

Abstract
Krüppel-like factor 4 (KLF4) is a zinc-finger-containing transcription factor, the expression of which is enriched in the postmitotic cells of the intestinal epithelium. KLF4 is a target gene of the tumor suppressor adenomatous polyposis coli (APC). We sought to determine the role of KLF4 in suppressing the tumorigenecity of RKO colon cancer cells, which do not express KLF4. We utilized an established system in RKO cells, in which an inducible promoter controls expression of KLF4. Four independent assays were used to assess the effects of KLF4 induction on tumor cells. We find that KLF4 overexpression reduces colony formation, cell migration and invasion, and in vivo tumorigenecity. The mechanism of action of KLF4 does not involve apoptosis. These findings, along with our previous findings that KLF4 induces G1/S arrest, suggest that KLF4 is a cell cycle checkpoint protein that can reduce tumorigenecity of colon cancer cells.
Keywords: Krüppel-like factor 4, tumorigenecity, RKO, colorectal cancer
Krüppel-like factor 4 (KLF4; formerly known as gutenriched Krüppel-like factor or GKLF) is a transcription factor with three carboxyl C2H2 zinc-fingers that exhibit homology to the Drosophila melanogaster segmentation gene product Krüppel (Shields et al., 1996). Since KLF4 was first described in 1996, there have been a variety of reports on its tissue distributions and functions. Expression of KLF4 appears to be enhanced primarily in the postreplicative epithelial cells of the intestine, skin, and thymus (Shields et al., 1996). Consistent with its tissue distribution, KLF4 has been shown to be imperative for terminal differentiation of keratinocytes of the skin (Segre et al., 1999) and goblet cells of the intestines (Katz et al., 2002). In the thymus, KLF4 is expressed during cortical differentiation suggesting a role in T-lymphocyte differentiation (Panigada et al., 1999). KLF4 is also highly expressed in the testes, specifically in the postmeiotic germ cells and somatic Sertoli cells, suggesting an important role in testicular differentiation (Behr and Kaestner, 2002). Finally, KLF4 is expressed in vascular endothelial cells (Yet et al., 1998), and may function to inhibit TGF-β-mediated differentiation of vascular smooth muscle cells (Adam et al., 2000).
Depending on cell type, KLF4 expression has been reported to be associated with both inhibition and induction of proliferation. In NIH 3T3 fibroblasts, increased KLF4 expression is associated with conditions that cause growth arrest such as serum deprivation or contact inhibition (Shields et al., 1996). Forced expression of KLF4 by transfection in the same cell line inhibits DNA synthesis. In mouse embryonic fibroblasts, KLF4 interacts with the tumor suppressor p53 and induces the cyclin-dependent kinase inhibitor p21 (Zhang et al., 2000). KLF4 expression is decreased in early intestinal adenomas, colonic adenomas, and colonic adenocarcinomas of mice and patients with hereditary and sporadic tumors, suggesting that decreased KLF4 expression may contribute to tumorigenesis (Dang et al., 2000; Shie et al., 2000b). Indeed, the tumor suppressor adenomatous polyposis coli (APC) regulates KLF4 via induction of CDX2 (Dang et al., 2001). CDX2 is a homeobox gene specifically expressed in the intestine, and is important for regulation of intestinal epithelial cell development and maintenance (Suh and Traber, 1996). In HT29 and RKO colon cancer cells, overexpression of KLF4 leads to repression of cyclin D1 (Shie et al., 2000a; Chen et al., 2001). In addition, KLF4 overexpression in RKO cells induces p21 (Chen et al., 2001). The mechanism by which KLF4 inhibits growth in colon cancer cells remains unclear. While studies in HT29 colon cancer cells reveal that KLF4 causes both G1/S arrest and apoptosis (Chen et al., 2000; Shie et al., 2000b), studies in RKO and HCT116 colon cancer cells show evidence of only G1/S arrest (Chen et al., 2001; Yoon et al., 2003). In the prostate, KLF4 expression is decreased in prostate cancer and benign prostate hypertrophy (Foster et al., 2000; Luo et al., 2002). KLF4 is also expressed in the esophageal squamous epithelium, where it activates the promoters of several differentiation genes: Epstein–Barr virus ED-L2, keratin 4, and keratin 19 (Jenkins et al., 1998; Brembeck and Rustgi, 2000). In vascular smooth muscle cells, KLF4 inhibits proliferation of redox-sensitive growth and induces p21, p27, p53, and retinoblastoma (Rb) (Nickenig et al., 2002). Contrary to these studies, all of which show a growth-inhibitory effect, KLF4 has transforming activity in RK3E primary rat kidney cells (Foster et al., 1999). In the same vein, KLF4 expression is increased in primary breast ductal carcinoma and oral squamous cancers, while KLF4 overexpression inhibits integrin expression and induces the tumor marker clusterin, both changes that enhance tumor progression and metastasis (Foster et al., 2000). It is unclear whether somatic mutations of KLF4 may contribute to the tumor phenotype in the last two studies.
To better understand the role of KLF4 in colorectal cancer, we developed an inducible system for KLF4 in the human colon cancer cell line RKO (Chen et al., 2001). RKO is a poorly differentiated colorectal cancer cell line that expresses wild-type APC, β-catenin, and p53. RKO cells, derived from sporadic tumors exhibiting microsatellite instability (Liu et al., 1995), are deficient in hMLH1 because of hypermethylation of the gene promoter region (Veigl et al., 1998). More recently, RKO cells have been found to have a mutated allele of CDX2 (da Costa et al., 1999) and undetectable levels of KLF4 (Dang et al., 2001).
