

Abstract
The gut-enriched Krüppel-like factor (GKLF) is a newly identified zinc finger-containing transcription factor. Recent studies indicate that GKLF binds to a core DNA sequence of 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′, which is found in an endogenous cis element, the basic transcription element (BTE) of the cytochrome P-450IA1 (CYP1A1) promoter. The present study characterizes the ability of GKLF to regulate CYP1A1 expression. By electrophoretic mobility gel shift assay (EMSA) and methylation interference assay, GKLF was found to bind BTE in a manner similar to several other transcription factors known to interact with BTE including Sp1 and BTEB. Cotransfection studies in Chinese hamster ovary cells showed that GKLF inhibited the CYP1A1 promoter in a dose- and BTE-dependent manner. The same experiments also revealed that BTE was responsible for a significant portion of the CYP1A1 promoter activity. EMSA of nuclear extracts from Chinese hamster ovary cells showed that Sp1 and Sp3 were two major proteins that interacted with BTE. Additional cotransfection studies showed that GKLF inhibited Sp1-mediated activation of the CYP1A1 promoter. In contrast, GKLF enhanced Sp3-dependent suppression of the same promoter. Moreover, the ability of GKLF to inhibit Sp1-dependent transactivation was in part due to physical interaction of the two proteins. These findings indicate that GKLF is a negative regulator of the CYP1A1 promoter in a BTE-dependent fashion and that this inhibitory effect is in part mediated by physical interaction with Sp1.
Regulation of gene expression is dependent on the functions of sequence-specific DNA-binding proteins called transcription factors. Proteins with the zinc finger motif constitute a significant portion of the transcription factor family, primarily because of the stable nature of interaction between a zinc finger and its cognate DNA-binding sequence (1–5). Among the zinc finger proteins the Cys2-His2 (C2H2) type, initially identified in the Xenopus laevis transcription factor TFIIIA (6), represents the most common zinc finger motif for DNA binding (7). Additional homology in the amino acid sequence of C2H2 zinc fingers is found in a family of proteins closely related to the Drosophila segmentation gene product Krüppel (8). Examples of Krüppel-like proteins include Sp1 (9), zif268/Egr-1 (10), EKLF1 (11), and WT-1 (12), which collectively exhibit a diverse range of regulatory functions in the cell.
The gut-enriched Krüppel-like factor (GKLF) (also called epithelial zinc finger) is a newly identified Krüppel-type protein with three C2H2 zinc fingers located in the carboxyl terminus (13–15). The amino acid sequence in the zinc finger region of GKLF is closely related to several Krüppel-like proteins including LKLF (16), EKLF (11), and BTEB2 (17). However, we recently showed that the amino acid sequence required for the nuclear localization of this group of proteins is more conserved in GKLF, LKLF, and EKLF than in BTEB2 (18). These findings suggest that the former three transcription factors belong to a distinct subfamily of closely related Krüppel proteins (18).
The in vivo expression of GKLF is enriched in epithelial cells of the gastrointestinal tract (13, 14) and skin (14) and in vascular endothelial cells (15). In vitro, expression of GKLF is induced in conditions that promote growth arrest such as serum deprivation and contact inhibition (13). In addition, constitutive expression of GKLF inhibits DNA synthesis (13). Taken together, these observations suggest that GKLF may have an important function in regulating growth and proliferation of specific epithelial and endothelial tissues.
The best studied “first-degree” relative of GKLF is EKLF, which is crucial for expression of the β-globin gene (11, 19, 20). Recent studies utilizing the gene targeting technique also showed that EKLF is essential for erythropoiesis (21, 22). EKLF acts by binding to and activating the CACCC element in the promoter of the β-globin gene (19, 20). Because of the close homology in the zinc finger sequence between GKLF and EKLF, GKLF was also found to interact with the β-globin CACCC motif (14, 15). However, whether GKLF regulates endogenous β-globin gene expression is unclear from these studies. By using an empirical approach called the target detection assay, we recently obtained a consensus, minimal essential binding sequence of 5′-(G/A)(G/A)GG(C/T)G(C/T)-3′ for GKLF (23). This sequence is similar but not identical to the CACCC element required for globin gene expression.
Another cis sequence that is similar to the empirically derived GKLF-binding sequence is the basic transcription element (BTE). This element is present in the promoter of a conserved family of genes encoding the cytochrome P-450 drug-metabolizing enzymes including CYP1A1 (24, 25). GKLF has been shown to interact with high affinity with BTE (14, 23). These findings raised the interesting question whether GKLF is involved in the regulation of the CYP1A1 promoter through BTE. Previous studies have shown that BTE is essential for the basal CYP1A1 promoter activity (24) and that it is the focus of interaction for a multitude of transcription factors including Sp1, BTEB, and BTEB2 (17, 26). Of interest is that several BTE-containing cytochrome P-450 genes including CYP1A1 are expressed in the epithelial cells of the intestinal tract (27–29) in a similar distribution to that found for GKLF (13, 14). Our current study therefore represents a continuing attempt to decipher further the function of GKLF in regulating expression of endogenous genes, using CYP1A1 as a potential “target” gene.
EXPERIMENTAL PROCEDURES
Materials
Purified human Sp1 was purchased from Promega (Madison, WI). A rabbit polyclonal anti-GKLF serum was described before (13, 18, 23). Antisera directed against human Sp1 and Sp3 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Oligonucleotides were synthesized at the Genetics Core Facility of the Johns Hopkins University School of Medicine. The sequences of the individual oligonucleotides in the sense orientation are as follows (shown in italics are linker sequences): BTE (17), 5′-gatcGAGAAGGAGGGCGTGC-CAACgatc-3′; GKLF (23), 5′-atgcAGGAGAAAGAAGGGCGTAGTATC-TActag-3′; Sp1 (Promega), 5′-ATTCGATCGGGGCGGGGCGAG-3′; AP2 (Promega), 5′-GATCGAACTGACCGCCCGCGGCCCGT-3′.
DNA Constructs
Complementary DNA constructs containing various regions of the coding sequence of GKLF were generated in the mammalian expression vector PMT3 (30). Included were GKLF-(1–483), containing the entire coding region, GKLF-(350–483), containing the carboxyl-terminal nuclear localization signal and the three zinc fingers, and GKLF-(1–401), containing the amino-terminal sequence including the nuclear localization signal but excluding the three zinc fingers (18, 23). Reporter constructs linking the chloramphenicol acetyl-transferase (CAT) reporter to various regions of the CYP1A1 promoter have been described (24). Included were the following: pMC6.3k, containing 6.3 kilobase pairs of the rat CYP1A1 promoter; pMC6.3kΔ-(96/53) that has an internal deletion between nucleotides −96 and −53 of the promoter; pMC6.3kΔ-(96/44) that has an internal deletion between nucleotides −96 and −44 of the promoter; pSV/MC53, containing the SV40 enhancer linked to 53 bp of the CYP1A1 promoter; and pSV/MC44, containing the SV40 enhancer linked to 44 bp of the CYP1A1 promoter. Two of these constructs (pMC6.3kΔ-(96/44) and pSV/MC44) do not contain BTE, which is located between nucleotide positions −53 and −44 of the CYP1A1 promoter (24). CMV-Sp1 and CMV-Sp3, expression constructs containing Sp1 and Sp3, respectively, were generously provided by Dr. G. Suske (31). pGST-Sp1ZnF and pGST-Sp1Q1, fusion constructs of glutathione S-transferase (GST) to aa residues 620–778 (containing the three zinc fingers) and 1–262 (containing one of the transactivation domains) of Sp1, respectively, were kindly provided by Dr. Y. Shi (32).
Production of Recombinant Protein
The production of bacterially expressed recombinant GKLF between aa residues 350–483 was described previously (23). This protein contains the nuclear localization signal and all three zinc fingers of GKLF and will be referred to as “recombinant GKLF” throughout the text. Briefly, the prokaryotic expression plasmid pET-16b (Novagen (Madison, WI)) containing aa residues 350–483 of GKLF was used to transform E. coli BL21(DE3) pLysS strain. Induction of recombinant protein production was achieved by the addition of 1 mm isopropyl-β-D-thiogalactopyranoside to logarithmically growing cells for 4 h. Cell lysates were prepared by treating pelleted bacteria with lysis buffer (20 mm Tris-HCl, pH 7.9, 0.5 m NaCl, 6 m urea, 5 mm imidazole, 1 μg g/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 20 μm phenylmethylsulfonyl fluoride) on ice for 30 min, followed by sonication and purification by a Ni2+-NTA-agarose column (Qiagen (Santa Clarita, CA)) equilibrated with lysis buffer. After extensive washing, bound proteins were eluted with the same buffer containing instead 1 m imidazole. The eluted protein was serially dialyzed against a solution of 10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 10 μm ZnCl2, 10% glycerol, and gradually decreasing concentrations of urea from 6 m to nil. The purity of the protein was approximately 90% as estimated by Coomassie Blue staining of the eluted protein resolved by denaturing polyacrylamide gel electrophoresis.
