

Abstract
Biometals have an important role in AD (Alzheimer's disease) and metal ligands have been investigated as potential therapeutic agents for treatment of AD. In recent studies the 8HQ (8-hydroxyquinoline) derivative CQ (clioquinol) has shown promising results in animal models and small clinical trials; however, the actual mode of action in vivo is still being investigated. We previously reported that CQ–metal complexes up-regulated MMP (matrix metalloprotease) activity in vitro by activating PI3K (phosphoinositide 3-kinase) and JNK (c-jun N-terminal kinase), and that the increased MMP activity resulted in enhanced degradation of secreted Aβ (amyloid β) peptide. In the present study, we have further investigated the biochemical mechanisms by which metal ligands affect Aβ metabolism. To achieve this, we measured the effects of diverse metal ligands on cellular metal uptake and secreted Aβ levels in cell culture. We report that different classes of metal ligands including 8HQ and phenanthroline derivatives and the sulfur compound PDTC (pyrrolidine dithiocarbamate) elevated cellular metal levels (copper and zinc), and resulted in substantial loss of secreted Aβ. Generally, the ability to inhibit Aβ levels correlated with a higher lipid solubility of the ligands and their capacity to increase metal uptake. However, we also identified several ligands that potently inhibited Aβ levels while only inducing minimal change to cellular metal levels. Metal ligands that inhibited Aβ levels [e.g. CQ, 8HQ, NC (neocuproine), 1,10-phenanthroline and PDTC] induced metal-dependent activation of PI3K and JNK, resulting in JNK-mediated up-regulation of metalloprotease activity and subsequent loss of secreted Aβ. The findings in the present study show that diverse metal ligands with high lipid solubility can elevate cellular metal levels resulting in metalloprotease-dependent inhibition of Aβ. Given that a structurally diverse array of ligands was assessed, the results are consistent with the effects being due to metal transport rather than the chelating ligand interacting directly with a receptor.
Keywords: Alzheimer's disease, amyloid, c-Jun N-terminal kinase (JNK), copper, metal complex, metalloprotease
Abbreviations: Aβ, amyloid β; AD, Alzheimer's disease; APP, amyloid precursor protein; BC, bathocuprione; BCS, bathocuproine sulfonate; BP, bathophenanthroline; BPS, bathophenanthroline disulfonate; CHO, Chinese-hamster ovary; CQ, clioquinol; DCF, 2′,7′-dichlorofluorescein; DCF-DA, DCF-diacetate; DFO, deferoximine; HRP, horseradish peroxidase; 8HQ, 8-hydroxyquinoline; ICP-MS, inductively coupled plasma MS; IRE, iron-responsive element; JNK, c-Jun N-terminal kinase; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase; MMP, matrix metalloprotease; MT, membrane-type; NC, neocuproine; PBST, PBS containing 0.05% (v/v) Tween 20; PDTC, pyrrolidine dithiocarbamate; PI3K, phosphoinositide 3-kinase; RFU, relative fluorescence unit; TM, tetrathiomolybdate
INTRODUCTION
Biometals such as copper (Cu), zinc (Zn) and iron (Fe) have an important role in several neurodegenerative diseases. Cu and Zn promote aggregation and enhance neurotoxicity of Aβ (amyloid β) peptide, a major constituent of amyloid plaques in brains of AD (Alzheimer's disease) patients [1–4]. The parental APP (amyloid precursor protein) also contains metal-binding domains for Cu and Zn [5–8]. However, the physiological role of metal binding to APP or Aβ is not certain.
Due to the important role of biometals in neurodegeneration, considerable focus has been placed on developing novel therapeutic approaches involving modulation of metal–protein interactions [9,10]. Early attempts to target metals in AD involved treatment of AD patients with the Fe-selective chelator DFO (deferoximine). Initial reports showed that DFO altered the rate of cognitive decline in AD patients [11], although it is unknown whether this was due to chelation of Fe or modulation of other metals. However, the low lipid solubility of DFO combined with its ability to induce severe anaemia precluded further development of DFO as a therapeutic agent.
More recently, the 8HQ (8-hydroxyquinoline) derivative 5-chloro-7-iodo-8-hydroxyquinoline or CQ (clioquinol; a disused antibiotic) was shown to ‘dissolve’ plaques in vitro by removal of metals [9]. Subsequent studies in animal models of AD demonstrated a marked reduction in amyloid plaque formation in brains of treated animals [9]. A small clinical trial of CQ demonstrated a significant slowing of cognitive decline in a subset of AD patients with a parallel reduction in plasma Aβ-(1–42) levels [12]. Despite this early success, the mechanism of action of CQ in vivo remains largely unknown. Importantly, hydrophobic metal ligands such as CQ have the potential to substantially alter cellular metal homoeostasis [13,14]. High levels of metals are released during synaptic activity in the brain [15,16], so it is feasible that CQ or other hydrophobic ligands may bind to these metals and transport them into adjacent cells. The potential affects of this on cell function generally, and APP and Aβ metabolism specifically, are not known.
Recently, we investigated the effects of CQ and Cu or Zn on Aβ metabolism in APP overexpressing CHO (Chinese-hamster ovary) cells [14]. We found that CQ greatly elevated cellular levels of Cu and Zn but not Fe. CQ-mediated increases in intracellular metal levels resulted in activation of PI3K (phosphoinositide 3-kinase), leading to downstream phosphorylation of GSK3 (glycogen synthase kinase 3) and potentiation of the MAPK (mitogen-activated protein kinase) JNK (c-Jun N-terminal kinase). The stimulation of this pathway culminated in up-regulation of cellular metalloprotease activity and degradation of extracellular Aβ. Whether CQ has a similar action in vivo is currently under investigation, although the concentration of CQ and metals in treated AD patients suggests that a similar mechanism is feasible [9,12,17,18].
Although our previous study revealed an important insight into the potential therapeutic mechanism of CQ, it also raised the question of whether the observed activation of PI3K and Aβ degradation may be a CQ-specific effect. We previously proposed that a ligand-mediated increase in intracellular metal bioavailability is the critical step in this process [14], raising the possibility that the metal ligand itself is important only in that it delivers bioavailable metals across the plasma membrane. To investigate this, we have performed an extensive study of a diverse range of metal ligands to determine their impact on metal uptake, MAPK signalling pathways and Aβ levels in APP-CHO cells.
EXPERIMENTAL
Materials
5,7-Diiodo-8HQ, CQ, 5,7-dichloro-8HQ, 5-chloro-8HQ, 8HQ, 8HQ-2-sulfonic acid, 4,8-dihydroxy-2-carboxylic acid-8HQ, 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline [BC (bathocuproine)], 4,7-diphenyl-1,10-phenanthroline [BP (bathophenanthroline)], 3,4,7,8-tetra-methyl-1,10-phenanthroline, 2,9-dimethyl-1,10-phenanthroline [NC (neocuproine)], 2-chloro-1,10-phenanthroline, 5-methyl-1,10-phenanthroline, 1,10-phenanthroline, 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline disulfonate [BCS (BC sulfonate)], 4,7-diphenyl-1,10-phenanthroline disulfonate [BPS (BP disulfonate)], PDTC (pyrrolidine dithiocarbamate), TM (tetrathiomolybdate), DFO, D-penicillamine and SP600125 were purchased from Sigma–Aldrich. GM6001, MMP (matrix metalloprotease) inhibitor-I, MMP-2 inhibitor-I and MMP-9 inhibitor-I were obtained from Merck Biosciences. Anti-APP 22C11 was obtained from Chemicon. Antibody 369 (epitope 656–695 of APP) was a gift from Dr Sam Gandy (Thomas Jefferson University, Philadelphia, PA, U.S.A.). Antibodies to total or phosphorylated-specific forms of Akt and JNK were obtained from Cell Signaling Technology.
APP-transfected CHO cells
APP-CHO cells were generated by expressing the 695 amino acid APP cDNA in the pIRESpuro2 expression vector (Clontech) as described previously [14]. Briefly, cells were transfected using Lipofectamine™ 2000 and cultured in RPMI 1640 medium supplemented with 1 mM glutamine and 10% fetal (v/v) bovine serum (all from Invitrogen). Transfected cells were selected and maintained using 7.5 μg/ml puromycin (Sigma–Aldrich).
Exposure of cells to metal ligands
APP-overexpressing cells were passaged at a ratio of 1:6 and grown in 6- or 12-well plates for 2–3 days before experiments. Lipid-soluble ligands (10 mM stock solutions in DMSO) and lipid-insoluble ligands (10 mM stock solution in distilled-deionized H2O) were added to a final concentration of 25 μM (unless stated otherwise) to serum-free RPMI medium supplemented with puromycin as above. Basal metal levels in the medium were 0.5, 1.3 and 2.1 μM for Cu, Zn or Fe respectively, as determined by ICP-MS (inductively coupled plasma MS). Additional metals were added to the medium (25 μM final concentration) and mixed by aspiration prior to addition to cells. Control cultures were treated with vehicle (DMSO or distilled-deionized H2O) alone. Inhibitors of JNK (SP600125) or metalloproteases (GM6001, MMP inhibitor-I, MMP-2 inhibitor-I or MMP-9 inhibitor-I) were prepared as 10 mM stock solutions in DMSO and added at the concentrations indicated. Cultures were incubated for 6 h, and conditioned medium was taken for measurement of Aβ levels by ELISA [14]. For immunoblotting, cells were harvested into Phosphosafe extraction buffer (Novagen) containing protease inhibitor cocktail (Calbiochem) and stored at −80 °C until use. Alternatively, cells were washed and harvested for analysis of metal levels by ICP-MS.
