


Abstract
Lon is an ATP-powered protease that binds DNA. However, the function of DNA binding by Lon remains elusive. Studies suggest that human Lon (hLon) binds preferentially to a G-rich single-stranded DNA (ssDNA) sequence overlapping the light strand promoter of mitochondrial DNA. This sequence is contained within a 24-base oligonucleotide referred to as LSPas. Here, we use biochemical and biophysical approaches to elucidate the structural properties of ssDNAs bound by hLon, as well as the thermodynamics of DNA binding by hLon. Electrophoretic mobility shift assay and circular dichroism show that ssDNAs with a propensity for forming parallel G-quartets are specifically bound by hLon. Isothermal titration calorimetry demonstrates that hLon binding to LSPas is primarily driven by enthalpy change associated with a significant reduction in heat capacity. Differential scanning calorimetry pinpoints an excess heat capacity upon hLon binding to LSPas. By contrast, hLon binding to an 8-base G-rich core sequence is entropically driven with a relatively negligible change in heat capacity. A considerable enhancement of thermal stability accompanies hLon binding to LSPas as compared to the G-rich core. Taken together, these data support the notion that hLon binds G-quartets through rigid-body binding and that binding to LSPas is coupled with structural adaptation.
INTRODUCTION
The ATP-dependent Lon protease is a member of the AAA+ family of proteins (ATPases associated with various cellular activities) (1), which is present in archaea, prokaryotes and eukaryotic mitochondria, peroxisomes and plastids (2–9). Lon has multiple cellular functions such as degrading abnormal polypeptides (2,4,10,11), as well as certain regulatory proteins (12–15) and metabolic enzymes (16–19), acting as a chaperone (20,21) and binding to nucleic acids (22–28). In addition, Lon may be important in maintaining genome integrity and expression, either by selectively degrading proteins involved in mtDNA metabolism, or by directly binding to DNA and/or RNA.
Recent work demonstrates that hLon binds to mtDNA in living cells and interacts preferentially with the control region for transcription initiation and mtDNA replication (28). It is possible that the sequence-specific binding of Lon to the mitochondrial genome may function to localize the protease at sites where it degrades protein substrates involved in mtDNA integrity. In bacteria, some of Lon's protein substrates are involved in genome metabolism, which manifests the role of Lon in maintaining genome stability. For example, in Caulobacter crescentus, Lon mediates the ATP-dependent turnover of the CcrM DNA methyltransferase, thereby regulating DNA replication (15). In Escherichia coli, Lon selectively degrades the transcription regulator SoxS only when this factor is not bound to DNA or to RNA polymerase (29). In yeast, results demonstrate that the TCA enzyme mitochondrial aconitase is present in mitochondrial nucleoids and functions in maintaining mtDNA, independent of its enzymatic activity (30–32). In light of observations that mildly oxidized mammalian mitochondrial aconitase is subject to Lon-mediated proteolysis, it is possible that Lon participates in nucleoid remodeling by degrading mtDNA-bound aconitase in response to metabolic changes and environmental stress (19,33). However, no data as yet support the localization of mammalian aconitase to mitochondrial nucleoids (34). The role of Lon in mitochondrial homeostasis is indispensable in normal fibroblasts; depletion of the protease using an antisense morpholino oligonucleotide leads to apoptosis and collapse of mitochondrial structure (35). Nevertheless, understanding Lon's physiological importance is enmeshed by its multiple functions as a protease and a DNA-binding protein. Thus, it remains unclear the extent to which mtDNA binding contributes to the essential role of Lon in mitochondrial biology.
