lipid loading of macrophages. (A, B) In the plasma membrane of normal cultured macrophage, adipophilin label is widely distributed throughout the membrane but upon lipid loading, the adipophilin becomes clustered in elevated domains in the plasma membrane. (C, D) Fractures that penetrate beneath the plasma membrane demonstrate that lipid droplets lie beneath the elevated adipophilin-rich plasma membrane domains. Bars: 0.2 µm
A related area to which FRIL has shed new light is the mechanism of milk fat globule secretion (26). Milk fat globule formation involves the trafficking of what is essentially a lipid droplet with an outer phospholipid monolayer (termed the secretory granule) to the cell surface; the granule is then enveloped by a portion of plasma membrane and released from the cell as a milk fat globule. The milk fat globule thus has a lipid bilayer derived from the plasma membrane lying externally to the phospholipid monolayer that encloses the lipid core (Fig. 9A). The molecular mechanism of the secretory process is proposed to involve formation of complexes between butyrophilin in the plasma membrane with cytosolic xanthine oxidoreductase; the resulting complexes are then believed to interact with adipophilin on the outer surface of the lipid droplet to enclose the secretory granule in plasma membrane.
Fig. 9.
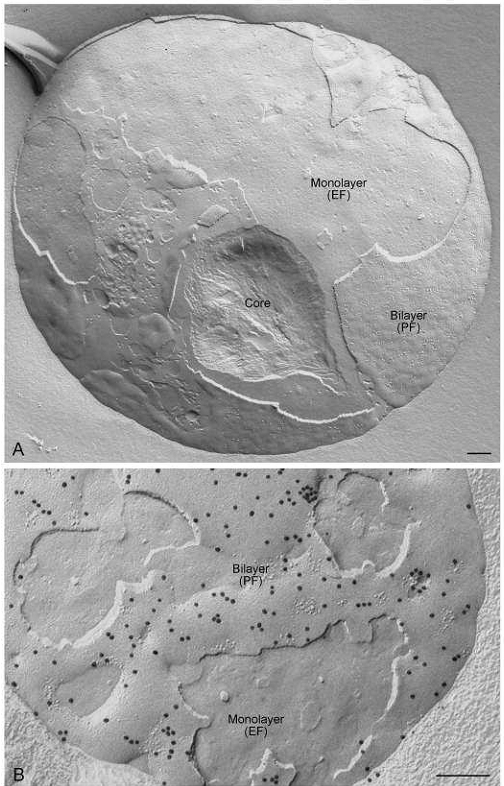
Freeze-fracture view illustrating the structure of a milk fat globule secreted from a human mammary epithelial cell. (A) The globule consists of a lipid droplet core surrounded by a phospholipid monolayer, which in turn is surrounded by a membrane bilayer derived from the plasma membrane which enwraps the droplet during secretion. These different structures are revealed as the fracture plane skips between them (bilayer seen in P face view; phospholipid monolayer in E face view; core seen as lipid is scooped out by cross fracture) (B) FRIL image demonstrating the distribution of adipophilin in the secreted milk fat globule. Abundant label is seen in the bilayer P face (PF). Bars: 0.2 µm
FRIL, however, demonstrates that the topological distribution of the relevant proteins would make the proposed mechanism impossible (Figs. 9-12). Adipophilin is actually found in the plasma membrane domains to which secretory granules are apposed in the mammary epithelial cell (26).
Fig. 12.
