


Abstract
Meta-predictors make predictions by organizing and processing the predictions produced by several other predictors in a defined problem domain. A proficient meta-predictor not only offers better predicting performance than the individual predictors from which it is constructed, but it also relieves experimentally researchers from making difficult judgments when faced with conflicting results made by multiple prediction programs. As increasing numbers of predicting programs are being developed in a large number of fields of life sciences, there is an urgent need for effective meta-prediction strategies to be investigated. We compiled four unbiased phosphorylation site datasets, each for one of the four major serine/threonine (S/T) protein kinase families—CDK, CK2, PKA and PKC. Using these datasets, we examined several meta-predicting strategies with 15 phosphorylation site predictors from six predicting programs: GPS, KinasePhos, NetPhosK, PPSP, PredPhospho and Scansite. Meta-predictors constructed with a generalized weighted voting meta-predicting strategy with parameters determined by restricted grid search possess the best performance, exceeding that of all individual predictors in predicting phosphorylation sites of all four kinase families. Our results demonstrate a useful decision-making tool for analysing the predictions of the various S/T phosphorylation site predictors. An implementation of these meta-predictors is available on the web at: http://MetaPred.umn.edu/MetaPredPS/.
INTRODUCTION
The past few years have seen a boom in the development of prediction programs in a wide range of life science areas. The 2006 Web Server issue of Nucleic Acids Research (1) alone introduced nearly 150 web servers, a considerable proportion of which are prediction servers developed over the past 2 years. In many key problem domains, multiple prediction programs coexist. While each prediction program has its unique virtues and strengths, often times none of them is perfect—every program makes false predictions under certain circumstances. When these programs make conflicting predictions, it is difficult for bench researchers—the users of the programs—to arrive at sensible decisions, thus the original intention of the programs is defeated. In several important problem domains, efforts have been made to assess and compare prediction programs independently (2–6). These efforts assist users to determine which programs to ‘trust more’ based on the types of problems they have. In this series of studies, we take one step further on this issue: in addition to making an independent assessment of the predicting performance of several prediction programs in the defined problem domains, we investigate strategies of combining the strengths of these predictors, called element predictors, to construct ‘meta-predictors’ whose performance may exceed that of any element predictor. In the previous study (7), we investigated the meta-prediction in the domain of protein subcellular localization problem. In the study described herein, we look at the meta-prediction in another important problem domain: the prediction of phosphorylation sites of four major families of serine/threonine (or S/T) protein kinases.
Protein phosphorylation is the transfer of a phosphate group from a high-energy phosphate donor such as ATP, to an amino acid such as serine (S), threonine (T) or tyrosine (Y) of a protein, mediated by a protein kinase. Protein phosphorylation is the most common form of post-translational modification of proteins; it plays vital roles in the regulation of a variety of important cellular processes including metabolism, signal transduction pathways, transcription, translation, cell growth and cell differentiation. Only about 2% of the human proteome encode protein kinases, but more than 30% of the proteome are affected by kinase-mediated phosphorylation (8). Nearly half of human kinases have been linked to cancers and other diseases (9).
Protein kinases are often classified into two broad categories based on the amino acid residues on which phosphorylation take place: S/T kinases and Y kinases. The most common S/T kinases are found in the four families: CDK, CK2, PKA and PKC. Kinases in these four families are responsible for about half of the known S/T kinase reactions taking place in eukaryotic organisms.
Because experimental identification of phosphorylation sites is labor-intensive and costly, accurate computational methods for predicting phosphorylation sites are very important. A large number of such computation programs have been developed. Some of these programs [including NetPhos (10), DISPHOS (11) and Berry et al.'s predicting programs (12)] are generic or non-specific predictors; i.e. they predict whether a candidate site is a phosphorylation site or not, but do not make predictions about which kinases are involved. Other programs [e.g. KinasePhos (13), Scansite (14), PHOSITE (15), PredPhospho (16), NetPhosK (17), PREDIKIN (18), GPS (19) and PPSP(20)] are kinase-specific prediction programs; they make predictions about whether a candidate site is a phosphorylation site of a certain kinase, or of a certain family or group of kinases.
In terms of data features used, a vast majority of these prediction programs (including NetPhos, NetPhosK, KinasePhos, Scansite, PHOSITE, PredPhospho, GPS and PPSP) extract and use sequence features from the candidate phosphorylation sites [peptides typically of length between 7 and 50 surrounding the phosphorylatable residues (S/T/Y)], while PREDIKIN takes advantage of structural data from which interactions between residues in the substrate protein and corresponding residues in the kinase are derived, and DISPHOS utilizes features related to predicted disordered protein regions and predicted secondary structures. In terms of underlying classification methods, some programs (e.g. Scansite) use scores calculated from position-specific score matrices (PSSM) as basis of making predictions, others apply clustering or segmentation methods (e.g. GPS and PHOSITE), or train hidden Markov models (HMM) (KinasePhos), logistic regression models (DISPHOS), artificial neural networks (ANN) (NetPhos, NetPhosK), support vector machines (SVM) (PredPhospho) or Bayesian-based models (PPSP) for making predictions.
In this study, we choose several of these prediction tools as element predictors, and explore meta-predicting strategies for the phosphorylation site predicting problem for the four major families of S/T kinases: CDK, CK2, PKA and PKC. After compiling unbiased phosphorylation site datasets for these kinase families, we use these datasets to assess the predicting performance of these element predictors. Subsequently, we look at several strategies to develop meta-predictors for these kinase families. We show that a generalized weighted voting strategy with parameters determined by restricted grid search produced meta-predictors with predicting performances surpassing those of all element predictors for all four kinase families. A web server implementing these meta-predictors has been established and is accessible at the URL http://MetaPred.umn.edu/MetaPredPS/.
MATERIALS AND METHODS
Compilation of MetaPS06 datasets
We compiled four unbiased datasets, each corresponding to one of the four major S/T kinase families: CDK, CK2, PKA and PKC, for the meta-prediction of the phosphorylation sites for these kinase families. These datasets are termed the MetaPS06 datasets. The compilation of these datasets consisted of four steps: (i) making an ‘include-all’ compilation of S/T phosphorylation sites; (ii) removing data used in the development of element predictors from the compilation; (iii) assigning class labels to the phosphorylation sites in the compilation and (iv) making four balanced datasets for the four kinase families (Figure 1).
Figure 1.