ICP-MS
Cells were treated with ligands and/or metals for 6 h and collected by centrifugation at 20000 g for 2 min in a Hermle microfuge (Labnet). Metal levels were determined in quadruplicate cell pellets by ICP-MS as described previously [14] and converted into the fold increase in metal compared with untreated controls.
Detection of metal-ligand redox activity by DCF (2′,7′-dichlorofluorescein) fluorescence
Redox activity of the compounds was assessed by monitoring their ability to generate hydrogen peroxide in the presence of Cu(II) or Fe(III) and ascorbate. Hydrogen peroxide production was measured using the fluorescent probe DCF-DA (DCF-diacetate; Sigma–Aldrich) as follows. HRP (horseradish peroxidase) was initially dissolved in Dulbecco's PBS (Sigma–Aldrich) at a concentration of 50 μM, then further diluted to a concentration of 2.5 μM and stored on ice until used. DCF-DA was initially dissolved in 500 μl of N2-purged DMSO (7 mM), then deacetylated with 500 μl of 50 mM NaOH for 30 min and diluted in PBS to a concentration of 500 μM. It was then wrapped in foil and stored on ice until used. Ascorbate was dissolved in 2H2O at a concentration of 3 mM, sonicated for 15 min, then diluted in PBS to a concentration of 25 μM. It was wrapped in foil and stored on ice until used. CuCl2 and FeCl3 were each dissolved in distilled-deionized H2O at a concentration of 10 mM, then diluted in PBS to a concentration of 2.5 μM. The ligands were dissolved in either DMSO or distilled-deionized H2O to a concentration of 10 mM and then diluted in PBS to a concentration of 12.5 μM.
To each well of a black clear-bottomed 96-well plate (Wallac), 50 μl each of metal, ligand, HRP, DCF and ascorbate solutions were added to yield a final working concentration of 0.5 μM metal, 2.5 μM ligand, 0.5 μM HRP, 100 μM DCF and 5 μM ascorbate (total volume 250 μl). The plate was loaded using the on-board dispenser of a Varioskan microplate reader (Thermo Electron), and the fluorescence of four replicates was measured over a period of 1 h at 37 °C (λex=485 nm, λem=535 nm), shaking for 5 s at 2 min intervals. DCF fluorescence was given as RFUs (relative fluorescence units).
Double-antibody capture ELISA for Aβ detection
Aβ levels were determined in culture medium using the DELFIA® Double Capture ELISA as described by White et al. [14]. 384-Well plates (Greiner) were coated with mAb (monoclonal antibody) G210 in coating buffer [15 mM Na2CO3 and 35 mM NaHCO3 (pH 9.6)] for Aβ-(1–40) detection. Plates were washed with PBST [PBS containing 0.05% (v/v) Tween 20] and blocked with 0.5% (w/v) casein. Biotinylated mAb WO2 [epitope at Aβ-(5–8)] and culture medium or Aβ peptide standards were added (50 μl) to each well and incubated overnight at 4 °C. Plates were washed with PBST and streptavidin-labelled Europium (PerkinElmer) was added. Plates were washed, enhancement solution (PerkinElmer) was added and plates were read in a WALLAC Victor2 plate reader with excitation at 340 nm and emission at 613 nm. Aβ-(1–40) peptide standards and samples were assayed in triplicate. Quadruplicate medium samples were each measured in triplicate and used to calculate the Aβ concentration (expressed as ng/ml) based on the standard curve generated on each plate. Although the combination of G210 and WO2 is aimed at specifically capturing Aβ-(1–40), the possibility of detecting N-terminally truncated species has not been ruled out. However, Western blot analysis of conditioned medium using WO2 failed to identify additional cross-reactive species [14].
Western blot analysis of protein expression and phosphorylation
Cell lysates (two–four replicates) prepared in Phosphosafe buffer were mixed with electrophoresis SDS sample buffer (Novex) and separated on 12% Novex SDS/PAGE Tris/glycine gels. Proteins were transferred on to PVDF membranes and blocked with skimmed milk solution in PBST before immunoblotting for total or phospho-specific proteins. Membranes were probed for 1 h at room temperature (22 °C) or overnight at 4 °C with antisera against C-terminal APP (antibody 369) or full-length APP (22C11) at 1:2000. Antibodies to Akt or phospho-Akt and JNK or phospho-JNK were used at 1:5000. Secondary HRP-conjugated goat anti-rabbit-IgG or rabbit anti-mouse-IgG were used at 1:5000 or 1:10000. Blots were developed using ECL Advance Chemiluminescence (Amersham Biosciences) and imaged on a GeneGnome Chemiluminescence Imager (Syngene). We found that the expression of total levels of JNK were unaffected by metal uptake in APP-CHO cells. We therefore used total JNK levels to assess equal sample loading and protein transfer for all immunoblots.
Cell adhesion assay
Cell adhesion to collagen type IV was determined using an assay kit from Merck Biosciences (Innocyte ECM Cell Adhesion Assay, Collagen type IV). Cells (quadruplicate samples) were treated with metal ligands (with or without inhibitors) for 4 h before harvesting with a rubber policeman into culture medium. Cells were dissociated by aspiration and re-plated on to collagen type IV and cultured for a further 2 h. The medium was discarded and cells were washed briefly with two changes of PBS before addition of calcein-AM for 1 h (at 37 °C). Cell adhesion was determined by fluorescent spectrophotometry on a WALLAC Victor2 plate reader with excitation/emission at 490 nm/535 nm.
MMP assay
Activity of MMP-2 in cell lysates was determined using the Enzolyte MMP fluorimetric assay kit (Anaspec). Briefly, cell lysates (in triplicate, freshly extracted without protease inhibitors) were incubated with MMP-specific peptide substrates as indicated in the manufacturer's protocol. The substrate used was 5-FAM-Pro-Leu-Ala-Nva-Dap(QXL520)-Ala-Arg-NH2 (MMP-2). Cleavage of substrate by MMP-2 removed the quenching effect of QXL520 on 5-FAM resulting in increased fluorescence at excitation/emmission of 490/535 nm. Cultures were also pre-treated with inhibitors of metalloproteases and assayed for MMP activity.
Statistical analysis
All results described in the graphical representations are means±S.E.M. unless stated. Results were analysed on raw data using a two-tailed Students' t test.
RESULTS
Modulation of cellular Cu and Zn levels by 8HQ and phenanthroline derivative metal ligands
To investigate the effects of metal ligands on Aβ metabolism we initially examined how different classes of 8HQ and phenanthroline ligands modulated metal levels in APP-CHO cells. The Clog P values were calculated by the program Chemdraw using the method described by Ghose and Crippen [19] (see Table 1 for details of ligands). The cells were treated with 25 μM ligand alone (uncomplexed) or together with 25 μM Cu, Zn or Fe(II) (metal-complexed). An equal metal-to-ligand ratio was used to reflect the high extracellular metal content in vivo [17] and this ratio was used in our previous study on CQ-Cu inhibition of Aβ [14]. Cells were treated for 6 h and metal levels were determined by ICP-MS in cell pellets. The ligands alone had little effect on cellular metal levels; however, when complexed to metal, a range of effects on Cu uptake were observed. Seven different 8HQ derivatives were examined with an estimated Clog P value ranging from 4.14 (high lipid solubility) down to −1.89 (low lipid solubility; 8HQ ligands are numbered #1–7, Table 1). As expected, the 8HQ derivatives with higher lipid solubility (Clog P value of 2.08–4.14) induced significant increases in cellular Cu (Figure 1A). Interestingly, the two ligands with the highest Clog P values [5,7-diiodo-8HQ (#1), 4.14; and 5-chloro-7-iodo-8HQ (#2), 3.75] induced Cu uptake of 125- and 200-fold respectively, whereas three ligands (#3–#5) with lower estimated lipid solubility induced a substantially higher level of Cu uptake (350–375-fold) (Figure 1A). In contrast, the two compounds with negative Clog P values [8HQ-2-sulfonic acid (#6) and 4,8-dihydroxy-2-carboxylic acid-8HQ (#7)] induced no increase in cellular Cu (Figure 1A). This phenomenon may reflect a ‘bell-shaped’ curve for metal uptake in whereas ligands with very high lipophilicity may be more likely to adhere non-specifically to membranes, debris or the culture vessel and therefore result in lower metal uptake into the cells. Ligands with moderate lipophilicity would induce strong metal uptake and those with low lipophilicity induce little or no uptake.
Table 1. List of metal ligands used to modulate cellular metal levels and Aβ metabolism.