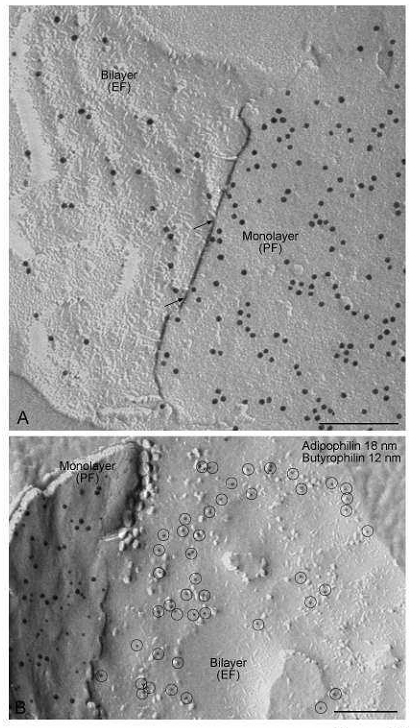
Labeling of butyrophilin. (A) Individual strings of butyrophilin label in the ridges on the bilayer E face (EF) are continuous at sites in which the fracture steps between the monolayer P face (PF) and the bilayer E face (arrows). Thus, the bilayer is strongly inflected toward the monolayer at these ridges; the ridges are sites of apposition between monolayer and bilayer. (B) Double labeling shows that butyrophilin is distributed differently from adipophilin in the globule envelope. Whereas butyrophilin labeling (12 nm gold) occurs on both the P face (PF) of the monolayer and on the E face of the bilayer (encircled), adipophilin (18 nm gold) is labeled on the P face of the monolayer, but not on the E face of the bilayer. Bars: 0.2 µ
Furthermore, adipophilin is localized in the bilayer surrounding the secreted milk fat globule (Fig. 9B) and in the monolayer enclosing the lipid droplet (Fig. 10). Xanthine oxidoreductase is diffusely distributed in the lipid droplet monolayer (Fig. 10). Intensive labeling of butyrophilin occurs on both the P face of the monolayer and the E face of the bilayer (Fig. 11, 12). Importantly, butyrophilin labeling on the bilayer E face occurs as a network of ridges that tightly appose and match the distribution of butyrophilin label in the monolayer (Fig. 12A). Double labeling shows that the distribution of butyrophilin differs from that of adipophilin. Whereas butyrophilin labeling occurs both on the P face of the monolayer and on the E face of the bilayer, adipophilin is labeled on the P face of the monolayer, but not on the E face of the bilayer (Fig. 12B). The findings from FRIL suggest that while adipophilin-rich domains in the plasma membrane may be linked to secretory granule positioning at the cell surface, butyrophilin-butyrophilin interactions between monolayer and bilayer are responsible for envelopment of the granule by the plasma membrane and its release from the cell (26).
Fig. 10.
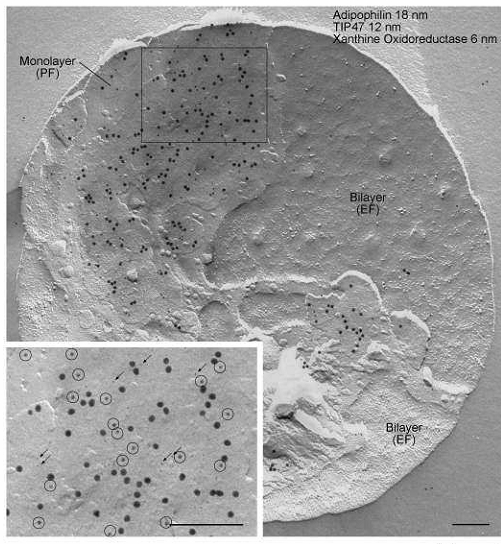
FRIL images demonstrating the triple immunogold labeling of adipophilin (18 nm), TIP47 (12 nm) and xanthine oxidoreductase in the monolayer P face (PF) of the milk fat globule. Label for all three proteins is absent in the bilayer E face (EF). Inset: A higher magnification view of the boxed area. TIP47 (12 nm, encircled) and xanthine oxidoreductase (6 nm, arrows). Note the specificity of labeling and complete lack of background. Bars: 0.2 µm
Fig. 11.