The production of GST-Sp1ZnF and GST-Sp1Q1 (32) protein was achieved using transformed DH5α strain of E. coli (Life Technologies, Inc.) after 2 h of induction with 1 mm isopropyl-β-D-thiogalactopyranoside. The bacterial pellet was collected by centrifugation and resuspended in a buffer containing 10 mm Tris-HCl, pH 7.8, 2 mm EDTA, 3% glycerol, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 20 μm phenylmethylsulfonyl fluoride. The bacteria were sonicated and then centrifuged to remove debris. The soluble fusion proteins present in the supernatant were stored in aliquots at −80°C.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed as described previously (23). 0.1 pmol of 32P-end-labeled oligonucleotides and 100 ng of purified protein were used in experiments involving recombinant GKLF or purified Sp1. In experiments that contained competitor DNA, a 10-fold molar excess unlabeled oligonucleotide over the labeled probe was included. Nuclear extracts from CHO cells were prepared by a modification of a published method (33). Briefly, 1 × 106 cells were washed twice with ice-cold PBS (phosphate-buffered saline), scraped in 5 ml of PBS, and collected by centrifugation at 400 × g for 5 min. The cell pellet was suspended in 4 packed cell volumes of buffer A containing 10 mm Tris-HCl, pH 7.8, 1.5 mm MgCl2, 10 mm KCl, and 0.5 mm dithiothreitol and left on ice for 10 min. Cells were then lysed by 10 strokes with a Dounce homogenizer. Nuclei were collected by centrifugation at 4,500 × g for 5 min, resuspended in 2 packed cell volumes of buffer B (20 mm Tris-HCl, pH 7.8, 1.5 mm MgCl2, 420 mm KCl, 0.5 mm dithiothreitol, and 20% glycerol), and rocked gently at 4°C for 1 h. After the complete lysis of nuclei, the suspension was centrifuged at 10,000 × g at 4°C for 30 min. The supernatant containing nuclear extracts was dialyzed exhaustively against buffer C (20 mm Tris-HCl, pH 7.8, 100 mm KCl, 0.2 mm EDTA, and 20% glycerol) and stored at −80°C. Ten μg of nuclear extracts were used for each EMSA reaction. In reactions that contained antibodies, 2 μg of affinity purified IgG were added to the mixture of nuclear extracts and DNA probe.
Methylation Interference Assay
Methylation interference assay was similar to that previously described (34). The synthetic BTE oligonucleotide was first labeled with [γ-32P]ATP using T4 polynucleotide kinase and annealed to the unlabeled complementary DNA strand. The singly end-labeled double-stranded oligonucleotide was partially methylated with a solution of 0.5% dimethyl sulfate in 50 mm sodium cacodylate and 0.1 mm EDTA. Ten pmol of partially labeled probe and 2.5 μg of purified recombinant GKLF were used in each EMSA reaction as described above. Following electrophoresis in a 6% nondenaturing polyacrylamide gel, bands corresponding to the shifted GKLF-BTE complex and the free, non-shifted BTE probe were excised and the DNA within the bands eluted by electrophoresis. The eluted DNA was then cleaved with 0.1 m piperidine at 95°C for 30 min. The cleaved products were resolved on a 12% denaturing polyacrylamide gel and visualized by autoradiography.
Transfection and Reporter Assays
Transient transfection of CHO cells by Lipofectin was described previously (18). The DNA contained a mixture of various amounts of effector and reporter constructs along with pCMV-SPORT-β-galactosidase (Life Technologies, Inc.) as an internal standard. The plasmid pBluescript (Stratagene (La Jolla, CA)) was added to bring the final DNA quantity up to 15 μg/10-cm dish. Cells were collected 24 h after transfection. CAT assays were performed as described before (35). Briefly, cell pellets were collected by centrifugation, resuspended in 100 mm Tris-HCl, pH 7.8, and lysed by repeated cycles of freezing and thawing. Extracts were incubated in 100 mm Tris-HCl, pH 7.8, 1 mm acetyl-CoA and 0.5 μCi of [14C]chloramphenicol (60 mCi/mmol (NEN Life Science Products)) at 37°C for 1 h. After extracting with ethyl acetate, the acetylated product and the substrate were resolved by thin layer chromatography using Silica Gel 60 (EM Science (Gibbstown, NJ)) in a solvent of 95% chloroform and 5% methanol. Following autoradiography of the developed gel plates, spots corresponding to the acetylated products and the substrate were excised and counted by liquid scintillation for quantification. β-Galactosidase activity was determined by the chemiluminescent assay (36) using Lumi-Gal 530 (Lumigen Inc. (Southfield, MI)) as a substrate. In all measurements the amount of extract used for the CAT assay was first adjusted after standardizing to the β-galactosidase activity.
GST Pull-down Experiments
GST pull-down experiments were performed as described previously (37). Five hundred μl of bacterial lysates containing GST-Sp1ZnF or GST-Sp1Q1 at an approximate concentration of 3 mg/ml were mixed with 250 μl of glutathione-Sepharose 4B beads (Amersham Pharmacia Biotech) equilibrated in PBS and rotated for 30 min at 25°C. After three successive washes with 1.5 ml of PBS, the beads were mixed with 50 μg of purified recombinant GKLF in a binding buffer containing 20 mm Tris-HCl, pH 8.0, 1 mm MgCl2, 2 mm ZnCl2, 0.1% dithiothreitol, 10% glycerol, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 20 μm phenylmethylsulfonyl fluoride, and rotated at 4°C for 60 min. The beads were collected by centrifugation at 500 × g for 5 min, and the supernatant was saved as the flow-through fraction. After washing the beads repeatedly with PBS, bound proteins were eluted with a buffer containing 50 mm Tris-HCl, pH 8.0, and 10 mm reduced glutathione for 20 min at 25°C. Two μg each of protein from the flow-through and the eluant fractions were examined by Western blot analysis for the presence of recombinant GKLF.
RESULTS
GKLF Interacts with the Basic Transcription Element (BTE) of the Rat CYP1A1 Promoter
A double-stranded oligonucleotide representing the sequence from nucleotide position −59 to −40, which contains BTE (24), of the rat CYP1A1 gene was synthesized and used as a probe in EMSA to determine whether it interacted with purified recombinant GKLF. As shown in Fig. 1, the presence of a single DNA-protein complex was observed when the probe was incubated with GKLF (lane 2) but not with control bovine serum albumin (lane 1). The presence of a 10-fold molar excess of unlabeled wild-type BTE oligonucleotide in the reaction competed efficiently for the formation of the complex (lane 3). Similarly, several mutant oligonucleotides including M2, M3, M8, and M9 were able to compete. In contrast, mutant oligonucleotides M4–M7 failed to compete with the probe for complex formation, indicating that the nucleotides mutated in these oligonucleotides were necessary for the binding of GKLF. Mutants M4–M7 contain the sequence 5′-GAGGCGT-3′ (boxed sequence, Fig. 1), which is the minimal essential binding site for GKLF (23). The results in Fig. 1, therefore, established that GKLF binds to BTE and that such binding occurs at a sequence consistent with the empirically determined binding site for GKLF.
Fig. 1. EMSA of wild-type and mutant BTE oligonucleotides with recombinant GKLF.
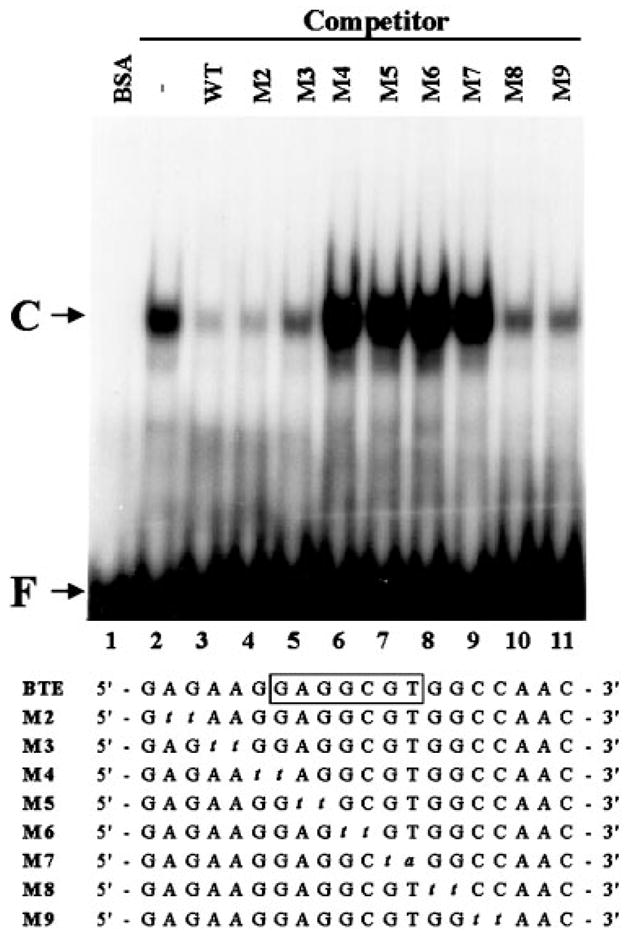
EMSA was performed as described under “Experimental Procedures.” 0.1 pmol of labeled BTE oligonucleotide and 100 ng of purified recombinant GKLF were used in each reaction except for lane 1 which contained 100 ng of bovine serum albumin (BSA) as control. Unlabeled competitor oligonucleotides were added in 10-fold molar excess over the probe. The sequences of the wild-type and the individual mutant oligonucleotides are shown. The boxed sequence in the wild-type BTE indicates the established minimal essential binding site of GKLF (23). The mutated bases are indicated by italic lower-case. WT is wild-type BTE. C denotes DNA-protein complex, and F denotes free DNA probe.
The Interaction between GKLF and BTE Is Similar to That between Sp1 and BTE
BTE is a cis element shown to interact with a number of transcription factors including Sp1, BTEB, and BTEB2 (17, 26). The nucleotides within BTE that are involved in contacting Sp1 are similar to those contacting BTEB (38). To determine whether GKLF also contacted these bases, methylation interference assay was performed using singly end-labeled double-stranded BTE oligonucleotides and recombinant GKLF. The results in Fig. 2 indicate that guanine residues in positions 9, 10, 12, 14, and 15 on the sense strand and those in positions 11, 16, 17, and 20 on the antisense strand were involved in contacting GKLF since their methylation resulted in an interference of binding. This pattern of methylation interference is very similar, if not identical, to that observed for Sp1 and BTEB (38), suggesting the three proteins bind to BTE in a similar fashion.