Our inducible system consists of RKO-EcR and RKO-EcR-KLF4 cell lines. The cell line RKO-EcR was generated by stably transfecting RKO cells with the pVgRXR plasmid (Invitrogen, Carlsbad, CA, USA), as previously described (Chen et al., 2001). The pVgRXR plasmid contains the Zeocin resistance gene and constitutively expresses a modified D. melanogaster ecdysone receptor (EcR) and the human retinoid X receptor (RXR). Upon addition of the insect hormone ecdysone (Ponasterone A), EcR and RXR heterodimerize to form a functional ecdysone receptor that transactivates the ecdysone-inducible promoter (EcRE). The RKO-EcR human colon cancer cell line does not harbor the EcRE insect promoter. Thus, addition of Ponasterone A to RKO-EcR cells will facilitate heterodimerization without subsequent gene induction (No et al., 1996). The cell line RKO-EcR-KLF4 was generated by stably transfecting RKO-EcR cells with the pAdLoxEGI-KLF4 plasmid, as previously described (Chen et al., 2001). The pAdLoxEGI-KLF4 plasmid contains the EcRE linked to an expression cassette containing the enhanced green fiuorescence protein (EGFP), followed by an internal ribosome entry site and the full-length coding region of KLF4 cDNA. Thus, addition of Ponasterone A to RKO-EcR-KLF4 cells induces heterodimerization of EcR and RXR, which bind and transactivate EcRE to induce EGFP and KLF4 gene expression.
We deduced that RKO-EcR-KLF4 cells would provide a background in which we can test the role of KLF4 in tumors, independent of aberrant APC or β-catenin signaling. We find that overexpression of KLF4 inhibits colony formation, migration, and invasion in vitro. In vivo, KLF4 overexpression has pronounced effects on tumorigenecity when grown as xenografts in athymic nude mice. To address the mechanisms by which KLF4 inhibits growth, we test for the evidence of apoptosis in RKO cells induced for KLF4 and find that overexpression of KLF4 does not induce apoptosis.
KLF4 overexpression decreases colony formation, migration, and invasion
To investigate the role of KLF4 in the tumor phenotype of RKO cells, we overexpressed KLF4 in RKO cells and tested in vitro tumor properties such as colony formation, migration, and invasion. Overexpression of KLF4 reduces colony formation in an anchorage-independent environment. Figure 1a shows the rate of colony formation in RKO-EcR-KLF4 cells induced for KLF4 overexpression to be 20 ± 9% of uninduced cells. In contrast, RKO-EcR cells do not exhibit any difference in colony formation when induced; thus the rate of colony formation in RKO-EcR cells is 100 ± 13%. When we compare the differences in colony formation rates between RKO-EcR-KLF4 and RKO-EcR cells, they are significant (P < 0.0001). Thus, KLF4 induction decreases anchorage independent colony formation by approximately 80%.
Figure 1.
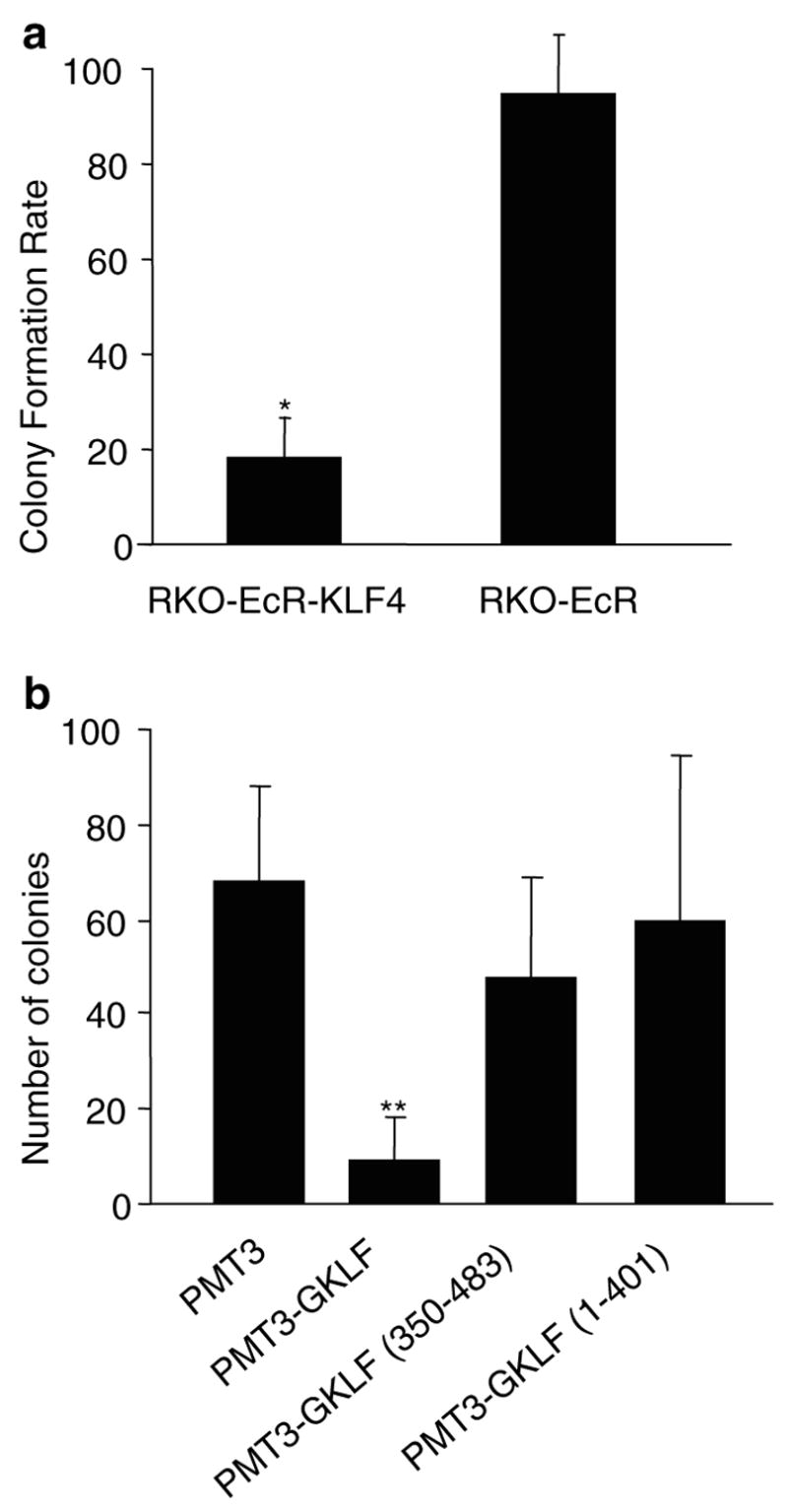