ND, not done as the Clog P calculation is not suitable for metal complexes. phen, phenanthroline.
| # | Compound | Structure | Clog P (solubility) |
|---|---|---|---|
| 8HQ derivatives | |||
| 1 | 5,7-diiodo-8HQ | 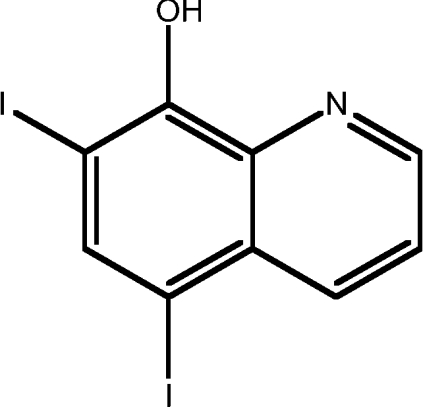 |
4.14 |
| 2 | CQ | 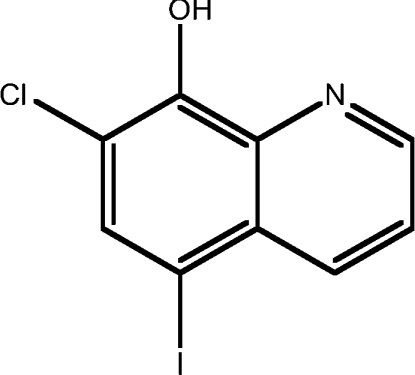 |
3.75 |
| 3 | 5,7-dichloro-8HQ | 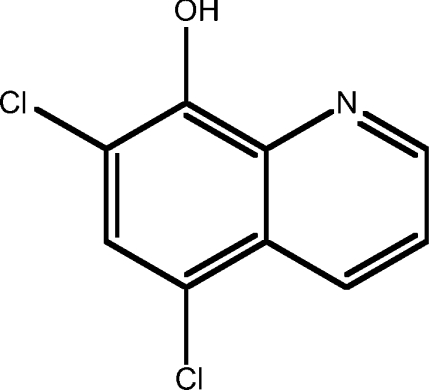 |
3.34 |
| 4 | 5-chloro-8HQ |  |
2.58 |
| 5 | 8HQ | 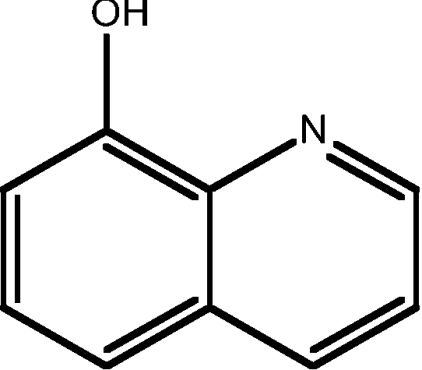 |
2.08 |
| 6 | 8HQ-2-sulfonic acid | 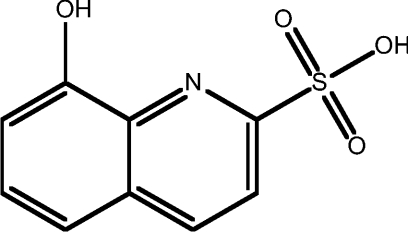 |
−0.32 |
| 7 | 4,8-dihydroxy-2-carboxylic acid-8HQ | 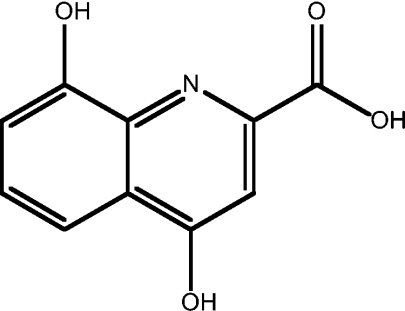 |
−1.89 |
| Phenanthroline derivatives | |||
| 8 | BC | 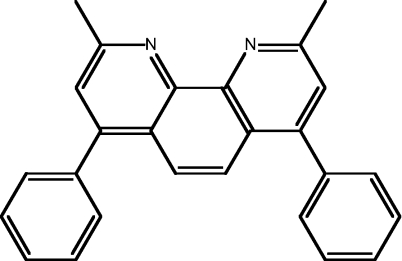 |
6.83 |
| 9 | BP | 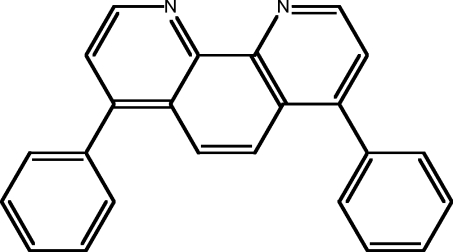 |
5.83 |
| 10 | 3,4,7,8-tetra-methyl-1,10-phen | 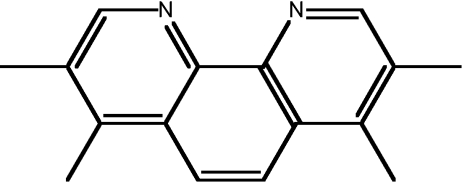 |
3.95 |
| 11 | NC | 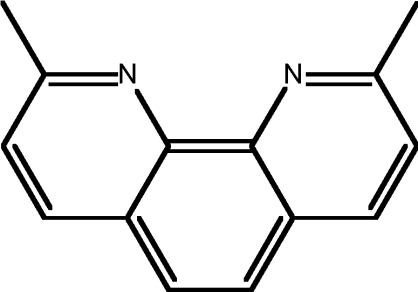 |
3.05 |
| 12 | 2-chloro-1,10-phen | 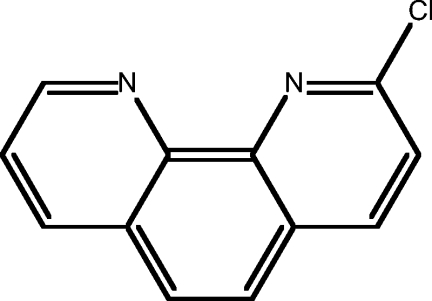 |
2.81 |
| 13 | 5-methyl-1,10-phen | 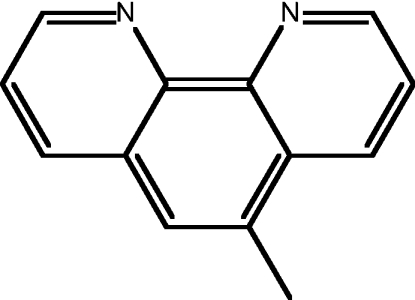 |
2.55 |
| 14 | 1,10-phen |  |
2.05 |
| 15 | BCS |  |
2.02 |
| 16 | BPS |  |
1.03 |
| Sulfur compounds | |||
| 17 | PDTC |  |
1.71 |
| 18 | D-penicillamine | 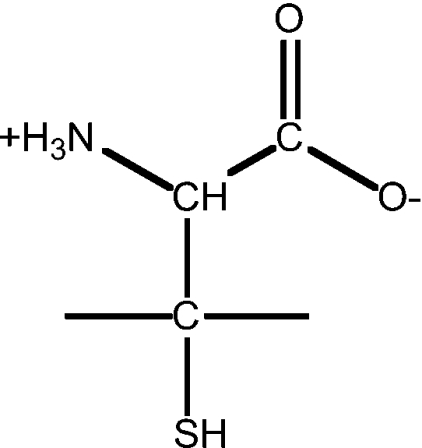 |
−0.4 |
| Other compounds | |||
| 19 | TM | ND | |
| 20 | DFO | 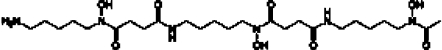 |
−0.5 |
Figure 1. Effect of metal ligands on Cu and Zn uptake into APP-CHO cells.

Cells were treated with 25 μM ligand–metal complexes for 6 h and metal levels were determined in cell pellets by ICP-MS. Metal uptake is given as the fold increase compared with vehicle-treated control. (A) Treatment of cells with 8HQ derivatives and Cu. (B) Treatment of cells with phenanthroline derivatives and Cu. (C) Treatment of cells with 8HQ derivatives and Zn. (D) Treatment of cells with phenanthroline derivatives and Zn. *P<0.01 and **P<0.05. phen, phenanthroline.
Similar results were observed in cells treated with nine phenanthroline derivatives (phenanthroline ligands are numbered #8–16; Table 1). The ligands with the highest Clog P values had a moderate effect on increasing Cu levels (135–175-fold uptake), whereas compounds with a Clog P value in the mid-range induced greater Cu uptake (Figure 1B). Interestingly, NC (#11) induced significantly greater uptake of Cu (325-fold) when compared with other ligands despite a Clog P value in the middle of the range of all the ligands tested (3.05 as compared with a range of 1.03–6.83 for all compounds; Table 1). This may again reflect a bell-shaped curve for metal uptake as seen with 8HQ ligands. The Clog P value of 3.05 may be optimal for metal uptake whereas higher Clog P values may result in non-specific interactions with the membrane that hinders uptake. The two sulfonated phenanthroline derivatives (#8 and #9) induced no increase in cellular Cu (Figure 1B). This was despite one of those ligands [BCS (#15)] having a Clog P almost identical with the non-sulfonated 1,10-phenanthroline (#14; 2.02 and 2.05 respectively; Table 1). These results demonstrate that, although Cu uptake by 8HQ and phenanthroline derivatives generally correlate with lipid solubility, additional structural modifications to the ligands, especially the addition of sulfonated groups, can greatly alter Cu uptake.