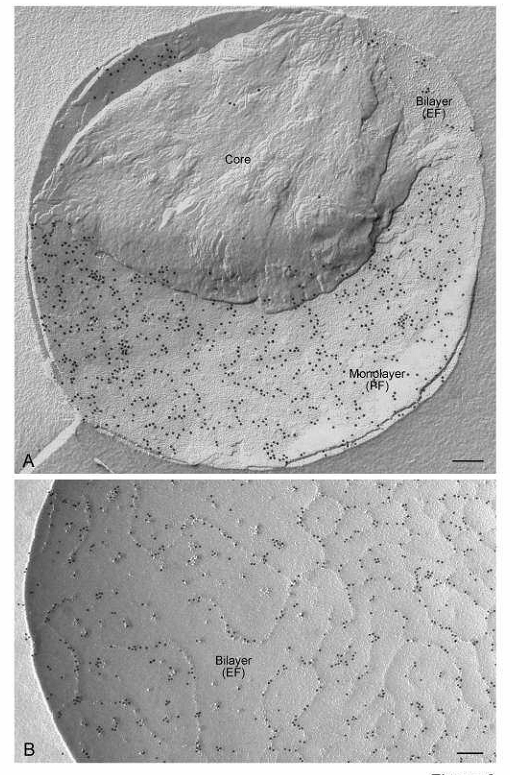
Distribution of butyrophilin in concavely fractured milk fat globules. Intensive labeling of butyrophilin occurs on both (A) the P face (PF) of the monolayer and (B) E face (EF) of the bilayer. Butyrophilin labeling on the bilayer E face occurs exclusively in a network of more or less prominent ridges. Bars: 0.2 µm
Acknowledgments
We are extremely grateful to all members of the Robenek and Severs laboratories for their contributions to the work. The authors acknowledge the continuing support of the Deutsche Forschungsgemeinschaft and the British Heart Foundation (PG/05/003/18157). Both authors contributed equally to the creation of this manuscript.
Protocols
Equipment
Balzers freeze-fracture machine (e.g., BAF 301) equipped with electron beam guns and quartz crystal thin film monitor.
Balzers gold/nickel alloy specimen carriers, flat-topped type.
Two liquid nitrogen bench dewars, one equipped with inner receptacle for primary coolant, and one with stainless steel specimen holding basket.
Fine forceps of the type used for electron microscopy
Binocular microscope
Porcelain dishes with wells for replica processing
Replica transfer implement - glass Pasteur pipette with tip melted to give a fine knob
Sonicator
Copper grids (200 mesh)
Transmission electron microscope
Reagents
Cell culture media and supplements as detailed below.
Phosphate buffered saline (PBS)
Tris-HCl buffer (Tris)
Glycerol
Ethanol
Cryogens: liquid nitrogen; nitrogen slush, Freon or propane.
Sodium dodecyl sulphate (SDS) solution: 5% SDS (Serva) in 10 mM Tris, 30mM sucrose
Blocking solution: 5% BSA in PBS
0.5% EM grade glutaraldehyde in PBS
Primary antibodies:
Adipophilin: mouse monoclonal antibody raised against a peptide matching amino acids 5-27 from the amino terminus of human adipophilin (AP 125; Progen Biotechnik, Heidelberg, Germany).
TIP47: guinea pig polyclonal antibody raised against peptide matching amino acids 1-16 from the amino terminus of human TIP47 (GP30; Progen Biotechnik, Heidelberg, Germany).
Perilipin: guinea pig polyclonal antibody raised against a synthetic peptide corresponding to the N-terminal region of human perilipin A and B (GP30; Progen Biotechnik, Heidelberg, Germany).
Xanthine oxidoreductase: rabbit polyclonal antibody raised against bovine xanthine oxidoreductase (R1119P; Acris Antibodies, Hiddenhausen, Germany)
Butyrophilin: polyclonal antibodies raised in rabbit against 2 sequences, one in the N-terminus and one in the C-terminus (26).
Caveolin-1: mouse monoclonal antibody raised against caveolin-1 (Clone 2297, BD Transduction Laboratories, Lexington, KY, USA).
Secondary antibody-gold conjugates
Goat anti-mouse, goat anti-rabbit and donkey anti-guinea pig coupled to 6nm, 12nm or 18nm colloidal gold (Jackson Immunoresearch, West Grove, PA, USA)
Biological Specimens used for Lipid Droplet Studies
Human THP-1 monocyte/macrophages
Culture human THP-1 monocytes (American Type Culture Collection, Manassas, VA, USA) in suspension at 37°C in a 5% CO2 atmosphere in RPMI 1640 medium with recommended supplements.
