

Abstract
PPARδ is the only member in the PPAR subfamily of nuclear receptors that is not a target of current drugs. Animal studies demonstrate PPARδ activation exerts many favorable effects, including reducing weight gain, increasing skeletal muscle metabolic rate and endurance, improving insulin sensitivity and cardiovascular function and suppressing atherogenic inflammation. These activities stem largely from the ability of PPARδ to control energy balance, reduce fat burden and protect against lipotoxicity caused by ectopic lipid deposition. Therefore, PPARδ represents a novel therapeutic target and the development of PPARδ agonists/modulators may be useful for treating the whole spectrum of metabolic syndrome.
Keywords: nuclear receptor, PPAR delta, obesity, energy balance, metabolic disease
1. PPARδ, a lipid sensing nuclear receptor
PPARδ (also called PPARβ), together with the other two siblings, PPARα and PPARγ, constitute the PPAR subfamily of the nuclear receptor superfamily [1-3]. These receptors control transcription through binding to specific DNA elements, consisting of two repeats of the core sequence AGGTCA separated by one nucleotide (DR-1), in the target gene promoters [4]. They have been shown to be activated by dietary fatty acids (FAs), particularly polyunsaturated FAs, and regulate various aspects of metabolic processes. However, it is the identification of the lipid-lowering fibrates and insulin sensitizer thiazolidinediones as ligands for PPARα and PPARγ, respectively, that sparks the extensive research in PPAR biology [5,6]. PPARδ is the only subtype that is not a target of current drugs. It is expressed in most metabolically active tissues. Early work has identified adipose differentiation-related protein (ADRP), a lipid droplet coating protein, as a PPARδ target gene and demonstrated that the ability of dietary fatty acids carried by very low-density lipoprotein to regulate ADRP expression is dependent on PPARδ, implicating that this receptor acts as a lipid sensor and is a potential therapeutic target to treat metabolic diseases [7]. Subsequent studies have revealed that PPARδ controls an array of metabolic genes involved in glucose homeostasis and fatty acid synthesis/storage, mobilization and catabolism in a tissue-specific manner (Table 1). Several synthetic ligands, namely GW501516, GW7042 and L165041, have also been developed, each exhibiting different efficacies in ameliorating symptoms of metabolic disorders [8-11]. This review will discuss the biological activity of PPARδ in energy balance and how this activity modulates cellular function as well as systemic metabolic homeostasis (Fig. 1). Potential therapeutic implications of PPARδ agonists in treating metabolic diseases demonstrated in animal models will be described. The effects of GW501516 will be the main focus of the discussion, as this compound is best characterized.
Table 1.
PPARδ in cellular function and metabolism
| Target cell/tissue | Function | Downstream signaling |
|---|---|---|
| Placenta giant cells, | differentiation | Akt1, PDK1, ILK, ADRP, etc [14] |
| Keratinocytes | survival | Akt1, PDK1, ILK [15] |
| Intestinal Paneth cells | differentiation | L-FABP, Ihh [35] |
| Adipocytes | fatty acid catabolism, Thermogenesis | CPT1, AOX, LCAD, UCP1/3, etc [17] |
| Skeletal muscle, Cardiac muscle | fatty acid oxidation, Oxidative metabolism, | CPT1, LCAD, UCP2/3, PDK4, COX II/IV, PGC-1α etc [18, 19, 22] |
| Macrophages | fatty acid metabolism inflammation | ADRP, CPT1, UCP2, ATGL, MCP-1/3, IL-1β, etc [26, 31] |
| Hepatocytes | lipogenesis, glucose metabolism | pentose phosphate pathway FAS, ACC2, etc [25] |
Fig. 1.