Similar effects were observed when cells were treated with the 8HQ derivatives as Zn complexes (Figures 1C and 1D). 8HQ derivatives with a high Clog P value induced high levels of Zn uptake, whereas three ligands with the lowest Clog P values induced no effect on cellular Zn (Figure 1C). However, as for Cu, there were substantial differences between two ligands with very similar Clog P values. 5-Chloro-8HQ (#4; Clog P value of 2.58) induced a 14-fold increase in Zn whereas 8HQ (#5; Clog P value of 2.08; Table 1) induced no change in Zn compared with Zn added alone (Figure 1C). The lack of Zn uptake by 8HQ was surprising; however, this could reflect the tetrahedral structure of 8HQ–Zn complexes rather than the square planar structure of 8HQ–Cu.
The results obtained with phenanthroline–Zn complexes did not reflect those observed with phenanthroline–Cu complexes shown in Figure 1(B). Only one phenanthroline derivative NC (#11; Clog P value of 3.05) induced a high level of Zn uptake (8.5-fold), with the other ligands having only a modest, or no effect on cellular Zn levels (Figure 1D). These findings show that although higher lipid solubility is a factor in increased Zn uptake by 8HQ compounds it has limited effect on Zn uptake by phenanthroline derivatives. In addition, other structural features of both ligand classes can have dramatic effects on metal uptake. Overall, 5,7-dichloro-8HQ (#3) and 5-chloro-8HQ (#4) were the most effective 8HQ ligands at increasing cellular levels of Cu and Zn, whereas NC (#11) was the only phenanthroline derivative with high efficacy at inducing uptake of both Cu and Zn.
Differential inhibition of secreted Aβ levels by 8HQ and phenanthroline derivatives complexed with Cu or Zn
As many 8HQ and phenanthroline derivative metal ligands induced substantial increases in cellular metal levels (Figure 1), we next examined the effects of this on secreted Aβ in culture medium as reported previously [14]. Diverse effects of ligands on Aβ levels were observed and although there was a general correlation between metal uptake and inhibition of Aβ, this was not observed with all ligands. The best correlation between Cu uptake and loss of Aβ was seen with the 8HQ derivatives. The three ligands that produced the highest Cu uptake [5,7-dichloro-8HQ (#3), 5-chloro-8HQ (#4) and 8HQ (#5)] (Figure 1) also induced the greatest loss of Aβ with levels being less than 5% of untreated control (Figure 2A). The two 8HQ derivatives that had no effect on Cu uptake [8HQ-2-sulfonic acid (#6) and 4,8-dihydroxy-2-carboxylic acid-8HQ (#7)] (Figure 1A) also had little effect on secreted Aβ levels (Figure 2A).
Figure 2. Effect of metal ligands on Aβ-(1–40) levels in APP-CHO cells.

Cells were treated with 25 μM ligand–metal complexes for 6 h and Aβ-(1–40) levels were determined by ICP-MS in conditioned medium. (A) Treatment of cells with 8HQ derivatives and Cu. (B) Treatment of cells with phenanthroline derivatives and Cu. (C) Treatment of cells with 8HQ derivatives and Zn. (D) Treatment of cells with phenanthroline derivatives and Zn. phen, phenanthroline.
In contrast, the pattern of effects with phenanthroline derivatives was more complex. Although NC (#11) induced a high level of Cu uptake (Figure 1) and a low level of Aβ (8%) (Figure 2B), the other phenanthroline derivatives that increased cellular Cu induced a variable decrease in Aβ. For example, BP (#9) induced a moderate increase in cellular Cu (165-fold) (Figure 1B) but reduced Aβ levels to 2.5% (Figure 2B), whereas 1,10-phenanthroline (#14) increased Cu by 250-fold (Figure 1B) and only decreased Aβ to 56% of the controls (Figure 2B).
In our Zn studies, 8HQ derivative ligands #1–#4 (Table 1) all increased cellular Zn (Figure 1C) and all greatly inhibited Aβ levels in the presence of Zn (Figure 2C). However, 8HQ (#5) had no measurable effect on cellular Zn levels yet reduced Aβ to 2% of controls. Similarly with phenanthroline derivatives, only small increases in Zn uptake were induced by BC (#8) and BP (#9) (Table 1 and Figure 1D), but these ligands greatly inhibited Aβ levels in culture medium (to 5 and 2% respectively; Figure 2D). Linear regression analysis of metal uptake compared with Aβ levels revealed a strong correlation between Cu uptake and Aβ (r=0.72). However, a lack of correlation was seen with Zn and Aβ (r=0.43). This may indicate subtle differences in the mechanism of Aβ production or degradation induced by Cu compared with Zn. Importantly, our findings demonstrate that some 8HQ and phenanthroline derivatives can induce substantial inhibition of secreted Aβ with relatively low levels of cellular metal uptake. This may be an essential consideration in developing anti-Aβ ligands that have low cellular cytotoxicity due to limited metal uptake.
Diverse ligand classes show similar effects on Aβ levels in cell culture
In addition to the 8HQ and phenanthroline derivative compounds, we tested several other known metal ligands, including the sulfur-containing ligands PDTC and D-penicillamine and the additional well-documented metal ligands TM and DFO (Table 1). Treatment of cultures with these compounds and Cu resulted in no significant increase in cellular Cu except with PDTC (Figure 3A). PDTC has previously been demonstrated to be a potent metal ionophore [20]. We then tested the effect of these ligands on Aβ levels and found that, as expected, only PDTC–Cu complexes inhibited Aβ levels (Figure 3B). This supports our findings that metal uptake is required to induce inhibition of secreted Aβ in cell culture.
Figure 3. Effect of alternative ligand–metal complexes on metal uptake and Aβ-(1–40) levels in APP-CHO cells.

Cells were treated with metal ligands with or without 25 μM metal for 6 h and metal uptake was determined by ICP-MS. Aβ-(1–40) levels were determined in conditioned medium by ELISA. (A) Cu uptake in cells after treatment with sulfur-containing or alternative metal ligands together with Cu. (B) Aβ-(1–40) levels in medium from cultures treated with TM, D-penicillsamine, DFO or PDTC. (C) Aβ-(1–40) levels in cells treated with low concentrations of CQ, 8HQ, NC, 1,10-phenanthroline or PDTC plus Cu. (D) Fe levels in cells treated with CQ, 8HQ, NC, 1,10-phenanthroline or PDTC and Fe(II). (E) Aβ-(1–40) levels in medium of cells treated with CQ, 8HQ, NC, 1,10-phenanthroline or PDTC and Fe(II). *P<0.01. d-pen, D-penicillamine; phen, phenanthroline.
To further examine the effects of diverse metal ligands on metal uptake and Aβ metabolism, we selected several key ligands and performed additional analysis. CQ was selected as the ‘prototype’ ligand based on our previous report [14]. 8HQ was selected as the ‘parent’ molecule of this ligand class. Similarly, NC (#11) was chosen as a well-studied member of the phenanthroline class [21] and 1,10-phenanthroline as the ‘parent’ compound. PDTC was selected as it is a different class of compound (sulfur-containing) and is also well-documented [20]. Initially we examined how each of the ligands affected secreted Aβ levels when added as Cu complexes at low concentrations. We observed a significant decrease in secreted Aβ with 0.1 μM of each ligand–Cu complex except 1,10-phenanthroline (Figure 3C). Each ligand also revealed a dose–response decrease in secreted Aβ levels. These findings establish that diverse metal ligands are potent inhibitors of Aβ production in vitro. We next examined whether each of these ligands had any effect on cellular Fe uptake and subsequent Aβ levels when added with Fe(II). As shown in Figure 3(D), none of the ligands showed any increase in cellular Fe above that seen with Fe alone. This was also reflected in the Aβ levels as none of the ligands showed substantial effects on Aβ in the presence of Fe (Figure 3E).
Metal–ligand complexes reveal limited redox activity
The metal–ligand complexes were examined for redox potential ability to mediate reduction of Cu(II) or Fe(III). This was important as increased redox activity by metal–ligand complexes has the potential to induce cytotoxic effects through generation of ROS (reactive oxygen species). Such effects may be detrimental to the development of the ligands as therapeutic compounds. To measure this, metal [Cu(II) or Fe(III)]–ligand complexes were incubated with the reductant ascorbate and hydrogen peroxide generation was determined using a DCF fluorescence assay. As shown in Figure 4(A), none of the compounds examined induced a higher level of hydrogen peroxide generation than Cu(II) alone indicating a lack of intrinsic redox activity. In contrast, three of the ligands induced Fe(III)-mediated hydrogen peroxide greater than Fe(III) alone (Figure 4B). 5,7-diiodo-8HQ (#1), 1,10-phenanthroline (#14) and BPS (#16) each revealed a relatively strong redox activity towards Fe(III). In addition, 5-chloro-8HQ (#4) and 5-methyl-1,10-phenanthroline (#13) also induced small but significant increases in hydrogen peroxide levels in the presence of Fe(III) (Figure 4B). These results demonstrate that some of the ligands have Fe(III)-dependent redox activity which could induce cytotoxic effects. However, the majority of ligands were redox inactive towards Fe(III) and none revealed activity towards Cu(II).
