

Abstract
The catalytic subunit of the human cytomegalovirus DNA polymerase is critical for the replication of the virus. In the present study, we report the expression and purification of a recombinant catalytic subunit of the human cytomegalovirus DNA polymerase expressed in bacteria which retains polymerase activity. As a first step towards elucidating the nature of the interaction between the enzyme, DNA and dNTPs, we have utilized endogenous tryptophan fluorescence to evaluate the binding of ligands to the enzyme. Using this technique, we demonstrate that the minimal DNA-binding site of the enzyme is 6 nt. We also report the first detailed study of the binding kinetics and thermodynamic parameters involved in the interaction between the enzyme, DNA and dNTPs. Our thermodynamic analyses indicate that the initial formation of the enzyme–DNA binary complex is driven by a favourable entropy change, but is also clearly associated with an unfavourable enthalpic contribution. In contrast, the interaction of dNTPs to the binary complex was shown to depend on a completely different mode of binding that is dominated by a favourable enthalpy change and associated with an unfavourable entropy change. In order to provide additional insights into the structural modifications that occur during catalysis, we correlated the effect of DNA and dNTP binding on protein structure using CD. Our results indicate that the enzyme undergoes a first conformational change upon the formation of the protein–DNA binary complex, which is followed by a second structural modification upon dNTP binding. The present study provides a better understanding of the molecular basis of DNA and dNTP recognition by the catalytic subunit of the human cytomegalovirus DNA polymerase.
Keywords: binding kinetics, cytomegalovirus, deoxynucleotide, DNA-binding activity, fluorescence spectroscopy, thermodynamics
Abbreviations: ANS, 8-anilinonaphthalene-l-sulfonic acid; dATPαS, deoxyadenosine 5′-[α-thio]triphosphate; Ni-NTA, Ni2+-nitrilotriacetate
INTRODUCTION
The human cytomegalovirus is a DNA virus that can cause a variety of important diseases in immunocompromised patients and in newborns [1]. These include gastrointestinal diseases, pneumonia and retinitis, whereas newborns born with cytomegalic inclusion disease can have symptoms such as microcephaly, hearing loss, liver damage, pneumonitis and mental retardation [2]. Although the incidence of cytomegalovirus infection has significantly been reduced in recent years due to the use of antiretroviral drugs, cytomegalovirus infections remain difficult to manage [3–6].
Human cytomegalovirus is a member of the Herpesviridae family, which includes an important number of human pathogens [1]. The DNA polymerase activity of the human cytomegalovirus has been shown to require two distinct polypeptides, a property shared with other viruses from the Herpesviridae family. The larger subunit, which is responsible for the catalytic DNA polymerase activity, is encoded by the cytomegalovirus UL54 gene [7–9], which harbours the conserved domains (I–VII) that are also found in other type-B DNA polymerases. The smaller accessory subunit is encoded by the UL44 gene [10] and has been shown to stimulate the polymerase activity of the catalytic subunit by increasing its processivity [10,11]. Moreover, the critical importance of this protein–protein interaction has been demonstrated in a previous study using short peptides that disrupt the physical interaction between the two subunits [12]. The presence of these peptides results in the successful inhibition of the polymerase activity, clearly highlighting the importance of the UL54–UL44 interaction. Using an in vitro translation system, a previous mutagenesis analysis has also identified a short region located in motif III (amino acids 804–807) of the enzyme that is important for the interaction between the enzyme and DNA [9].
As a critical component of the viral replication machinery, the cytomegalovirus DNA polymerase is the specific target for a number of antiviral drugs currently used to inhibit the replication of the virus [13]. These include ganciclovir, cidofovir and foscarnet that are currently approved for the treatment of cytomegalovirus infections in immunocompromised patients. Both ganciclovir and cidofovir are nucleoside analogues that act as competitive inhibitors of the DNA polymerase and function as DNA chain terminators [14,15], while foscarnet is an analogue of PPi that acts non-competitively with respect to the deoxynucleotides [16]. The precise mechanism of action of this drug remains largely speculative although it has been proposed that the PPi analogue could bind close to the active centre of the enzyme, thereby inhibiting the PPi exchange reaction [17]. Although the use of ganciclovir, cidofovir and foscarnet can reduce the viral burden, the antiviral therapy has been plagued by the emergence of drug-resistant viral strains [3–6].
By comparison with other viral DNA polymerases, characterization studies of the cytomegalovirus enzyme have been very limited. Based on clinical studies of drug-resistant isolates, most studies have focused on the identification of specific residues involved in the binding to the anti-cytomegalovirus drugs [4,6,19–22]. Moreover, biochemical studies have been limited to the identification of the kinetic parameters (Km, kcat) of the polymerase reaction [13,22]. This lack of biochemical information on the enzymatic activity of the protein stems from the fact that previous studies have mostly used proteins synthesized by in vitro translation systems or insect cells infected with baculoviruses. Our understanding of the DNA synthesis by the cytomegalovirus DNA polymerase remains largely incomplete. One area that requires additional characterization concerns how the enzyme interacts with DNA and dNTPs. In the present paper, we report the expression and purification of a recombinant catalytic subunit (UL54) of the human cytomegalovirus protein expressed in bacteria which retains polymerase activity. Using fluorescence spectroscopy and CD studies, we describe a detailed thermodynamic study on the formation of the enzyme–DNA binary complex, which provides insights into the relative enthalpic and entropic contributions to the binding energy, as well as on the effects of DNA binding on both the structure and stability of the protein. Moreover, we have also characterized the binding of DNA molecules of various lengths and various sequences to determine both the minimal length for stable binding and the specificity of the DNA–enzyme interaction. Finally, we also monitored the binding of dNTPs to the enzyme–DNA complex, and completed a structural analysis of the protein in order to gain insights into the structural modifications that occur during catalysis. Such analyses are crucial in order to understand the molecular basis of the interaction between the catalytic subunit of the cytomegalovirus DNA polymerase and its ligands.
MATERIALS AND METHODS
Protein expression and purification
A plasmid for the expression of the catalytic subunit of the cytomegalovirus DNA polymerase was generated by inserting a fragment of the cytomegalovirus UL54 gene in the pET-28a expression plasmid (Novagen). This fragment encodes amino acids 293–953 of the protein, and was cloned between the NdeI and HindIII cloning sites of the pET-28a plasmid. In this context, the protein is fused in frame with an N-terminal peptide containing six tandem histidine residues, and expression of the His6-tagged protein is driven by a T7 RNA polymerase promoter. The resulting recombinant plasmid (pET-UL54) was transformed into Escherichia coli BL21(DE3) and a 1000 ml culture of E. coli BL21(DE3)/pET-UL54 was grown at 37 °C in Luria–Bertani medium containing 30 μg/ml kanamycin until the D600 (attenuance) reached 0.5. The culture was adjusted to 0.4 mM IPTG (isopropyl β-D-thiogalactoside) and 2% (v/v) ethanol, and the incubation continued at 37 °C for 3 h. The cells were then harvested by centrifugation at 5000 g for 5 min, and the pellet was stored at −80 °C. All subsequent procedures were performed at 4 °C. Thawed bacteria pellets were resuspended in 50 ml of buffer A [50 mM Tris/HCl, pH 7.5, 150 mM NaCl and 10% (w/v) sucrose] and cell lysis was achieved by the addition of lysozyme and Triton X-100 to final concentrations of 50 μg/ml and 0.1% respectively. The lysates were sonicated to reduce viscosity, and any insoluble material was removed by centrifugation at 13000 rev./min for 45 min using a Sorval SS-34 rotor. The soluble extract was applied to a 5 ml column of Ni-NTA (Ni2+-nitrilotriacetate)–agarose (Qiagen) that had been equilibrated with buffer A containing 0.1% Triton X-100. The column was washed with the same buffer and then eluted stepwise with buffer B [50 mM Tris/HCl, pH 8.0, 0.1 M NaCl and 10% (v/v) glycerol] containing 50, 100, 200, 500 and 1000 mM imidazole. The polypeptide composition of the column fractions was monitored by SDS/PAGE. The recombinant protein was retained on the column and recovered in the 200 and 500 mM imidazole fractions. Following dialysis against buffer C (50 mM Tris/HCl, pH 8.0, 50 mM NaCl, 2 mM dithiothreitol, 10% glycerol and 0.05% Triton X-100), the purified preparation was stored at −80 °C. The protein concentration was determined using the Bio-Rad dye binding method with BSA as the standard.