Procedure of compiling the MetaPS06 dataset.
Step 1. Making ‘include-all’ compilation of S/T phosphorylation sites
The ‘include-all’ compilation of S/T phosphorylation sites was assembled from three different data sources: (i) Phospho.ELM (21), Version 5.0 (released in May 2006) (ii) PhosphoSite (22), obtained in July 2006 and (iii) Swiss-Prot Release 51.1 (release date: 14 November 2006). The data from Phospho.ELM and PhosphoSite include both non-specific phosphorylation sites (for which no information is provided about the kinases mediating the phosphorylation processes) and kinase-specific phosphorylation sites (for which the names of the kinases or of the kinase groups/families are provided). Only kinase-specific phosphorylation sites were included in the compilation. The S/T phosphorylation site data from Swiss-Prot were extracted by searching in the ‘FT’/‘MOD_RES’ field of the annotation using keywords ‘phosphoserine’ and ‘phosphothreonine’. Non-specific phosphorylation sites were eliminated, as well as sites whose annotations contain the following words: ‘by similarity’, ‘potential’, ‘probably’ or ‘partial’.
In summary, 2195, 3159 and 1145 S/T phosphorylation sites were retrieved from these three data sources, respectively. These data were merged, resulting in 5626 non-redundant S/T phosphorylation sites. The term ‘phosphorylation site’ used here refers to phosphorylation by a single kinase. A ‘physical’ S/T site may be phosphorylated by several kinases. If so, it is considered as several phosphorylation sites, one for each kinase.
Step 2. Removing data used in the development of element predictors
To make unbiased datasets, all data used in the development of any element predictors must be excluded (7). In the original report of each element prediction program, the data used in the development of the program is described. For most element prediction programs (GPS, KinasePhos, NetPhosK, PPSP and PredPhospho), Phospho.ELM (or its predecessor PhosphoBase) was used as the major data source (Table 1). The latest version of Phospho.ELM used in the development of these programs was the release of September 2004 (used in the development of GPS and PPSP). In the Phospho.ELM dataset we obtained (the May 2006 release), all entries annotated prior to 2005 are labeled with an annotation date of 31 December 2004. We removed all phosphorylation site data that originated from, or overlapped with, any pre-2005 Phospho.ELM entries from our data.
Table 1.
Summary of the 15 element predictors
| Element predictor | Refs. | URLs |
|---|---|---|
| GPS | (19,25) | http://973-proteinweb.ustc.edu.cn/gps/gps_web/predict.php |
| KinasePhos_90 | (13,24) | http://kinasephos.mbc.nctu.edu.tw/ |
| KinasePhos_95 | ||
| KinasePhos_100 | ||
| KinasePhos_bitscore | ||
| NetPhosK_0.3 | (17) | http://www.cbs.dtu.dk/services/NetPhosK/ |
| NetPhosK_0.5 | ||
| NetPhosK_0.7 | ||
| PPSP_highsens | (20) | http://bioinformatics.lcd-ustc.org/PPSP/ |
| PPSP_balanced | ||
| PPSP_highspec | ||
| PredPhospho | (16) | http://pred.ngri.re.kr/PredPhospho.htm |
| Scansite_low | (14) | http://scansite.mit.edu/motifscan_seq.phtml |
| Scansite_medium | ||
| Scansite_high |
For data features and classification methods, see text.
For three element prediction programs—KinasePhos, NetPhosK and PredPhospho—Swiss-Prot was listed as one of the data sources. The latest version of Swiss-Prot used was Release 45.0, 25 October 2004 (used in the development of KinasePhos). All phosphorylation site data originated from, or overlapped with, any Swiss-Prot entries bearing an initial entry date prior to 25 October 2004 were eliminated from our phosphorylation site data.
Three element prediction programs—GPS, PPSP and NetPhosK—used small numbers of phosphorylation sites obtained through manual curation of the literature. The curated data used in the development of GPS and PPSP were acquired from the authors of these programs through personal communications, and these data were eliminated from the phosphorylation site compilation. We were unable to obtain the manually curated data used in the development of NetPhosK. However, considering that this program was released comparatively early (prior to December 2003), there is good chance that the small number of manually curated phosphorylation sites used in the development of this program were gathered in the Phospho.ELM effort which took place in the following year, and were excluded from the compilation together with other earlier Phospho.ELM data.
Scansite is different from all other element prediction programs in that data obtained from oriented peptide library and phage display experiments were used in its development. These data are completely independent of the data compilation we made. Because no overlap exists between these two types of data, no data exclusion was necessary.
At the end of this step, 3252 S/T phosphorylation sites remained in the compilation.
Step 3. Assigning class labels to phosphorylation sites
Next, each phosphorylation site remaining in the compilation was assigned one of five class labels—four of which corresponding to the four kinase families: CDK, CK2, PKA and PKC, and ‘other’, denoting all other S/T kinase families. The class labels were assigned based on the annotations from the original data sources, according to a kinase name index table (available as Supplementary Table 1). The kinase name index table was created based primarily on KinBase (23), with a small number of manually added entries. Following the assignment of class labels, some redundant entries were emerged. After these redundant entries were removed, 2489 phosphorylation sites remained in the compilation. They include 294 CDK phosphorylation sites, 229 CK2 phosphorylation sites, 360 PKA phosphorylation sites, 348 PKC phosphorylation sites and 1258 phosphorylation sites labeled with the ‘other’ class label.
Step 4. Making of four balanced datasets
The task of phosphorylation site prediction consists of four separate two-class classification problems, one for each of the four major kinase families. For each of the four classification problems, a dataset needs to be prepared that include both ‘positive data’ and ‘negative data’. A good phosphorylation site predictor should be able to differentiate true phosphorylation sites for a given kinase family from phosphorylation sites of other families as well as from non-phosphorylation sites (occurrences of S/T in proteins that are not substrates of any S/T kinases). Thus, the ‘negative data’ should include two different types of data: the true phosphorylation sites of other kinase families, and non-phosphorylation sites.
A collection of ‘non-phosphorylation sites’ was compiled: from the same set of proteins where the known phosphorylation sites reside in, 1400 occurrences of S/T were picked for which no phosphorylation has been reported to have taken place. This collection of S/T sites likely includes some true phosphorylation sites that had not yet been identified. However, in the absence of a better method of making non-phosphorylation sites, this way of preparing the non-phosphorylation site data is an accepted practice in phosphorylation site prediction (15,24).