Effect of KLF4 induction on colony formation. (a) Anchorage-independent colony formation. For each cell type (RKO-EcR- KLF4 or RKO-EcR), the colony formation rate was calculated by dividing the number of spherical colonies in each Ponasterone A-treated well (induced condition) by the number of colonies in the corresponding ethanol-treated well (uninduced condition), and multiplied by 100. Treatment of RKO-EcR-KLF4 cells with 5 μm Ponasterone A results in overexpression of KLF4 (Chen et al., 2001). Two-tailed Student’s t-test was used to determine whether the differences in rates of colony formation between the two different cell types, RKO-EcR-KLF4 versus RKO-EcR were statistically significant. Each value is the average of three triplicate dishes, *P < 0.0001. RKO-EcR-KLF4 and RKO-EcR cells were treated with either 5 μm Ponasterone A or equal volume of ethanol (the vehicle in which Ponasterone A is suspended), suspended in 0.33% agar in complete growth medium, and plated on six-well plates layered with 0.5% agar in complete growth medium at a concentration of 2.0 × 104 cells/well. Cells were allowed to grow for 2 weeks and spherical colonies counted. Complete growth medium consisted of Dubelco’s minimal essential media (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml of streptomycin, and 150 μg/ml Zeocin (Invitrogen). Ponasterone A was obtained from Invitrogen as a powder and resuspended as per the manufacturer’s directions in 100% ethanol. (b) Colony suppression assays with different KLF4 residues. RKO cells were cotransfected (50 : 1) with 980 ng/six-well dish of PMT3, PMT3- KLF4 (full-length KLF4), PMT3-KLF4 (350–483), which contains the C-terminal nuclear localizing signal and three zinc-fingers, or PMT3- KLF4 (1–401), which contains the N-terminal region including the nuclear localization signal but excluding the zinc-fingers and 20 ng/six-well dish of pBabe Puro as previously described (Geiman et al., 2000). 2 days following transfection, cells were fed complete media containing 0.75 μg/ml puromycin (Sigma, St Louis, MO, USA) for 2 weeks. Resistant colonies of cells were stained with 0.1% crystal violet and counted using Scion image software for Windows (www.scioncorp.com). Each value represents the mean number of colonies for six wells. **P < 0.01 by two-tailed Student’s t-test between PMT3- and PMT3- KLF4-transfected cells. All transfections were performed using the Lipofectamine reagent protocol (Invitrogen) on 10–15% confluent RKO cells
To address whether the effects seen are indeed secondary to overexpression of KLF4, we performed colony suppression assays in RKO cells stably cotransfected with pBabe Puromycin and either full-length or the carboxyl or amino terminal portions of KLF4. Figure 1b shows that full-length KLF4 significantly suppresses colony formation by at least 50%, when compared to control plasmid (P < 0.01). The C- and the N-terminal portions of KLF4 do not significantly mediate growth suppression.
Another property of tumors is their ability to migrate and invade. To investigate the effect of KLF4 on migration, we induced KLF4 overexpression and performed migration assays. Figure 2a shows that after 6 h, the number of KLF4-induced RKO-EcR-KLF4 cells that have migrated across a transwell filter is 20 ± 11% of uninduced cells. In contrast, the number of induced RKO-EcR cells that migrate across the filter is 94 ± 15% of uninduced cells. The 80% difference in migration rates between RKO-EcR-KLF4 and RKO-EcR cells is significant (P < 0.0001). Similarly, KLF4 overexpression significantly inhibits the rates of cell invasion. Figure 2b shows that 86 ± 2.3% of RKO-EcR-KLF4 cells induced for KLF4 for 6 h invade through a matrigel-coated filter compared to similar uninduced cells. This rate is significantly less than the rate of invasion for RKO-EcR cells (100 ± 4.5%, P < 0.01).