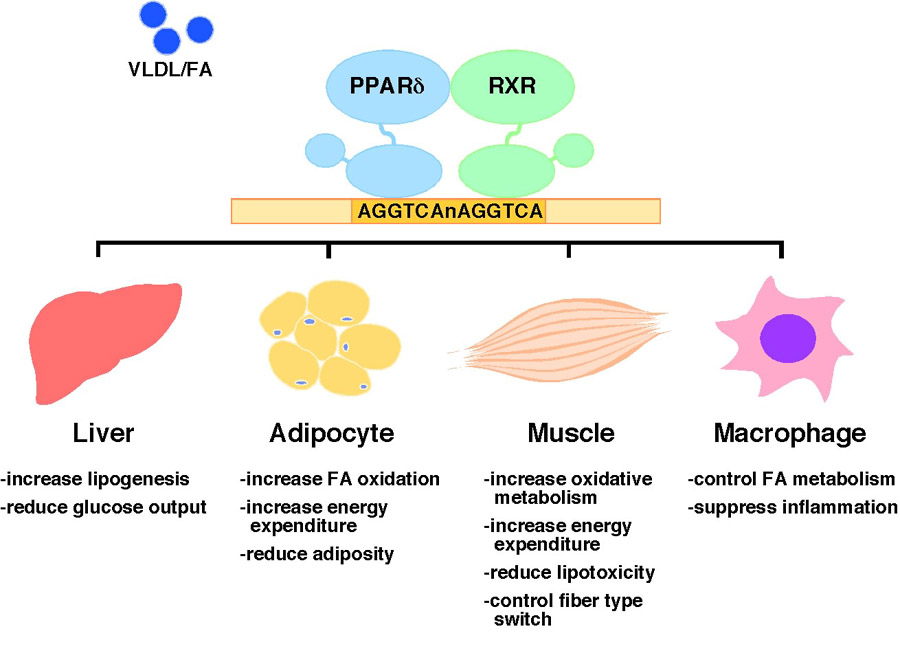
Transcriptional regulation of metabolic homeostasis by PPARδ. PPARδ/RXR heterodimers bind to the DR-1 type response elements with a core sequence AGGTCA on target gene promoters and turn on transcription upon ligand activation. Dietary fatty acids (FAs) carried by very low-density lipoprotein (VLDL) have been shown to activate PPARδ. PPARδ controls many metabolic programs in glucose and fatty acid homeostasis through direct transcriptional regulation, while its activity in suppressing inflammatory response is believed to be indirect. Pharmacological activation of PPARδ exerts many therapeutic effects, including reducing hepatic glucose production, increasing fatty acid catabolism in adipocyte and muscle and lowering the inflammatory status. In animal models, these effects lead to attenuation of metabolic syndrome, such as obesity, dyslipidemia and insulin resistance.
2. PPARδ and metabolic control
2.1 Lipid storage and mobilization in placenta
Attempts to delineate PPARδ function through the generation of knockout mice had generated limited information. Two targeting lines were created, one had the deletion in the DNA binding domain (DBD) and another in the C-terminus of ligand binding domain, both of which resulted in embryonic lethality [12,13]. A small number of PPARδ−/− pups that survived to term exhibited growth retardation and reduced abdominal fat mass. Genotyping of embryos at different gestation stages revealed that PPARδ−/− embryos died at around day 10.5 due to placental defects, in which the connection between placentas and the material deciduas was abnormally loose [12]. This phenotype was reproduced in the third knockout lines, also created by deletion in the DBD [14]. A close examination of the defect uncovered that PPARδ is required for the differentiation of functional trophoblast giant cells, which play an important role in processes such as material deciduas remodeling and secretion of placental hormones. PPARδ does so by activation of the phosphatidylinositol 3-kinase (PI3K)/Akt1 signaling pathway. Similar regulation of Akt1 signaling in cell survival is also observed in keratinocytes and intestinal epithelial cells (see below) [15,16]. As the PI3K/Akt signaling is critical in many cellular metabolic processes, this finding is in line with the role of PPARδ as a metabolic sensor controlling vital cell functions. Interestingly, the expression of ADRP in placenta appears to be tightly associated with giant cell differentiation and is regulated through both direct transcriptional control and PI3k/Akt1 activation mediated by PPARδ. Although the function of ADRP in lipid metabolism is not completely understood, it is proposed to be involved in storing and perhaps compartmentalizing fatty acids to be used for normal fetal development [14].