Polymerization assay
A DNA substrate (P20/T60) consisting of heteropolymeric oligonucleotides was used to monitor the polymerase activity of the enzyme. The substrate consisted of a primer (5′-TAATACGACTCACTATAGGG-3′) and a template (5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCCGACCCGCCCTATAGTGAGTCGTA-3′). Equimolar concentrations of the oligonucleotides were heated at 95 °C for 5 min and cooled at room temperature (21 °C). Standard reaction mixtures (50 μl) containing 25 mM Tris/HCl (pH 8.0), 10 mM MgCl2, 50 mM NaCl, 0.2 μM [α-32P]ATP, 500 mM of each dNTP, 5 μM of DNA template and purified DNA polymerase (0.6 μM) were incubated for 30 min at 37 °C. The reaction products were analysed by electrophoresis through a 10% (w/v) polyacrylamide gel containing 8 M urea. The radiolabelled DNA products were located by autoradiography.
Fluorescence measurements
Fluorescence was measured using a Hitachi F-2500 fluorescence spectrophotometer. Excitation was performed at a wavelength of 290 nm. Background emission was eliminated by subtracting the signal from either buffer alone or buffer containing the appropriate quantity of substrate.
The extent to which ligands (DNA or dNTPs) bind to the enzyme was determined by monitoring the fluorescence emission of a fixed concentration of proteins and titrating with a given ligand. The binding can be described by eqn (1),
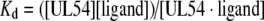 |
(1) |
where Kd is the apparent dissociation constant, [UL54] is the concentration of the protein, [UL54·ligand] is the concentration of complexed protein and [ligand] is the concentration of unbound ligand.
The proportion of ligand-bound protein as described by eqn (1) is related to the measured fluorescence emission intensity by eqn (2),
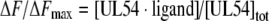 |
(2) |
where ΔF is the magnitude of the difference between the observed fluorescence intensity at a given concentration of ligand and the fluorescence intensity in the absence of ligand, ΔFmax is the difference at infinite [ligand], and [UL54]tot is the total protein concentration.
If the total ligand concentration, [ligand]tot, is in large molar excess relative to [UL54]tot, then it can be assumed that [ligand] is approximately equal to [ligand]tot. Eqns (1) and (2) can then be combined to give eqn (3),
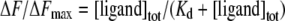 |
(3) |
The Kd values were determined from a non-linear least-squares regression analysis of titration data by using eqn (3).
CD spectroscopy measurements
CD measurements were performed with a Jasco J-810 spectropolarimeter. The samples were analysed in quartz cells with path lengths of 1 mm. Far-UV and near-UV wavelength scans were recorded from 200 to 250 nm and from 250 to 340 nm respectively. All the CD spectra were corrected by subtraction of the background for the spectrum obtained with either buffer alone or buffer containing the ligand. The ellipticity results were expressed as mean residue ellipticity, [θ], in degrees·cm2·dmol−1.
Equilibrium unfolding experiments
Thermal transitions were monitored by following the change in CD ellipticity of the protein (1 μM) at 222 nm. The samples were heated from 10 to 95 °C, at a heating rate of 1 °C/min. The ellipticity results were expressed as mean residue ellipticity, [θ], in degrees·cm2·dmol−1. The fraction of unfolded protein at each temperature was determined by calculating the ratio [θ222]/[θ222]d, where [θ222]d is the molar ellipticity for the completely unfolded enzyme.
ANS (8-anilinonaphthalene-l-sulfonic acid)-binding measurements
Binding of ANS was evaluated by measuring the fluorescence enhancement of ANS (50 μM) upon excitation at a wavelength of 380 nm. The emission spectra were integrated from 400 to 600 nm.
RESULTS
Expression, purification and DNA polymerase activity of the protein
In order to investigate the initial binding of deoxynucleotides to the catalytic subunit of the human cytomegalovirus DNA polymerase (UL54), we intended to purify a large quantity of the enzyme. Previous biochemical studies performed with the enzyme have either used a coupled in vitro transcription/translation system [22,23] or insect cells infected with recombinant baculoviruses [18,24] to generate the full-length protein (1242 amino acids). Since a limited amount of protein is obtained from these systems, an expression plasmid was created to express the recombinant protein in E. coli. Because of the large size of the enzyme, we focused on the region encompassing amino acids 293–953 of the protein. This domain contains the consensus sequences of the DNA polymerase type-B family [25] as well as most of the residues [16,17] that have been associated with drug resistance in UL54 (Figure 1). The truncated enzyme was therefore expressed in E. coli with an N-terminal His6-tagged epitope and purified from soluble bacterial extracts by Ni-NTA–agarose chromatography. The purified protein was located mainly in the 200 and 500 mM imidazole eluates (Figure 2A). All subsequent experiments were performed with the protein found in the 500 mM fraction, as it displayed the highest level of purity. The concentration of protein in this fraction was estimated to be 130 ng/ml. Immunoblotting analysis using a monospecific anti-His antibody also confirmed the identity of the protein (Figure 2B).
Figure 1. Functional domains of the cytomegalovirus DNA polymerase.

The conserved domains (I–VII) found in other type-B DNA polymerases are shown. These domains, as initially determined by Wang et al. [43], have been defined as follows according to the amino acid numbering of the enzyme: IV (379–421), II (696–742), VI (771–790), III (805–845), I (905–919), VII (962–970) and V (978–988). The consensus region of the DNA polymerase type-B family is also indicated (293–953). Additionally, the regions that have been shown to be critical for drug resistance are also shown along with the specific amino acids that have been shown to be associated with drug resistance.
Figure 2. Purification and enzymatic activity of the purified cytomegalovirus DNA polymerase.

(A) The peptide compositions of various purification fractions were analysed by SDS/PAGE. The gels were fixed and stained with Coomassie Blue dye. Lane 1, Ni-NTA–agarose flow-through; lane 2, Ni-NTA–agarose wash; lane 3, 50 mM imidazole eluate; lane 4, 100 mM imidazole eluate; lane 5, 200 mM imidazole eluate; lane 6, 500 mM imidazole eluate; lane 7, 1000 mM imidazole eluate. The positions and sizes (in kDa) of the size markers are indicated on the left. (B) The purified cytomegalovirus protein (500 mM imidazole fraction) was analysed by immunoblotting using a monospecific anti-His antibody. (C) Polymerization assays performed with a heteropolymeric primer/template DNA substrate (P20/T60). The reaction mixtures (50 μl) containing 20 mM Tris/HCl (pH 7.5), 10 mM MgCl2, 50 mM NaCl, 5 μM of primer/template DNA substrate, 0.2 mM [α-32P]ATP and 500 mM of dGTP, dCTP and dTTP were incubated for 30 min at 37 °C in the presence of 0.6 μM of either the full-length protein expressed in a coupled transcription/translation system (lane 2) or recombinant enzyme (lane 3). A negative control (vector alone) synthesized with the coupled transcription/translation system was also used in the assay (lane 1). The reaction products were analysed by electrophoresis through a 10% polyacrylamide gel containing 8 M urea. An autoradiogram of the gel is shown, and the positions of the size markers are also indicated. (D) Increasing amounts of enzyme were added to the heteropolymeric primer/template DNA substrate (P20/T60). The reaction mixtures containing 5 μM of primer/template DNA substrate, 0.6 μM of enzyme, 1 mM [α-32P]ATP and 1 mM of dGTP, dCTP and dTTP were incubated for 30 min at 37 °C. The reaction products were analysed by liquid-scintillation counting following the precipitation of the DNA reaction products. (E) Polymerization assays were performed in the presence of increasing concentrations of foscarnet, and the incorporation of radiolabelled ATP was quantified by liquid-scintillation counting.