Figure 2.
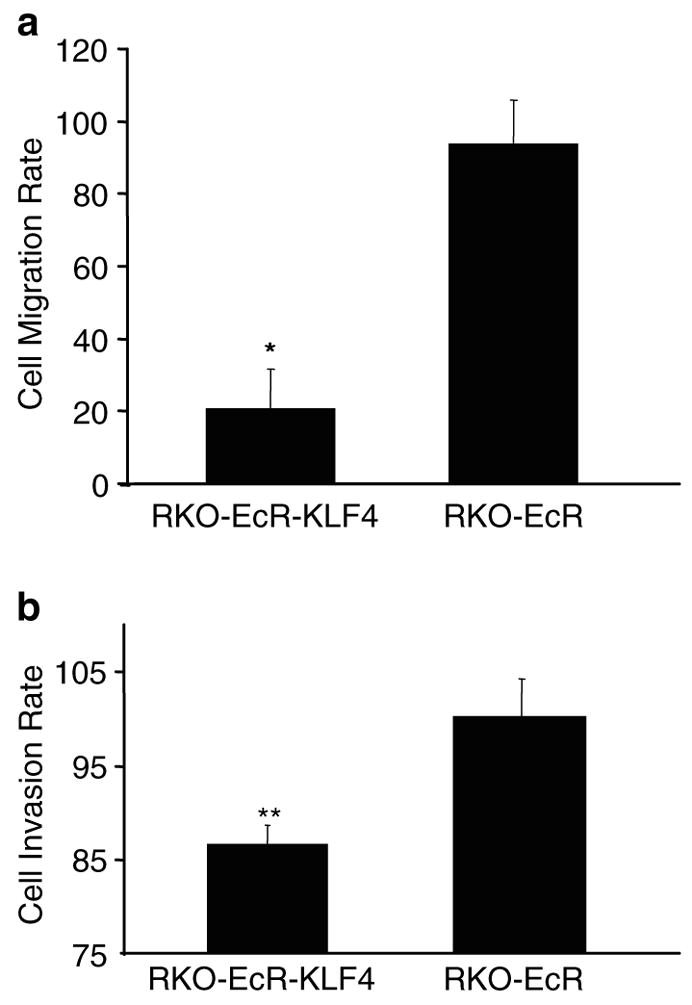
Effect of KLF4 induction on cell migration and invasion. For each cell type, RKO-EcR-KLF4 or RKO-EcR, cell migration and invasion rates were calculated by dividing the number of migrating or invading cells in the induced condition by the number in the corresponding uninduced condition, and multiplied by 100. KLF4 is induced only in RKO-EcR-KLF4 cells treated with Ponasterone A. Means and standard deviations of cell migration and invasion rates were calculated and plotted. Two-tailed Student’s t-test was used to determine whether the differences in rates of migration or invasion between the two different cell types, RKO-EcR-KLF4 versus RKO-EcR, were statistically significant. All migration and invasion assays were performed in triplicate wells and repeated twice. *P < 0.0001, **P < 0.01. (a) Cell migration was assayed in six-well plates with an 8.0 μm pore size polycarbonate membrane (Transwell, Costar, Cambridge, MA, USA) as previously described (Pouliot et al., 2001). Briefly, the membranes were rinsed twice with phosphate-buffered saline (PBS) and allowed to equilibrate in DMEM over 24 h. Thereafter, the lower chamber was filled with 2.6 ml of complete media (DMEM, antibiotics, and 10% FBS). The upper chamber was filled with 1.5 ml of serum-free medium and 1.0 × 105 RKO-EcR or RKO-EcR-KLF4 cells treated with either 5 μm Ponasterone A or equal volume of ethanol. The plates were incubated at 37°C. After 6 h, cells in the culture medium from the lower chamber were collected and strongly adherent cells on the underside of the membrane and bottom of the lower chamber were detached with trypsin and pooled. The cells were pelleted and the number of migrated cells counted on a hemocytometer. To assess for cell proliferation, the cells on the upper chamber were detached with trypsin and counted on a hemocytometer. The total number of cells at the end of 6 h was roughly equivocal to the number plated. (b) Tumor cell invasion was assayed using six-well plates with an 8.0 μm pore size polycarbonate membrane coated with Matrigel Matrix (BD Biosciences, Chicago, IL, USA) as previously described (Rozic et al., 2001). Briefly, the lower chamber was filled with 2.6 ml of complete media (DMEM, antibiotics, and 10% FBS). The upper chamber was filled with 1.5 ml of serum-free medium and 1 × 105 RKO-EcR or RKO-EcR-KLF4 cells treated with either 5 μm Ponasterone A or equal volume of ethanol. The plates were incubated at 37°C. After 6 h, cells remaining on the upper surface of the membrane were removed with a cotton swab. The membranes were then fixed and stained with the Diff Quik staining kit (Dade AG, Dudingen, Switzerland). Membranes were removed from transwells, mounted on glass slides, and cells counted under a light microscope (an average of five random nonoverlapping fields at 400 × magnification)
These findings indicate that full-length KLF4 potently inhibits colony formation, migration, and invasion in RKO colon cancer cells in vitro. Of note, we find that in vitro colony suppression by KLF4 is diminished after 6 weeks of induction, as are migration and invasion after 24 h of induction (data not shown). These findings are associated with decreased EGFP expression and may reflect overgrowth of untransfected cells (Chen et al., 2001). To control for this, all studies are performed with the same passage of cells.