2.2 adipocyte energy homeostasis
As mentioned earlier, the surviving PPARδ−/− mice had reduced fat mass. Given that PPARγ regulates the adipocyte differentiation program, a role of PPARδ in adipogenesis was suspected. However, adipocyte-specific PPARδ−/− mice showed no difference in sizes of both epidydimal white and interscapular brown fat pads, suggesting that PPARδ is not required for adipocyte differentiation [12]. The function of PPARδ in adipose tissue was realized later in transgenic mice expressing an active form of PPARδ (VP16 activation domain fused to the N-terminus of PPARδ, VP16-PPARδ) driven by the ap2 promoter [17]. This manipulation bypassed the use of ligand and achieved constitutive PPARδ activation in adipocytes, resulting in mice that were lean and had a reduction in body fat composition. Remarkably, these animals were protected from obesity induced by high fat diets or by deletion of the leptin receptor gene. Gene expression analyses demonstrated that in brown fat, PPARδ activation induced genes in fatty acid oxidation, including carnitine palmitoyltransferase 1 (CPT1), acyl-CoA oxidase (AOX) and long chain acyl-CoA dehydrogenase (LCAD), and in thermogenesis/energy dissipation, including uncoupling protein 1 (UCP1) and UCP3. The expression change in white fat was less evident. However, UCP-1, which was not present in white adipocyte of wild type mice, was drastically up-regulated in transgenic animals, indicating that increased PPARδ activity led to a brown fat-like phenotype in white adipose tissue (WAT). Indeed, the cell sizes of WAT were significantly decreased in transgenic mice. Consistent with the observed gene regulation in vivo, PPARδ activation in cultured 3T3-L1 adipocytes or C2C12 myotubes by ligand treatment also increased the β-oxidation rate. Collectively, these data suggest that PPARδ increases fat combustion and this metabolic control may provide a means to regulate adiposity.
2.3 oxidative metabolism and muscle function
In skeletal muscle, PPARδ is expressed at a higher level in soleus muscle, which consists of the fatigue resistant, type I fibers that are rich in mitochondria and use oxidative metabolism for energy production. Conversely, PPARδ is detected at a lower level in gastrocnemius muscle, which contains a mixture of the oxidative type I as well as glycolytic type II fibers [18]. The increased fatty acid catabolism by PPARδ in myotubes indicates that this receptor may be a critical metabolic regulator in muscle. To test this hypothesis, a similar transgenic approach was conducted in mice to induce PPARδ activation in muscle. Remarkably, over-expression of either wild type PPARδ or VP16-PPARδ in muscle resulted in fiber type switching, with a 2-fold increase in type I fibers in gastrocnemius muscle [18,19]. This was accompanied by induction of mitochondria numbers and genes of oxidative metabolism, fatty acid catabolism and type I fiber markers. This phenotype resembles that of muscle-specific over-expression of peroxisome proliferators-activated receptor γ co-activator 1α (PGC-1α), indicating a functional interaction between PPARδ and PGC-1α in muscle [20]. As a consequence of enrichment in type I muscle fibers and enhanced oxidative capacity, these transgenic mice were lean and had an improved metabolic status and increased running endurance tested in oxygen-infused treadmills. The role of PPARδ in skeletal muscle function was further validated in muscle-specific PPARδ−/− mice, which exhibited reciprocal phenotypes, including reduction in type I muscle fibers, depressed expression of genes in fatty acid uptake, catabolism, energy uncoupling and mitochondrial electron transport chain, increased weight gain and a mild defect in glucose metabolism when challenged with a high fat diet [21]. Interestingly, the expression of PGC-1α in muscle appeared to be under the control of PPARδ, supporting the proposal that PGC-1α serves as a co-activator of PPARδ to control mitochondria biogenesis and muscle fiber type plasticity [17,18,21].