The ability of the purified enzyme to catalyse the synthesis of DNA was initially analysed in a gel-based assay using a defined heteropolymeric primer/template (P20/T60) substrate. As shown in Figure 2(C), the purified enzyme had the ability to perform multiple deoxynucleotide incorporation events. However, as reported previously with the native enzyme [22], the recombinant protein was not highly processive as evidenced by the numerous shorter elongation products that were also synthesized. Additionally, we also used a coupled in vitro transcription/translation system to generate the full-length protein and compare its activity with the recombinant truncated enzyme. As shown in Figure 2(C), both proteins displayed similar polymerization patterns upon incubation with dNTPs.
A second polymerization assay was used to quantify the specific activity of the recombinant enzyme. In this assay, the purified protein was added to the same primer/template combination, but the incorporation of radiolabelled dATP was monitored by liquid-scintillation counting following the precipitation of the DNA reaction products. As can be seen in Figure 2(D), the extent of dATP incorporation during a 30 min reaction was proportional to the amount of input UL54 protein. A specific activity of 120 nmol·min−1·μg−1 was evaluated from this experiment. Foscarnet, an antiviral used for the treatment of human cytomegalovirus infections and a potent inhibitor of the cytomegalovirus DNA polymerase activity [3,13,19], was shown to inhibit the polymerization activity of the purified protein. As shown in Figure 2(E), the addition of 2.7 μM of foscarnet resulted in 50% inhibition of the polymerase reaction.
Intrinsic fluorescence properties of the cytomegalovirus DNA polymerase
We and others have previously shown that the binding of DNA to free enzymes can result in a significant decrease in protein emission fluorescence [26–32]. We therefore intended to use fluorescence spectroscopy to characterize the binding of DNA to the cytomegalovirus DNA polymerase. The fluorescence emission spectrum of the purified enzyme in standard buffer at 22 °C is shown in Figure 3(A). The purified protein used in our assays harbours a single tryptophan residue (Trp-780), which provided a significant fluorescence signal upon excitation at 290 nm. The emission maximum of the enzyme (338 nm) is blue-shifted relative to that of free L-tryptophan, which under the same conditions is observed to be at 350 nm. The λmax of tryptophan is highly sensitive to the polarity of the microenvironment in which its indole side chain is located. Blue shifts of protein emission spectra have been ascribed to shielding of the tryptophan residues from the aqueous phase [33]. This shielding is the result of the protein's three-dimensional structure. Accordingly, denaturation of the enzyme with 8 M urea results in a red shift of λmax towards 350 nm (Figure 3A). The molar intensity of the fluorescence emission spectrum of the purified cytomegalovirus DNA polymerase was also evaluated to determine whether significant protein aggregation, or the loss of protein from solution through adhesion, could influence the binding parameters. As shown in Figure 3(A), an increase in fluorescence was observed with increasing concentrations of the enzyme. A linear change of 0.03 fluorescence intensity units/nM of protein was observed over the range examined. This relatively small change can be attributed to minor losses of proteins from solution through adhesion. All subsequent binding experiments were therefore performed at a protein concentration of 1 μM, with the assumption that the DNA-binding assays were not complicated by the presence of an aggregation equilibrium.
Figure 3. Titration of the cytomegalovirus DNA polymerase with DNA.

(A) Background-corrected fluorescence emission spectra of the enzyme. 1, Purified protein in 50 mM Tris/HCl and 50 mM potassium acetate (pH 7.5); 2, Purified protein after a 2 h exposure to an 8 M solution of urea at 25 °C. Fluorescence spectra were recorded at an excitation wavelength of 290 nm. The molar fluorescence of the protein is also displayed in the right-hand corner. Various concentrations of the purified protein were assayed in 50 mM Tris/HCl and 50 mM potassium acetate (pH 7.5). Excitation was performed at 290 nm, and emission was monitored at 338 nm. (B) Increasing concentrations of a DNA substrate of 30 nt encompassing the cytomegalovirus immediate-early promoter sequence were added to a 1 μM solution of the enzyme in binding buffer (20 mM Tris/HCl, pH 7.5, 50 mM NaCl and 10 mM MgCl2) and the emission spectrum was scanned from 310 to 440 nm. (C) A saturation isotherm can be generated from these results by plotting the change in fluorescence intensity at 338 nm as a function of added DNA. A double-reciprocal plot of the saturation isotherm is shown in the right-hand corner. (D) Thermodynamic parameters of the interaction between DNA and the cytomegalovirus DNA polymerase. Binding reactions were performed at various temperatures, and the respective association constants were evaluated. A van't Hoff plot for the interaction between DNA and the protein is shown. The effect of temperature on the association constant was evaluated at pH 7.5. (E) Kinetic analysis of real-time binding of DNA to the protein. A 1 μM solution of the enzyme was incubated with 100 μM DNA. Emission was monitored for 40 s at 338 nm, and excitation was performed at 290 nm.
DNA-binding activity
In order to monitor the formation of the enzyme–DNA binary complex, increasing concentrations of DNA were added to the purified cytomegalovirus DNA polymerase, and the fluorescence intensity was monitored. We initially monitored the binding of a DNA substrate of 30 nt encompassing the cytomegalovirus immediate-early promoter sequence. We observed that the binding of DNA to a fixed concentration of the enzyme resulted in a modification of the intensity of the intrinsic fluorescence of the protein. Typical emission spectra are shown in Figure 3(B). The addition of increasing amounts of DNA produced a decrease in the fluorescence intensity. Approx. 80% of the intrinsic protein fluorescence was accessible to the quencher DNA substrate (Figure 3C). A double-reciprocal plot of the saturation isotherm, generated by plotting the change of fluorescence intensity as a function of added DNA, indicated a Kd value of 47.5 μM (Figure 3C).
Fluorescence spectroscopy was also used to evaluate the Gibbs free energy of binding (ΔG), as well as both the enthalpy (ΔH) and the entropy changes (ΔS) associated with the binding of DNA to the enzyme. Evaluation of these thermodynamic parameters yields important insights into the nature of the DNA-binding reaction. Our binding studies indicate that the ΔG for the interaction of DNA with the enzyme was −24.8 kJ/mol. The precise enthalpic and entropic contributions to the free energy of binding were then determined by measuring the initial binding of DNA to the enzyme as a function of temperature. Analysis of a van't Hoff plot for the interaction between DNA and the enzyme (Figure 3D) revealed that binding of the DNA substrate is entropically favoured (TΔS=32.7 kJ/mol), with a minor contribution coming from an unfavourable enthalpy change (ΔH=7.9 kJ/mol). The implications of these thermodynamic findings will be further discussed in the Discussion section.