Fig. 2. Methylation interference assay of BTE by recombinant GKLF.
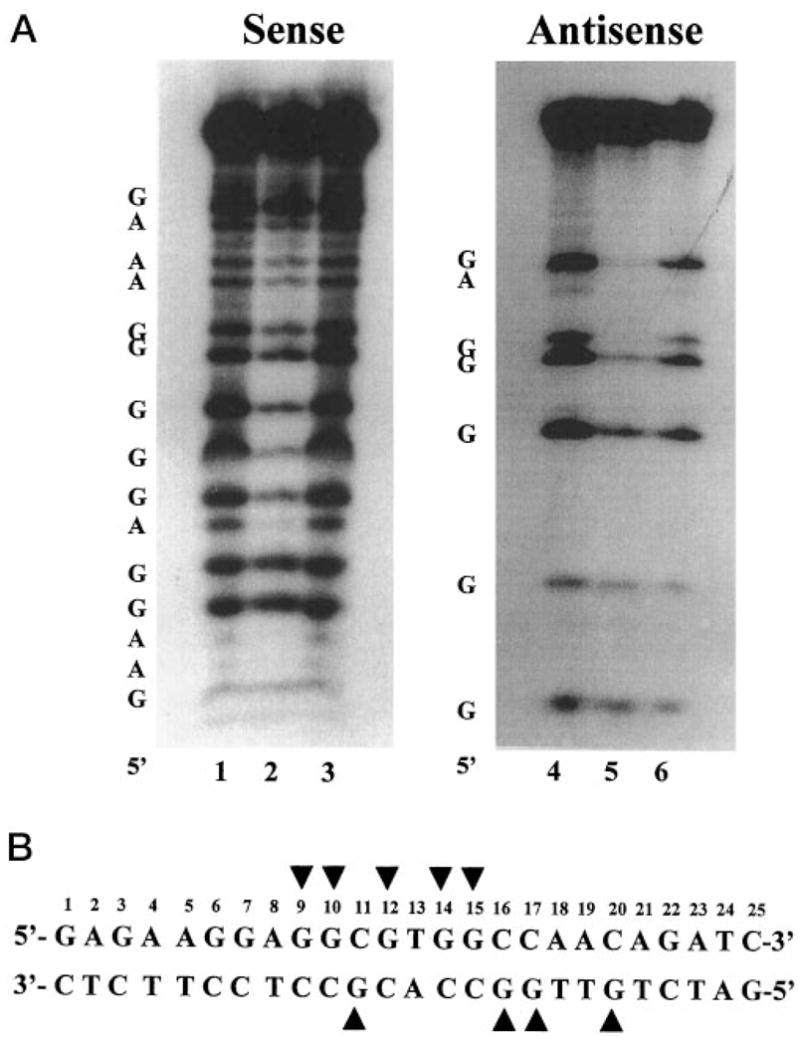
Methylation interference assay was performed as described under “Experimental Procedures.” The double-stranded BTE oligonucleotide was labeled at the 5′ end of either the sense or antisense strand. A, is the result of the interference assay. The 5′ end of each strand is shown. The DNA included in lanes 1, 3, 4, and 6 was derived from the unbound free probe, whereas the DNA included in lanes 2 and 5 was from the bound probe. Methylated guanine and adenine residues are identified. B, shows the guanine residues (arrowheads) within the BTE, which when methylated resulted in an interference of binding to GKLF.
GKLF Suppresses the Activity of the CYP1A1 Promoter
To determine whether GKLF might influence the activity of the CYP1A1 promoter, cotransfection experiments were performed in CHO cells using a CYP1A1 promoter-linked reporter gene and an expression plasmid encoding either full-length or truncated forms of GKLF. The reporter pMC6.3k contains 6.3 kilobase pairs of the rat CYP1A1 promoter linked to CAT (24) and exhibited high basal activities in transfected cells (shown in the 1st lanes of each panel in Fig. 3). The presence of a cotransfected expression plasmid containing full-length GKLF (PMT3-GKLF-(1–483)) caused a decrease in the reporter activity in a dose-dependent manner (Fig. 3A). A similar suppressive effect was observed when cells were cotransfected with a plasmid containing a truncated form of GKLF that retained its zinc finger region (PMT3-GKLF-(350–483)) (Fig. 3B). In contrast, another truncated form of GKLF in which the three zinc fingers were deleted had no such suppressive effect on the CYP1A1 promoter (PMT3-GKLF-(1–401)) (Fig. 3C). This lack of effect was not due to failure of this zinc finger-minus form of GKLF to localize to the nucleus since the nuclear localization signal of GKLF is retained in this particular construct (18).
Fig. 3. Cotransfection of CHO cells with a CYP1A1 promoter-linked CAT reporter and GKLF expression constructs.
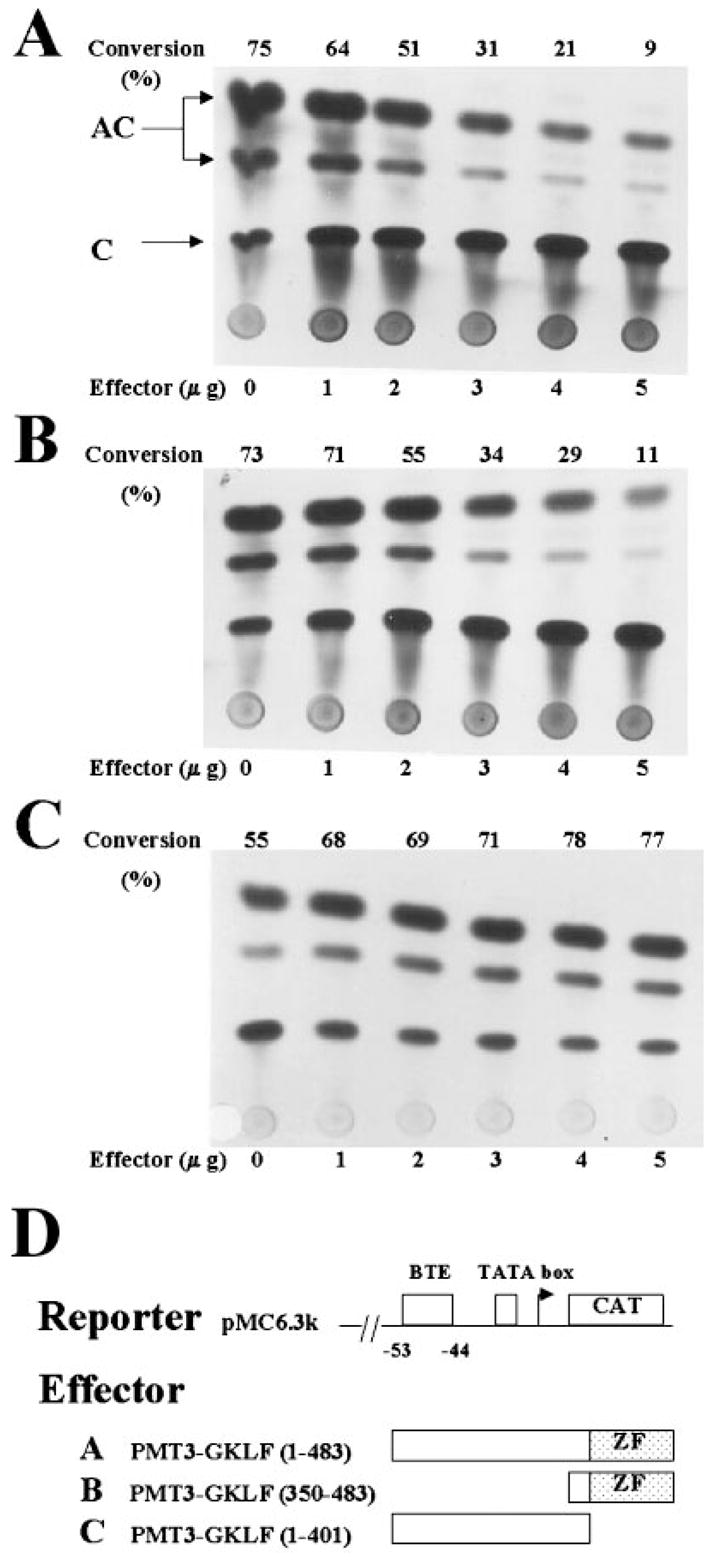
Transient transfections of CHO cells were performed with 5 μg/10-cm dish of pMC6.3k that contains 6.3 kilobase pairs of the rat CYP1A1 promoter linked to the CAT reporter (24) and increasing amounts of the mammalian expression plasmid PMT3 (30) containing either the full-length (effector A) or two truncated forms of GKLF (effectors B and C). The amount of extracts used to determine the CAT activity was first standardized to the β-galactosidase activity derived from the internal control, pCMV-SPORT-β-galactosidase. C denotes the substrate chloramphenicol, and AC denotes acetylated forms of chloramphenicol. The number at the top of each panel represents the percentage of substrate conversion ((AC/AC + C) × 100). ZF denotes the zinc fingers of GKLF.