Add 100 µM phorbol 12-myristate 13-acetate to the medium for 3 days to stimulate the cells to differentiate into adherent macrophages.
Add 50-100 µg/ml acetylated low density lipoprotein (AcLDL) at day 2 for 24 – 48 hours to induce lipid droplet formation.
3T3-L1 cells (adipocytes)
Maintain 3T3-L1 cells in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine fetal calf serum (FCS), 2 mM glutamine, 100 µg/ml penicillin and 100 µg/ml streptomycin, at °C in a 5% CO2 atmosphere.
To induce differentiation of 3T3-L1 cells to adipocytes, incubate confluent cell monolayers in DMEM supplemented with 10% FCS, 0.5 mM isobutylmethylxanthine, 10 µg/ml insulin, and 10 µM dexamethasone for 72 h.
To induce lipid droplet formation, change medium to 50 µg/ml acLDL in DMEM supplemented with 10 µg/ml insulin and 10% FCS for4 - 48 hours.
Human milk
Gently centrifuge fresh milk (e.g., 4,000 rpm in an Eppendorf bench centrifuge model number 5415D).
Collect supernatant containing suspended milk fat globules (and occasional mammary epithelial cells)
Mounting and freezing the specimens
-
Prepare equipment for freezing the specimens either by:
- pouring liquid nitrogen into dewar flask and then dispensing primary coolant (e.g., Freon or propane) into inner receptacle; or,
- pouring liquid nitrogen into insulated container and placing under vacuum (subjection to several cycles of evacuation may be necessary) to produce nitrogen slush (i.e., liquid nitrogen cooled below its boiling point).
Pour liquid nitrogen into a second bench dewar equipped with stainless steel specimen collecting basket
Place several cleaned gold/nickel alloy flat-topped specimen carriers under binocular microscope in readiness for mounting the samples.
-
For cell samples, scrape cells from culture dish using rubber policeman or take suspensions of cells directly, and either:
- Place sample in 30% glycerol in PBS, gently centrifuge to concentrate (12,000 x g, 30 seconds), and mount on the specimen carriers; or,
- Place a droplet of 30% glycerol in PBS directly on the carriers, pellet cells without initial glycerol treatment and add fragments of cell pellet to the glycerol on the carriers.
For milk samples, place the supernatant directly on the specimen carrier in a droplet of 30% glycerol in PBS.
Inspect mounted specimens using the binocular microscope ensuring that the sample appears as a concentrated hemispherical mound on the flat top of the carrier. Carefully remove excess liquid from the specimen if necessary using the tips of fine forceps.
If using a primary coolant (Freon or propane), melt the frozen coolant using a metal rod. If using nitrogen slush, remove the coolant from the vacuum. Grip the flange of the carrier using fine forceps and plunge the mounted specimen into the coolant, and then immediately transfer to liquid nitrogen in second bench dewar equipped with holding basket. Repeat so that all specimens are frozen quickly in succession. Once the specimens have been frozen, they may be transferred for longer term storage in a liquid nitrogen refrigerator or processed immediately for freeze fracture.
Preparing freeze-fracture replicas
Set up the electron beam guns for oblique (38°) platinum/carbon evaporation (electrode 1) and vertical carbon only evaporation (electrode 2). Check that the microtome knife is clean and undamaged; replace if necessary.
Pump out vacuum chamber, test guns and thin film monitor, and switch the specimen table on to cooling.
When the specimen table has reached -150°C, organize frozen mounted specimens adjacent to door of freeze-fracture machine, in readiness for insertion.
Bring chamber of freeze-fracture machine to atmospheric pressure, remove any condensation on the cold table using liquid Freon, rapidly insert frozen specimens onto the cold table, close the chamber door, and evacuate.
As the chamber is pumping out, switch on cooling of the microtome, and adjust temperature of the table to bring the specimens to -100°C.
When the knife has completed cooling (i.e., the lowest temperature attainable with the liquid nitrogen supply lines) and the vacuum has reached 5 x 10-6 mbar, fracture the specimens (now at -100°C) by microtome knife; take one or more sweeps with the knife while observing with the binocular microscope.