Although the function and mechanism of Lon-mediated proteolysis is understood in detail, the physiological relevance and structural dynamics of nucleic acid binding by Lon remain unclear. In vitro studies demonstrate that both bacterial and mammalian Lon show a propensity for binding GT-rich oligonucleotides. Bacterial Lon binds to double-stranded DNA, whereas mouse and human Lon bind to ssDNA (22–25). Observations that the DNA-binding and enzymatic activities of bacterial and mammalian Lon are coordinately regulated and that hLon binds to mtDNA in cells, support the notion that the association of Lon to DNA is physiologically relevant (25,36). In vitro experiments have shown that hLon specifically binds to a ssDNA sequence that is referred to as LSPas, which overlaps the light strand promoter anti-sense or G-rich strand mtDNA (23). Additional in vitro data also demonstrate that hLon binds G-rich oligonucleotides corresponding to regions present throughout the heavy strand of mtDNA but shows the highest affinity for LSPas (25,28). An hLon-binding consensus has been suggested by results obtained using a combination of in vitro and cell-based assays along with bioinformatics (28). Furthermore, in cultured mammalian cells, hLon has been shown to associate with sites distributed primarily within one-half of the genome and preferentially with the control region for mtDNA replication and transcription encompassing LSPas (28). In oxidatively stressed cells, hLon binding to mtDNA is reduced. However, it is unknown whether oxidative stress directly divests hLon of its DNA-binding property, or whether hLon binding is blocked by the oxidative modification of mtDNA-binding sites per se, or by other proteins recruited to repair these damaged sites.
Previous studies on the binding of hLon to ssDNA provided limited insight into the specificity, stability and dynamics of this interaction. It has been suggested that hLon may recognize G-rich sequences that form guanine tetraplexes, or quadruplexes, which are four-stranded structures based on the hydrogen-bonded guanine tetrad, or G-quartet (25). G-quartet DNA is a high-ordered structure that contains polyanionic groups, and thus might interact with basic proteins in a nonspecific manner. In this study, we demonstrate that mtDNA sequences bound by hLon form parallel G-quadruplexes. Results show that hLon selectively binds to DNA sequences that have a propensity for forming G-quartets. However, the presence of G-quartets does not confer specificity for hLon association. Human Lon complexed with LSPas exhibits a large negative heat capacity change, which is a thermodynamic signature of sequence-specific interactions. By contrast, the interaction between hLon and other G-rich sequences examined leads to little or no significant change in heat capacity, and is primarily entropy driven. Taken together, these energetic data show that the affinity of hLon for ssDNA is determined by the propensity of the ssDNA sequence to form G-quartets, whereas the sequence specificity of hLon binding hinges on the extent of structural adaptation and release of interfacial solvent molecules between the protease and its DNA ligand.
MATERIALS AND METHODS
Expression and purification of human Lon protease
The expression plasmid of human lon was transformed into Rosetta 2 competent E. coli cells (Novagen), and recombinant hLon was expressed and purified as previously described (25) with some modifications. Briefly, transformed cells were grown at 37°C in medium containing 3% tryptone, 2% yeast extract, 1% MOPS (pH 7.2), 100 µg/ml ampicillin and 34 µg/ml chloramphenicol. At mid-log phase (OD600 ∼ 0.6–0.8), hLon production was induced by the addition of 1 mM IPTG for 1.5 h. The cells were harvested and resuspended in lysis buffer [50 mM Tris–HCl, pH 8.0, 300 mM NaCl, 10 mM imidazole, 10 mM β-Me, 20% glycerol (v/v) and 1% Triton X-100]. The lysate was sonicated on ice, and centrifuged at 48 000g at 4°C for 1 h. The supernatant was filtered (0.22 µm) and then applied to a HiTrap HP column (Amersham) equilibrated with Buffer A [20 mM Tris–HCl, pH 8.0, 300 mM NaCl, 10 mM MgCl2, 10% glycerol (v/v) and 40 mM imidazole]. After serial washes with Buffer A and Wash Buffer B (the same as A but containing 80 mM imidazole), hLon was eluted in Elution Buffer (the same as Buffer A but containing 400 mM imidazole). Purified hLon was concentrated with Amicon-50000 (Millipore), and dialyzed extensively against a buffer containing 20 mM HEPES–NaOH (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 0.1 mM DTT and 20% glycerol (v/v) unless specified otherwise. The purification steps described above were conducted at 4°C to avoid autodigestion of hLon. To exclude protein aggregates, the sample was centrifuged and filtered through a 0.22 µm-syringe filter. The protein concentration was determined by the Bradford method using bovine serum albumin (BSA) as a standard (Bio-Rad) (37).