To determine whether the suppressive effect of GKLF on the CYP1A1 promoter was mediated by BTE, additional cotransfection experiments were performed using reporters with various internal deletions in the CYP1A1 promoter sequence. Two such deletion constructs analyzed were pMC6.3kΔ-(96/53) and pMC6.3kΔ-(96/44), with the latter known to lack any BTE-mediated activity (24). As shown in Fig. 4, whereas full-length GKLF suppressed the activities of both pMC6.3kΔ-(96/53) and wild-type pMC6.3k reporters, it failed to suppress that of pMC6.3kΔ-(96/44). The truncated form of GKLF, PMT3-GKLF-(1–401), failed to suppress any reporters. These results indicate that the suppressive effect of GKLF on the CYP1A1 promoter is specifically mediated by BTE.
Fig. 4. Cotransfection of CHO cells with various CYP1A1 reporter and GKLF effector constructs.
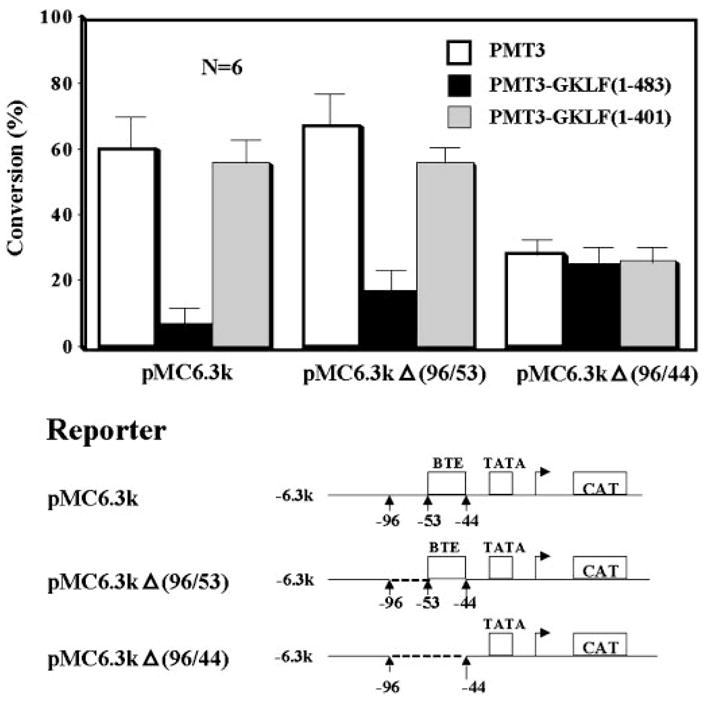
Transfection conditions were essentially the same as those described in Fig. 3. The amount of reporter and effector plasmids used was 5 μg/10-cm dish each. A description of the reporter constructs can be found under “Experimental Procedures.” PMT3 indicates the vector alone. Shown are the means ± S.E. of six independent experiments, each of which was carried out in duplicate.
The BTE-specific nature of the effect of GKLF on the CYP1A1 promoter was substantiated by studying two additional promoter-reporter constructs, pSV/MC53 and pSV/MC44. pSV/MC53 contained 53 bp and pSV/MC44 contained 44 bp of the CYP1A1 promoter linked to the SV40 virus enhancer. They differed from each other by the presence and absence of BTE, respectively (24). As shown in Fig. 5, PMT3-GKLF-(1–483) inhibited the reporter activity of pSV/MC53 in a dose-dependent fashion (Fig. 5A) but not that of pSV/MC44 (Fig. 5B). When additional effector constructs were analyzed by similar transfection experiments, the inhibitory effect on the pSV/MC53 construct was observed for both full-length GKLF and GKLF that retained the zinc finger region (PMT3-GKLF-(350–483)) (Fig. 6). As in Fig. 3, GKLF lacking zinc fingers (PMT3-GKLF-(1–401)) had no effect on pSV/MC53. In no cases did any of the effector constructs exhibit any effect on pSV/MC44, which lacked BTE (Fig. 6).
Fig. 5. Cotransfection of CHO cells with minimal CYP1A1 promoter-reporters and the full-length GKLF-expressing construct.
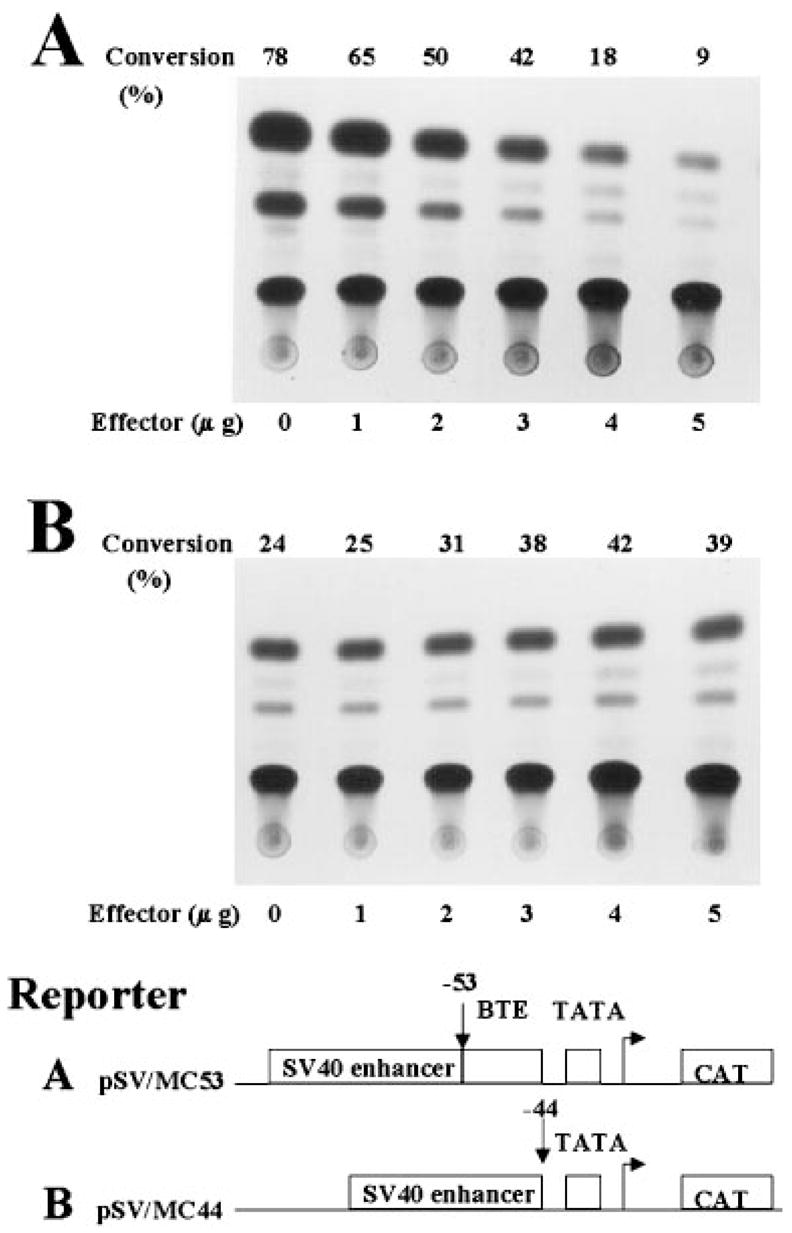
A description of the two reporter constructs (A and B) was provided under “Experimental Procedures” and “Results.” Five μg/10-cm dish of the reporter and increasing amounts of the full-length GKLF construct, PMT3-GKLF-(1–483), were used in the experiments.
Fig. 6. Cotransfection of CHO cells with minimal CYP1A1 promoter-reporters and expression constructs containing various forms of GKLF.
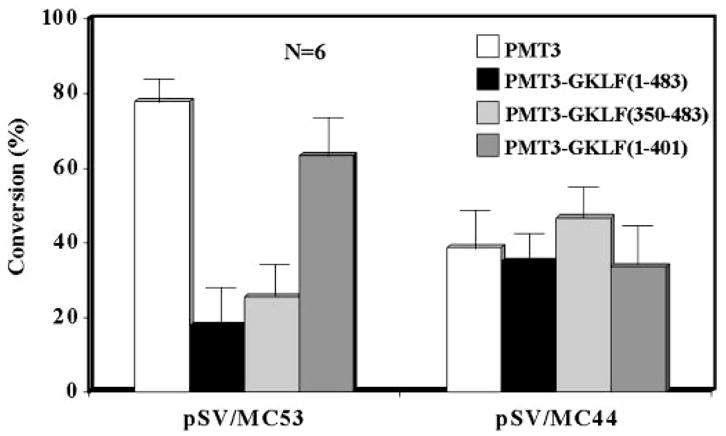
The experimental conditions were similar to those described in Fig. 5. The amount of DNA used for all constructs was 5 μg/10-cm dish. Shown are the means ± S.E. of substrate conversion from six independent experiments. Each experiment was carried out in duplicate.
Sp1 and Sp3 Are the Major BTE Binding Transcription Factors in CHO Cells
In addition to the findings that GKLF suppressed the activity of the CYP1A1 promoter, results of the preceding transfection experiments also indicated that BTE was responsible for a significant fraction of the CYP1A1 promoter activity. This was demonstrated by the observation that deletion of BTE from the promoter resulted in approximately a 50% reduction in the promoter activity (Figs. 4 and 6). To determine which transcription factor(s) activated BTE in the transfected cells, EMSA was performed on BTE with nuclear extracts obtained from CHO cells. As shown in Fig. 7, multiple bands representing DNA-protein complexes were formed (lane 1). The addition of a 10-fold molar excess cognate BTE competitor oligonucleotide abolished many of the complexes (lane 2). Similarly, the presence of a competitor oligonucleotide representing the consensus Sp1-binding site decreased the formation of a number of complexes (lane 3). In contrast, the addition of a competitor oligonucleotide representing either an AP2-binding sequence or the minimal essential binding sequence for GKLF (23) failed to influence complex formation (lanes 4 and 5, respectively). These results suggest that the primary BTE-binding proteins in CHO cells include Sp1 and Sp1-related proteins.