For each of the four datasets, all phosphorylation sites in the data compilation (prepared in previous steps) with the corresponding class labels were included as the ‘positive set’. All phosphorylation sites bearing any other class labels (the three other families, and ‘other’), and the 1400-sample non-phosphorylation sites data were lumped together. All sites in the lumped set overlapping with any sites in the ‘positive set’ were removed, after which, the ‘negative set’ was constructed by picking samples at random from the lumped set. The size of the negative set was set to be 1.5 times that of the positive set, following the established practice in the field (16). The final, balanced datasets for the classification of CDK, CK2, PKA and PKC phosphorylation sites consist of 294 positive and 441 negative samples, 229 positive and 343 negative samples, 360 positive and 540 negative samples and 348 positive and 552 negative samples, respectively. These datasets are available as Supplementary Table 2.
Table 2.
Weight combinations, permutations and possible weighted sum values in the restricted grid search parameter selection scheme
| Weight combinationsa | Number of corresponding weight permutations |
|---|---|
| 1 × 1 | 15 |
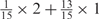 |
1365 |
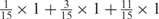 |
2730 |
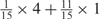 |
15 015 |
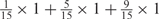 |
2730 |
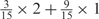 |
1365 |
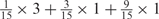 |
60 060 |
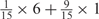 |
45 045 |
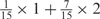 |
1365 |
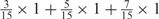 |
2730 |
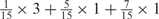 |
60 060 |
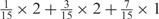 |
90 090 |
 |
270 270 |
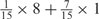 |
45 045 |
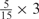 |
455 |
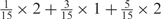 |
90 090 |
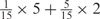 |
135 135 |
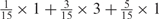 |
60 060 |
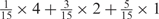 |
675 675 |
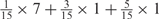 |
360 360 |
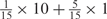 |
15 015 |
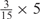 |
3003 |
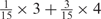 |
225 225 |
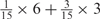 |
420 420 |
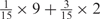 |
75 075 |
 |
1365 |
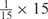 |
1 |
| Possible weighted | 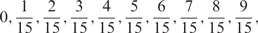 |
| sum values | 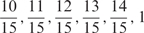 |
aWeight combinations are denoted as the sum of each weight value multiplied by the number of weights taking the weight value, with weight value 0 omitted. For instance, ‘1 × 1’ represents cases where one weight takes the value 1, and the other 14 weights taking the value 0; and ‘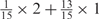 ’ represent cases where 2 of the 15 weights take the value
’ represent cases where 2 of the 15 weights take the value 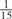 , 1 weight takes the value
, 1 weight takes the value 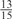 , and the remaining 12 weights take the value 0. Each weight combination corresponds to one or more weight permutations. For instance, for weight combination ‘1 × 1’, the weight value 1 can be taken by each of the 15 weights, thus it corresponds to
, and the remaining 12 weights take the value 0. Each weight combination corresponds to one or more weight permutations. For instance, for weight combination ‘1 × 1’, the weight value 1 can be taken by each of the 15 weights, thus it corresponds to 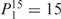 weight permutations. Similarly, for weight combination ‘
weight permutations. Similarly, for weight combination ‘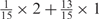 ’, there are
’, there are 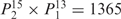 corresponding weight permutations.
corresponding weight permutations.
Selection of element predictors
Because we focused on predicting phosphorylation sites for the four major S/T kinase families—CDK, CK2, PKA and PKC—in this study, non-specific phosphorylation site prediction programs, including NetPhos and DISPHOS, were excluded. PHOSITE was excluded because its implementation is not available. PREDIKIN requires kinase sequences as input, thus it was also excluded.
Six programs remained in the list of element predicting programs included in this study. A total of 12 element predictors are derived from these programs (Table 1). Each prediction program is discussed below:
GPS (19,25)
GPS uses the 7-mer peptide sequence surrounding the center S/T position (three amino acids on each side of S/T) as its features. It calculates a similarity score between each pair of 7-mer sequences in the training dataset based on the BLOSUM62 substitution matrix, and performs a clustering of the training sequences. The prediction is made based on the average similarity score calculated between the test sequence and all sequences belonging to a given kinase family.
KinasePhos (13,24)
KinasePhos uses the 9-mer peptide sequence surrounding the center S/T (four amino acids on each side of S/T) as its features, and constructs profile HMM for making predictions about phosphorylation sites. Several options are provided in the prediction program, including three given levels of specificity: 90%, 95% and 100%, and an option ‘by default HMM bit score’. These four options are regarded as four separate element predictors for the meta-prediction problem, and they are termed KinasePhos_90, KinasePhos_95, KinasePhos_100 and KinasePhos_bitscore, respectively.
NetPhosK (17)
NetPhosK uses the 15-mer or 17-mer peptide sequence surrounding the center S/T position as features, and it trains ANN models for making prediction about phosphorylation sites. Two options are provided in the prediction server: ‘prediction without filtering’ and ‘prediction with ESS filtering’. We chose the ‘prediction without filtering’ option because the ESS filtering would take prohibitively long time to compute for the scale of prediction task we had. Furthermore, the NetPhosK server allows the setting of a ‘threshold’ value. Although a wide range of threshold values are provided for selection (between 0 and 0.95), we found that threshold values below 0.3 lead to very low specificity (close to 0), and threshold values above 0.7 give rise to very low sensitivity in the prediction results. Thus we chose to use these three threshold levels: 0.3, 0.5 and 0.7 for making predictions, and they are regarded as three separate element predictors, namely, NetPhosK_0.3, NetPhosK_0.5 and NetPhosK_0.7, respectively, in this study.
PPSP (20)
PPSP defines features using the 9-mer peptide sequence surrounding the S/T position, and a Bayesian-based model was constructed for making predictions about phosphorylation sites. The PPSP prediction server provides three options: high sensitivity, balanced and high specificity. These three options are regarded as three separate element predictors for our purposes, and they are referred to as PPSP_highsens, PPSP_balanced and PPSP_highspec, respectively.
PredPhospho (16)
PredPhospho uses peptide sequences of variable lengths (7-mer through 51-mers) centered on the S/T position, as its features; it trains SVM for making its predictions about phosphorylation sites.