Evaporate platinum/carbon from electrode 1, immediately after the final sweep of the knife (settings: 1,800V, 70-80 mA) to deposit a film of average 2nm thickness determined with the quartz crystal monitor.
Evaporate carbon from electrode 2 (settings: 2,400V, 120-140 mA) to deposit a film of 24nm thickness determined with the quartz crystal monitor.
Bring chamber of freeze-fracture machine to atmospheric pressure, remove replicated specimens with fine forceps and place under PBS in a well of a porcelain spotting dish.
SDS treatment and immunolabeling of replicas
Place spotting dish under the binocular microscope and, using fine forceps, pluck visible cellular material from the replica.
Dispense 5% SDS in 10 mM Tris/30mM sucrose (pH 8.3) into a separate porcelain spotting dish. Use a glass tip to transfer the replica from the PBS into the SDS, with observation under the binocular microscope.
Gently agitate the SDS solution using a shaking machine. Cover replica and leave in SDS for 20-24 hours at room temperature. From time to time, place under the binocular microscope and continue tugging away any visible strands of biological material using forceps.
Pipette PBS into an unused well of the dish, and transfer replica into the PBS. Wash for 1 hour.
For immunolabeling, transfer replica into successive wells of dish at room temperature as follows: i) blocking solution (15 minutes), ii) primary antibody in blocking solution (1 hour), iii) PBS, three changes (total 30 minutes), iv) secondary antibody in blocking solution (1 hour), v) PBS, three changes (total 30 minutes), vi) 0.5% glutaraldehyde in PBS (10 minutes), vii) distilled water, two changes. Controls are conducted in parallel (e.g., secondary antibody alone to check for non-specific binding to the replica, irrelevant primary antibody etc).
Mount replicas on electron microscope grids, and examine in transmission electron microscope.
Guidelines
Note that the above protocol is that used for the results presented here and is illustrative of the key principles of the technique. However, it should be borne in mind that the precise details of treatment times, concentration of reagents, choice of buffers, blocking solution ingredients, etc, will vary according to the preferences of different laboratories and types of specimen processed. Before starting out, the intending practitioner is advised to compare the various published protocols and select one that has been successfully used for similar types of sample to those in hand.
The gold/nickel alloy specimen carriers must be scrupulously clean to ensure that the sample adheres strongly and is not dislodged by the microtome blade upon fracturing. Cleaning is done by sonication, ethanol treatment and, after drying, storing in a sealed container.
The entire operation of placing the cells in glycerol, mounting and freezing has to be done rapidly, i.e., in less than 2 minutes. Once the sample has been frozen, transfer to the liquid nitrogen dewar with holding basket must be swift to avoid warming and slow re-freezing of the specimen.
As an alternative to direct glycerol treatment of non-fixed specimens, brief fixation in formaldehyde may be used prior to the glycerol treatment. The formaldehyde should be prepared freshly from paraformaldehyde and used at a concentration of 1-4%. Use minimal fixation times (e.g., 10 minutes). It is worth checking beforehand by immunofluorescence light microscopy that the targeted epitopes can be successfully labeled after formaldehyde fixation.
Freon is the traditional primary cryogen used for freezing specimens for freeze-fracture electron microscopy but its use is now discouraged owing to deleterious effects on the ozone layer; the alternatives are to use propane as a primary cryogen (cooled with liquid nitrogen) or nitrogen slush (liquid nitrogen cooled below its boiling point by subjecting to cycles of evacuation). When using nitrogen slush, the nitrogen will gradually melt and reach its boiling point, at which point it no longer works as an effective cryogen and fresh slush has to be prepared.
Note that the protocol for preparing freeze-fracture replicas described here is based on the use of the Balzers BAF 300 machine, in which frozen specimens are mounted on the cold table inside the vacuum chamber rather than externally on a detachable cold table for insertion into a cold stage in the machine. For a detailed protocol on the latter variation, see Severs (1).