Fig. 7. EMSA of BTE oligonucleotides using CHO cell nuclear extracts.
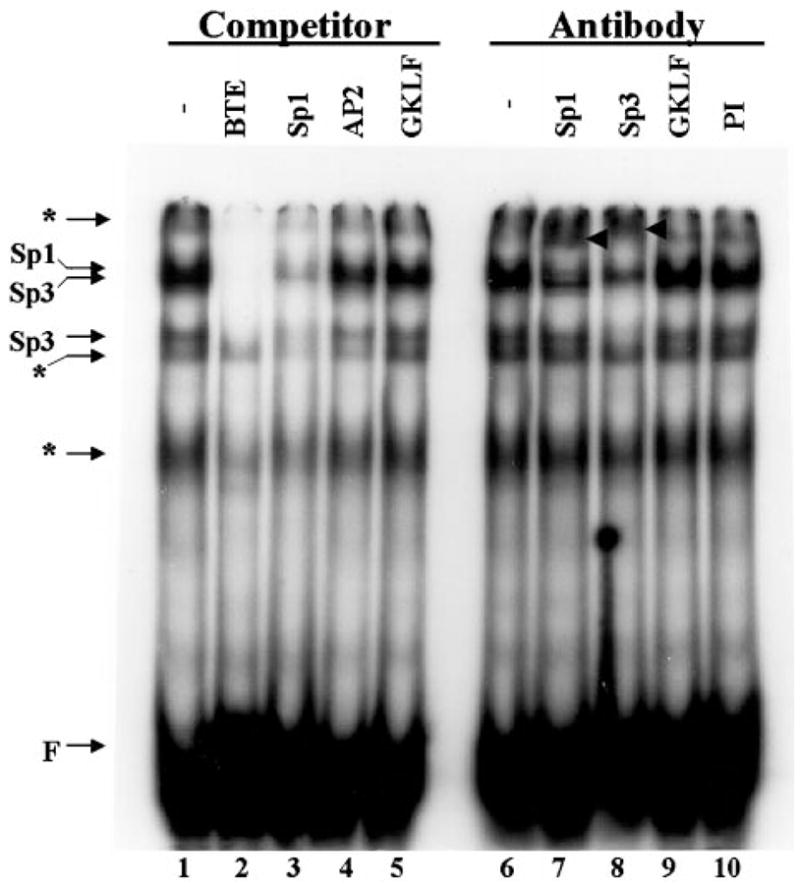
EMSA was performed using 0.1 pmol of labeled BTE oligonucleotides and 10 μg of nuclear extracts obtained from CHO cells. Where indicated, competitor oligonucleotides (see “Experimental Procedures”) were added at a 10-fold molar excess of the labeled probe. In experiments involving antibodies, 2 μg of the stated antibody were added to the reaction. PI denotes pre-immune serum for GKLF. Super-shifted bands in lanes 7 and 8 are labeled with arrowheads. The positions of Sp1 and Sp3 in the DNA-protein complexes are indicated. The asterisks indicate either nonspecific complexes (since they competed poorly) or complexes containing as yet unidentified proteins.
To establish further the identities of the BTE-binding proteins in the nuclear extracts, EMSA was performed in the presence of antibodies directed against specific transcription factors. Whereas the formation of the complexes was unaffected by an antibody directed against GKLF (lane 9) or by preimmune serum (lane 10), the addition of anti-Sp1 (lane 7) and anti-Sp3 (lane 8) interrupted the formation of specific complexes with the resultant formation of “supershifted” bands (arrowheads, lanes 7 and 8). These results indicate that CHO cells contained both Sp1 and Sp3 that bound to BTE but little or no endogenous GKLF, as evidenced by the failure of the minimal essential GKLF-binding sequence to compete and by the lack of GKLF antibody to disrupt complex formation.
GKLF Abrogates Sp1-dependent Activation and Enhances Sp3-dependent Suppression of the CYP1A1 Promoter
To determine whether Sp1 and Sp3 affected the CYP1A1 promoter and whether GKLF modulated Sp1- and Sp3-dependent activity of the same promoter, cotransfection experiments were conducted in CHO cells using the pMC6.3k reporter and various combinations of expression plasmids containing Sp1, Sp3, and GKLF. Fig. 8 shows that Sp1 caused an increase in the CYP1A1 promoter activity in a dose-dependent fashion (lanes 1–6). The inductive effect of Sp1 was abrogated by the presence of increasing amounts of the expression construct containing full-length GKLF (Fig. 8, lanes 7–12). In contrast to Sp1, Sp3 caused an inhibition of the same promoter in a dose-dependent fashion (Fig. 9, lanes 1–6). The addition of GKLF to the cotransfection further increased the inhibition of the CYP1A1 promoter (Fig. 9, lanes 7–12). These observations indicate that Sp1 and Sp3 modulate the CYP1A1 promoter in a reciprocal manner. They also show that GKLF abrogates Sp1-dependent activation and enhances Sp3-dependent suppression of the CYP1A1 promoter.
Fig. 8. Contransfection of CHO cells with a CYP1A1 promoter-reporter construct and expression plasmids containing Sp1 and GKLF.
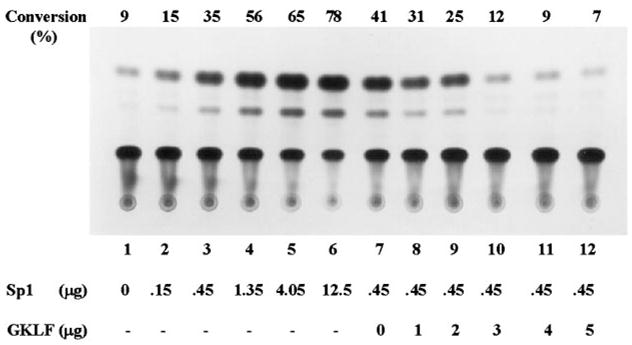
CHO cells were transiently transfected with 5μg/10-cm dish of pMC6.3k and increasing amounts of CMV-Sp1 (lanes 1–6) or increasing amounts PMT3-GKLF in the presence of 0.45 μg/10-cm dish of CMV-Sp1. Quantification of substrate conversion was performed as in previous figures. The amount of cell extracts used for the CAT assay was intentionally lowered by 5-flod to demonstrate the inductive effect of the promoter by Sp1. Shown is a representative result from three independent experiments.
Fig. 9. Cotransfection of CHO cells with a CYP1A1 promoter-reporter and expression plasmids containing Sp3 and GKLF.
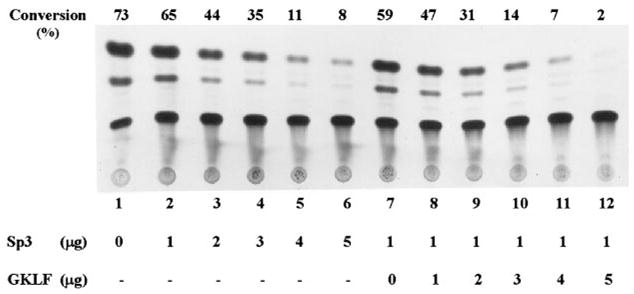
Cotransfection experiments were performed as described in Fig. 8 with the exception that CMV-Sp3 was substituted for CMV-Sp1. The amount of cell extracts used for the CAT assay was similar to those used in Figs. 4 and 6. Shown is a representative result from two independent experiments.
Recombinant GKLF and Sp1 Exhibit Similar Affinities for BTE and GKLF Competes with Sp1 for Binding to BTE
We then compared the binding affinities of GKLF and Sp1 to BTE using EMSA. Equal molar quantities of recombinant GKLF or purified human Sp1 were incubated with increasing amounts of a labeled BTE oligonucleotide. The intensity of the shifted complex was quantified with a densitometer and plotted against the probe concentration (Fig. 10). By Scatchard analysis (not shown), it was estimated that the dissociation constant (Kd) was 0.7 nM between Sp1 and BTE and 1.6 nM between BTE and GKLF. The estimated Kd of Sp1 to BTE is consistent with previous measurements of Sp1 with its binding site, which ranged between 0.5 and 3 nM (38–40).
Fig. 10. Kinetics of binding between Sp1 and GKLF to BTE.