KLF4 overexpression decreases in vivo tumor formation
To investigate the role of KLF4 on in vivo tumor growth, we implanted athymic nude mice with RKO-EcR-KLF4 cells on the right flank, RKO-EcR cells on the left flank, and treated the mice with intraperitoneal Ponasterone A or placebo over the next 3 weeks. Figure 3a shows that 3 weeks after tumor implantation, tumor volumes are smaller in tumors overexpressing KLF4 (RKO-EcR-KLF4+PA) when compared to the other three control-treated tumors (mean volumes of 0.35 cm3 compared to 0.8–1.07 cm3). The differences were significant between RKO-EcR-KLF4 and RKO-EcR tumors from mice treated with Ponasterone A (P < 0.05).
Figure 3.
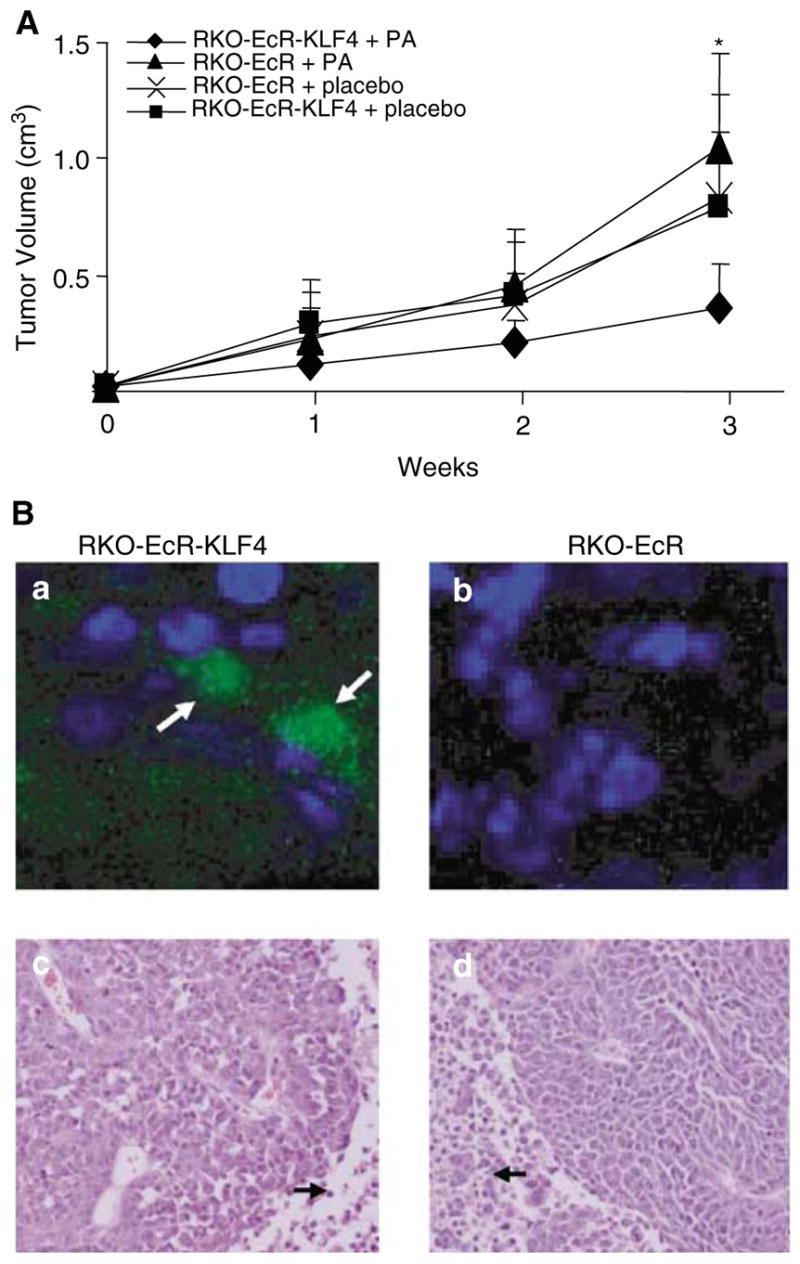
Effect of KLF4 induction on in vivo tumor growth. (a) In vivo tumor growth of RKO-EcR and RKO-EcR-KLF4 cells treated with Ponasterone A (PA) or placebo in athymic mice. KLF4 is induced only in RKO-EcR-KLF4 cells treated with PA. The other three conditions are negative controls. 1.0 × 106 RKO-EcR and RKO-EcR-KLF4 cells were harvested, treated accordingly with 5 μm PA or ETOH, and inoculated subcutaneously in the flanks of 6-week-old female athymic nu/nu mice (Charles River Labs, Wilmington, MA, USA). Each mouse served as its own control, since RKO-EcR cells were injected on the left flank and RKO-EcR-KLF4 cells on the right flank. Mice were subsequently treated twice per week with intraperitoneal injections of either 5.0 mg PA dissolved in DMSO and mixed with 100–150 μl of sesame oil or equal volume of DMSO mixed with sesame oil as previously reported (No et al., 1996). Tumor sizes in two dimensions were measured weekly, and volumes were calculated with the formula (L × W2) × 0.5, where L is the length and W is the width. At the end of 3 weeks, mice were euthanized (because of tumor burden) and the tumor xenografts harvested for imaging. Mice were housed in barrier environments, with food and water provided ad libitum as approved by the Johns Hopkins Animal Care and Use Committee. Means and standard deviations of tumor volumes were calculated and plotted. Two-tailed Student’s t-test was used to determine statistical significance between groups. *P < 0.05 between RKO-EcR-KLF4 (-♦-) and RKO-EcR (-▲-) in mice treated with PA. Each experimental group was composed of 10 mice and was repeated once (total N = 20 for each group). (b) Top panel: confocal photomicrographs of 3-week-old explanted (a) RKO-EcR-KLF4 and (b) RKO-EcR xenografts from mice treated with PA, which induces KLF4 and EGFP in RKO-EcR-KLF4 cells only. Nuclei are stained blue with Hoechst solution. Arrows point to EGFP in a subset of RKO-EcR-KLF4 cells. Tumor explants from mice were frozen in OCT compound (Tissue-Tek) and sectioned onto glass slides with a cryostat. Sections were then fixed in 0.05% glutaraldehyde in PBS for 10 min and stained with 0.1% Hoechst 33258 (Sigma) in solution with 3.7% formaldehyde