The use of fluorescence spectroscopy also allowed us to monitor the kinetics of the DNA binding to the cytomegalovirus DNA polymerase (Figure 3E). The progress of the binding reaction was followed for 30 s upon addition of a saturating concentration of DNA. The results show that there is a rapid exponential decrease in fluorescence intensity following the addition of DNA substrate. An apparent association rate of 3.6 μM−1·s−1 was estimated from the data. Half-maximal quenching was observed at approx. 0.1 s, while maximal quenching was achieved after 0.3 s of incubation with DNA, and remained constant thereafter. The exponential decrease in fluorescence observed following the addition of DNA was not due to photobleaching, as similar results were obtained when the protein was incubated away from the light source (results not shown).
Minimal DNA length for stable binding
We next sought to determine the minimal length of DNA required for stable binding of the cytomegalovirus DNA polymerase. Fluorescence spectroscopy assays were therefore performed with DNA substrates ranging from 30 to 5 nt (Figure 4A). Our results indicate that the enzyme could bind efficiently to DNA substrates of 20, 15, 12, 10, 9, 8, 7 and 6 nt in length (Table 1). However, we could not detect the binding of the enzyme to the DNA substrate of 5 nt, and no accurate binding constants could be efficiently and repeatedly determined for this DNA substrate. Based on these results, we conclude that the minimal DNA-binding site size of the cytomegalovirus is 6 nt.
Figure 4. Characterization of the interaction between DNA and the catalytic subunit of the cytomegalovirus DNA polymerase.

(A) Affinity of the enzyme for DNA of 30 nt (■), 20 nt (▼), 15 nt (●), 12 nt (◇), 10 nt (Δ), 9 nt (X), 8 nt (*), 7 nt (♦) and 6 nt (▲). Increasing concentrations of DNA were added to a 1 μM solution of the enzyme and the emission spectrum was scanned from 310 to 440 nm upon excitation at 290 nm. Saturation isotherms can be generated from these results by plotting the change in fluorescence intensity at 338 nm as a function of added DNA. (B) Interaction of the enzyme with DNA of various lengths. The apparent Kd values were plotted as a function of substrate length. (C) The specificity of binding was also monitored as a function of the length of the various DNA molecules used in the binding assays. (D) The binding of various substrates was monitored by fluorescence spectroscopy. Increasing concentrations of a DNA substrate of 30 nt encompassing the cytomegalovirus immediate-early promoter sequence (■), a DNA substrate corresponding to the complement of the cytomegalovirus immediate-early promoter sequence (▼), a DNA substrate of 30 nt of random sequence (●), and an RNA substrate of 30 nt corresponding to cytomegalovirus immediate-early promoter sequence (◇) were used in the binding assays.
Table 1. Dissociation constants (Kd), association constants (Ka) and free energies of binding (ΔG) for the interaction of UL54 with DNA.
| Ligand | Length (nt) | Kd (mM) | Ka (M−1) | ΔG (J/mol) | Ligand structure |
|---|---|---|---|---|---|
| Promoter-30 | 30 | 48 | 21050 | −24660 | 5′-GGGAGCTCGTTTAGTGAACCGTCAGATCTC-3′ |
| Promoter-20 | 20 | 61 | 16420 | −24050 | 5′-GGGAGCTCGTTTAGTGAACC-3′ |
| Promoter-15 | 15 | 93 | 10720 | −22990 | 5′-GGGAGCTCGTTTAGT-3′ |
| Promoter-12 | 12 | 130 | 7681 | −22170 | 5′-GGGAGCTCGTTT-3′ |
| Promoter-10 | 10 | 115 | 8719 | −22480 | 5′-GGGAGCTCGT-3′ |
| Promoter-9 | 9 | 97 | 10320 | −22900 | 5′-GGGAGCTCG-3′ |
| Promoter-8 | 8 | 99 | 10070 | −22840 | 5′-GGGAGCTC-3′ |
| Promoter-7 | 7 | 95 | 10500 | −22940 | 5′-GGGAGCT-3′ |
| Promoter-6 | 6 | 94 | 10680 | −22980 | 5′-GGGAGC-3′ |
| Promoter-5 | 5 | No binding detected | 5′-GGGAG-3′ | ||
| Rv-Promoter-30 | 30 | 49 | 20450 | −24590 | 5′-GAGATCTGACGGTTCACTAAACGAGCTCCC-3′ |
| ds-Promoter-30 | 30 | 42 | 23580 | −24950 | 5′-GGGAGCTCGTTTAGTGAACCGTCAGATCTC-3′ 3′- CCCTCGAGCAAATCACTTGGCAGTCTAGAG-5′ |
| Random-30 | 30 | 61 | 16390 | −24040 | 5′-TCAGTAACTTAGCCTTGTCTTGAATCATCC-3′ |
| RNA-30 | 30 | 59 | 17010 | −24130 | 5′-GGGAGCUCGUUUAGUGAACCGUCAGAUCUC-3′ |
Assuming that each nucleotide within the minimal DNA-binding site makes the same number of contacts with the enzyme, the apparent Kd should in theory display a small hyperbolic decrease, since each additional nucleotide can provide a new binding position. This is precisely what was observed (Figure 4B), as the ΔG for substrates ranging from 10 to 20 nt changed linearly with a slope of 157 J·mol−1·nt−1. Beyond that length, the ΔΔG was smaller. Moreover, we observed that the specific constant of binding (Bmax/Kd), which takes into account both the strength and the maximal level of binding, showed that the binding efficiency increased dramatically up to a length of 20 nt, and levelled off thereafter (Figure 4C).
The specificity of the protein–DNA interaction was also monitored by using various nucleic acids substrates. Binding of the enzyme to the DNA substrate was not sequence-specific as similar dissociation constants were obtained for DNA substrates (30 nt) of various sequences (Figure 4D and Table 1). Moreover, similar binding parameters were determined when a double-stranded DNA substrate was used in the binding assays. Surprisingly, similar binding parameters were also obtained for an RNA substrate of 30 nt, indicating that the enzyme did not efficiently discriminate between RNA and DNA (Figure 4D and Table 1).
Binding of dNTPs
Fluorescence spectroscopy has been shown to be a powerful technique to monitor the binding of nucleotides/deoxynucleotides to enzymes [6,31,32]. Therefore we intended to use fluorescence spectroscopy to characterize the binding of deoxynucleotides to the binary complex of the cytomegalovirus DNA polymerase bound to DNA. In order to monitor the binding of deoxynucleotides to the enzyme–DNA complex, increasing concentrations of dATP were added to the purified cytomegalovirus DNA polymerase bound to DNA, and the fluorescence intensity was monitored. We observed that the binding of dATP to a fixed concentration of the enzyme–DNA complex resulted in a modification of the intensity of the intrinsic fluorescence of the protein. Typical emission spectra are shown in Figure 5(A). The addition of increasing amounts of dATP produced a decrease in the fluorescence intensity. Approx. 30% of the intrinsic protein fluorescence was accessible to the quencher dATP substrate. A double-reciprocal plot of the saturation isotherm, generated by plotting the change of fluorescence intensity as a function of added dATP, indicated a Kd value of 15 μM (Figure 5A). The binding of the three other deoxynucleotides was also evaluated. As shown in Table 2, the protein displayed similar binding activity for all four dNTPs, although dTTP appeared as a slightly better substrate as judged by the ΔG. It should be noted that the dNTP binding activity was absolutely dependent on the presence of a bivalent cation. No binding could be detected in the absence of metal ions (results not shown).
Figure 5. Titration of the cytomegalovirus DNA polymerase with dATP.