Figure 4. DCF fluorescence measurement of hydrogen peroxide generation by metal–ligand complexes.

(A) Ligands were incubated with Cu(II) and hydrogen peroxide generation (redox activity) was determined by DCF fluorescence. All samples contained ascorbate and HRP. Ligand and/or metal were added as indicated. No ligand induced higher levels of hydrogen peroxide than Cu(II) alone indicating a lack of redox activity towards Cu(II). (B) Ligands were incubated with Fe(III) and hydrogen peroxide generation was determined by DCF fluorescence. All samples contained ascorbate and HRP. Ligand and/or metal were added as indicated. Several ligands induced a significant increase in hydrogen peroxide compared with Fe(III) alone (*P<0.001). phen, phenanthroline.
Diverse metal ligand–Cu complexes induce JNK-dependent loss of Aβ
To determine how the metal–ligand complexes induced a loss of secreted Aβ, we first examined the effect of CQ, our ‘prototype’ ligand, on APP expression and metabolism. As reported previously [14], treatment of APP-CHO cells with CQ alone inhibited cellular APP expression (Figure 5A). Treatment with Cu or Zn had no significant effect on APP expression in the 6 h incubation period. Co-treatment with CQ and Cu induced no greater loss of APP expression compared with CQ alone (Figure 5A), consistent with our previous findings [14]. Co-treatment with CQ and Zn induced a greater loss of APP expression compared with CQ alone (Figure 5A), although this effect was variable between cultures (results not shown). Interestingly, treatment of cells with Fe(II) plus CQ restored the loss of APP expression observed with CQ alone (Figure 5A). This effect is consistent with the previous study that CQ can inhibit APP expression by preventing Fe interaction with the APP mRNA IRE (iron-responsive element) [22]. Addition of Fe either quenched this effect by binding to CQ or promoted APP expression via the IRE on APP mRNA [22]. However, as shown previously [14], these results demonstrated that the loss of secreted Aβ was not due to inhibition of APP expression as treatment with CQ alone or CQ plus Cu both inhibited APP levels but only CQ plus Cu decreased Aβ levels. Furthermore, we found that treatment with CQ induced a loss of secreted APP from conditioned medium which corresponded closely to the loss of APP expression (Figure 5B). Again, co-treatment with CQ and Cu induced a similar loss of secreted APP compared with CQ alone (Figure 5B). These findings are consistent with our previous finding that CQ did not induce a loss of secreted Aβ through significant perturbation of APP processing. This was supported by our results showing that CQ or CQ and Cu had no effect on expression of C-terminal APP fragments in treated cells (Figure 5B) and did not affect activity of APP-cleaving enzymes [14]. We also examined whether Aβ accumulated in APP-CHO cells treated with CQ and Cu for 6 h. However, cellular Aβ could not be consistently detected in lysates by Western blot analysis or immunoprecipitation in treated cells or untreated controls (results not shown). This may reflect the cell-type used (CHO cells) and the short incubation time (6 h). Given that secreted Aβ levels were readily detected by ELISA or Western blot analysis [14] after 6 h, we would have anticipated detectable levels of intracellular Aβ if the loss of secreted Aβ was due to intracellular accumulation. However, the findings strongly suggested an alternative mechanism for the loss of secreted Aβ induced by CQ and other lipid-soluble metal ligands.
Figure 5. Effect of CQ and metals on APP metabolism.

(A) APP-CHO cells were treated with CQ and Cu, Zn or Fe(II) for 6 h. CQ alone or CQ and Cu or Zn induced a decrease in cellular APP expression. Co-treatment with Fe(II) inhibited the loss of APP expression induced by CQ. (B) Treatment of APP-CHO cells with CQ or CQ plus Cu induced a reduction in secreted APP (sAPP) levels in conditioned medium after 6 h. The loss of sAPP corresponded to the loss of cellular APP. Treatment with CQ, Cu or CQ and Cu had no effect on expression of APP C-terminal fragments (CTFs).
In our previous study, we found that CQ and Cu induced activation of PI3K (increased phospho-Akt levels) and subsequent stimulation of JNK activity [14]. This was a critical step in the inhibition of secreted Aβ levels by CQ–Cu complexes. To determine whether this mechanism was involved with other metal ligands, we treated cells with ligand–Cu complexes and analysed cellular Akt and JNK phosphorylation. As with CQ and Cu, we found that 8HQ–Cu, NC–Cu, 1,10-phenanthroline–Cu and PDTC–Cu all induced strong activation (phosphorylation) of Akt and JNK (Figures 6A–6D). Each of these complexes also increased cellular metal and reduced Aβ levels. The compounds that had no effect on cellular metal uptake or Aβ levels also failed to induce robust activation of PI3K or JNK (BPS, TM and D-penicillamine; Figures 6D and 6E), although TM did induce mild activation of Akt. To confirm that JNK activation was involved in loss of Aβ, cultures were co-treated with the JNK inhibitor SP600125 (25 μM). We have shown previously that this concentration of inhibitor blocked JNK activation and restored secreted Aβ levels in cultures treated with CQ–Cu [14]. Exposure of APP-CHO cells to ligand–Cu complexes and SP600125 completely prevented the loss of Aβ in conditioned medium induced by CQ–Cu, 8HQ–Cu, NC–Cu, 1,10-phenanthroline–Cu and PDTC–Cu (Figure 6F). As the JNK inhibitor could potentially affect APP or Aβ metabolism, we examined this in CQ–Cu-treated cells. However, as shown in Figure 6(G), SP600125 did not restore the loss of APP expression induced by treatment with CQ–Cu. In addition, no obvious changes to processing of C-terminal APP fragments were observed. These findings strongly suggested that the ability of SP600125 to restore Aβ levels was not through modulation of APP processing but via an alternative pathway such as the inhibition of Aβ-degrading metalloproteases as reported previously [14].
Figure 6. Activation of PI3K and JNK by diverse ligand–metal complexes in APP-CHO cells.

Cells were treated with ligands plus Cu for 6 h and activation (phosphorylation) of JNK was determined by Western blot analysis in cell lysates. (A) Activation of PI3K (phospho-Akt formation) by Cu and 8HQ, NC, PDTC and 1,10-phenanthroline. Some activation was observed with TM plus Cu but not with BPS or D-penicillamine plus Cu. (B) Activation of JNK by CQ or 8HQ and Cu. (C) JNK activation by NC or 1,10-phenanthroline and Cu. (D) Activation of JNK by PDTC and Cu but not by BPS and Cu. (E) No activation of JNK by TM or D-penicillamine and Cu. (F) Treatment of cells with an inhibitor of JNK activation (SP600125; 25 μM) prevented the loss of Aβ-(1–40) induced by CQ, 8HQ, NC, 1,10-phenanthroline or PDTC and Cu. *P<0.01. (G) Treatment of cells with the JNK inhibitor, SP600125 inhibited JNK activation but did not restore the loss of APP expression induced by CQ–Cu complexes. The inhibitor also did not alter C-terminal APP processing. CTF, C-terminal fragment; d-pen, D-penicillamine; phen, phenanthroline.
Diverse metal ligand–Cu complexes induce activation of metalloproteases
In our previous study, CQ–Cu induced significant loss of cell adhesion to collagen type IV matrix [14]. This was mediated through up-regulation of cellular metalloprotease activity by metal uptake. We therefore examined whether other metal ligand–Cu complexes induced similar effects on metalloprotease activity. Cells were treated with CQ–Cu, 8HQ–Cu, NC–Cu, 1,10-phenanthroline–Cu or PDTC–Cu complexes and cell adhesion was determined on collagen type IV. We found that each of the metal complexes tested induced a significant decrease in cell adhesion whereas the metal-free ligands had no effect (Figure 7A). In contrast, ligands that failed to increase metal uptake (TM, D-penicillamine and BPS) had a limited effect on cell adhesion when added to cells in the presence of Cu; BPS actually increased adhesion (Figure 7B). To confirm that these effects were due to up-regulation of matrix-degrading metalloproteases, activity of MMP-2 was determined in cell lysates. The free metal ligands (8HQ, NC and PDTC) alone had little effect on MMP-2 activity; however, when added as Cu complexes, substantial increases in MMP-2 activity were seen (Figure 7C). In each case, co-treatment with the broad-spectrum metalloprotease inhibitor or an MMP-2 inhibitor prevented the increased MMP-2 activity. These results demonstrate that diverse metal ligands can induce Cu-dependent up-regulation of cellular metalloproteases.
Figure 7. Effect of metal ligands on cell adhesion and MMP activity in APP-CHO cells.