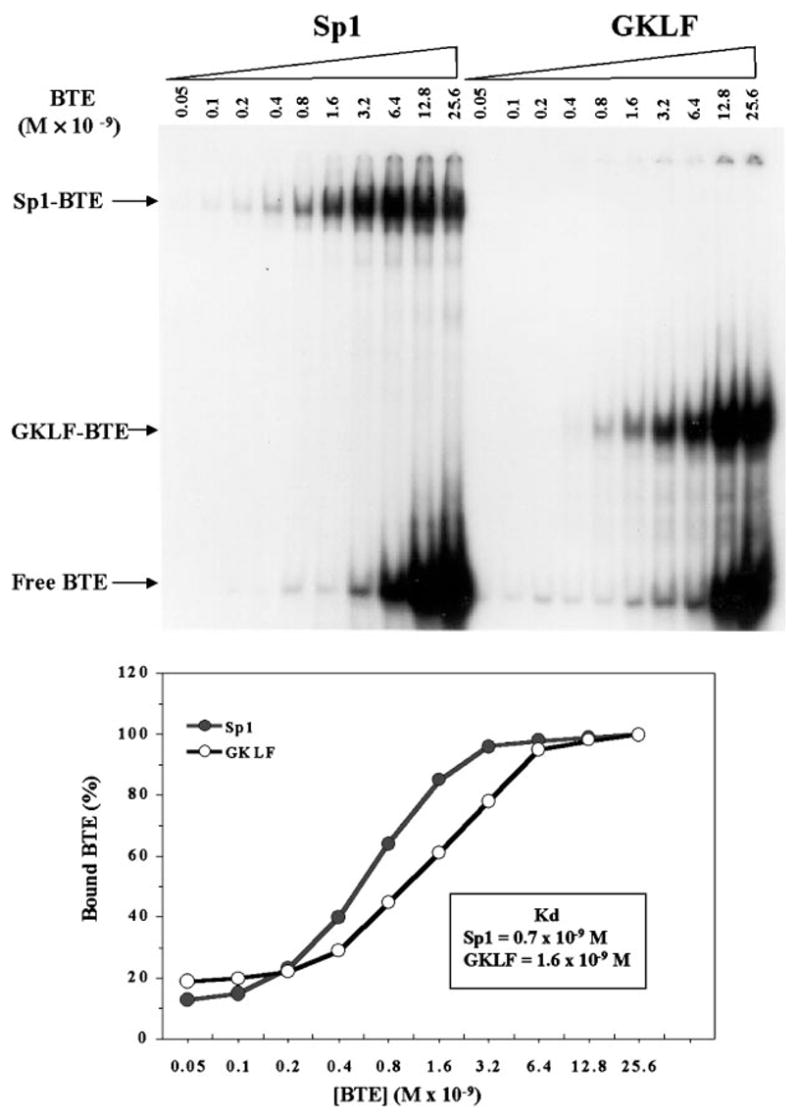
EMSA was performed using increasing amounts of labeled BTE probe and 200 ng of purified human Sp1 or 60 ng of recombinant GKLF (top panel). Quantification of the intensity of the shifted bands was performed by densitometric measurement. One hundred percent bound was defined as the band that had the highest intensity for each protein (bottom panel).
To examine further the relationship among GKLF, Sp1, and BTE, EMSA was performed using a mixture of recombinant GKLF and purified human Sp1. As demonstrated in Fig. 11, the presence of GKLF inhibited the binding of Sp1 to the BTE DNA in a dose-dependent fashion (lanes 11–15). As the purity of the two protein preparations was similar to each other, a unit quantity of recombinant GKLF was approximately 4 times in molar quantity when compared with the same amount of Sp1. Lane 12 therefore contained a close to equal molar quantity of GKLF and Sp1, which resulted in an approximately 50% decrease of binding of Sp1 to BTE due to the presence of GKLF.
Fig. 11. Competition of binding of BTE to Sp1 by GKLF.
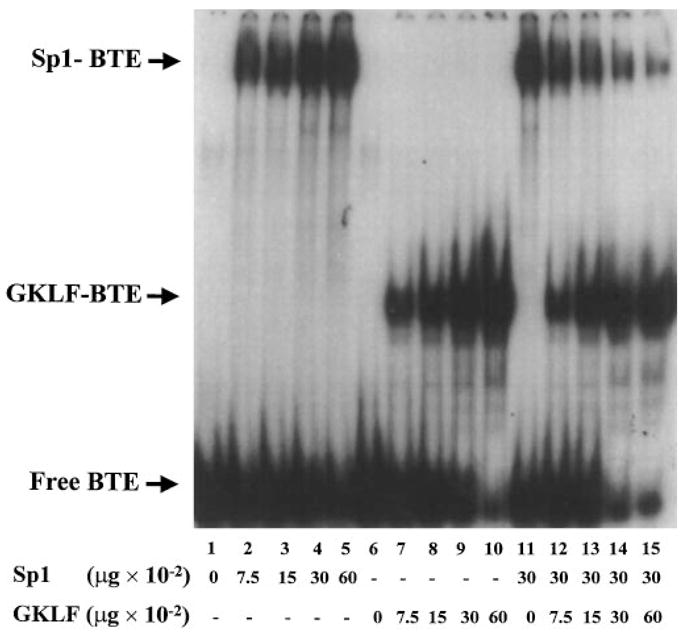
EMSA was performed as before. 0.1 pmol of the labeled BTE oligonucleotide was incubated with increasing amounts of purified human Sp1 or recombinant GKLF (lanes 1–5 and lanes 6–10, respectively). In addition, the probe was incubated with 30 ng of purified Sp1 and increasing amounts of recombinant GKLF (lanes 11–15).
GKLF and Sp1 Physically Interact with Each Other
Although the result in Fig. 10 indicated that the Kd of GKLF to BTE was at least twice that of Sp1, GKLF was nonetheless able to compete with Sp1 for binding to BTE at a similar molar ratio (Fig. 11). These findings suggested that there might be an additional mechanism by which GKLF affected the binding of Sp1 to BTE. Protein-protein interaction is one such potential mechanism. To determine whether GKLF and Sp1 could physically interact with each other, GST pull-down experiments were performed using recombinant GKLF and two GST fusion proteins containing two different regions of Sp1. As can be seen in Fig. 12, GKLF was retained by the glutathione-Sepharose 4B beads only in the presence of GST-Sp1ZnF, which contains the carboxyl-terminal 159 aa residues of Sp1 including its three zinc fingers (lane 7) (32). In contrast, GKLF was recovered only in the flow-through fractions of beads that retained either GST alone (lane 3) or GST-Sp1Q1 (lane 5), a portion of Sp1 that contains a glutamine-rich transactivation domain (32). These results therefore provide strong evidence for a direct physical interaction between GKLF and Sp1, which may potentially involve the zinc finger regions of the two proteins.
Fig. 12. GST pull-down of GKLF by Sp1.
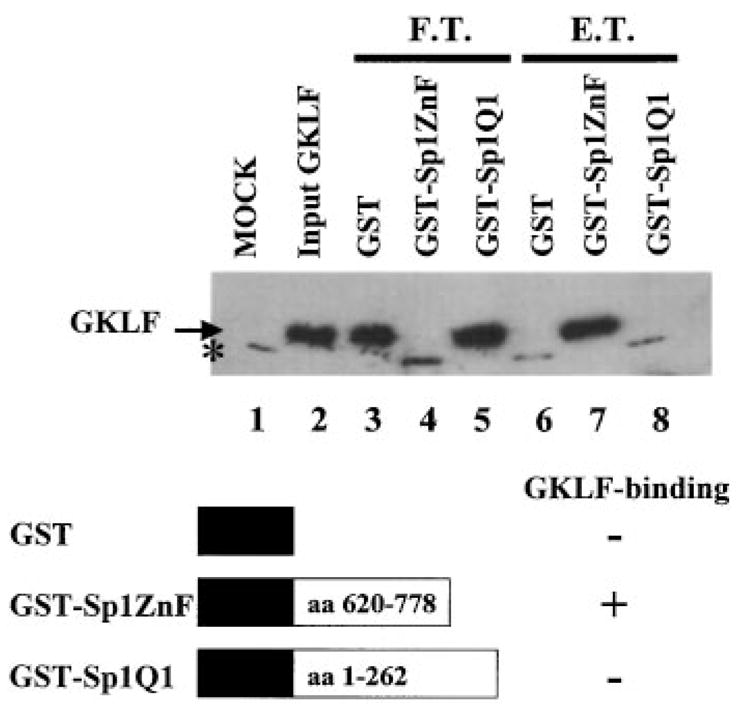
The GST pull-down experiments are described under “Experimental Procedures.” Western blot analysis was performed on various protein fractions using a polyclonal rabbit GKLF antibody (13). Lanes 1 and 2 contained input lysates from mock- (vector alone-) transformed and GKLF-transformed bacteria, respectively. Lanes 3–5 contained the flow-through (F.T.) fractions of GKLF that did not bind to the glutathione-Sepharose 4B beads containing GST, GST-Sp1ZnF, and GST-Sp1Q1, respectively. Lanes 6–8 contained the eluant (E.T.) fractions of GKLF that bound to the beads and were eluted with 1 mm reduced glutathione. The location of GKLF is indicated and has a molecular mass of 24 kDa. The asterisk denotes a nonspecific cross-reacting material that was present in the lysates of mock-transformed bacteria.
DISCUSSION
GKLF was initially identified by low-stringency library screening with the zinc finger region of zif268/Egr-1 (13). Recent studies suggest that GKLF is one of three members of a subfamily of closely related Krüppel proteins (18). The other two members are LKLF (16) and EKLF (11). Despite a significant degree of conservation in the zinc finger region (13) and in the nuclear localization signal (18) among these three proteins, their tissue distributions and presumed physiological functions are quite different. EKLF is expressed primarily in erythroid tissues including bone marrow and spleen (11). Gene targeting experiments indicated that EKLF-deficient mice were defective in erythropoiesis during fetal development (21, 22). LKLF, found primarily in lung and spleen (16), is necessary to maintain the quiescent state of single-positive T lymphocytes. T cells from mice deficient in LKLF exhibited a spontaneously activated phenotype and died in the peripheral lymphoid organs from Fas ligand-induced apoptosis (41). In addition, LKLF is required for the maintenance of vascular integrity during fetal development (42). The physiological function of GKLF is less well defined although its epithelial (13, 14) and endothelial (15) nature of expression suggests that it may have a role in regulating tissue-specific gene expression. In addition, the growth arrest-associated nature of GKLF expression points to a potential function in the regulation of cell proliferation.