and 0.5% Nonidet P-40 in PBS. Sections were then washed in Tris-buffered saline with 0.1% Triton X-100 (TBST) for 5 min, twice in distilled water for 2 min, mounted with Fluorescent Mounting Medium (Dako, Carpinteria, CA, USA), and visualized under confocal microscopy. (b) Bottom panel: light photomicrographs ( × 20) of 3-week old explanted (c) RKO-EcR-KLF4 and (d) RKO-EcR xenografts from mice treated with PA. Representative sections from both tumor types demonstrate poorly differentiated carcinoma. Essentially no inflammatory infiltrate is seen and the tumor is homogenous in appearance. Scattered mitoses and occasional apoptotic bodies are present. The central portions of the neoplasms (arrows) show tumor necrosis. Tumor explants from mice were fixed in 4% paraformaldehyde, paraffin embedded, sectioned, and stained with hematoxylin and eosin by the Johns Hopkins Comparative Pathology Department
To assess whether tumor xenografts still express the plasmid containing EGFP and KLF4 after 3 weeks, we examined RKO-EcR-KLF4 and RKO-EcR tumor explants from animals treated with Ponasterone A. Figure 3b, top panel, shows that a subset of cells in the RKO-EcR-KLF4 explants still expresses EGFP, which implies continued concomitant KLF4 expression. Figure 3b, bottom panel, shows that the tumors are composed of a homogenous population of poorly differentiated carcinoma, with evidence of central necrosis. These results show that KLF4 overexpression in RKO cells markedly reduces in vivo tumor growth.
KLF4 overexpression does not induce apoptosis
To further investigate the mechanism by which KLF4 inhibited growth, we studied the effects of KLF4 overexpression on the cell cycle and apoptosis. FACS analyses of induced RKO-EcR-KLF4 cells confirm the previous findings of G1/S arrest (Chen et al., 2001). Again, our FACS analyses show no evidence of apoptosis (Chen et al., 2001). To confirm these findings, we performed two additional assays for apoptosis by looking for morphological changes in induced cells with Hoechst nuclear stain and by detection of a ladder pattern on genomic DNA gel electrophoresis. Figure 4a is a representative of our results with Hoechst nuclear staining. In RKO-EcR-KLF4 cells treated with Ponasterone A to induce KLF4, we see no evidence of nuclear condensation, chromatin fragmentation, apoptotic bodies, or plasma membrane disruption. In contrast, RKO-EcR-KLF4 cells treated with 5-FU, a known proapoptotic agent (Rigas et al., 2002), exhibit these morphological changes. Figure 4b shows results of electrophoresis of genomic DNA from RKO-EcR and RKO-EcR-KLF4 cells treated with Ponasterone A, ethanol, or 5-FU. Genomic DNA from cells treated with 5-FU form a ladder, a finding associated with apoptosis. In contrast, KLF4 induction with Ponasterone A and negative controls do not show evidence of a DNA ladder. These results show that KLF4 does not induce apoptosis.
Figure 4.