Cells were treated with metal ligands and Cu for 6 h and cell adhesion was determined on a collagen type IV matrix. MMP-2 activity was measured by fluorometric assay in cell lysates. (A) Cell adhesion after treatment with CQ, 8HQ, NC, 1,10-phenanthroline or PDTC with or without Cu. (B) Cell adhesion after treatment with TM, D-penicillamine or BPS with or without Cu. (C) MMP-2 activity in cells after treatment with 8HQ, NC or PDTC and Cu. In each treatment, the broad-spectrum metalloprotease inhibitor GM6001 or the MMP-2 inhibitor prevented MMP activation (*P<0.01). d-pen, D-penicillamine; phen, phenanthroline.
Finally, we confirmed that activation of metalloproteases by ligand–metal complexes was responsible for loss of cell adhesion and a reduction in secreted Aβ as shown previously for CQ–Cu [14]. Loss of cell adhesion induced by 8HQ–Cu, NC–Cu or PDTC–Cu was prevented by co-treatment with the broad-spectrum metalloprotease inhibitor GM6001 and the specific MMP-2 inhibitor (Figure 8A). Similarly, these inhibitors prevented the reduction in secreted Aβ levels induced by 8HQ–Cu, NC–Cu and PDTC–Cu demonstrating that diverse metal ligands can induce metal uptake, triggering metalloprotease activity and increased Aβ degradation (Figure 8B). These results provide important support for the hypothesis that metal–ligand complexes may be useful for inhibiting Aβ accumulation in AD.
Figure 8. Effect of JNK and MMP inhibitors on loss of cell adhesion and secreted Aβ levels in APP-CHO cells.

Cells were treated with 8HQ, NC or PDTC and Cu for 6 h in the presence or absence of 25 μM SP600125 (JNK inhibitor), 20 μM GM6001 (broad-spectrum MMP inhibitor), 25 μM MMP inhibitor-I (broad-spectrum MMP inhibitor), 10 μM MMP-2 inhibitor-I or 10 μM MMP-9 inhibitor-I. (A) Inhibition of JNK or MMPs blocked the loss of cell adhesion induced by each metal ligand complexed with Cu (*P<0.01). (B) Inhibition of MMPs prevented the loss of secreted Aβ-(1–40) induced by each metal ligand complexed with Cu (*P<0.01).
DISCUSSION
The present study is the first to describe the systematic comparison of diverse metal ligands for their effects on metal uptake and Aβ metabolism. In the present study we have shown that different classes of metal ligands have various effects on cellular metal uptake and accumulation of Aβ in cell culture. Generally, more lipid-soluble metal ligands elevated cellular Cu and Zn levels consistent with previous reports [13,23–25] and induced a reduction in levels of secreted Aβ. In contrast, metal ligands with a low lipid solubility had little effect on metal levels or secreted Aβ; however, there were interesting exceptions to these trends in both 8HQ and phenanthroline classes of ligands.
These results confirm that the effects of CQ–Cu and CQ–Zn on Aβ metabolism reported previously [14] were not mediated by CQ itself, but instead were induced irrespective of the class or structure of the metal ligand used. 8HQ, phenanthroline or alternative ligands such as PDTC all induced a reduction in secreted Aβ levels in the presence of Cu. Therefore it is the complexed metal (i.e. Cu or Zn) and not the carrier ligand that modulates Aβ metabolism. This is consistent with previous reports that different metal ligands can induce analogous effects on APP expression [22,26].
These results are important for the future development of metal-ligand-based therapeutic intervention in AD. Our finding that Cu complexes of CQ, 8HQ, NC, 1,10-phenanthroline and PDTC all significantly reduced secreted Aβ levels shows that diverse metal-ligand classes could be examined for in vivo therapeutic efficacy against Aβ deposition [9,12,14]. Numerous derivatives of these compounds could be screened for anti-Aβ activity combined with low toxicity. This could be done relatively easily using cell-based approaches as reported in the present study. For example, our studies revealed that BC (#8) induced a relatively small increase in intracellular Zn but reduced Aβ levels by 95%. These assays could allow the detection of compounds that can induce a dramatic loss of Aβ with little change to cellular metal levels, thus reducing the potential for adverse side effects from excess metal. This may also help to avoid complications of treatment with metal ligands as was observed in some patients treated with CQ. Increased occurrence of sub acute-myelo-optic-neuropathy was originally linked to CQ treatment, although recent epidemiological studies have questioned this association [27–29]. Current evidence suggests that metal ligands such as CQ could potentially modulate vitamin B12 availability leading to adverse side affects. This could be directly related to either metal complexation by ligands or through alteration of intracellular metal levels. Whatever the process, further development of ligands that induce limited effects on metal homoeostasis are likely to offer safer therapeutic outcomes.
As we previously reported, our results did not support a role for increased intracellular Aβ accumulation as the mechanism for loss of detectable cellular Aβ. No evidence of accumulated Aβ could be seen in cell lysates and we found no evidence of altered APP metabolism other than a reduction in overall levels of cellular and secreted APP induced by CQ or CQ and Cu. Furthermore, we reported previously that CQ–Cu complexes also induced loss of extracellular synthetic Aβ added to culture medium and this was inhibited by metalloprotease inhibitors [14]. Although these findings are consistent with a role for elevated MMP activity and cleavage of secreted Aβ in cultures treated with metal–ligand complexes, we cannot be certain whether this is occurring in the medium or at the cell surface. Additionally, although MMP activation was observed in cells treated with a number of different metal–Cu complexes and inhibition by MMP inhibitors blocked this effect, there may still be additional mechanisms involved in Aβ processing that are specific for each ligand–metal complex. However, the investigation of such a potential diverse range of metabolic effects was outside the scope of the present study.
We previously reported that CQ–Cu complexes induced activation of PI3K and JNK [14]. In the present study we showed that a diverse range of metal ligands also induced PI3K and JNK activation and this correlated with Cu uptake and loss of Aβ induced by the same ligands (8HQ, NC, 1,10-phenanthroline and PDTC). Conversely, ligands that failed to affect metal levels or Aβ also had no effect on JNK activity (TM, D-penicillinamine and BPS). Our results strongly suggest that activation of JNK by CQ–Cu and the other ligands that elevated cellular Cu resulted in decreased extracellular Aβ levels through up-regulation of MMP activity. This was supported by the fact that inhibition of MMPs prevented the loss of secreted Aβ and we have reported previously that inhibition of JNK prevented up-regulation of MMP activity by CQ–Cu [14]. Furthermore, we found no evidence of altered cellular APP metabolism after treatment with the JNK inhibitor. Previous studies have shown an important role for JNK activation in altering synaptic plasticity [30] and Aβ-mediated neurotoxicity [31,32]. As metal–ligand complexes are able to activate JNK, there is the potential that these complexes could modulate such effects in neurons in vivo. However, studies on neuronal cells are needed to fully understand how metal–ligand complexes alter synaptic plasticity and Aβ neurotoxicity and whether co-treatment with JNK inhibitors affects these factors in metal-treated cells.
Our findings are consistent with previous reports that lipid-soluble metal ligands can induce activation of protein kinases such as JNK and that metals have been shown to activate MMPs [33–36]. Although the process of MAPK activation has not been fully addressed, there is considerable evidence that this can be mediated through activation of PI3K by metals such as Cu and Zn [37]. We have shown in the present study and previously [14] that metal–ligand complexes activate PI3K resulting in phosphorylation of its downstream target Akt. This subsequently controls activation of MAPKs such as ERK (extracellular-signal-regulated kinase) and JNK. Although it is not yet known how metals activate PI3K, there is evidence to suggest that this could be mediated through up-regulation of receptor tyrosine kinase activity [38]. As non-lipophilic ligands do not stimulate these pathways, there may be a requirement for partial membrane penetration by the complexes to achieve receptor activation. This could be responsible for the reduction in Aβ seen in cultures treated with 1,10-phenanthroline–Zn or 8HQ–Zn complexes when no increase in cellular Zn was observed (Figures 1 and 2). This is supported by the observation that ligand–Zn complexes can also activate JNK with little or no metal uptake ([14] and A. R. White, unpublished work). If true, this would mean that the large increases in metal uptake induced by many of the lipophilic compounds is unnecessary and it is only the partial membrane penetration that is required for induction of signalling pathways leading to inhibition of Aβ. Therefore assays could be developed to screen for ligand–metal complexes that have the ability to bind to and penetrate the membrane but not facilitate dramatic increases in potentially toxic metals. Whether these effects occur in vivo has not been established, although Malm et al. [39] recently reported activation of PI3K/Akt by PDTC in APP/PS1 transgenic mice. Interestingly, they did not find altered Aβ levels in the brains of treated animals. However, it is possible that there were changes in levels of specific soluble oligomeric forms of Aβ without an overall reduction in Aβ levels. Alternatively, metal–ligand complexes, as used in the present study, may induce different effects on Aβ compared with free ligands.