Scansite (14)
Scansite organizes peptide sequence features obtained from an oriented peptide library and phage display experiments into position-specific scoring matrices (PSSM). For a potential phosphorylation site, the PSSM score is calculated and the prediction is made based on whether the score exceeds a pre-set threshold value. Three options are provided in the Scansite prediction program, each representing a different stringency levels: high stringency, medium stringency and low stringency. They are regarded as three separate element predictors for the purposes of the meta-prediction problem, namely, Scansite_high, Scansite_medium and Scansite_low.
Obtaining and pre-processing prediction results of element predictors
All six online prediction servers take protein sequences as their input. Prediction jobs were submitted to each of the prediction servers using locally developed Perl scripts with the specified prediction options. The prediction result pages were parsed and processed with another set of Perl scripts.
For every potential phosphorylation site (or every occurrence of S/T in the protein sequence), certain numerical scores were produced by the element prediction programs. These scores are of different mathematical meanings for different prediction programs. For example, the score produced by KinasePhos is the HMM bit score, the score produced by PPSP represents the risk difference in the Bayesian decision model and the score produced by Scansite is the calculated PSSM score. In addition to the numerical score, each element prediction program also makes a binary determination about whether a site is a phosphorylation site for a given family of kinases. The numerical scores produced by the prediction programs contain richer information than the binary determination, which might lead to improved predicting performance in meta-predictors. However, it is difficult to compare these scores across prediction programs due to their different meanings. In this study, we ignored the numerical scores, and only used the binary determinations.
Performance measures
In the protein subcellular localization prediction problem (7), only a small fraction of samples (about 3%) are categorized into multiple subcellular compartments. For the phosphorylation site prediction problem, however, a substantial proportion of phosphorylation sites (>20%) are known to be phosphorylated by multiple kinases. Therefore it is more proper to treat the phosphorylation site prediction for the four major S/T kinase families as four separate two-class classification problems, than considering it as a single four-class classification problem, as was done for the protein subcellular localization prediction problem. For two-class classification problems, commonly used performance measures for predictors include sensitivity, specificity, accuracy and Matthew's correlation coefficient (MCC) (7). Sensitivity and specificity indicate the predictor's abilities to curb false-negative and false-positive predictions, respectively, and these two measures need to be examined together to obtain an overall evaluation of the predictor, because a predictor with very high sensitivity but very low specificity (or vice versa) is not very useful. Accuracy can indicate the performance of a predictor on its own. However, the accuracy of a predictor can vary considerably with the ratio of positive samples and negative samples in the dataset. MCC is much less susceptible to this problem; it is the most widely used prediction measure for two-class prediction programs.
The area under the receiver operating characteristic (or ROC) curve was used to measure the performance of a predicting program or strategy for which multiple configurations or options are possible. For each configuration or option, the sensitivity and specificity were evaluated, the ROC curve [sensitivity versus (1-specificity) curve] was plotted, and the area underneath this curve was calculated.
Combinatorial strategy
The ‘combination approach’ or ‘consensus approach’ is a simple meta-prediction strategy that has been used in two-class classification problems in other problem domains (5,26). In this approach, the meta-predictor makes predictions by applying logic AND operations to all predictions made by element predictors. In other words, a positive prediction is made by the meta-predictor for a particular sample only if all element predictors make positive predictions on the same sample.
Simple voting strategies: unweighted voting, unreduced weighted voting and reduced weighted voting
The unweighted voting, unreduced weighted voting and reduced weighted voting strategies were described in the previous study (7). For multi-class prediction problems such as the protein subcellular localization prediction problem, the prediction can be made by picking from the multiple classes the one that gives rise to the highest score, and no score threshold needs to be set. For two-class prediction problems such as the phosphorylation site prediction problem, however, there is the need to set a score threshold. Generally, a linear voting-based two-class classifier makes a positive prediction if the following condition is satisfied:
| 1 |
where N is the number of element predictors (which is equal to 15 for unreduced voting for the phosphorylation site meta-predicting problem, and it will take a smaller integer value for reduced voting schemes); wj is the weight of the jth element predictor—for unweighted voting, wj = 1 for all js; Pj indicates the prediction made by the jth element predictor, Pj = 1 if a positive prediction is made, and Pj = 0 if otherwise; and T is the threshold score.
For a simple majority voting, the threshold T should be set as the half of the sum of all weights for the element predictors. That is,
| 2 |
Restricted grid search
In a weighted voting strategy denoted by Equation (1), there are N + 1 parameters—N weight parameters (wj) and the threshold parameter T that need to be determined. Our task is to select proper values for these N + 1 (=16) parameters that would result in a classier with a good predicting performance (in terms of MCC or accuracy).
Generally, two approaches can be taken to determine these parameter values: optimization and grid search (or exhaustive search). A large number of optimization algorithms are available which can be applied to find a set of ‘optimal parameters’ that maximize the GCC or accuracy. Properly chosen optimization algorithms can be very efficient in run time performance. However, optimization methods may find a local, rather than a global, optimum. Grid search is not susceptible to the local optimum problem, but can be very costly in running time. Effective searching of 16 parameters (15 weight and 1 threshold parameters) is a rather challenging task.
We developed a restricted grid search scheme to select the values of these 16 parameters. First, we decided that the weight of any element predictors can only take one of the following 9 values: 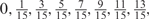 and 1. Second, we required that the sum of the weights of all 15 element predictors equal 1. With these two restrictions, the number of possible weight combinations is limited to 27, corresponding to 2 659 764 weight permutations, as is summarized in Table 1. With Pj taking one of two possible values: 0 and 1, the weighted sum
and 1. Second, we required that the sum of the weights of all 15 element predictors equal 1. With these two restrictions, the number of possible weight combinations is limited to 27, corresponding to 2 659 764 weight permutations, as is summarized in Table 1. With Pj taking one of two possible values: 0 and 1, the weighted sum 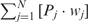 can produce a total of 16 different values (Table 2). T could be any of these 16 values. With this scheme, the search space for the 16 parameters was effectively limited to a manageable size of (2 659 764 × 16 = 42 556 224). The grid search of the 16 parameters was conducted on each of the four MetaPS06 datasets with 10-fold cross-validation.
can produce a total of 16 different values (Table 2). T could be any of these 16 values. With this scheme, the search space for the 16 parameters was effectively limited to a manageable size of (2 659 764 × 16 = 42 556 224). The grid search of the 16 parameters was conducted on each of the four MetaPS06 datasets with 10-fold cross-validation.