Cardiomyocytes is another muscle type that utilizes fatty acid as a main energy source. An intact fatty acid catabolic capacity is therefore critical to maintain normal heart function. Knowing its function in skeletal muscle, it is not surprising that PPARδ also serves as a key regulator of fatty acid catabolism in the heart. Cardiomyocyte-restricted PPARδ−/− resulted in decreased expression of CPT1, LCAD, AOX and UCP3 and a reduced β-oxidation rate, leading to lipid accumulation in the heart [22]. Assessment of cardiac performance demonstrated that rates of cardiac contractility and relaxation were reduced in cardiomyocyte PPARδ−/− mice. These mice eventually developed cardiac hypertrophy and cardiomyopathy and died between 4 to 10 months of age. This study demonstrates that in the heart, PPARδ regulates oxidative metabolism not only for energy production but also for protection against lipotoxicity.
3. PPARδ and metabolic diseases
3.1 Obesity and dyslipidemia
The incidence of obesity has increased considerably, partly caused by energy surplus and sedentary life styles. Accompanied with it are metabolic abnormalities, including high blood pressure, inflammation, insulin resistance, dyslipidemia (high levels of triglyceride (TG) and low-density lipoprotein cholesterol (LDL-c) and low levels of high-density lipoprotein cholesterol (HDL-c), atherosclerosis and cancer [23]. Given that forced PPARδ activation in transgenic models protects against obesity and induces an exercise-like metabolic status, the development of PPARδ modulators is of interest in treating metabolic syndrome. In fact, a high affinity synthetic agonist, GW501516, has been shown to reduce weight gain and decrease circulating TG in high fat fed or ob/ob mice [18,24]. This effect is believed to be mediated by increased peripheral fatty acid catabolism. In obese rhesus monkeys, this agonist at doses up to 3 mg/kg body weight/day ameliorated dyslipidemia, lowering TG and LDL-c and increasing HDL-c, while weight gain was not affected [8]. The most impressive effect in this study appeared to be the 79% increase in HDL-c, which correlated with an increase in HDL particles and in levels of apolipoproteins, including apoA-I, apoA-II and apoC-III. The HDL-c raising activity was attributed to the induction of a cholesterol efflux pump ABCA1 by PPARδ demonstrated in monocytic cells and fibroblasts. GW501516 and another agonist L165041 also increased HDL-c in db/db mice, albeit to a lesser extent (30∼40%) [10,25]. Interestingly, in mouse macrophages, it has been shown that both ABCA1 expression and ABCA1 mediated cholesterol efflux were unaffected by GW501516 treatment or PPARδ gene deletion [26]. It is unclear whether the mechanistic discrepancy is due to species differences or other mechanisms, such as increased apoA-I levels, are responsible for the HDL-c raising effect of PPARδ agonists. In addition, whether PPARδ activation could reduce adiposity in humans remains to be determined.
3.2 Insulin resistance and hepatosteatosis
The effect of GW501516 on glucose homeostasis has been studied in several mouse models of insulin resistance/type II diabetes. In high fat fed C57BL/6 mice, co-administration of this PPARδ agonist at 3 mg/kg/day for 2 months increased the metabolic rate, reduced fatty liver and decreased lipid accumulation and increased mitochondrial biogenesis in muscle. Circulating insulin levels also declined, whereas the improvement in glucose tolerance and insulin sensitivity determined by the glucose and insulin tolerant test (GTT and ITT) was moderate [24]. Nevertheless, the effect appeared to be dose-dependent, as at 10 mg/kg/day, the ability of GW501516 to lower blood glucose levels and enhance glucose tolerance became apparent [18]. Interestingly, in addition to improving glucose homeostasis, GW501516 treatment normalized pancreatic islet hypertrophy and increased glucose-stimulated insulin secretion in ob/ob mice [24]. GW501516 did not induce insulin release in isolated islets [27], suggesting the increased insulin secretion was secondary to the improved islet function. The molecular mechanism underlying the enhanced glucose metabolism was proposed to be mediated by increased fatty acid catabolism in muscle. In fact, gene expression analyses revealed that levels of LCAD, UCP2/3, CPT1, PGC-1α and glucose transport 4 (GLUT4) were up-regulated in muscles of ligand treated animals.