(A) Increasing concentrations of dATP were added to a 1 μM solution of the enzyme in binding buffer (20 mM Tris/HCl, pH 7.5, 50 mM NaCl and 10 mM MgCl2) and the emission spectrum was scanned from 310 to 440 nm. A double-reciprocal plot of the saturation isotherm is shown in the right-hand corner. (B) The effect of increasing ionic strength on the apparent dissociation constant of the enzyme for dATP was investigated. Increasing concentrations of KCl were added to the binding reactions to generate the desired ionic strengths. (C) Thermodynamic parameters of the interaction between dATP and the cytomegalovirus DNA polymerase. Binding reactions were performed at various temperatures, and the respective association constants were evaluated. A van't Hoff plot for the interaction between dATP and the protein is shown. The effect of temperature on the association constant was evaluated at pH 7.5. (D) Kinetic analysis of real-time binding of dATP to the protein. A 1 μM solution of the enzyme was incubated with 1 mM dATP. Emission was monitored for 30 s at 338 nm, and excitation was performed at 290 nm.
Table 2. Dissociation constants (Kd), association constants (Ka) and free energies of binding (ΔG) for the interaction of UL54 with dNTPs.
| Ligand | Kd (mM) | Ka (M−1) | ΔG (J/mol) |
|---|---|---|---|
| dATP | 15 | 66667 | −27519 |
| dGTP | 14 | 71429 | −27690 |
| dCTP | 16 | 62500 | −27360 |
| dTTP | 7 | 42857 | −29408 |
Fluorescence spectroscopy was also used to evaluate a number of different parameters involved in the interaction between dNTPs and the enzyme–DNA complex. A critical component of the energetics of protein–ligand binding is the interaction of protein residues with the electrostatic field of the ligand. In order to evaluate the contribution of electrostatic interactions to the dNTP-binding activity, binding assays were performed in the presence of increasing ionic strength. The contribution of specific electrostatic interactions to the binding of dATP to the cytomegalovirus DNA polymerase–DNA complex was investigated by monitoring the salt dependence of the binding process. As shown in Figure 5(B), the dATP–enzyme interaction was attenuated at elevated KCl concentrations. The experimentally observed equilibrium constant was clearly dependent on the ionic strength of the solution. The equilibrium binding experiments showed that the apparent Kd at 500 mM KCl was 63 μM, more than 4-fold higher than at 10 mM KCl. This change in binding affinity corresponds to a ΔΔG of 3.6 kJ/mol. Evaluation of the Gibbs energy due to electrostatic interactions (ΔΔGES) by extrapolation of the binding reaction to an ionic strength of 1.0 M, the standard state where electrostatic interactions are effectively eliminated, revealed that 12.9% of the binding energy is derived from electrostatic interactions. These findings indicate that hydrogen-bonding and/or electrostatic interactions with the triphosphate moiety contribute to the binding of dNTPs to the enzyme–DNA complex.
Fluorescence spectroscopy was also used to evaluate the ΔG, as well as both the ΔH and the ΔS associated with the binding of dATP to the enzyme. Evaluation of these thermodynamic parameters yields important insights into the nature of the dATP binding reaction. Our binding studies indicate that the ΔG for the interaction of dATP with the enzyme was −28.6 kJ/mol. The precise enthalpic and entropic contributions to the free energy of binding were then determined by measuring the initial binding of dATP to the enzyme as a function of temperature. The binding reaction was shown to be connected with a high enthalpy of association, ΔH=−56.5 kJ/mol. Analysis of a van't Hoff plot for the interaction between dATP and the enzyme (Figure 5C) revealed that the TΔS value for the binding reaction was −27.9 kJ/mol, clearly indicating that the initial binding step is primarily driven by enthalpy, with an unfavourable entropic contribution. Moreover, these results indicate that the reaction is spontaneous at low temperatures but tends to reverse at higher temperatures. Accordingly, the binding of dATP could not be observed repeatedly at temperatures higher than 70 °C (results not shown). This can most probably be attributed to the denaturation of the protein that occurs at higher temperatures, and the concomitant destabilization of the architecture of the enzyme. The implications of these thermodynamic findings will be further discussed in the Discussion section.
Fluorescence spectroscopy was also used to monitor the kinetics involved in the binding of deoxynucleotides to the enzyme–DNA binary complex (Figure 5D). The binding reaction was followed for 30 s upon addition of a saturating concentration of dATP. Our results indicate that there is a rapid exponential decrease in fluorescence intensity following the addition of dATP corresponding to an apparent association rate of 0.4 μM−1·s−1. Half of the binding reaction was observed at approx. 0.5 s, while maximal binding was attained after 1.5 s of incubation with dATP, and remained constant thereafter. As was the case with the DNA-binding assays, the exponential decrease in fluorescence observed following the addition of dATP was not due to photobleaching, as similar results were obtained when the protein was incubated away from the light source (results not shown).
Effect of DNA binding on the structure of the enzyme
Limited information is available on the structure of the human cytomegalovirus DNA polymerase. Structural studies have been hindered by the absence of a defined crystal structure. Therefore structural information has been obtained through the use of modelling studies [34] based on other solved polymerases for which crystallographic information is available. As a consequence, we next sought to investigate whether the binding of DNA to the cytomegalovirus DNA polymerase results in a modification of the protein architecture. Near-UV CD spectra can provide useful information on the structural features of a protein, reflecting the environments of the aromatic amino acid side chains and giving information about the tertiary structure of a protein. In an effort to determine whether the binding of DNA results in the modification of the cytomegalovirus DNA polymerase structure, near-UV CD spectra were recorded both in the presence and the absence of DNA. Analysis of the near-UV CD spectra of the protein in both the absence and presence of DNA was performed from 250 to 400 nm. As can be seen in Figure 6(A), a significant reduction of the amplitude of the signal is observed over the 250–300 nm region when the protein is incubated with DNA. Overall, the CD spectra suggest that the protein undergoes conformational changes upon the formation of the enzyme–DNA binary complex.
Figure 6. Structural consequences of ligand binding to the cytomegalovirus DNA polymerase.

(A) Near-UV CD spectra were recorded for the unliganded protein (black line), the protein–DNA binary complex (thick line), and the transition state of the enzyme (protein–DNA–dATPαS) (dotted line). Formation of the protein–DNA binary complex was induced by incubating the enzyme (1 μM) with 100 μM DNA (primer template P20/T60). The transition state was generated by adding 1 mM of dATPαS to the binary complex. The spectra were recorded from 250 to 400 nm, and the average of three wavelength scans is presented. (B) Binding of ANS to the cytomegalovirus DNA polymerase during urea denaturation. The unliganded protein (1 μM) (■), binary complex (□) and transition state (○) were unfolded with various concentrations of urea at 22 °C for 1 h. Fluorescence emission was monitored after ANS addition (50 μM) at an excitation wavelength of 380 nm. The integrated fluorescence area between 400 and 600 nm was evaluated. (C) Thermal denaturation of the enzyme. Thermal denaturation was recorded for the unliganded protein (◇), the protein–DNA binary complex (□) or the transition state (○). CD spectra were recorded at a constant wavelength of 222 nm from 10 to 90 °C at a protein concentration of 1 μM. (D) The affinity of the enzyme for the substrate of 30 nt encompassing the cytomegalovirus immediate-early promoter sequence was also monitored in the absence (■) or presence of 1 mM foscarnet (□).
In order to gain additional insights into the structural modifications that occur upon DNA binding, we investigated the binding of a structural fluorescent reporter to the enzyme. The exposure of the hydrophobic area of the cytomegalovirus DNA polymerase was evaluated by measuring the binding of ANS to the protein. ANS is a reporter of exposed hydrophobic surfaces on proteins that binds with high affinity to hydrophobic patches, which results in an enhancement of ANS intrinsic fluorescence [35]. Our results revealed that the unliganded protein binds very weakly to ANS, probably reflecting limited hydrophobic regions at the surface of the protein (Figure 6B). A very modest modification of the ANS fluorescence is observed when the protein is incubated in the presence of saturating concentrations of DNA (Figure 6B). Increasing the urea concentrations did not modify the emission intensity. Overall, these results indicate that the conformational change that is observed upon DNA binding does not involve significant hydrophobic exposure on the surface of the enzyme.