RESULTS
Performance assessment of element predictors
The predicting performance of each of the 15 element predictors was assessed using these unbiased MetaPS06 datasets. As is shown in Table 3, the element predictors vary considerably in predicting performance. For the four kinase families CDK, CK2, PKA and PKC, the element predictors that offers the best predicting performance are PredPhospho (accuracy: 0.853, MCC: 0.708), NetPhosK_0.5 (accuracy: 0.871, MCC: 0.730), PredPhospho (accuracy: 0.827, MCC: 0.642) and PPSP_balanced (accuracy: 0.743, MCC: 0.477), respectively.
Table 3.
Predicting performance of element predictors
| Element predictor | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| CDK | ||||
| GPS | 0.908 | 0.800 | 0.844 | 0.695 |
| KinasePhos_90 | 0.884 | 0.717 | 0.784 | 0.589 |
| KinasePhos_95 | 0.799 | 0.837 | 0.822 | 0.632 |
| KinasePhos_100 | 0.571 | 0.923 | 0.782 | 0.542 |
| KinasePhos_bitscore | 0.912 | 0.685 | 0.776 | 0.588 |
| NetPhosK_0.3 | 1 | 0 | 0.400 | N/Aa |
| NetPhosK_0.5 | 0.639 | 0.748 | 0.705 | 0.387 |
| NetPhosK_0.7 | 0.065 | 0.998 | 0.624 | 0.188 |
| PPSP_highsens | 0.983 | 0.075 | 0.438 | 0.128 |
| PPSP_balanced | 0.905 | 0.796 | 0.839 | 0.687 |
| PPSP_highspec | 0.054 | 0.982 | 0.611 | 0.100 |
| PredPhospho | 0.898 | 0.823 | 0.853 | 0.708 |
| Scansite_low | 0.667 | 0.884 | 0.797 | 0.571 |
| Scansite_medium | 0.405 | 0.971 | 0.744 | 0.479 |
| Scansite_high | 0.153 | 0.993 | 0.657 | 0.290 |
| CK2 | ||||
| GPS | 0.699 | 0.895 | 0.816 | 0.613 |
| KinasePhos_90 | 0.581 | 0.904 | 0.774 | 0.523 |
| KinasePhos_95 | 0.476 | 0.950 | 0.760 | 0.504 |
| KinasePhos_100 | 0.266 | 0.985 | 0.698 | 0.386 |
| KinasePhos_bitscore | 0.594 | 0.901 | 0.778 | 0.530 |
| NetPhosK_0.3 | 0.961 | 0.525 | 0.699 | 0.506 |
| NetPhosK_0.5 | 0.755 | 0.948 | 0.871 | 0.730 |
| NetPhosK_0.7 | 0.245 | 1.000 | 0.698 | 0.403 |
| PPSP_highsens | 0.930 | 0.227 | 0.509 | 0.208 |
| PPSP_balanced | 0.742 | 0.933 | 0.857 | 0.700 |
| PPSP_highspec | 0.048 | 1.000 | 0.619 | 0.171 |
| PredPhospho | 0.594 | 0.959 | 0.813 | 0.616 |
| Scansite_low | 0.576 | 0.983 | 0.820 | 0.640 |
| Scansite_medium | 0.380 | 0.997 | 0.750 | 0.512 |
| Scansite_high | 0.135 | 1.000 | 0.654 | 0.293 |
| PKA | ||||
| GPS | 0.817 | 0.809 | 0.812 | 0.618 |
| KinasePhos_90 | 0.722 | 0.843 | 0.794 | 0.569 |
| KinasePhos_95 | 0.650 | 0.887 | 0.792 | 0.560 |
| KinasePhos_100 | 0.361 | 0.952 | 0.716 | 0.405 |
| KinasePhos_bitscore | 0.775 | 0.804 | 0.792 | 0.573 |
| NetPhosK_0.3 | 0.878 | 0.724 | 0.786 | 0.590 |
| NetPhosK_0.5 | 0.694 | 0.874 | 0.802 | 0.583 |
| NetPhosK_0.7 | 0.483 | 0.959 | 0.769 | 0.525 |
| PPSP_highsens | 0.967 | 0.231 | 0.526 | 0.270 |
| PPSP_balanced | 0.850 | 0.806 | 0.823 | 0.645 |
| PPSP_highspec | 0.008 | 0.998 | 0.602 | 0.048 |
| PredPhospho | 0.808 | 0.839 | 0.827 | 0.642 |
| Scansite_low | 0.644 | 0.917 | 0.808 | 0.596 |
| Scansite_medium | 0.422 | 0.981 | 0.758 | 0.515 |
| Scansite_high | 0.158 | 0.991 | 0.658 | 0.288 |
| PKC | ||||
| GPS | 0.718 | 0.753 | 0.739 | 0.466 |
| KinasePhos_90 | 0.649 | 0.789 | 0.733 | 0.441 |
| KinasePhos_95 | 0.480 | 0.864 | 0.710 | 0.378 |
| KinasePhos_100 | 0.129 | 0.977 | 0.638 | 0.211 |
| KinasePhos_bitscore | 0.687 | 0.722 | 0.708 | 0.404 |
| NetPhosK_0.3 | 0.716 | 0.695 | 0.703 | 0.403 |
| NetPhosK_0.5 | 0.491 | 0.841 | 0.701 | 0.358 |
| NetPhosK_0.7 | 0.333 | 0.935 | 0.694 | 0.348 |
| PPSP_highsens | 0.954 | 0.274 | 0.546 | 0.289 |
| PPSP_balanced | 0.741 | 0.743 | 0.743 | 0.477 |
| PPSP_highspec | 0.006 | 1.000 | 0.602 | 0.059 |
| PredPhospho | 0.598 | 0.805 | 0.722 | 0.412 |
| Scansite_low | 0.411 | 0.866 | 0.684 | 0.315 |
| Scansite_medium | 0.170 | 0.946 | 0.636 | 0.189 |
| Scansite_high | 0.069 | 0.994 | 0.624 | 0.179 |
Predicting performance assessed on MetaPS06 datasets. Element predictors having the best predicting performance are shown in italic.
aMCC is undefined.
Combinations of element predictors
We examined the predicting performance of all combinatorial meta-predictors constructed with 2–6 element predictors, and the combinatorial meta-predictors yielding the best predicting performance are shown in Table 4. For CDK, four combinatorial meta-predictors achieved slightly better predicting performance than that of the best element predictor (PredPhospho). For PKC, two combinatorial meta-predictors achieved slightly better predicting performance than that of the best element predictor (PPSP_balanced). However, for the other two kinase families, CK2 and PKA, the combination approach did not produce meta-predictors with satisfactory predicting performances.