Although these studies demonstrate a potential insulin sensitizing activity of PPARδ agonists, precaution needs to be taken in data interpretation. Notably, long-term treatment with GW501516 reduces weight gain in mice, which could be responsible for most observed effects [18,24]. Furthermore, although GW501516 has a relative high affinity for PPARδ, the local concentrations in certain tissues may be high enough to activate PPARα or PPARγ [8]. These concerns were partially addressed in a study, in which GW501516 was given to mice that had already developed obesity and insulin resistance, at 2 mg/kg/day for 2 weeks [25]. In diet induced obese and db/db mice, this regimen effectively lowered blood glucose and insulin and increased glucose tolerance before changes in weight gain and circulating lipid parameters surfaced. This activity was completed abolished in PPARδ−/− mice, demonstrating PPARδ dependency. PPARδ−/− mice also exhibited other metabolic defects, including a lower metabolic rate and respiratory exchange ratio and glucose intolerance. The insulin sensitizing activity of GW501516 was further characterized by the euglycemic-hyperinsulinemic clamp in db/db mice. Ligand treatment enabled insulin to suppress free fatty acid release and lowered hepatic glucose production at the basal state and during clamp. Interestingly, while the calculated insulin stimulated glucose disposal was enhanced by ligand, the total glucose disposal rates were similar between GW501516 and vehicle treated group. Given that the muscle is the major site of glucose disposal, these results suggest that the metabolic effects in the liver mediated by PPARδ activation occur prior to notable changes in the muscle. Consistent with this idea, DNA array analyses demonstrated that most expression differences were in the liver, where a cluster of lipogenic genes, including fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC), were induced by ligand treatment. At the first glance, these observations seem to contradict the known PPARδ activity. It is proposed that the glucose lowering effect is mediated, in part, by metabolizing glucose for lipogenesis in the liver. In fact, there was a moderate increase in TG levels in livers of ligand treated mice. The newly made fats are consumed through increased fatty acid oxidation in muscle at a later stage, as long-term ligand administration reduces hepatic lipid content.
The combined hepatic and muscular effects of PPARδ appear to constitute a “fatty acid futile cycle”, resulting in improved glucose and lipid metabolism. However, the lipogenic activity in the liver can be a concern as hepatosteatosis is common in diabetic patients. The effect of GW501516 on steatosis has been examined in a mouse model of non-alcholic steaohepatitis (NASH) induced by a diet deficient in methionine and choline [28]. This diet induced massive lipid accumulation and inflammatory cell infiltration in the liver. Co-administration of GW501516 at 10 mg/kg/day for 5 weeks reduced hepatic TG and numbers of fatty droplets and inflammatory cells. Furthermore, the expression of pro-inflammatory markers, such as monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor α (TNFα), interleukin-1β (IL-1β) and IL-6 was down-regulated by ligand treatment, consistent with the ability of PPARδ agonists to suppress inflammation in macrophages (see below). Collectively, these findings suggest that PPARδ is a potential therapeutic target for insulin resistance and hepatic steatosis.
3.3 Atherosclerosis
Macrophage derived foam cells are a major component of atherosclerotic lesions [29,30]. The formation of foam cells is facilitated by elevated LDL-c and inflammatory response within the vessel wall. Studies in cultured macrophages have demonstrated that PPARδ activation by GW501516 did not affect cholesterol accumulation or efflux, while it suppressed levels of inflammatory mediators, such as MCP-1, MCP-3 and IL-1β [26], suggesting PPARδ could be atheroprotective. However, LDL receptor (LDLR)−/− mice reconstituted with PPARδ−/− bone marrow developed less lesions than those with wild type bone marrow [26]. This unexpected result was attributed to a receptor activation-like phenotype in PPARδ−/− macrophages, due to a unique property of unliganded PPARδ to interact with repressors and co-repressors in the macrophage. The suppression of inflammatory genes (trans-repression), such as MCP-1, by PPARδ activation is mediated partly by ligand induced release of a transcriptional repressor, BCL-6, which then binds to and suppresses the promoters of several cytokines and chemokines. PPARδ−/− results in BCL-6 availability similar to that of ligand activation. On the other hand, for direct transcriptional targets such as ADRP, unliganded PPARδ actively represses transcription and PPARδ−/− relieves the repression leading to increased basal expression, again, similar to that of ligand activation [31].