Among the three aforementioned Krüppel-like proteins, only EKLF has a clearly established target gene. EKLF is a crucial transcription factor for β-globin gene expression and exerts its effect through a CACCC element present in the β-globin promoter (19, 20). Although both GKLF (14, 15) and LKLF (16) have also been found to interact with the CACCC element, no definitive endogenous genes have been identified as the targets for either protein to date. It is within the context of identifying target genes that we conducted a study to empirically determine the binding sequence for GKLF (23). That study not only established a minimal essential binding site of GKLF, it also identified CACCC and BTE as two naturally occurring cis elements that conform with the binding sequence of GKLF. Our current effort is therefore focused on establishing the relationship between GKLF- and BTE-mediated gene transcription.
BTE was selected for the present study because it is the focal point onto which multiple transcription factors converge to exert their effects. Along with the present study, BTE has been found to interact with no fewer than five Krüppel-like factors including Sp1 (26, 38), Sp3 (this study), BTEB (26, 38), BTEB2 (17), and GKLF (14, this study). An additional significance of BTE in transcriptional regulation is demonstrated by its presence in the promoter of a plethora of genes that belong to the cytochrome P-450 superfamily (43). Examples include, but are not limited to, CYP1A1 (24, 25), CYP1A2 (44, 45), CYP2B1 (25, 46), CYP2B2 (25, 46), CYP3A4 (47), CYP3A5 (48), CYP3A16 (49), and CYP11A (50). In many of these genes BTE was proven to be a crucial determinant of their promoter activity. Moreover, at least one BTE-binding protein, Sp1, has been shown to physically interact with transcription factors binding to other regions of the CYP1A1 promoter and to exert a cooperative influence on the drug-inducible expression of this gene (51). These studies suggest that BTE is likely a crucial cis element involved in the coordinated expression of a large number of related genes. Of additional interest is that many cytochrome P-450 genes including CYP1A1, CYP1A2, CYP2B1, and CYP2B2 (27–29) are expressed in the epithelium of the gastrointestinal tract in a similar distribution to that of GKLF (13, 14). These observations raise the distinct possibility that GKLF is a major contributor to the intestinal epithelial expression of these physiologically important cytochrome P-450 genes.
By base-specific mutational analysis and methylation interference assay (Figs. 1 and 2, respectively), recombinant GKLF appears to interact with BTE in a very similar, if not identical, manner to Sp1 and BTEB (38). The dissociation constants of the three proteins in binding to DNA also appear to be reasonably similar to one another (Fig. 10; Ref. 38). These results suggest that GKLF, Sp1, and BTEB exhibit similar affinities with respect to BTE binding. It is of interest to note that GKLF binds poorly to a consensus Sp1-binding site (23). Conversely, Sp1 and Sp3 bind poorly to the empirically determined GKLF-binding site (Fig. 7). To this end, BTE seems to represent a “composite” site to which all these proteins exhibit a high affinity. These features increase the attractiveness of BTE as a “model” element in studying the increasingly complex mechanism by which it mediates gene expression.
The results of our cotransfection experiments firmly established that GKLF suppresses CYP1A1 promoter activity in a BTE-dependent fashion. Moreover, this inhibitory effect appears to be exerted at the expense of Sp1, which has an activating effect on the same promoter. This behavior of GKLF is reminiscent of that observed for BTEB (26). In that study, BTEB exhibited a similar suppressive effect on the CYP1A1 promoter in a BTE-dependent manner. In addition, BTEB inhibited Sp1-mediated activation of the same promoter. However, this suppressive effect was dependent on the context of the DNA, BTEB was an activator when multiple copies of BTE were present in the promoter. Our laboratory has made a similar observation. When two tandem copies of either a consensus GKLF-binding site (23) or BTE2 were used to drive a reporter, GKLF increased the reporter activity. The mechanism for this pleiotropic effect is unclear at this time although there are examples of other bifunctional transcription factors including Sp3 (31, 52, 53) and YY1 (54, 55) that exhibit a similar DNA-dependent positive and negative effect. GKLF was also found to contain distinct domains that mediate repression and transactivation similar to Sp3 and YY1 (15, 56, 57).3 It is therefore possible that conformational changes resulting from protein-protein and/or protein-DNA interactions may be the basis for the pleiotropic effect of GKLF.
Although a number of possible mechanisms may exist that are responsible for the suppression of the CYP1A1 promoter by GKLF, the most likely explanation is that the suppression is based on a competitive mechanism. This is demonstrated by the inhibition of binding of Sp1 to BTE in the presence of recombinant GKLF (Fig. 11). This passively accomplished repression may be augmented by GKLF once it is bound to the promoter via an intrinsic repressive domain within GKLF (15). The cooperative nature of suppression exerted by GKLF and Sp3 on the CYP1A1 promoter (Fig. 9) further supports the latter hypothesis. A similar finding on the competitive binding to BTE by different proteins has previously been observed between Sp1 and BTEB (38). Taken together, GKLF and BTEB appear to exhibit a very similar behavior with regard to their effects on a BTE-driven promoter. To the contrary, BTEB2, a more GKLF-related Krüppel protein than BTEB (13, 18), is an activator of BTE-driven promoters (17). These findings further increase the complexity by which BTE is utilized as a basal promoter element to drive gene expression.
The competitive effect of GKLF on the binding of Sp1 to BTE appears to be further augmented by a direct physical interaction between the two proteins (Fig. 12). Interestingly, in neither our study nor a previous report (38) was a stable ternary complex observed between GKLF, Sp1, and BTE or between BTEB, Sp1, and BTE. In contrast, the interaction between the bifunctional Krüppel protein YY1, Sp1, and a YY1-binding sequence resulted in the formation of a ternary complex (32). The reason for the difference is unclear, although in the latter case the interaction of YY1 and Sp1 led to a cooperative induction of their target promoter. In a different system YY1 was shown to repress Sp1-mediated transcription and that such repression does not depend on physical interaction between YY1 and Sp1 (58). Additional studies are therefore necessary to delineate the exact effect of interaction between specific proteins on regulating promoter activity.
GKLF is expressed in a tissue-selective manner (13, 14). Its interaction with a “housekeeping” transcription factor such as Sp1 is reminiscent of other situations in which tissue-specific and ubiquitous transcription factors interact to control tissue-specific gene expression. Examples include the interaction between MyoD1 and E12/E47 during myogenesis (59) and between Oct-2 and Oct-1 during lymphogenesis (60). To this end, the expanding repertoire of proteins that interact with Sp1 increases its importance and versatility in regulating gene expression. Examples of other important cellular or viral proteins that are capable of physically interacting with Sp1 include YY1 (32), Sp3 (61), Rb (62), p53 (63), v-Rel (64), C/EBPβ (65), GATA-1 (66), AhR.Arnt (51), Tat (67), and Tax (68). As in the case of GKLF, many of these proteins either have tissue-specific functions or are involved in crucial cellular functions such as growth and differentiation. The further examination of the involvement of Sp1 in the context of selective protein-protein interaction may further increase the understanding of the mechanisms regulating tissue-specific gene expression.
Acknowledgments
We thank Drs. Suske and Shi for plasmids. We also thank Deborah Geiman and Channing Mahatan for helpful discussion and technical assistance.
Footnotes
This work was supported in part by Grants DK44484 and DK52230 from the National Institutes of Health (to V. W. Y.).
The abbreviations used are: EKLF, erythroid Krüppel-like factor; BTE, basic transcription element; BTEB2, basic transcription factor element binding protein 2; CAT, chloramphenicol acetyltransferase; CHO, Chinese hamster ovary; CYP1A1, cytochrome P-450IA1; EMSA, electrophoretic mobility shift assay; GKLF, gut-enriched Krüppel-like factor; GST, glutathione S-transferase; LKLF, lung Krüppel-like factor; PBS, phosphate-buffered saline; aa, amino acid; bp, base pair(s).
W. Zhang and V. W. Yang, unpublished observations.
D. E. Geiman and V. W. Yang, unpublished observations.
Note Added in Proof—While this manuscript was under review, Jenkins et al. (69) demonstrated that GKLF was able to transactivate the human keratin 4 and Epstein-Barr virus ED-L2 promoters via a CACCC-like element.