Oligonucleotides
The oligonucleotides used in this study were purchased from ScinoPharm Biotech (Taiwan), and the sequences are listed in Table 1. DNA concentrations were determined spectrophotometrically using the extinction coefficients at 260 nm calculated from nearest-neighbor approximation (38).
Table 1.
The DNA sequences used in this study
| Sequence | Location of nucleotidesa | Strand | |
|---|---|---|---|
| LSPas | 5′-AATAATGTGTTAGTTGGGGGGTGA-3′ | 455–432 | Noncoding |
| LSPas18 | 5′-GTGTTAGTTGGGGGGTGA-3′ | 449–432 | Noncoding |
| TG6T | 5′-TGGGGGGT-3′ | 441–434 | Noncoding |
| LSPs | 5′-TCACCCCCCAACTAACACATTATT-3′ | 455–432 | Coding |
| LSPs18 | 5′-TCACCCCCCAACTAACAC-3′ | 449–432 | Coding |
| AC6A | 5′-ACCCCCCA-3′ | 441–434 | Coding |
| LS1as | 5′-GGCGTAGGTTTGGTCTAGGGT-3′ | 7114–7134 | Noncoding |
| LS2as | 5′-GTAGAGGGGGTGCTATAGGGT-3′ | 8262–8282 | Noncoding |
| LS3as | 5′-GTATGGGGGTAATTATGGTGG-3′ | 8395–8415 | Noncoding |
| LS4as | 5′-GGAGGGGGGTTGTTAGGGGGT-3′ | 10 936–10 956 | Noncoding |
| LS5as | 5′-CGAGGGTGGTAAGGATGGGGG-3′ | 12 387–12 407 | Noncoding |
| LS6as | 5′-GGGGAGGGGTGTTTAAGGGGGT-3′ | 15 526–15 546 | Noncoding |
| LSPasGA | 5′-AATAATGTGTTAGTTGGAAGGTGA-3′ | Mutant | |
| 18-mer ssDNA | 5′-CTCTTCTCCTCTCTTTCC-3′ | Control |
aThe locations of nucleotides of these sequences on human mitochondrial DNA are numbered.
Polyacrylamide gel electrophoresis
The DNA oligonucleotides were analyzed by native and denaturing gel electrophoresis on a SE600 with a water-circulating system (Amersham, NJ, USA). The samples were prepared at a concentration of 50 µM by diluting the stock solutions with TB buffer [89 mM Tris–borate, pH 8.0, 100 mM NaCl and 5 mM MgCl2] in the presence (denaturing) or absence (native) of 7 M urea. Prior to loading, the samples were heated at 95°C for 10 min and gently cooled down to room temperature. DNA electrophoresis was performed on 20% polyacrylamide gels [1 : 19 (w/w) bisacrylamide: acrylamide] containing either native or denaturing TB buffer and run at constant power of 25 W. The nucleic acids were detected by UV shadowing by placing the gel on fluorescein-coated silica (Merck) (39).
Electrophoresis mobility shift assay (EMSA)
5′-Fluorescein-labeled LSPas or LSPasGA (1 µM) was incubated with hLon (6.4 µM) in a 50 µl reaction containing 20 mM HEPES–NaOH (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 0.1 mM DTT and poly (dI-dC)·(dI-dC) (50 ng/reaction) (Amersham), in the presence and absence of 50-fold-excess unlabeled DNA competitor. After incubation for 20 min at 25°C or specified otherwise, the reaction mixtures were resolved on 6% native PAGE containing TGE buffer [50 mM Tris, pH 8.5, 0.38 M glycine, 2 mM EDTA and 0.05 mM β-ME] run at a constant power of 8 W. The electrophoresis temperature was controlled at 20°C with water circulation. The gels were scanned with Typhoon 9200 Variable Mode Imager (Amersham) at blue-excited fluorescence mode (488 nm).