The loss-of-function study was confounded by the repressor sequestering activity of unliganded PPARδ. Receptor activation, however, is expected to inhibit lesion progression, since PPARδ agonists exhibit HDL-c raising and anti-inflammatory activities. The effect of a less characterized PPARδ agonist, GW0742, on atherogenesis has been examined in LDLR−/− mice. At a high dose of 60 mg/kg/day, GW0742 treatment for 10 weeks was able to reduce lesions in aortic valves by 55% [9], whereas it had no effect at either 5 or 6 mg/kg/day [9,32]. Although the expression of inflammatory markers in aortas was down-regulated by GW0742, levels of insulin and HDL-c were unchanged and weight gain was increased, which were in sharp contrast to the activities of GW501516 discussed earlier. The reason for the discrepancy in the therapeutic effects between the two agonists may be explained by differences in their efficacies. In cell-based transactivation assays, GW0742 has an EC50 of 30 nM, whereas the EC50 of GW501516 is 1 nM [8,9]. It has yet to be determined whether GW501516 can increase HDL-c in mouse models of atherosclerosis, such as LDLR−/− or apoE−/− mice, and inhibit lesion progression.
3.4 Inflammatory bowel disease and colorectal cancer
Inflammatory bowel disease (IBD), a collective term for Crohn's disease and ulcerative colitis, is characterized by sustained activation of mucosal immune response against normal constituents of luminal microflora, leading to long-term damage of gastrointestinal function [33,34]. The etiology of IBD is not completely understood. Defects in the barrier function of intestinal epithelium and the mucosal immune system are likely involved in the pathogenesis of this disease. PPARδ is abundantly expressed in the gastrointestinal tract. When PPARδ−/− mice were subjected to experimental colitis induced by dextran sodium sulfate (DDS), they exhibited more severe symptoms in weight loss, shortened colon length and colitis scores compared to wild type controls, indicating that PPARδ may play a role in the etiology of IBD [35]. The mechanism underlying the protective function of PPARδ remains elusive but is likely mediated by its ability to modulate the function and immune response of endothelium. PPARδ−/− mice have been shown to have reduced numbers of Paneth cells, which are one of the four epithelial cell types and are responsible for the antimicrobial activity in the mouse intestine [35]. As a result, PPARδ−/− mice were found to have altered populations of the gut microflora. It is proposed that PPARδ is downstream of the Wnt-β-catenin/TCF4 pathway in regulating the proliferation and differentiation of Paneth cells from stem cells. These findings suggest that PPARδ may regulate microflora homeostasis and intestinal epithelium integrity thereby alleviating the symptoms of experimental colitis.
The potential beneficial effect of PPARδ in epithelial cell renewal in IBD could turn out to be a double-edged sword, as uncontrolled Wnt-β-catenin/TCF4 signaling through mutations in the adenomatous polyposis coli (APC) tumor suppressor gene causes colorectal cancer [36,37]. The role of PPARδ in colorectal carcinogenesis has been under heated debate. It has been hypothesized that the Wnt-β-catenin/TCF4 pathway activates the expression of PPARδ to stimulate cell proliferation [38]. Supporting this hypothesis were the findings that GW501516 treatment increased while deletion of PPARδ decreased adenoma growth in APCmin mice [16,39]. PPARδ activation was shown to promote epithelial cell survival through the Akt signaling pathway. A similar anti-apoptotic property of PPARδ has been reported in keratinocytes during wound healing [15,16]. In sharp contrast, two other studies demonstrated that deletion of PPARδ increased intestinal polyp formation in APCmin mice [40,41]. Furthermore, The expression of PPARδ was suppressed, rather than enhanced by APC inactivation. The apparent difference between these studies was the use of two different PPARδ knockout lines, both of which, however, have been shown to have no detectable PPARδ protein [13,14]. Undoubtedly, more studies, including the use of intestine specific knock out mice, are needed before any conclusion regarding the role of PPARδ in tumorigenesis can be reached.