References
- 1.Klug A, Rhodes D. Cold Spring Harb Symp Quant Biol. 1987;52:473–482. doi: 10.1101/sqb.1987.052.01.054. [DOI] [PubMed] [Google Scholar]
- 2.Berg JM. J Biol Chem. 1990;265:6513–6516. [PubMed] [Google Scholar]
- 3.Berg JM. Annu Rev Biophys Biophys Chem. 1990;19:405–421. doi: 10.1146/annurev.bb.19.060190.002201. [DOI] [PubMed] [Google Scholar]
- 4.Klug A, Schwabe JW. FASEB J. 1995;9:597–604. [PubMed] [Google Scholar]
- 5.Berg JM, Shi Y. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 6.Pelham HR, Brown DD. Proc Natl Acad Sci U S A. 1980;77:4170–4174. doi: 10.1073/pnas.77.7.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoovers JM, Mannens M, John R, Bliek J, van Heyningen V, Porteous DJ, Leschot NJ, Westerveld A, Little PFR. Genomics. 1992;12:254–263. doi: 10.1016/0888-7543(92)90372-y. [DOI] [PubMed] [Google Scholar]
- 8.Schuh R, Aicher W, Gual U, Cote S, Preiss A, Maier D, Seifert E, Nauber U, Shroder C, Kemler R, Jackle H. Cell. 1986;47:1025–1032. doi: 10.1016/0092-8674(86)90817-2. [DOI] [PubMed] [Google Scholar]
- 9.Kadonaga JT, Carner KR, Masiarz FR, Tjian R. Cell. 1987;51:1079–1090. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- 10.Christy BA, Lau LF, Nathans D. Proc Natl Acad Sci U S A. 1988;85:7857–7861. doi: 10.1073/pnas.85.21.7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller IJ, Bieker JJ. Mol Cell Biol. 1993;13:2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 13.Shields JM, Christy RJ, Yang VW. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. J Biol Chem. 1996;271:31384–31390. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 15.Yet SF, McA’Nulty MM, Folta SC, Yen HW, Yoshizumi M, Hsieh CM, Layne MD, Chin MT, Wang H, Perrella MA, Jain MK, Lee ME. J Biol Chem. 1998;273:1026–1031. doi: 10.1074/jbc.273.2.1026. [DOI] [PubMed] [Google Scholar]
- 16.Anderson KP, Kern CB, Crable SC, Lingrel JB. Mol Cell Biol. 1995;15:5957–5965. doi: 10.1128/mcb.15.11.5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sogawa K, Imataka H, Yamasaki Y, Kusume H, Abe H, Fujii-Kuriyama Y. Nucleic Acids Res. 1993;21:1527–1532. doi: 10.1093/nar/21.7.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shields JM, Yang VW. J Biol Chem. 1997;272:18504–18507. doi: 10.1074/jbc.272.29.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartzog GA, Myers RM. Mol Cell Biol. 1993;13:44–56. doi: 10.1128/mcb.13.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bieker JJ, Southwood CM. Mol Cell Biol. 1995;15:852–860. doi: 10.1128/mcb.15.2.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 22.Perkins AC, Sharpe AH, Orkin SH. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 23.Shields JM, Yang VW. Nucleic Acids Res. 1998;26:796–802. doi: 10.1093/nar/26.3.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanagida A, Sogawa K, Yasumoto KI, Fujii-Kuriyama Y. Mol Cell Biol. 1990;10:1470–1475. doi: 10.1128/mcb.10.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujii-Kuriyama Y, Imataka H, Sogawa K, Yasumoto K-I, Kikuchi Y. FASEB J. 1992;6:706–710. doi: 10.1096/fasebj.6.2.1537460. [DOI] [PubMed] [Google Scholar]
- 26.Imataka H, Sogawa K, Yasumoto K-I, Kikuchi Y, Sasano K, Lobayashi A, Hayami M, Fujii-Kuriyama Y. EMBO J. 1992;11:3663–3671. doi: 10.1002/j.1460-2075.1992.tb05451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traber PG, Chianale J, Florence R, Kim K, Wojcik E, Gumucio JJ. J Biol Chem. 1988;263:9449–9455. [PubMed] [Google Scholar]
- 28.Traber PG, Wang W, Yu L. Am J Physiol. 1992;263:G215–G223. doi: 10.1152/ajpgi.1992.263.2.G215. [DOI] [PubMed] [Google Scholar]
- 29.Traber PG, McDonnell WM, Wang W, Florence R. Biochim Biophys Acta. 1992;1171:167–175. doi: 10.1016/0167-4781(92)90117-i. [DOI] [PubMed] [Google Scholar]
- 30.Swick AG, Janicot M, Cheneval-Kastelic T, McLenithan JC, Lane MD. Proc Natl Acad Sci U S A. 1992;89:1812–1816. doi: 10.1073/pnas.89.5.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hagen G, Muller S, Beato M, Suske G. EMBO J. 1994;13:3843–3851. doi: 10.1002/j.1460-2075.1994.tb06695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JS, Galvin KM, Shi Y. Proc Natl Acad Sci U S A. 1993;90:6145–6149. doi: 10.1073/pnas.90.13.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hendrickson W, Schleif R. Proc Natl Acad Sci U S A. 1985;82:3129–3133. doi: 10.1073/pnas.82.10.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorman CM, Moffat LF, Howard BH. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beale EG, Deeb EA, Handley RS, Akhavan-Tafti H, Shaap AP. BioTechniques. 1992;12:320–323. [PubMed] [Google Scholar]
- 37.Kaelin WG, Pallas DC, DeCaprio JA, Kaye FJ, Livingston DM. Cell. 1991;64:521–532. doi: 10.1016/0092-8674(91)90236-r. [DOI] [PubMed] [Google Scholar]
- 38.Sogawa K, Kikuchi Y, Imataka H, Fujii-Kuriyama Y. J Biochem (Tokyo) 1993;114:605–609. doi: 10.1093/oxfordjournals.jbchem.a124224. [DOI] [PubMed] [Google Scholar]
- 39.Harrington MA, Jones PA, Imagawa M, Karin M. Proc Natl Acad Sci U S A. 1988;85:2066–2070. doi: 10.1073/pnas.85.7.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letovsky J, Dynan WS. Nucleic Acids Res. 1989;17:2639–2653. doi: 10.1093/nar/17.7.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo CT, Veselits ML, Leiden JM. Science. 1997;277:1986–1990. doi: 10.1126/science.277.5334.1986. [DOI] [PubMed] [Google Scholar]
- 42.Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. Genes Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalez IC, Gotoh O, Okuda K, Nebert DW. DNA Cell Biol. 1993;12:1–51. doi: 10.1089/dna.1993.12.1. [DOI] [PubMed] [Google Scholar]
- 44.Quattrochi LC, Tukey RH. Mol Pharmacol. 1989;36:66–71. [PubMed] [Google Scholar]
- 45.Chung I, Bresnick E. Mol Pharmacol. 1995;47:677–685. [PubMed] [Google Scholar]
- 46.Suwa Y, Mizukami Y, Sogawa K, Fujii-Kuriyama Y. J Biol Chem. 1985;260:7980–7984. [PubMed] [Google Scholar]
- 47.Hashimoto H, Toide K, Kitamura R, Fujita M, Tagawa S, Itoh S, Kamataki T. Eur J Biochem. 1993;218:585–595. doi: 10.1111/j.1432-1033.1993.tb18412.x. [DOI] [PubMed] [Google Scholar]
- 48.Jounaidi Y, Guzelian PS, Maurel P, Vilarem MJ. Biochem Biophys Res Commun. 1994;205:1741–1747. doi: 10.1006/bbrc.1994.2870. [DOI] [PubMed] [Google Scholar]
- 49.Itoh S, Abe Y, Kubo A, Okuda M, Shimoji M, Nakayama K, Kamataki T. Biochim Biophys Acta. 1997;1350:155–158. doi: 10.1016/s0167-4781(96)00215-1. [DOI] [PubMed] [Google Scholar]
- 50.Venepally P, Waterman MR. J Biol Chem. 1995;270:25402–25410. doi: 10.1074/jbc.270.43.25402. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi A, Sogawa K, Fujii-Kuriyama Y. J Biol Chem. 1996;271:12310–12316. doi: 10.1074/jbc.271.21.12310. [DOI] [PubMed] [Google Scholar]
- 52.Majello B, De Luca P, Lania L. J Biol Chem. 1997;272:4021–4026. doi: 10.1074/jbc.272.7.4021. [DOI] [PubMed] [Google Scholar]
- 53.Hagen G, Dennig J, Preisβ A, Beato M, Suske G. J Biol Chem. 1995;270:24989–24994. doi: 10.1074/jbc.270.42.24989. [DOI] [PubMed] [Google Scholar]
- 54.Shrivastava A, Calame K. Nucleic Acids Res. 1994;22:5151–5155. doi: 10.1093/nar/22.24.5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi Y, Lee JS, Galvin KM. Biochim Biophys Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 56.Dennig J, Beato M, Suske G. EMBO J. 1996;15:5659–5667. [PMC free article] [PubMed] [Google Scholar]
- 57.Austen M, Luscher B, Luscher-Firzlaff JM. J Biol Chem. 1997;272:1709–1717. doi: 10.1074/jbc.272.3.1709. [DOI] [PubMed] [Google Scholar]
- 58.Galvin KM, Shi Y. Mol Cell Biol. 1997;17:3723–3732. doi: 10.1128/mcb.17.7.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Blanco MA, Clerc RG, Sharp PA. Genes Dev. 1989;3:739–745. doi: 10.1101/gad.3.6.739. [DOI] [PubMed] [Google Scholar]
- 61.Bigger CB, Melnikova IN, Gardner PD. J Biol Chem. 1997;272:25976–25982. doi: 10.1074/jbc.272.41.25976. [DOI] [PubMed] [Google Scholar]
- 62.Udvadia AJ, Templeton DJ, Horowitz JM. Proc Natl Acad Sci U S A. 1995;92:3953–3957. doi: 10.1073/pnas.92.9.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohlsson C, Kley N, Werner H, LeRoith D. Endocrinology. 1998;139:1101–1107. doi: 10.1210/endo.139.3.5832. [DOI] [PubMed] [Google Scholar]
- 64.Strom AC, Forsberg M, Lillhager P, Westin G. Nucleic Acids Res. 1996;24:1981–1986. doi: 10.1093/nar/24.11.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee YH, Williams SC, Baer M, Sterneck E, Gonzalez FJ, Johnson PF. Mol Cell Biol. 1997;17:2038–2047. doi: 10.1128/mcb.17.4.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Merikam M, Orkin SH. Mol Cell Biol. 1995;15:2437–2447. doi: 10.1128/mcb.15.5.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamine J, Chinnadurai G. J Virol. 1992;66:3932–3936. doi: 10.1128/jvi.66.6.3932-3936.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trejo SR, Fahl WE, Ratner L. J Biol Chem. 1997;272:27411–27421. doi: 10.1074/jbc.272.43.27411. [DOI] [PubMed] [Google Scholar]
- 69.Jenkins TD, Opitz OG, Okano J, Rustgi AK. J Biol Chem. 1998;273:10747–10754. doi: 10.1074/jbc.273.17.10747. [DOI] [PubMed] [Google Scholar]