If the SDS treatment does not remove sufficient material from the replica, it is worth trying longer treatment periods, concentrations ranging from 2-6% and/or heating up to 55°C
References
- Freeze-fracture electron microscopy. Severs NJ. Nature Protocols. 2007;2:547–576. doi: 10.1038/nprot.2007.55. [DOI] [PubMed] [Google Scholar]
- Severs NJ. Freeze-fracture cytochemistry: an explanatory survey of methods. In "Rapid Freezing, Freeze Fracture, and Deep Etching" (NJ Severs, DM Shotton eds.), 1995, pp. 173-208. Wiley-Liss, New York.
- Freeze-fracture replica electron microscopy combined with SDS digestion for cytochemical labeling of integral membrane proteins - Application to the immunogold labeling of intercellular junctional complexes. Fujimoto K. J Cell Sci. 1995;108:3443–3449. doi: 10.1242/jcs.108.11.3443. [DOI] [PubMed] [Google Scholar]
- SDS-digested freeze-fracture replica labeling electron microscopy to study the two-dimensional distribution of integral membrane proteins and phospholipids in biomembrane: practical procedure, interpretation and application. Fujimoto K. Histochem Cell Biol. 1997;107:87–96. doi: 10.1007/s004180050092. [DOI] [PubMed] [Google Scholar]
- Label-fracture: a method for high resolution labeling of cell surfaces. Pinto da Silva P, Kan FWK. J Cell Biol. 1984;99:1156–1161. doi: 10.1083/jcb.99.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Label-fracture of cell surfaces by replica staining. Andersson Forsman C, Pinto da Silva P. J Histochem Cytochem. 1998;36:1413–1418. doi: 10.1177/36.11.2459187. [DOI] [PubMed] [Google Scholar]
- Pinto da Silva P. Visual thinking of biological membranes: from freeze-etching to label-fracture. In "Immunogold labeling methods in cell biology" (A Verkleij, JLN Leunissen eds.), 1989, pp. 179-197. CRC Press, Boca Raton.
- Pinto da Silva P, Andersson Forsman C, Fujimoto K. Fracture-flip: nanoanatomy and topochemistry of cell surfaces. In "Cells and tissues: a three-dimensional approach by modern techniques in microscopy" (P Motta ed.), 1989, pp. 49-56. Alan R. Liss, New York. [PubMed]
- Kan FWK, Pinto da Silva P. Label-fracture cytochemistry. In "Colloidal Gold. Principles, Methods and Applications" (MA Hayat ed.), 1989, pp. 175-201. Academic Press, Orlando.
- Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Proc Natl Acad Sci USA. 1998;95:11981–11986. doi: 10.1073/pnas.95.20.11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Direct immunogold labeling of connexins and aquaporin-4 in freeze-fracture replicas of liver, brain and spinal cord: factors limiting quantitative analysis. Rash JE, Yasumura T. Cell Tiss Res. 1999;296:307–321. doi: 10.1007/s004410051291. [DOI] [PubMed] [Google Scholar]
- Connexin36 vs Connexin32, “Miniature” neuronal gap junctions and limited electrotonic coupling in rodent suprachiasmatic nucleus. Rash JE, Olson CO, Pouliot WA, Davidson KGV, Yasumura T, Furman CS, Royer S, Kamasawa N, Nagy JI, Dukek FE. Neuroscience. 2007;149:350–371. doi: 10.1016/j.neuroscience.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunia I, Recouvreur M, Nicolas P, Kumar NM, Bloemendal H, Benedetti EL. Sodium dodecyl Sulphate-freeze-fracture immunolabeling of gap junctions. In "Methods in Molecular Biology: Connexin Methods and Protocols, Vol 154" (R Bruzzone, C Giaume eds.), 2001, pp. 33-55. Humana Press, Totowa. [DOI] [PubMed]
- Abundance and ultrastructural diversity of neuronal gap junctions in the OFF and ON sublaminae of the inner plexiform layer of rat and mouse retina. Kamasawa N, Furman CS, Davidson KG, Sampson JA, Magnie AR, Gebhardt BR, Kamasawa M, Yasumura T, Zumbrunnen JR, Pickard GE, Nagy JI, Rash JE. Neuroscience. 2006;142:1093–1117. doi: 10.1016/j.neuroscience.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KG, Nagy JI, Rash JE. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Nagy JI, Dudek FE, Rash JE. Brain Res Rev. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Ultrastructural localization of connexins (Cx36, Cx43, Cx45), glutamate receptors and aquaporin-4 in rodent olfactory mucosa, olfactory nerve and olfactory bulb. Rash JE, Davidson KG, Kamasawa N, Yasumura T, Kamasawa M, Zhang C, Michaels R, Restrepo D, Otterson OP, Olson CO, Nagy JI. J Neurocytol. 2005;34:307–341. doi: 10.1007/s11068-005-8360-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Johnson TJA, Dinchuk JE, Levinson SR. Labeling intramembrane particles in freeze-fracture replicas. In "Freeze-fracture Studies of Membranes" (SW. Hui ed.), 1989, pp. 41-59. CRC Press, Boca Raton.