4. Conclusions
Adipocyte hypertrophy and dysfunction in obesity results in ectopic fatty acid accumulation leading to lipotoxicity in tissues such as pancreas (β cells), muscle and heart, which is believed to be one of the major causes of metabolic diseases [42,43]. Work from animal models suggests that PPARδ activation reduces fat burden exerting many favorable activities, including reducing weight gain, increasing skeletal muscle metabolic rate and endurance, improving insulin sensitivity and cardiovascular function and suppressing atherogenic inflammation. Therefore, the development of PPARδ agonists/modulators may be useful for treating the whole spectrum of metabolic syndrome. Nevertheless, the relevance of these findings to human pathophysiology remains to be determined. Recent epidemiology studies show evidence suggesting that PPARδ polymorphisms are associated with body mass index, fasting glucose levels and insulin resistance [44,45]. The result of the first human trial with PPARδ agonist has been reported [46]. In this study with a small cohort, healthy volunteers were given placebo or GW501516 at 2.5 mg or 10 mg once daily for 2 weeks while hospitalized and sedentary. No toxicity was observed during this treatment period. Both regimens reduced circulating TG and prevented the decline of HDL-c and apoA-I levels due to lack of physical activity. There was no information regarding weight gain. These studies, although limited, provide certain validation for results from animal work. A critical question to be addressed is whether PPARδ is oncogenic, even though the carcinogenic activity of other PPAR agonists appear to be specific to rodents. Lastly, metabolic dysregulation is now recognized as a state of low grade, chronic inflammation [23]. Consistent with this idea, free fatty acids have been shown to induce inflammatory response through toll-like receptor 4 [47]. Furthermore, analogous to the macrophage/endothelium interaction at the vascular wall, the crosstalk between adipocytes and adipose resident macrophages is proposed to play an important role in metabolic dysregulation and insulin resistance [48,49]. Future studies designed to dissect the function of PPARδ in these processes will undoubtedly identify novel therapeutic pathways to control the progression of metabolic diseases.
Acknowledgments
We thank P. Olson for valuable comments and J. Gound and C. Lenhart for administrative assistance. S.M. Reilly is supported by the NIEHS training grant (T32 #ES07155). This paper was supported by American Heart Association and American Diabetes Association (C.-H.L.).
Abbreviations
- PPAR
peroxisome proliferator-activated receptor
- FA
fatty acid
- ADRP
adipose differentiation-related protein
- AOX
acyl-CoA oxidase
- CPT1
carnitine palmitoyltransferase 1
- LCAD
long chain acyl-CoA dehydrogenase
- PGC-1β
peroxisome proliferators-activated receptor γ co-activator 1β
- GLUT4
glucose transport 4
- FAS
fatty acid synthase
- ACC
acetyl-CoA carboxylase
- NASH
non-alcholic steaohepatitis
- IBD
Inflammatory bowel disease
- COX II
cytochrome oxidase II
- PDK4
pyruvate dehydrogenase kinase 4, PDK1, 3-phosphoinositide-dependent 3-kinase
- ILK
integrin-linked kinase
- ATGL
adipose triglyceride lipase
- L-FABP
liver fatty acid binding protein
- Ihh
Indian hedgehog
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 2.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–7. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 3.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SS, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–22. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 1995;270:12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 7.Chawla A, et al. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci U S A. 2003;100:1268–73. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliver WR,, Jr., et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98:5306–11. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham TL, Mookherjee C, Suckling KE, CN AP, Patel L. The PPARdelta agonist GW0742X reduces atherosclerosis in LDLR(−/−) mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 10.Leibowitz MD, et al. Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett. 2000;473:333–6. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- 11.Kang K, Hatano B, Lee CH. PPARdelta Agonists and Metabolic Diseases. Curr Atheroscler Rep. 2007;9:72–7. doi: 10.1007/BF02693931. [DOI] [PubMed] [Google Scholar]