Table 4.
Predicting performance of combinatorial meta-predictors
| Number of element predictors in combination | Element predictors included in best combinatorial meta-predictor | Accuracy | MCC |
|---|---|---|---|
| CDK | |||
| 2 | GPS, PredPhospho | 0.859 | 0.717 (0.36)* |
| 3 | GPS, NetPhosK_0.3, PredPhospho | 0.859 | 0.717 (0.36)* |
| 4 | GPS, NetPhosK_0.3, PPSP_highsens, PredPhospho | 0.859 | 0.716 (0.38)* |
| 5 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, PredPhospho | 0.856 | 0.708 (0.50)* |
| 6 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, PredPhospho, Scansite_low | 0.799 | 0.575 |
| Best element predictor (PredPhospho) | 0.853 | 0.708 | |
| CK2 | |||
| 2 | NetPhosK_0.3, PPSP_balanced | 0.857 | 0.700 |
| 3 | GPS, NetPhosK_0.3, PPSP_balanced | 0.832 | 0.652 |
| 4 | GPS, NetPhosK_0.3, PPSP_highsens, Scansite_low | 0.808 | 0.621 |
| 5 | GPS, KinasePhos_90, NetPhosK_0.3, PPSP_highsens, Scansite_low | 0.778 | 0.565 |
| 6 | GPS, KinasePhos_90, NetPhosK_0.3, PPSP_highsens, PredPhospho, Scansite_low | 0.748 | 0.508 |
| Best element predictor (NetPhosK_0.5) | 0.871 | 0.730 | |
| PKA | |||
| 2 | NetPhosK_0.3, PredPhospho | 0.827 | 0.638 |
| 3 | GPS, NetPhosK_0.3, PPSP_highsens | 0.82 | 0.625 |
| 4 | GPS, NetPhosK_0.3, PPSP_highsens, PredPhospho | 0.819 | 0.619 |
| 5 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, PredPhospho | 0.81 | 0.599 |
| 6 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, PredPhospho, Scansite_low | 0.784 | 0.555 |
| Best element predictor (PredPhospho) | 0.827 | 0.642 | |
| PKC | |||
| 2 | NetPhosK_0.3, PPSP_balanced | 0.76 | 0.489 (0.37)* |
| 3 | GPS, NetPhosK_0.3, PPSP_balanced | 0.757 | 0.485 (0.41)* |
| 4 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, | 0.739 | 0.448 |
| 5 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, PredPhospho | 0.717 | 0.405 |
| 6 | GPS, KinasePhos_bitscore, NetPhosK_0.3, PPSP_highsens, PredPhospho, Scansite_low | 0.676 | 0.319 |
| Best element predictor (PPSP_balanced) | 0.741 | 0.477 |
Predicting performance assessed on MetaPS06 datasets. For each l (2 ≤ l ≤ 6), the composition and predicting performance of the best meta-predictors composed of l element predictors are shown, together with the predicting performance of the best element predictor. Meta-predictors having predicting performance exceeding that of the best element predictor are shown in italic.
*P-values in Fisher's Z-transformation test (compared with the MCC of the best element predictor) are shown in parentheses.
Simple voting strategies: unweighted voting, unreduced weighted voting and reduced weighted voting
We used the unweighted voting, unreduced weighted voting and reduced weighted voting strategies in constructing meta-predictors for the phosphorylation site problem. Unlike a multi-class prediction problem, a score threshold needs to be set for two-class phosphorylation site prediction problems. This score threshold is set as the half of the sum of all weights for the element predictors (see Materials and Methods).
As shown in Table 5, the prediction performance of no unweighted voting meta-predictor exceeded that of the best element predictors. For the CDK and PKA kinase families, unreduced weighted voting meta-predictors (using the accuracy values of the element predictors as the weights) achieved slightly improved prediction over than that of the best element predictors. For the CK2 and PKC kinase families, none of the unreduced weighted meta-predictors produced satisfactory predicting performance. For the CDK, PKA and PKC kinase families, the best reduced weighted voting meta-predictors improved accuracy between 1.0% and 3.1%, and MCC between 1.8% and 4.4%, compared to those of the best element predictors. However, for the CK2 kinase family, no reduced weighted voting meta-predictor offered a satisfactory predicting performance (Table 5).
Table 5.
Predicting performance of unweighted voting, best unreduced weighted voting and best reduced weighted voting meta-predictors
| Predictor | Accuracy | MCC |
|---|---|---|
| CDK | ||
| Best element predictor (PredPhospho) | 0.853 | 0.708 |
| Unweighted voting Meta-predictor | 0.853 | 0.699 |
| Best unreduced weighted voting Meta-predictor | 0.857 | 0.711 (0.45)* |
| Best reduced weighted voting Meta-predictor | 0.863 | 0.726 (0.24)* |
| CK2 | ||
| Best element predictor (NetPhosK_0.5) | 0.871 | 0.730 |
| Unweighted voting Meta-predictor | 0.809 | 0.617 |
| Best unreduced weighted voting Meta-predictor | 0.844 | 0.675 |
| Best reduced weighted voting Meta-predictor | 0.867 | 0.722 |
| PKA | ||
| Best element predictor (PredPhospho) | 0.827 | 0.642 |
| Unweighted voting Meta-predictor | 0.820 | 0.620 |
| Best unreduced weighted voting Meta-predictor | 0.837 | 0.669 (0.16)* |
| Best reduced weighted voting Meta-predictor | 0.839 | 0.675 (0.11)* |
| PKC | ||
| Best element predictor (PPSP_balanced) | 0.741 | 0.477 |
| Unweighted voting Meta-predictor | 0.733 | 0.433 |
| Best unreduced weighted voting Meta-predictor | 0.744 | 0.500 |
| Best reduced weighted voting Meta-predictor | 0.772 | 0.521 (0.11)* |
Predicting performance assessed on MetaPS06 datasets. Meta-predictors having predicting performance exceeding that of the best element predictor are shown in italic.
*P-values in Fisher's Z-transformation test (compared with the MCC of the best element predictor) are shown in parentheses.