- Spatial relationship of C-terminal domains of dystrophin and ß-dystroglycan in cardiac muscle support a direct molecular interaction at the plasma membrane interface. Stevenson S, Rothery S, Cullen MJ, Severs NJ. Circ Res. 2005;82:961–971. doi: 10.1161/01.res.82.1.82. [DOI] [PubMed] [Google Scholar]
- High-resolution en-face visualization of the cardiomyocyte plasma membrane reveals distinctive distributions of spectrin and dystrophin. Stevenson SA, Cullen MJ, Rothery S, Coppen SR, Severs NJ. Eur J Cell Biol. 2005;84:961–971. doi: 10.1016/j.ejcb.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Adipophilin-enriched domains in the ER membrane are sites of lipid droplet biogenesis. Robenek H, Hofnagel O, Buers I, Robenek MJ, Troyer D, Severs NJ. J Cell Sci. 2003;119:4215–4224. doi: 10.1242/jcs.03191. [DOI] [PubMed] [Google Scholar]
- Cholesterol transporter caveolin-1 transits the lipid bilayer during intracellular cycling. Robenek MJ, Schlattmann K, Zimmer KP, Plenz G, Troyer D, Robenek H. FASEB J. 2003;17:1940–1942. doi: 10.1096/fj.03-0008fje. [DOI] [PubMed] [Google Scholar]
- Lipids partition caveolin-1 from ER membranes into lipid droplets: updating the model of lipid droplet biogenesis. Robenek MJ, Severs NJ, Schlattmann K, Plenz G, Zimmer KP, Troyer D, Robenek H. FASEB J. 2004;18:866–868. doi: 10.1096/fj.03-0782fje. [DOI] [PubMed] [Google Scholar]
- Spatial integration of TIP47 and adipophilin in macrophage lipid bodies. Robenek H, Lorkowski S, Schnoor M, Troyer D. J Biol Chem. 2005;280:5789–5794. doi: 10.1074/jbc.M407194200. [DOI] [PubMed] [Google Scholar]
- Lipid droplets gain PAT family proteins by interaction with specialized plasma membrane domains. Robenek H, Robenek MJ, Buers I, Lorkowski S, Hofnagel O, Troyer D, Severs NJ. J Biol Chem. 2005;280:26330–26338. doi: 10.1074/jbc.M413312200. [DOI] [PubMed] [Google Scholar]
- Butyrophilin controls milk fat globule secretion. Robenek H, Hofnagel O, Buers I, Lorkowski S, Schnoor M, Robenek MJ, Heid H, Troyer D, Severs NJ. Proc Natl Acad Sci USA. 2006;103:10385–10390. doi: 10.1073/pnas.0600795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze-etching nomenclature. Branton D, Bullivant S, Gilula NB, Karnovsky MJ, Moor H, Muhlethaler K, Northcote DH, Packer L, Satir B, Satir P, Speth V, Staehelin LA, Steere RL, Weinstein RS. Science. 1975;190:54–56. doi: 10.1126/science.1166299. [DOI] [PubMed] [Google Scholar]