Structural and mechanistic implications
Because our CD assays indicated conformational changes upon DNA binding to the cytomegalovirus DNA polymerase, this raises the possibility that the formation of the catalytic active site of the enzyme involves additional structural rearrangements. We therefore intended to use spectroscopic approaches to gain insights into the structural modifications that occur during catalysis. A heteropolymeric DNA substrate (P20/T60) and a non-hydrolysable dATP analogue, dATPαS (deoxyadenosine 5′-[α-thio]triphosphate), were used to mimic the transition state (enzyme–DNA–dATP–Mg2+). The non-hydrolysable dATPαS analogue was used since the presence of Mg2+, DNA and the classic dATP substrate would result in catalysis and incorporation of nucleotides to the DNA chain. Prior control experiments demonstrated that dATPαS is not efficiently used by the cytomegalovirus DNA polymerase in the presence of Mg2+ ions (results not shown). As can be seen in Figure 6(A), analysis of the near-UV CD spectra revealed that structural changes occur when both dATP and Mg2+ ions were added to the enzyme–DNA complex, indicating that the catalytic centre of the enzyme undergoes structural rearrangements upon formation of the transition state.
In an effort to gain insights into the reaction chemistry, the stability of the enzyme bound to various ligands was assessed by CD spectroscopy. Thermal denaturation assays were performed both in the presence and absence of ligands, and unfolding of the enzyme was evaluated by monitoring the changes in the α-helix content of the protein (222 nm). Thermal denaturation of the unliganded protein initially revealed a midpoint of thermal transition (Tm) of 56.2 °C (Figure 6C). Formation of the enzyme–DNA binary complex had no significant effect on the midpoint of thermal transition (Figure 6C and Table 3), and evaluation of the thermodynamic parameters of unfolding revealed similar values for the unliganded protein or the protein bound to DNA (Table 3). However, denaturation studies performed on the transition state (enzyme–DNA–dATP–Mg2+) indicated a reduced stability, reflecting the transient nature of the intermediate (Figure 6C and Table 3). Note that all of the denaturation assays revealed monophasic unfolding curves, suggestive of a two-state unfolding model. No intermediate state could be detected during the unfolding process.
Table 3. Thermodynamic unfolding parameters measured by thermal denaturation.
The thermodynamic parameters of unfolding ΔHVH (van't Hoff enthalpy of denaturation) and ΔS (entropy of denaturation) were determined by thermal denaturation of the unliganded cytomegalovirus DNA polymerase and the protein bound to various ligands. Temperatureinduced denaturations were monitored by CD spectroscopy at 222 nm. 1 kcal=4.184 kJ.
| Protein | Tm (°C) | ΔHVH (kcal/mol) | ΔS (kcal·mol−1·K−1) |
|---|---|---|---|
| UL54 | 56.2 | −82.0 | −0.25 |
| UL54·DNA | 56.8 | −56.4 | −0.17 |
| UL54·DNA·dATPαS·Mg2+ | 54.6 | −86.3 | −0.26 |
Finally, we sought to investigate whether the DNA binding activity of the UL54 could be influenced by the presence of foscarnet. Foscarnet is an analogue of PPi that is used for the treatment of cytomegalovirus infections. This antiviral acts non-competitively with respect to dNTPs but the precise mechanism of action of this drug remains elusive. However, it has previously been proposed that the PPi analogue could bind close to the active centre of the enzyme, thereby inhibiting the PPi exchange reaction. We therefore intended to monitor whether the binding of foscarnet could influence the binding of DNA to the UL54 protein. Fluorescence spectroscopy was used to measure the binding of DNA in the presence of foscarnet because initial experiments revealed that foscarnet had no influence on the intrinsic fluorescence properties of the UL54 protein (results not shown). The enzyme was therefore incubated with increasing concentrations of DNA in the presence or absence of 1 mM foscarnet. As shown in Figure 6(D), the presence of foscarnet had no influence on the DNA binding activity of the enzyme, as similar Kd values were determined in the absence (47.5 μM) or presence (45.8 μM) of foscarnet. This result clearly indicates that the binding of the enzyme to the DNA substrate is not influenced by the presence of foscarnet. Note that a similar conclusion was obtained using EMSAs (electrophoretic mobility-shift assays) performed with a radiolabelled DNA substrate (results not shown).
DISCUSSION
The human cytomegalovirus DNA polymerase is a critical enzyme for the replication of the virus [7–9,13]. As a specific target for antiviral chemotherapy, it plays a key role in the treatment of infected individuals [14–17]. However, characterization of the cytomegalovirus DNA polymerase has been very limited, and a number of critical questions about the reaction chemistry remain to be addressed. These include (i) what are the thermodynamic parameters that are involved in the formation of the enzyme–DNA binary complex of DNA? (ii) What are the conformational changes that occur during the binding of DNA to the enzyme? (iii) What is the minimal length of the DNA substrate required for stable binding? (iv) Is the binding sequence-specific? and (v) what are the thermodynamic and structural implications of dNTP binding? In this regard, the multidimensional properties of fluorescence spectroscopy and CD provide accurate sensitivity to monitor numerous aspects of protein–ligand interactions.
In the present study, spectroscopic approaches were used to allow, for the first time, a precise quantification of the kinetic and thermodynamic parameters associated with the binding of DNA to the cytomegalovirus DNA polymerase. Although these binding parameters do not provide a complete picture of the binding activity, they can suggest which features are likely to be important for the interaction, and provide a framework to construct an accurate model of the complex. For instance, changes in both the magnitude and the sign of ΔH and ΔS, in conjunction with structural data generated by spectroscopy, can provide crucial information about the structural alterations that accompany ligand binding in terms of (i) changes in salvation state; (ii) interactions between ligand and protein, such as electrostatic and hydrophobic interactions; and (iii) changes in conformation/dynamics induced by ligand binding. Our thermodynamic analyses indicate that the formation of the protein–DNA binary complex is an entropy-driven process (TΔS=32.7 kJ/mol). Such entropic contributions, which have been observed in other enzymes involved in DNA binding, are generally associated with conformational changes and/or from the release of water molecules to the bulk solvent upon DNA binding [36]. Our CD assays clearly demonstrated the existence of conformational changes in the cytomegalovirus DNA polymerase following the binding of DNA. Interestingly, hydrophobic interactions are also associated with a relatively small ΔH (as compared with ΔG) and a positive ΔS. Since the parameters for the UL54–DNA interaction fit these characteristics, it is tempting to speculate that hydrophobic interactions are also an important determinant for the formation of the complex. In accordance with this hypothesis, examination of the ionic strength dependence for the interaction between DNA and the protein reveals that the affinity of the enzyme for RNA is only slightly affected by an increased ionic strength (results not shown). Taken together with the thermodynamic parameters, these results suggest that hydrophobic and stacking interactions probably play a more significant role in the formation of the enzyme–DNA complex than ionic contacts, which would be expected to be completely disrupted upon an increase in ionic strength of the solvent.
In comparison with the DNA-binding analyses, our thermodynamic studies of the interaction between the enzyme and dNTPs revealed a completely different mode of binding. Binding of dNTPs to the enzyme–DNA binary complex was dominated by a favourable ΔH, but is also clearly associated with an unfavourable ΔH. Favourable negative ΔS are generally associated with contributions from hydrogen bonds, van der Waal's interactions or ionic interactions [36], while the unfavourable negative ΔS are associated with the exposure of hydrophobic surfaces to the surface of the protein, and/or to a decrease in conformational flexibility [37]. Although our salt-dependence assays clearly established the importance of electrostatic interactions for the binding of dATP to the protein–DNA binary complex, both our fluorescence spectroscopy and CD studies indicate that aromatic residues move to a more hydrophobic environment upon binding, suggesting that a decrease in conformational flexibility is probably encountered upon dATP binding.