Circular dichroism (CD) spectroscopy
The CD experiments were carried out on a J-715 spectropolarimeter (Jasco, MD, USA) equipped with a temperature controller. A water-jacketed cuvette of 0.1-cm path length was used. The cell-holder chamber was constantly purged with a stream of dry nitrogen gas. For CD measurements, the DNA samples (50 µM) were heated to 95°C and cooled to room temperature prior to analysis. The data were recorded between 210 and 320 nm at a scanning rate of 20 nm/min. After background correction, the data are presented as molar ellipticity [θ]M (deg·dmol−1·cm2) using
| 1 |
Mr is the sample molecular weight (g/mole), c is the concentration in g/ml, and l is the path length in cm.
The far-UV CD spectra of hLon (2.5 µM) were measured in 20-mM ammonium acetate (pH 7.5) and 20% glycerol. The data were collected between 200 and 250 nm at a rate of 20 nm/min. The CD measurements of hLon in the presence of different DNA oligonucleotides were carried out under identical conditions. After background subtraction (the buffer with and without the corresponding amount of DNA), the CD intensity was presented as mean residue ellipticity [θ]MRE in deg·dmol−1·cm2, which was obtained by averaging [θ]M out the number of amino acid residues in hLon.
Isothermal titration calorimetry (ITC)
The measurements of binding thermodynamics between hLon and DNA were performed isothermally on a VP-ITC MicroCalorimeter (MicroCal, MA, USA). To minimize the artifacts resulting from small differences in buffer composition, DNA (titrant) and hLon (titrand) were prepared with the identical lot of buffer consisting of 50 mM HEPES–NaOH (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 10% glycerol (v/v) and 0.01% sodium azide. The samples were filtered (0.22 µm) and degassed immediately before use. The calorimetric cell was loaded with hLon at 30 µM in a volume of 1.43 ml; LSPas or TG6T at a concentration of 350 µM in the syringe was programmed to titrate into the reaction cell (12 µl/injection) with 6-min spacing intervals. The stirring rate of the injector was 300 r.p.m. The heat responses caused by the dilution of hLon were found negligible by titrating the buffer against hLon and vice versa. The ITC measurements of DNA against buffer were also carried out separately. After corrected for the dilution effects of DNA, the binding isotherms of hLon and DNA were normalized and fitted with Equations (2) and (3) by nonlinear least square minimization procedures (40).
| 2 |
Δqi is the corrected heat response in the ith step of titration, and n stands for the number of identical and independent binding sites for titrant to titrand. [titrand]tot is the total concentration of titrand in the sample cell at a constant volume of Vcell. ΔH represents the apparent enthalpy change obtained from single experiment.
The ℜ in Equation (2) is given by
 |
3 |
where Yi is the degree of saturation defined by Yi=[titranti]bound/[titrand]tot · [titranti]tot means the accumulative concentration of titrant in the sample cell until injection i.
The free energy and entropy changes of binding were obtained using the relationship ΔG = − RT·lnKa = ΔH−TΔS. In addition, the heat capacity changes of association between hLon and DNA were given by ΔCp = ∂(ΔH)/∂T, where ΔH is the binding enthalpy, and T is the corresponding experiment temperature on ITC.