Weighted voting with restricted grid search parameter selection
The weighted voting strategy with the weights set by the MCC or accuracy values of the element predictors did not render meta-predictors with satisfactory predicting performance for all four kinase families. We thus explored the more general form of the weighted voting strategy, with the weights of element predictors determined from the data.
A grid search, with carefully devised restricted search space, allows the search to be executed in a manageable amount of time (Table 2 and Materials and Methods). The grid search of the 16 parameters was conducted on each of the four MetaPS06 datasets with 10-fold cross-validation. As is shown in Table 6, for all four kinase families, the weighted voting meta-predictors obtained by grid search parameter selection exhibits outstanding predicting performance which not only exceeds that of the best element predictors, but also surpasses that of any combinatorial or reduced voting meta-predictors constructed described above. The meta-predictors achieved an increase in accuracy of between 1.1% and 4.3%, and an increase in MCC of between 2.2% and 8.1% compared to the best element predictor for each kinase family. For the CK2, PKA and PKC kinase families, the meta-predictors demonstrated significantly higher MCC values than that of the best of the element predictors (P < 0.05, Fisher's Z-transformation test, see Table 6). Moreover, an ROC-based comparison indicated that the meta-predictors had higher ROC areas than those of any element predicting programs for all four kinase families (Table 7). The minimal and maximal improvements in accuracy and MCC (across the four kinase families) achieved by the meta-predictors over the best predictor for each element predicting program are presented separately in Table 8.
Table 6.
Predicting performance of weighted voting meta-predictors with restricted grid search of parameters
| Sensitivity | Specificity | Accuracy | MCC | |
|---|---|---|---|---|
| CDK | 0.912 | 0.832 | 0.864 | 0.730 (0.19)* |
| CK2 | 0.878 | 0.904 | 0.893 | 0.779 (0.027)* |
| PKA | 0.883 | 0.828 | 0.850 | 0.699 (0.014)* |
| PKC | 0.773 | 0.791 | 0.784 | 0.558 (0.011)* |
Predicting performance assessed on MetaPS06 datasets.
*P-values in Fisher's Z-transformation test (compared with the MCC of the best element predictor) are shown in parentheses.
Table 7.
Areas under the ROC curves for the six element predicting programs and the weighted voting meta-predictor with restricted grid search
| CDK | CK2 | PKA | PKC | |
|---|---|---|---|---|
| GPS | 0.8761 | 0.8130 | 0.8446 | 0.7574 |
| KinasePhos | 0.8713 | 0.7508 | 0.8234 | 0.7440 |
| NetPhosK | 0.7767 | 0.9307 | 0.8749 | 0.7581 |
| PPSP | 0.8721 | 0.8767 | 0.8860 | 0.7994 |
| PredPhospho | 0.8670 | 0.7791 | 0.8537 | 0.7149 |
| Scansite | 0.7584 | 0.7734 | 0.7656 | 0.6397 |
| Max of the six category | GPS 0.8761 | NetPhosK 0.9307 | PKA 0.8896 | PPSP 0.7994 |
| Meta-predictor (weighted voting with restricted grid search) | 0.8956 | 0.9313 | 0.8946 | 0.8247 |
Table 8.
Minimal and maximal improvements in accuracy and MCC achieved by the weighted voting meta-predictor with restricted grid search over the best predictor of each element predicting program
| Minimal improvement in accuracy | Maximal improvement in accuracy | Minimal improvement in MCC | Maximal improvement in MCC | |
|---|---|---|---|---|
| GPS | 0.020 | 0.077 | 0.035 | 0.166 |
| KinasePhos | 0.042 | 0.115 | 0.098 | 0.249 |
| NetPhosK | 0.022 | 0.159 | 0.049 | 0.343 |
| PPSP | 0.025 | 0.041 | 0.043 | 0.081 |
| PredPhospho | 0.011 | 0.080 | 0.022 | 0.163 |
| Scansite | 0.042 | 0.100 | 0.103 | 0.243 |
Minimal and maximal improvements in accuracy and MCC were calculated across the four datasets for CDK, CK2, PKA and PKC kinase families.
The parameters selected in the four final weighted voting meta-predictors are shown in Table 9. For each of the four meta-predictors, at least eight non-zero weight parameters were selected. At least one non-zero weights were used for all but two element predictors (NetPhosK_0.7 and PPSP_highspec) in the four final meta-predictors, indicating that the good performance achieved by these meta-predictors was due to their ability to harness the combined strengths of multiple element predictors.
Table 9.
Parameters selected by the restricted grid search in the weighted voting meta-predictors
| GPS | Kinase Phos_90 | Kinase Phos_95 | Kinase Phos_100 | Kinase Phos_bit score | NetPhos K_0.3 | NetP hosK_0.5 | NetP hosK_0.7 | PPSP_ highsens | PPSP_ balanced | PPSP_ highspec | Pred Phospho | Scansite_ low | Scansite_ medium | Scansite_ high | T | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDK | 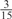 |
0 | 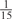 |
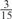 |
0 | 0 | 0 | 0 | 0 | 0 | 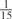 |
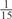 |
0 | 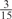 |
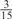 |
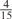 |
| CK2 | 0 | 0 | 0 | 0 | 0 | 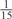 |
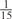 |
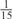 |
0 | 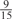 |
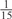 |
 |
0 | 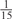 |
0 | 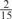 |
| PKA | 0 | 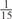 |
0 | 0 | 0 | 0 | 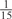 |
0 | 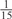 |
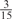 |
0 | 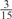 |
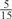 |
0 | 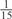 |
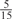 |
| PKC | 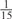 |
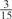 |
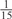 |
0 | 0 | 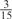 |
0 | 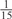 |
0 | 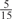 |
0 | 0 | 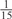 |
0 | 0 | 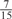 |
A web server implementing the four final meta-predictors was established and is accessible at http://MetaPred.umn.edu/MetaPredPS/.