- 12.Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, Boland R, Evans RM. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:303–8. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters JM, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol Cell Biol. 2000;20:5119–28. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadra K, Anghel SI, Joye E, Tan NS, Basu-Modak S, Trono D, Wahli W, Desvergne B. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor beta/delta. Mol Cell Biol. 2006;26:3266–81. doi: 10.1128/MCB.26.8.3266-3281.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–33. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, et al. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc Natl Acad Sci U S A. 2006;103:19069–74. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–70. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 18.Wang YX, et al. Regulation of Muscle Fiber Type and Running Endurance by PPARdelta. PLoS Biol. 2004;2, E294 doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. Faseb J. 2003;17:2299–301. doi: 10.1096/fj.03-0269fje. [DOI] [PubMed] [Google Scholar]
- 20.Lin J, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 21.Schuler M, et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4:407–14. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Cheng L, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–50. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 23.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka T, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003;100:15924–9. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee CH, et al. PPAR{delta} regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, Curtiss LK. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science. 2003;302:453–7. doi: 10.1126/science.1087344. [DOI] [PubMed] [Google Scholar]
- 27.Kharroubi I, Lee CH, Hekerman P, Darville MI, Evans RM, Eizirik DL, Cnop M. BCL-6: a possible missing link for anti-inflammatory PPAR-delta signalling in pancreatic beta cells. Diabetologia. 2006;49:2350–8. doi: 10.1007/s00125-006-0366-5. [DOI] [PubMed] [Google Scholar]
- 28.Nagasawa T, et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–91. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 29.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–16. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 30.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 31.Lee CH, Kang K, Mehl IR, Nofsinger R, Alaynick WA, Chong LW, Rosenfeld JM, Evans RM. Peroxisome proliferator-activated receptor {delta} promotes very low-density lipoprotein-derived fatty acid catabolism in the macrophage. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0510815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li AC, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–76. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–29. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 34.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–33. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 35.Hollingshead HE, Morimura K, Adachi M, Kennett MJ, Billin AN, Willson TM, Gonzalez FJ, Peters JM. PPARbeta/delta Protects Against Experimental Colitis Through a Ligand-Independent Mechanism. Dig Dis Sci. 2007 doi: 10.1007/s10620-006-9644-9. [DOI] [PubMed] [Google Scholar]
- 36.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 37.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 38.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–45. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat Med. 2004;10:245–7. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- 40.Reed KR, Sansom OJ, Hayes AJ, Gescher AJ, Winton DJ, Peters JM, Clarke AR. PPARdelta status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene. 2004;23:8992–6. doi: 10.1038/sj.onc.1208143. [DOI] [PubMed] [Google Scholar]
- 41.Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat Med. 2004;10:481–3. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- 42.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119:S10–6. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinberg JM. Lipotoxicity. Kidney Int. 2006;70:1560–6. doi: 10.1038/sj.ki.5001834. [DOI] [PubMed] [Google Scholar]
- 44.Aberle J, Hopfer I, Beil FU, Seedorf U. Association of peroxisome proliferator-activated receptor delta +294T/C with body mass index and interaction with peroxisome proliferator-activated receptor alpha L162V. Int J Obes (Lond) 2006;30:1709–13. doi: 10.1038/sj.ijo.0803345. [DOI] [PubMed] [Google Scholar]
- 45.Shin HD, et al. Genetic polymorphisms in peroxisome proliferator-activated receptor delta associated with obesity. Diabetes. 2004;53:847–51. doi: 10.2337/diabetes.53.3.847. [DOI] [PubMed] [Google Scholar]
- 46.Sprecher DL, et al. Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler Thromb Vasc Biol. 2007;27:359–65. doi: 10.1161/01.ATV.0000252790.70572.0c. [DOI] [PubMed] [Google Scholar]
- 47.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW,, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]