Additionally, metal ligands can be potent inhibitors of metalloproteases [40,41]. Even though the level of ligands added to the cells is not high (25 μM), their efficacy may be enhanced by the use of serum-free medium containing only low levels of trace metals (0.5 μM Cu and 1.3 μM Zn). The ligand-mediated inhibition of MMP activity could partially explain the reduced efficacy in promoting Aβ degradation by some of the ligands [2-chloro-1,10-phenanthroline (#12) and 1,10-phenanthroline (#14)].
Recently, Ahmad et al. [42] reported that overexpression of MT (membrane-type) MMPs resulted in cleavage and degradation of APP. Whether MT-MMPs are activated in APP-CHO cells treated with CQ–Cu or other metal ligands has not been established. It is feasible that up-regulated MT-MMPs could result in activation of MMP-2 (leading to degradation of extracellular secreted Aβ) and also result in MT-MMP-mediated degradation of APP. This remains to be tested.
Loss of cell adhesion correlates closely with loss of secreted Aβ for the ligands tested. This is most probably due to the fact that MMP activation by ligand–metal complexes is responsible for degradation of secreted Aβ and the extracellular matrix resulting in loss of adhesion [43,44]. Cell adhesion assays could therefore be used as a rapid and simple assay for preliminary screening of metal ligands that promote MMP-mediated degradation of Aβ.
The ability of diverse ligands to increase metal uptake and inhibit Aβ levels generally correlated with a higher lipid solubility as estimated using Clog P calculation (Table 1 and Figures 1 and 2). However, there were some exceptions to this trend. For example, NC induced a very high level of metal uptake despite a Clog P value in the mid-range of the phenanthroline derivatives tested. Although the reason for this is uncertain, the 2,9-dimethyl substituent on the 1,10-phenanthroline subtly alters the chemical properties and for reasons of stearic hindrance the square planar co-ordination normally associated with Cu(II) complexes is not favoured. This is the reason that this ligand is considered a Cu(I)-specific ligand as the tetrahedral structure is favoured and could promote Cu(I) uptake across the cell membrane. However, as NC induced high uptake of both Cu and Zn, it may reflect the fact that a Clog P value of approx. 3 might be ideal for metal movement across a membrane. It should also be noted that from our results, we were unable to establish for certainty what proportion of the metals are being transported into the cell and how much remains bound to the cell membrane. Phenanthroline-based complexes are charged and therefore tend to have reduced membrane permeability [45]. With these compounds, a large proportion of the metal may remain at the cell surface or alternatively, if transported into the cell as a phenanthroline–metal complex, could have reduced excretion. However, the actual movement of the metal will depend on the combination of phenanthroline derivative and metal (Cu or Zn). In contrast, the 8HQ derivatives form neutral complexes with Cu and Zn and therefore may have higher membrane permeability [45]. It would be interesting to determine whether a greater percentage of the cell-associated metal is internalized in 8HQ–metal-treated cells, although the increased permeability may also result in greater excretion of the metal complex from the cell, thus reducing intracellular metal levels. Whether active transport processes are involved in this is not known. As phenanthroline, 8HQ and alternative ligands such as PDTC all activate analogous pathways (PI3K and JNK) in our system, this may suggest that cellular uptake of complexes is less important than membrane association. However, this remains to be tested. Another potential difference between ligands could be redox-mediated affects on cell metabolism. However, this is unlikely as there was no correlation between kinase activation and redox activity of the complexes and we reported previously that antioxidants and free radical scavengers had no effect on kinase activation and changes to Aβ levels induced by CQ–Cu complexes [14]. Furthermore, many of the Zn complexes induced substantial loss of Aβ but Zn is effectively redox inactive in cells.
The level of metal uptake did not always correlate with loss of Aβ. Some ligands induced high metal uptake with a modest effect on Aβ [e.g. 5,7-diiodo-8HQ (#1) and 1,10-phenanthroline (#14)] whereas others induced a low level of metal uptake but greatly decreased Aβ [8HQ (#5) and BC (#8)]. In addition, there were some ligands that induced greatly disparate levels of metal uptake despite very similar Clog P values [e.g. 5-chloro-8HQ (#4), 8HQ (#5), NC (#11) and 2-chloro-1,10-phenanthroline (#12) in the presence of Zn; and 1,10-phenanthroline (#14) and BCS (#15) in the presence of Cu]. This is likely to reflect the fact that additional structural factors affect metal transport across the cell membrane by metal ligands [46]. Addition of sulfur groups can greatly alter the permeability of metal ligands and there are significant differences in uptake of charged (i.e. 1,10-phenanthroline–metal) and neutral (8HQ–metal) complexes [45]. Moreover, there may be differences in the subcellular location where the metals are released by the ligands.
In conclusion, the present study is the first comprehensive comparative study of how metal ligands modulate metal uptake and Aβ metabolism in vitro. Our findings have revealed that diverse classes of metal ligands can have similar effects on Aβ turnover with a number of lipid-soluble ligands dramatically reducing extracellular levels of Aβ when complexed to Cu or Zn. This was achieved through activation of JNK and up-regulation of MMP activity resulting in Aβ degradation. The present study forms an important base for further development of metal–ligand-mediated therapeutic intervention in AD. Our assay system could be developed for rapid screening of combinatorial libraries for metal ligands that alter cell adhesion (indicative of MMP activation) and decrease Aβ levels by increasing MMP activity. This would greatly improve the chances of identifying highly efficacious compounds that could promote Aβ clearance in vivo [12]. In this respect, we identified several ligands in the present study [8HQ–Zn (#5), BP–Cu/Zn (#9) and BC–Zn (#8)] that potently inhibit Aβ with minimal change to cellular metal uptake. These compounds should be assessed further for their Aβ inhibitory action. In addition to identification of potential therapeutic agents for treatment of AD, our studies have also provided further insights into the role of metals and metal ligands in modulation of cell signal transduction and Aβ metabolism.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (NH&MRC). A. R. W. and A. F. H. were supported by RD Wright Fellowships from the NH&MRC. K. J. B. was supported by an NH&MRC Senior Research Fellowship.
References
- 1.Bush A. I., Pettingell W. H., Multhaup G., d Paradis M., Vonsattel J. P., Gusella J. F., Beyreuther K., Masters C. L., Tanzi R. E. Rapid induction of Alzheimer A β amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 2.Glenner G. G., Wong C. W. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 3.Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atwood C. S., Scarpa R. C., Huang X., Moir R. D., Jones W. D., Fairlie D. P., Tanzi R. E., Bush A. I. Characterization of copper interactions with Alzheimer amyloid β peptides: identification of an attomolar-affinity copper binding site on amyloid β1–42. J. Neurochem. 2000;75:1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x. [DOI] [PubMed] [Google Scholar]
- 5.Bush A. I., Multhaup G., Moir R. D., Williamson T. G., Small D. H., Rumble B., Pollwein P., Beyreuther K., Masters C. L. A novel zinc(II) binding site modulates the function of the βA4 amyloid protein precursor of Alzheimer's disease. J. Biol. Chem. 1993;268:16109–16112. [PubMed] [Google Scholar]
- 6.Hesse L., Beher D., Masters C. L., Multhaup G. The β A4 amyloid precursor protein binding to copper. FEBS Lett. 1994;349:109–116. doi: 10.1016/0014-5793(94)00658-x. [DOI] [PubMed] [Google Scholar]
- 7.Multhaup G., Schlicksupp A., Hesse L., Beher D., Ruppert T., Masters C. L., Beyreuther K. The amyloid precursor protein of Alzheimer's disease in the reduction of copper(II) to copper(I) Science. 1996;271:1406–1409. doi: 10.1126/science.271.5254.1406. [DOI] [PubMed] [Google Scholar]
- 8.Barnham K. J., McKinstry W. J., Multhaup G., Galatis D., Morton C. J., Curtain C. C., Williamson N. A., White A. R., Hinds M. G., Norton R. S., et al. Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J. Biol. Chem. 2003;278:17401–17407. doi: 10.1074/jbc.M300629200. [DOI] [PubMed] [Google Scholar]