DISCUSSION
Combination and reduced voting strategies
We found that a weighted voting strategy with parameters selected by a grid search scheme produced satisfactory meta-predictors whose performance exceeds that of all element predictors for all four kinase families. In contrast, the combination strategy and the reduced weighted voting strategy (with weights set by the accuracy or MCC of the element predictors) failed to yield meta-predictors with satisfactory predicting performance for at least one of the kinase families. The combination strategy has been successfully applied in making two-class predictions in other problem domains, including the predictions of secreted proteins (5) and transmembrane proteins (26). By applying a logic AND operation to the predictions made by the element predictors, this strategy, in essence, attempts to achieve improved specificity at the cost of reduced sensitivity. This strategy is expected to work effectively for cases where element predictors have relatively high sensitivity but lower specificity values. The reduced weighted voting strategy (with weights set by the accuracy or MCC of the element predictors) has produced good meta-predictors in the protein subcellular localization prediction problem (7), but this strategy fails to yield meta-predictors with expected performance in the prediction of phosphorylation sites for the CK2 kinase family. The exact reason for this failure is not clear, although stronger correlation among the element predictors may play a role. Multiple element predictors from a single prediction program are expected to be more highly correlated than those from different programs. In the protein subcellular localization prediction problem, only one of the eight prediction programs provided multiple element predictors. In the phosphorylation site prediction problem, however, four of the six prediction programs did so.
A general weighted voting strategy
Weighted voting with weight parameters selected by grid search is a more general form of the weighted voting strategy. It does not assume that element predictors with better performance will contribute more to the performance of the meta-predictors. Rather, the weights of all element predictors are determined directly from the data through exhaustive search. This flexible approach not only results in meta-predictors with better predicting performance than combinatorial meta-predictors and reduced voting meta-predictors in the phosphorylation site prediction problem, but it is also expected to work effectively for a wide range of other problem domains. Grid search of large numbers of parameters (16 in this particular case) is very costly in running time. The key to this weighted voting scheme is the ability to restrict the search space to a manageable size without compromising the effectiveness of the search. If an unrestricted grid search is conducted with 16 parameters, each of which is searched in four steps (that is, each parameter is allowed to take four possible values), the total number of parameter permutations is 416 ≈ 42 billion. A grid search of this scale would take about 14 months to complete with a computer equipped with an Intel Core 2 DUO processor. With the carefully devised restricted search scheme developed in this study, the grid search of the 16 parameters—15 of which were searched in 9 steps, and the 16th in 16 steps) was completed in only about 10 h for each of the four kinase families.
Limitation of voting-based strategies
A limitation imposed by voting-based meta-prediction strategies is that they require the output produced by different element predictors (which is taken as input of the element predictors) to be compatible with one another. We are working on decision tree- and SVM-based meta-prediction strategies to overcome this problem. It is hoped that these new strategies, which can take advantage of more versatile output of element predictors, will lead to more effective meta-predictors applicable in a wider range of problem domains.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Dr W. Pan at the University of Minnesota for inspiring discussions and Dr Q. Zhu at the University of Houston for helpful assistance. We also thank the Supercomputing Institute, University of Minnesota for computational resources. T.L. acknowledges the support of NIH (1R21CA126209) and Minnesota Medical Foundation. X.G. acknowledges the support of NIH/GM/AI (R43 GM076941, 1R21CA126209) and the R. A. Welch Foundation (E-1270). Funding to pay the Open Access publication charges for this article was provided by NIH/NCI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Editorial. Web Server issue. Nucleic Acids Res. 2006;34:W1. doi: 10.1093/nar/gkq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- 3.Cuthbertson JM, Doyle DA, Sansom MS. Transmembrane helix prediction: a comparative evaluation and analysis. Protein Eng. Des. Sel. 2005;18:295–308. doi: 10.1093/protein/gzi032. [DOI] [PubMed] [Google Scholar]
- 4.Kapp EA, Schutz F, Connolly LM, Chakel JA, Meza JE, Miller CA, Fenyo D, Eng JK, Adkins JN, et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5:3475–3490. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 5.Klee EW, Ellis LB. Evaluating eukaryotic secreted protein prediction. BMC Bioinformatics. 2005;6:256. doi: 10.1186/1471-2105-6-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Kang S, Tang C, Ellis LB, Li T. Meta-prediction of protein subcellular localization with reduced voting. Nucleic Acids Res. 2007;35:e96. doi: 10.1093/nar/gkm562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinna LA, Ruzzene M. How do protein kinases recognize their substrates? Biochim. Biophys. Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 9.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 10.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 11.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry EA, Dalby AR, Yang ZR. Reduced bio basis function neural network for identification of protein phosphorylation sites: comparison with pattern recognition algorithms. Comput. Biol. Chem. 2004;28:75–85. doi: 10.1016/j.compbiolchem.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Huang HD, Lee TY, Tzeng SW, Wu LC, Horng JT, Tsou AP, Huang KT. Incorporating hidden Markov models for identifying protein kinase-specific phosphorylation sites. J. Comput. Chem. 2005;26:1032–1041. doi: 10.1002/jcc.20235. [DOI] [PubMed] [Google Scholar]
- 14.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koenig M, Grabe N. Highly specific prediction of phosphorylation sites in proteins. Bioinformatics. 2004;20:3620–3627. doi: 10.1093/bioinformatics/bth455. [DOI] [PubMed] [Google Scholar]
- 16.Kim JH, Lee J, Oh B, Kimm K, Koh I. Prediction of phosphorylation sites using SVMs. Bioinformatics. 2004;20:3179–3184. doi: 10.1093/bioinformatics/bth382. [DOI] [PubMed] [Google Scholar]
- 17.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 18.Brinkworth RI, Breinl RA, Kobe B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc. Natl Acad. Sci. USA. 2003;100:74–79. doi: 10.1073/pnas.0134224100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou FF, Xue Y, Chen GL, Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem. Biophys. Res. Commun. 2004;325:1443–1448. doi: 10.1016/j.bbrc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Xue Y, Li A, Wang L, Feng H, Yao X. PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics. 2006;7:163. doi: 10.1186/1471-2105-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diella F, Cameron S, Gemund C, Linding R, Via A, Kuster B, Sicheritz-Ponten T, Blom N, Gibson TJ. Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 23.Milanesi L, Petrillo M, Sepe L, Boccia A, D’Agostino N, Passamano M, Di Nardo S, Tasco G, Casadio R, et al. Systematic analysis of human kinase genes: a large number of genes and alternative splicing events result in functional and structural diversity. BMC Bioinformatics. 2005;6(Suppl. 4):S20. doi: 10.1186/1471-2105-6-S4-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005;33:W226–W229. doi: 10.1093/nar/gki471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005;33:W184–W187. doi: 10.1093/nar/gki393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia JX, Ikeda M, Shimizu T. ConPred_elite: a highly reliable approach to transmembrane topology predication. Comput. Biol. Chem. 2004;28:51–60. doi: 10.1016/j.compbiolchem.2003.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.