The absence of a crystal structure of the cytomegalovirus DNA polymerase makes it difficult to precisely locate the conformational changes that occur upon DNA and dNTP binding. However, based on the crystal structure of the closely related bacteriophage RB69 DNA polymerase [38], a three-dimensional modelling of the cytomegalovirus DNA polymerase has recently been proposed, which can provide useful information on the location of specific residues of the cytomegalovirus DNA polymerase [34]. Interestingly, crystallographic studies of many DNA polymerases have revealed similar structural features that are similar to a partially opened right hand with thumb, palm and finger domain [39–42]. The thumb domain associates with the DNA template, the palm harbours the essential carboxylate residues that are involved in catalysis, and the fingers domain is involved in the interaction with the incoming nucleotide [39–42]. Moreover, the crystal structures of various conformations of the RB69 DNA polymerase have been elucidated, and have provided crucial information on the structural rearrangements that occur during catalysis. For instance, a comparison of the RB69 DNA polymerase crystals with and without DNA indicated structural modifications upon DNA binding. These DNA-induced changes are mainly confined to the thumb domain of the enzyme. Upon DNA binding, the entire thumb domain is translocated relative to its position in the unliganded enzyme, closing down around the DNA, and conferring a more ‘open’ conformation on the protein. Analysis of the RB69 polymerase crystal structure also indicated that the thumb domain predominantly contacts the phosphodiester backbone of the DNA. Based on our nucleic acid binding assays, we demonstrated that the cytomegalovirus UL54 protein does not discriminate between DNA and RNA, consistent with the binding of the enzyme to the phosphodiester backbone of nucleic acids. It is therefore tempting to speculate that the UL44 accessory subunit, which associates with UL54, is critical for the specificity of the DNA polymerase for the DNA substrate/template. Such an interaction between UL54 and UL44 might also be crucial for sequence-specific binding, since the binding of the UL54 protein to DNA is clearly not sequence-specific.
Interestingly, the Kd values that were determined in the present study for the various dNTPs (Table 1) are significantly higher than the Km values that were previously evaluated [14,22,34] from polymerization assays (ranging from 0.16 to 0.8 μM). Similar Km values were also obtained with the purified protein used in the present study (results not shown). A likely explanation for the observation that the Km values are smaller than the respective Kd values is that the uptake of the occupied binding site for dNTPs is probably fast relative to the reappearance of the unoccupied binding site during catalysis. Moreover, it is interesting to note that substrate-independent Vmax has previously been observed for the UL54 subunit [22]. This suggests that a substrate-independent step in catalysis is rate-limiting. We envision that this is probably the reappearance of the unoccupied binding site for dNTPs during catalysis, which is consistent with a small Km/Kd ratio.
Crystal structures have now been elucidated for a number of DNA polymerases bound to various ligands (DNA, dNTPs and metal ions). A general model for DNA polymerization has been generated by comparison of these different structures [39–42]. The initial step involves the binding of the DNA polymerase to the DNA substrate, which causes the thumb domain of the enzyme to close around the DNA, leading to a more ‘open’ conformation to the protein. The next step involves the binding of the dNTP to this complex. This causes a conformational change in the fingers domain of the enzyme, which rotates towards the catalytic centre of the enzyme, conferring a more ‘closed’ catalytically competent conformation on the protein [39–42]. It is interesting to notice that such conformational changes have now been observed for the UL54 catalytic subunit of the cytomegalovirus DNA polymerase. Using spectroscopic approaches, the present study demonstrated that the protein undergoes structural modifications upon formation of the enzyme–DNA complex, which is followed by another conformational change upon dNTP binding. Although the complete understanding of the mechanisms underlying the replication of cytomegalovirus is still largely incomplete, characterization of the individual biochemical steps involved in catalysis should provide the basis for further studies in this direction. We believe that structural measurements performed on purified proteins should enable us to correlate conformation and kinetic parameters of ligand recognition, and add another dimension to investigate the individual biochemical steps involved in the replication of cytomegalovirus.
Acknowledgments
We thank Dr Jay A. Nelson (Oregon Health and Sciences University, Portland, OR, U.S.A.) for generously providing the initial plasmid harbouring the UL54 gene. We are also very grateful to Audrey Dubé, Dominique Lévesque and François Bachand for technical assistance. This work was supported by a grant from the Canadian Institutes for Health Research. M. B. is a New Investigator Scholar from the Canadian Institutes for Health Research.
References
- 1.Britt W. J., Alford C. A. Philadelphia: Lippincott-Raven Publishers; 1996. Fields Virology. [Google Scholar]
- 2.Trincado D. E., Scott G. M., White P. A., Hunt C., Rasmussen L., Rawlinson W. D. Human cytomegalovirus strains associated with congenital and perinatal infections. J. Med. Virol. 2000;61:481–487. doi: 10.1002/1096-9071(200008)61:4<481::aid-jmv11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 3.Chou S. W. Cytomegalovirus drug resistance and clinical implications. Transplant. Infect. Dis. 2001;3:20–24. doi: 10.1034/j.1399-3062.2001.00004.x. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert C., Boivin G. Human cytomegalovirus resistance to antiviral drugs. Antimicrob. Agents Chemother. 2005;49:873–883. doi: 10.1128/AAC.49.3.873-883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Springer K. L., Weinberg A. Cytomegalovirus infection in the era of HAART: fewer reactivations and more immunity. J. Antimicrob. Chemother. 2004;54:582–586. doi: 10.1093/jac/dkh396. [DOI] [PubMed] [Google Scholar]
- 6.Weinberg A., Jabs D. A., Chou S., Martin B. K., Lurain N. S., Forman M. S., Crumpacker C. Mutations conferring foscarnet resistance in a cohort of patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis. J. Infect. Dis. 2003;187:777–784. doi: 10.1086/368385. [DOI] [PubMed] [Google Scholar]
- 7.Ertl P. F., Powell K. L. Physical and functional interaction of human cytomegalovirus DNA polymerase and its accessory protein (ICP36) expressed in insect cells. J. Virol. 1992;66:4126–4133. doi: 10.1128/jvi.66.7.4126-4133.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heilbronn R., Jahn G., Burkle A., Freese U. K., Fleckenstein B., zur Hausen H. Genomic localization, sequence analysis, and transcription of the putative human cytomegalovirus DNA polymerase gene. J. Virol. 1987;61:119–124. doi: 10.1128/jvi.61.1.119-124.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye L. B., Huang E. S. In vitro expression of the human cytomegalovirus DNA polymerase gene: effects of sequence alterations on enzyme activity. J. Virol. 1993;67:6339–6347. doi: 10.1128/jvi.67.11.6339-6347.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mar E. C., Patel P. C., Huang E. S. Human cytomegalovirus-associated DNA polymerase and protein kinase activities. J. Gen. Virol. 1981;57:149–156. doi: 10.1099/0022-1317-57-1-149. [DOI] [PubMed] [Google Scholar]