- 9.Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., Barnham K. J., Volitakis I., Fraser F. W., Kim Y., et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 10.Lee J. Y., Friedman J. E., Angel I., Kozak A., Koh J. Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human β-amyloid precursor protein transgenic mice. Neurobiol. Aging. 2004;25:1315–1321. doi: 10.1016/j.neurobiolaging.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Crapper McLachlan D. R., Dalton A. J., Kruck T. P., Bell M. Y., Smith W. L., Kalow W., Andrews D. F. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 12.Ritchie C. W., Bush A. I., Mackinnon A., Macfarlane S., Mastwyk M., MacGregor L., Kiers L., Cherny R., Li Q. X., Tammer A., et al. Metal–protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 13.Treiber C., Simons A., Strauss M., Hafner M., Cappai R., Bayer T. A., Multhaup G. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer's disease. J. Biol. Chem. 2004;279:51958–51964. doi: 10.1074/jbc.M407410200. [DOI] [PubMed] [Google Scholar]
- 14.White A. R., Du T., Laughton K. M., Volitakis I., Sharples R. A., Xilinas M. E., Hoke D. E., Holsinger R. M., Evin G., Cherny R. A., et al. Degradation of the Alzheimer disease amyloid β-peptide by metal-dependent up-regulation of metalloprotease activity. J. Biol. Chem. 2006;281:17670–17680. doi: 10.1074/jbc.M602487200. [DOI] [PubMed] [Google Scholar]
- 15.Kardos J., Kovacs I., Hajos F., Kalman M., Simonyi M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989;103:139–144. doi: 10.1016/0304-3940(89)90565-x. [DOI] [PubMed] [Google Scholar]
- 16.Frederickson C. J., Suh S. W., Silva D., Frederickson C. J., Thompson R. B. Importance of zinc in the central nervous system: the zinc-containing neuron. J. Nutr. 2000;130:1471S–1483S. doi: 10.1093/jn/130.5.1471S. [DOI] [PubMed] [Google Scholar]
- 17.Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 18.Daniel K. G., Chen D., Orlu S., Cui Q. C., Miller F. R., Dou Q. P. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7:R897–R908. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghose A. K., Crippen G. M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987;27:21–35. doi: 10.1021/ci00053a005. [DOI] [PubMed] [Google Scholar]
- 20.Furuta S., Ortiz F., Zhu Sun X., Wu H. H., Mason A., Momand J. Copper uptake is required for pyrrolidine dithiocarbamate-mediated oxidation and protein level increase of p53 in cells. Biochem. J. 2002;365:639–648. doi: 10.1042/BJ20011251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gyulkhandanyan A. V., Feeney C. J., Pennefather P. S. Modulation of mitochondrial membrane potential and reactive oxygen species production by copper in astrocytes. J. Neurochem. 2003;87:448–460. doi: 10.1046/j.1471-4159.2003.02029.x. [DOI] [PubMed] [Google Scholar]
- 22.Rogers J. T., Randall J. D., Cahill C. M., Eder P. S., Huang X., Gunshin H., Leiter L., McPhee J., Sarang S. S., Utsuki T., et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer's amyloid precursor protein transcript. J. Biol. Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 23.Tjalve H., Stahl K. Effect of 5-chloro-7-iodo-8-hydroxy-quinoline (clioquinol) on the uptake and distribution of nickel, zinc and mercury in mice. Acta Pharmacol. Toxicol. 1984;55:65–72. doi: 10.1111/j.1600-0773.1984.tb01963.x. [DOI] [PubMed] [Google Scholar]
- 24.Chen S. H., Liu S. H., Liang Y. C., Lin J. K., Lin-Shiau S. Y. Death signaling pathway induced by pyrrolidine dithiocarbamate-Cu2+ complex in the cultured rat cortical astrocytes. Glia. 2000;31:249–261. doi: 10.1002/1098-1136(200009)31:3<249::aid-glia60>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 25.Zhu B. Z., Chevion M. Copper-mediated toxicity of 2,4,5-trichlorophenol: biphasic effect of the copper(I)-specific chelator neocuproine. Arch. Biochem. Biophys. 2000;380:267–273. doi: 10.1006/abbi.2000.1919. [DOI] [PubMed] [Google Scholar]
- 26.Borchardt T., Camakaris J., Cappai R., Masters C. L., Beyreuther K., Multhaup G. Copper inhibits β-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion. Biochem. J. 1999;344:461–467. [PMC free article] [PubMed] [Google Scholar]
- 27.Tateishi J. Subacute myelo-optico-neuropathy: clioquinol intoxication in humans and animals. Neuropathology. 2000;20:S20–S24. doi: 10.1046/j.1440-1789.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- 28.Yassin M. S., Ekblom J., Xilinas M., Gottfries C. G., Oreland L. Changes in uptake of vitamin B12 and trace metals in brains of mice treated with clioquinol. J. Neurol. Sci. 2000;173:40–44. doi: 10.1016/s0022-510x(99)00297-x. [DOI] [PubMed] [Google Scholar]
- 29.Chen D., Cui Q. C., Yang H., Barrea R. A., Sarkar F. H., Sheng S., Yan B., Reddy G. P., Dou Q. P. Clioquinol, a therapeutic agent for Alzheimer's disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007;67:1636–1644. doi: 10.1158/0008-5472.CAN-06-3546. [DOI] [PubMed] [Google Scholar]
- 30.Li X. M., Li C. C., Yu S. S., Chen J. T., Sabapathy K., Ruan D. Y. JNK1 contributes to metabotropic glutamate receptor-dependent long-term depression and short-term synaptic plasticity in the mice area hippocampal CA1. Eur. J. Neurosci. 2007;25:391–396. doi: 10.1111/j.1460-9568.2006.05300.x. [DOI] [PubMed] [Google Scholar]
- 31.Yao M., Nguyen T. V., Pike C. J. β-Amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 2005;25:1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao M., Nguyen T. V., Pike C. J. Estrogen regulates Bcl-w and Bim expression: role in protection against β-amyloid peptide-induced neuronal death. J. Neurosci. 2007;27:1422–1433. doi: 10.1523/JNEUROSCI.2382-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LaRochelle O., Gagne V., Charron J., Soh J. W., Seguin C. Phosphorylation is involved in the activation of metal-regulatory transcription factor 1 in response to metal ions. J. Biol. Chem. 2001;276:41879–41888. doi: 10.1074/jbc.M108313200. [DOI] [PubMed] [Google Scholar]
- 34.Ostrakhovitch E. A., Lordnejad M. R., Schliess F., Sies H., Klotz L. O. Copper ions strongly activate the phosphoinositide-3-kinase/Akt pathway independent of the generation of reactive oxygen species. Arch. Biochem. Biophys. 2002;397:232–239. doi: 10.1006/abbi.2001.2559. [DOI] [PubMed] [Google Scholar]
- 35.Adamson I. Y., Vincent R., Bakowska J. Differential production of metalloproteinases after instilling various urban air particle samples to rat lung. Exp. Lung Res. 2003;29:375–388. doi: 10.1080/01902140303753. [DOI] [PubMed] [Google Scholar]
- 36.Samet J. M., Dewar B. J., Wu W., Graves L. M. Mechanisms of Zn2+-induced signal initiation through the epidermal growth factor receptor. Toxicol. Appl. Pharmacol. 2003;191:86–93. doi: 10.1016/s0041-008x(03)00219-9. [DOI] [PubMed] [Google Scholar]
- 37.Barthel A., Ostrakhovitch E. A., Walter P. L., Kampkotter A., Klotz L. O. Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: Mechanisms and consequences. Arch. Biochem. Biophys. 2007;463:175–182. doi: 10.1016/j.abb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Wu W., Silbajoris R. A., Whang Y. E., Graves L. M., Bromberg P. A., Samet J. M. p38 and EGF receptor kinase-mediated activation of the phosphatidylinositol 3-kinase/Akt pathway is required for Zn2+-induced cyclooxygenase-2 expression. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;289:L883–L889. doi: 10.1152/ajplung.00197.2005. [DOI] [PubMed] [Google Scholar]
- 39.Malm T. M., Iivonen H., Goldsteins G., Keksa-Goldsteine V., Ahtoniemi T., Kanninen K., Salminen A., Auriola S., Van Groen T., Tanila H., Koistinaho J. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting β-amyloid burden. J. Neurosci. 2007;27:3712–3721. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sagara Y., Ito A., Omura T. Partial purification of a metalloprotease catalyzing the processing of adrenodoxin precursor in bovine adrenal cortex mitochondria. J. Biochem. (Tokyo) 1984;96:1743–1752. doi: 10.1093/oxfordjournals.jbchem.a135007. [DOI] [PubMed] [Google Scholar]
- 41.Cheung P. Y., Sawicki G., Wozniak M., Wang W., Radomski M. W., Schulz R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. 2000;101:1833–1839. doi: 10.1161/01.cir.101.15.1833. [DOI] [PubMed] [Google Scholar]
- 42.Ahmad M., Takino T., Miyamori H., Yoshizaki T., Furukawa M., Sato H. Cleavage of amyloid-β precursor protein (APP) by membrane-type matrix metalloproteinases. J. Biochem. (Tokyo) 2006;139:517–526. doi: 10.1093/jb/mvj054. [DOI] [PubMed] [Google Scholar]
- 43.Roher A. E., Kasunic T. C., Woods A. S., Cotter R. J., Ball M. J., Fridman R. Proteolysis of A β peptide from Alzheimer disease brain by gelatinase A. Biochem. Biophys. Res. Commun. 1994;205:1755–1761. doi: 10.1006/bbrc.1994.2872. [DOI] [PubMed] [Google Scholar]
- 44.Hofmann U. B., Houben R., Brocker E. B., Becker J. C. Role of matrix metalloproteinases in melanoma cell invasion. Biochimie. 2005;87:307–314. doi: 10.1016/j.biochi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Gaeta A., Hider R. C. The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. Br. J. Pharmacol. 2005;146:1041–1059. doi: 10.1038/sj.bjp.0706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaiser S. M., Escher B. I. The evaluation of liposome-water partitioning of 8-hydroxyquinolines and their copper complexes. Environ. Sci. Technol. 2006;40:1784–1791. doi: 10.1021/es051908y. [DOI] [PubMed] [Google Scholar]