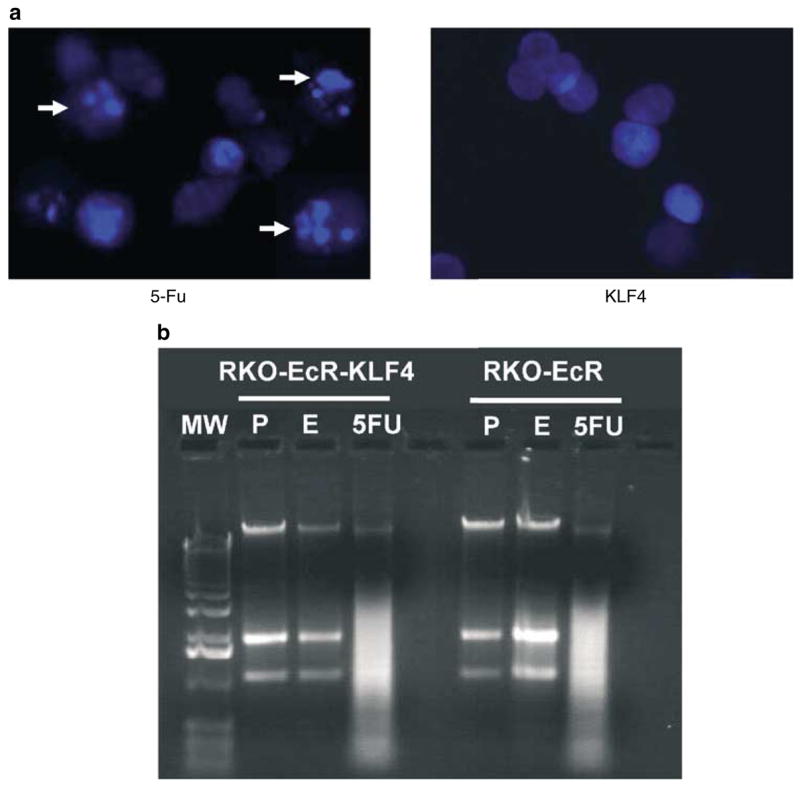
Effect of KLF4 induction on apoptosis. (a) Fluorescence photomicrograph of RKO-EcR-KLF4 cells treated with 5-FU to induce apoptosis or Ponasterone A to induce KLF4. Cells treated with 5-FU show chromatin condensation, nuclear fragmentation, and apoptotic bodies (arrows), all morphological hallmarks of apoptosis. For these studies, RKO-EcR-KLF4 cells were treated with 5 μm Ponasterone A or 50 μg/ml 5-fluorouracil (5-FU, Sigma) for 3 days, fixed with a solution containing 0.1% Hoechst, 3.7% formaldehyde, and 0.5% Nonidet P-40 in PBS and visualized under fluorescence microscopy. Both floating and adherent cells were collected for evaluation. (b) Representative electrophoresis of genomic DNA from RKO-EcR and RKO-EcR-KLF4 cells treated with 5 μm Ponasterone A (P), ethanol (E), or 5-FU for 72 h. Lane 1 depicts the 1kb molecular weight (MW) marker. With 5-FU treatment, the typical ladder pattern, often associated with apoptosis, is seen. Genomic DNA was obtained from 2.0 × 106 RKO-EcR and RKO-EcR-KLF4 cells treated with 5 μm Ponasterone A, equal volume ethanol, or 50 μg/ml 5-FU for 3 days using the Apoptotic DNA Ladder Kit (Roche, Basel, Switzerland). DNA was extracted from both floating and adherent cells. DNA was visualized in a 2.0% agarose gel with 0.1 μg/ml ethidium bromide. Similar results were obtained in cells treated with 5 or 10 μm Ponasterone A; thus only results with 5 μm treatments are shown
In summary, our data show that within the context of colon cancer, overexpression of KLF4 inhibits colony formation, migration, invasion, and in vivo tumorigenecity. Moreover, these antiproliferative effects are mediated by cell cycle control rather than apoptosis. Our system, using RKO colon cancer cells that do not express native KLF4, provides a background in which the function of KLF4 can be tested. The decrease in KLF4 in RKO cells is proposed to be secondary to mutated CDX2 and interrupted transcription of the KLF4 gene (Dang et al., 2001); thus the KLF4 transcription machinery is likely intact and functional. Our conclusion that KLF4 overexpression inhibits tumorigenecity in colon cancer is further supported by recent results showing that overexpression of KLF4 also induces G1/S arrest in HCT116 colon cancer cells, which express native KLF4 (Yoon et al., 2003). The antitumor effects of KLF4 in RKO cells require the full-length protein, which is consistent with previous results in other cell types, showing that full-length KLF4 is required for transactivation and colony suppression (Geiman et al., 2000; Dang et al., 2002). Finally, we find no evidence that KLF4 induces apoptosis in RKO cells. While previous reports have suggested that KLF4 induces apoptosis in HT29 colon cancer cells, we note that this was within the context of interferon-gamma treatment (Chen et al., 2000). It is unlikely that the lack of apoptosis is due to insufficient doses of Ponasterone A and the degree of KLF4 induction as experiments with higher doses reveal no difference in our assays.
Of note, we find that in our inducible system, the population of stably transfected cells as followed by EGFP decreases over time. This is likely secondary to growth arrest of cells overexpressing KLF4 and selective growth of a subpopulation of untransfected cells. In vitro, this is reflected by diminished growth suppression after prolonged periods of KLF4 induction. However, in vivo growth suppression remains sustained, actually becoming more marked with time. This observation suggests that KLF4 overexpression may also affect the tumor microenvironment in addition to epithelial cell growth in vivo, and would be consistent with previous reports of KLF4 localization and function in smooth muscle and vascular endothelial cells (Yet et al., 1998; Adam et al., 2000; Nickenig et al., 2002). Thus, again, context appears to contribute to KLF4 function. Additional studies are underway to further define the role of KLF4 in in vivo tumorigenesis.
Acknowledgments
We thank Leslie Metzger (Johns Hopkins Oncology Center Cell Imaging Core Facility), Boris Baibakov, and Olga Kovbasnjuk (Johns Hopkins Gastroenterology Confocal Facility) for assistance with FACS analysis and microscopy. This work is supported by the National Institutes of Health Grants DK59970 (DTD), DK52230, and CA84197 (VWY). DTD is a recipient of the Research Scholar Award from the American Gastroenterology Association and the Basic Research Award from the Glaxo Institute for Digestive Health. VWY is a recipient of the Georgia Cancer Coalition Distinguished Cancer Clinician Scientist Award.