- 11.Weiland K. L., Oien N. L., Homa F., Wathen M. W. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res. 1994;34:191–206. doi: 10.1016/0168-1702(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 12.Loregian A., Rigatti R., Murphy M., Schievano E., Palu G., Marsden H. S. Inhibition of human cytomegalovirus DNA polymerase by C-terminal peptides from the UL54 subunit. J. Virol. 2003;77:8336–8344. doi: 10.1128/JVI.77.15.8336-8344.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crumpacker C. S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992;92:3S–7S. doi: 10.1016/0002-9343(92)90329-a. [DOI] [PubMed] [Google Scholar]
- 14.Xiong X., Smith J. L., Kim C., Huang E. S., Chen M. S. Kinetic analysis of the interaction of cidofovir diphosphate with human cytomegalovirus DNA polymerase. Biochem. Pharmacol. 1996;51:1563–1567. doi: 10.1016/0006-2952(96)00100-1. [DOI] [PubMed] [Google Scholar]
- 15.Xiong X., Smith J. L., Chen M. S. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob. Agents Chemother. 1997;41:594–599. doi: 10.1128/aac.41.3.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eriksson B., Oberg B., Wahren B. Pyrophosphate analogues as inhibitors of DNA polymerases of cytomegalovirus, herpes simplex virus and cellular origin. Biochim. Biophys. Acta. 1982;696:115–123. doi: 10.1016/0167-4781(82)90018-5. [DOI] [PubMed] [Google Scholar]
- 17.Derse D., Bastow K. F., Cheng Y. Characterization of the DNA polymerases induced by a group of herpes simplex virus type I variants selected for growth in the presence of phosphonoformic acid. J. Biol. Chem. 1982;257:10251–10260. [PubMed] [Google Scholar]
- 18.Griffiths P. D. Cytomegalovirus therapy: current constraints and future opportunities. Curr. Opin. Infect. Dis. 2001;14:765–768. doi: 10.1097/00001432-200112000-00016. [DOI] [PubMed] [Google Scholar]
- 19.Chou S., Lurain N. S., Thompson K. D., Miner R. C., Drew W. L. Viral DNA polymerase mutations associated with drug resistance in human cytomegalovirus. J. Infect. Dis. 2003;188:32–39. doi: 10.1086/375743. [DOI] [PubMed] [Google Scholar]
- 20.Cihlar T., Fuller M. D., Cherrington J. M. Characterization of drug resistance-associated mutations in the human cytomegalovirus DNA polymerase gene by using recombinant mutant viruses generated from overlapping DNA fragments. J. Virol. 1998;72:5927–5936. doi: 10.1128/jvi.72.7.5927-5936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith I. L., Cherrington J. M., Jiles R. E., Fuller M. D., Freeman W. R., Spector S. A. High-level resistance of cytomegalovirus to ganciclovir is associated with alterations in both the UL97 and DNA polymerase genes. J. Infect. Dis. 1997;176:69–77. doi: 10.1086/514041. [DOI] [PubMed] [Google Scholar]
- 22.Tchesnokov E. P., Gilbert C., Boivin G., Gotte M. Role of helix P of the human cytomegalovirus DNA polymerase in resistance and hypersusceptibility to the antiviral drug foscarnet. J. Virol. 2006;80:1440–1450. doi: 10.1128/JVI.80.3.1440-1450.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cihlar T., Fuller M. D., Cherrington J. M. Expression of the catalytic subunit UL54 and the accessory protein UL44 of human cytomegalovirus DNA polymerase in a coupled in vitro transcription/translation system. Protein Expression Purif. 1997;11:209–218. doi: 10.1006/prep.1997.0781. [DOI] [PubMed] [Google Scholar]
- 24.Loregian A., Appleton B. A., Hogle J. M., Coen D. M. Residues of human cytomegalovirus DNA polymerase catalytic subunit UL54 that are necessary and sufficient for interaction with the accessory protein UL44. J. Virol. 2004;78:158–167. doi: 10.1128/JVI.78.1.158-167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito J., Brathwaite D. K. Compilation and alignment of DNA polymerase sequences. Nucleic Acids Res. 1991;19:4045–4057. doi: 10.1093/nar/19.15.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bougie I., Bisaillon M. Initial binding of the broad spectrum antiviral nucleoside ribavirin to the hepatitis C virus RNA polymerase. J. Biol. Chem. 2003;278:52471–52478. doi: 10.1074/jbc.M308917200. [DOI] [PubMed] [Google Scholar]
- 27.Bougie I., Charpentier S., Bisaillon M. Characterization of the metal ion binding properties of the hepatitis C virus RNA polymerase. J. Biol. Chem. 2003;278:3868–3875. doi: 10.1074/jbc.M209785200. [DOI] [PubMed] [Google Scholar]
- 28.Bougie I., Parent A., Bisaillon M. Thermodynamics of ligand binding by the yeast mRNA-capping enzyme reveals different modes of binding. Biochem. J. 2004;384:411–420. doi: 10.1042/BJ20041112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flowers S., Biswas E. E., Biswas S. B. Conformational dynamics of DnaB helicase upon DNA and nucleotide binding: analysis by intrinsic tryptophan fluorescence quenching. Biochemistry. 2003;42:1910–1921. doi: 10.1021/bi025992v. [DOI] [PubMed] [Google Scholar]
- 30.Painter G. R., Wright L. L., Hopkins S., Furman P. A. Initial binding of 2′-deoxynucleoside 5′-triphosphates to human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 1991;266:19362–19368. [PubMed] [Google Scholar]
- 31.Pan J. Y., Sanford J. C., Wessling-Resnick M. Effect of guanine nucleotide binding on the intrinsic tryptophan fluorescence properties of Rab5. J. Biol. Chem. 1995;270:24204–24208. doi: 10.1074/jbc.270.41.24204. [DOI] [PubMed] [Google Scholar]
- 32.Zhou T., Rosen B. P. Tryptophan fluorescence reports nucleotide-induced conformational changes in a domain of the ArsA ATPase. J. Biol. Chem. 1997;272:19731–19737. doi: 10.1074/jbc.272.32.19731. [DOI] [PubMed] [Google Scholar]
- 33.Eftink M. R., Ghiron C. A. Exposure of tryptophanyl residues in proteins: quantitative determination by fluorescence quenching studies. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 34.Shi R., Azzi A., Gilbert C., Boivin G., Lin S. X. Three-dimensional modeling of cytomegalovirus DNA polymerase and preliminary analysis of drug resistance. Proteins. 2006;64:301–307. doi: 10.1002/prot.21005. [DOI] [PubMed] [Google Scholar]
- 35.Labowicz J. R. 2nd Edn. New York: Kluwer/Plenum; 1999. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 36.Beaudette N. V., Langerman N. The thermodynamics of nucleotide binding to proteins. Crit. Rev. Biochem. 1980;9:145–169. doi: 10.3109/10409238009105433. [DOI] [PubMed] [Google Scholar]
- 37.Tame J. R. H., O'Brien R., Ladbury J. E. London: John Wiley and Sons; 1998. Biocalorimetry: Applications of Calorimetry in Biological Sciences. [Google Scholar]
- 38.Franklin M. C., Wang J., Steitz T. A. Structure of the replicating complex of a polα family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 39.Brautigam C. A., Steitz T. A. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 40.Delarue M., Poch O., Tordo N., Moras D., Argos P. An attempt to unify the structure of polymerases. Protein Eng. 1990;6:461–467. doi: 10.1093/protein/3.6.461. [DOI] [PubMed] [Google Scholar]
- 41.Ollis D. L., Brick P., Hamlin R., Xuong N. G., Steitz T. A. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 42.Savino C., Federici L., Johnson K. A., Vallone B., Nastopoulos V., Rossi M., Pisani F. M., Tsernoglou D. Insights into DNA replication: the crystal structure of DNA polymerase B1 from the archaeon Sulfolobus solfataricus. Structure. 2004;12:2001–2008. doi: 10.1016/j.str.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 43.Wang T. S., Wong S. W., Korn D. Human DNA polymerase α: predicted functional domains and relationships with viral DNA polymerases. FASEB J. 1989;3:14–21. doi: 10.1096/fasebj.3.1.2642867. [DOI] [PubMed] [Google Scholar]