References
- Adam PJ, Regan CP, Hautmann MB, Owens GK. J Biol Chem. 2000;275:37798–37806. doi: 10.1074/jbc.M006323200. [DOI] [PubMed] [Google Scholar]
- Behr R, Kaestner KH. Mech Dev. 2002;115:167–169. doi: 10.1016/s0925-4773(02)00127-2. [DOI] [PubMed] [Google Scholar]
- Brembeck FH, Rustgi AK. J Biol Chem. 2000;275:28230–28239. doi: 10.1074/jbc.M004013200. [DOI] [PubMed] [Google Scholar]
- Chen X, Johns DC, Geiman DE, Marban E, Dang DT, Hamlin G, Sun R, Yang VW. J Biol Chem. 2001;276:30423–30428. doi: 10.1074/jbc.M101194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Shie J, Tseng C. FEBS Lett. 2000;477:67–72. doi: 10.1016/s0014-5793(00)01764-6. [DOI] [PubMed] [Google Scholar]
- da Costa LT, He TC, Yu J, Sparks AB, Morin PJ, Polyak K, Laken S, Vogelstein B, Kinzler KW. Oncogene. 1999;18:5010–5014. doi: 10.1038/sj.onc.1202872. [DOI] [PubMed] [Google Scholar]
- Dang DT, Bachman KE, Mahatan CS, Dang LH, Giardiello FM, Yang VW. FEBS Lett. 2000;476:203–207. doi: 10.1016/s0014-5793(00)01727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang DT, Mahatan CS, Dang LH, Agboola IA, Yang VW. Oncogene. 2001;20:4884–4890. doi: 10.1038/sj.onc.1204645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang DT, Zhao W, Mahatan CS, Geiman DE, Yang VW. Nucleic Acids Res. 2002;30:2736–2741. doi: 10.1093/nar/gkf400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, Ruppert JM. Cancer Res. 2000;60:6488– 6495. [PubMed] [Google Scholar]
- Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie- Bell P, Grizzle W, Hayes MR, Broker TR, Chow LT, Ruppert JM. Cell Growth Differ. 1999;10:423–434. [PubMed] [Google Scholar]
- Geiman DE, Ton-That H, Johnson JM, Yang VW. Nucleic Acids Res. 2000;28:1106–1113. doi: 10.1093/nar/28.5.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TD, Opitz OG, Okano J, Rustgi AK. J Biol Chem. 1998;273:10747–10754. doi: 10.1074/jbc.273.17.10747. [DOI] [PubMed] [Google Scholar]
- Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, Kaestner KH. Development. 2002;129:2619–2628. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, Papadopolous N, Peltomaki P, de la Chapelle A, Hamilton SR, Kinzler KW, Vogelstein B. Nat Genet. 1995;9:48–55. doi: 10.1038/ng0195-48. [DOI] [PubMed] [Google Scholar]
- Luo J, Dunn T, Ewing C, Sauvageot J, Chen Y, Trent J, Isaacs W. Prostate. 2002;51:189–200. doi: 10.1002/pros.10087. [DOI] [PubMed] [Google Scholar]
- Nickenig G, Baudler S, Muller C, Werner C, Werner N, Welzel H, Strehlow K, Bohm M. FASEB J. 2002;16:1077–1086. doi: 10.1096/fj.01-0570com. [DOI] [PubMed] [Google Scholar]
- No D, Yao TP, Evans RM. Proc Natl Acad Sci USA. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigada M, Porcellini S, Sutti F, Doneda L, Pozzoli O, Consalez GG, Guttinger M, Grassi F. Mech Dev. 1999;81:103–113. doi: 10.1016/s0925-4773(98)00237-8. [DOI] [PubMed] [Google Scholar]
- Pouliot N, Nice EC, Burgess AW. Exp Cell Res. 2001;266:1–10. doi: 10.1006/excr.2001.5197. [DOI] [PubMed] [Google Scholar]
- Rigas A, Dervenis C, Giannakou N, Kozoni V, Shiff SJ, Rigas B. Cancer Invest. 2002;20:657–665. doi: 10.1081/cnv-120002491. [DOI] [PubMed] [Google Scholar]
- Rozic JG, Chakraborty C, Lala PK. Int J Cancer. 2001;93:497–506. doi: 10.1002/ijc.1376. [DOI] [PubMed] [Google Scholar]
- Segre JA, Bauer C, Fuchs E. Nat Genet. 1999;22:356– 360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Nucleic Acids Res. 2000a;28:2969–2976. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie JL, Chen ZY, O’Brien MJ, Pestell RG, Lee ME, Tseng CC. Am J Physiol Gastrointest Liver Physiol. 2000b;279:G806–G814. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- Shields JM, Christy RJ, Yang VW. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh E, Traber PG. Mol Cell Biol. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, Markowitz SD. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yet SF, McA’Nulty MM, Folta SC, Yen HW, Yoshizumi M, Hsieh CM, Layne MD, Chin MT, Wang H, Perrella MA, Jain MK, Lee ME. J Biol Chem. 1998;273:1026–1031. doi: 10.1074/jbc.273.2.1026. [DOI] [PubMed] [Google Scholar]
- Yoon HS, Chen X, Yang VW. J Biol Chem. 2003;278:2101–2105. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, Biggs JR, Kraft AS, Yang VW. J Biol Chem. 2000;275:18391–18398. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]