Differential scanning calorimetry (DSC)
DSC measures heat capacity (Cp) as a function of temperature. The experiments were carried out on a VP-DSC MicroCalorimeter (MicroCal, MA, USA). The sample preparation steps were similar to those in ITC, including the buffer composition. The sample cell was loaded with a solution containing hLon at 25 µM in the presence or absence of DNA (37.5 µM). The reference cell was loaded with the buffer. The heat capacity was scanned from 10°C to 110°C at a rate of 50°C/h. Scanning of the buffer against buffer as well as the DNA samples against buffer were performed in separate experiments. The heat capacity of the buffer effect was corrected from each thermogram according to the following equations (41).
| 4 |
| 5 |
| 6 |
∂Cp/∂T represents the slope of heat capacity in the pretransition region of sample. Denotations of S, P, L and PL stand for the solvent, protein, DNA and mixture of protein/DNA, respectively. The V-bars are the partial-specific volumes (ml/mg) of each component.
The effect of DNA binding on the heat capacity of hLon may be substantial, which is reflected on the pretransition Cp rather than the unfolding Cp because their interactions are mostly noncovalent. The pretransition Cp can be described by the following equation:
| 7 |
where Cp is the partial heat capacity, and Cp,0 is the partial heat capacity of the native state; Xi represents the population of solute in the ith state, and ΔHX,i is the enthalpy difference between the ith state and the native state (42). The heat capacities of proteins in the native state examined by DSC have always been found to increase linearly with temperature; they comprise not only an intrinsic variable of structural flexibility but also an instrumental artifact (42,43). To better assess the effect of DNA binding on hLon, we evaluated the difference in (∂Cp/∂T) between hLon and the DNA-bound hLon [Equation (8)].
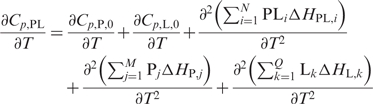 |
8 |
Both ∂Cp,P,0/∂T and ∂Cp,L,0/∂T are nearly zero because they represent the temperature dependence of the intrinsic heat capacities in the native state. Thus, the difference in ∂Cp,PL/∂T is primarily accountable for the structural flexibility of the protein, DNA and the complex.
RESULTS
The candidate-binding site of human Lon forms a parallel G-quartet structure in vitro
Recent work suggests that human Lon binds to mtDNA in living cells and preferentially associates with the control region (CR) of the mitochondrial genome (28). The putative mtDNA-binding target of hLon within CR overlaps the light strand promoter (LSP) and corresponds to the previously characterized oligonucleotide sequence referred to as LSPas. LSPas is a 24-base sequence located on the heavy (or antisense) strand of mtDNA (5′-AATAATGTGTTAGTTGGGGGGTGA-3′). Notably, the mtDNA heavy strand has a higher content of guanine and thymine residues, in contrast to the complementary light strand (44). LSPas contains six contiguous guanine bases, rendering a high-order structure demonstrated by electrophoretic analysis using both native and denaturing gels (Figure 1). Two forms of LSPas were observed (Figure 1A, lane 2): a monomeric form that migrated slightly slower than the complementary LSPs CA-containing oligonucleotide (lane 1) and a high-order structure that migrated slower than the double-stranded LSP (compare lanes 2 and 3). Similar migration differences were also observed for an 18-mer counterpart (LSPas18) that lacks six bases from the 5′ end of LSPas but retains the G-rich core sequence. The slower migrating forms of both LSPas and LSPas18 remained unchanged on denaturing polyacrylamide gels containing urea (Figure 1B, lanes 2 and 5, respectively), suggesting the presence of a structural motif, which was later identified by CD (Figure 2). Similarly, an 8-mer TG6T migrated much slower than its complementary AC6A (Figure S1, lanes 5 and 7, respectively). TG6T was unaffected by heat treatment at 120°C for 2 h, whereas the slower migrating form of LSPas was disrupted after heat treatment (Figure S1, compare lane 5 with 6, and lane 1 with 2). The susceptibility to heat denaturation may be attributed to the length of the ssDNA oligonucleotide, in addition to the number of bases flanking the G-rich core sequence.
Figure 1.

Electrophoretic mobility of G-rich and control oligonucleotides. Oligonucleotides were analyzed under (A) native and (B) denaturing conditions using 20% polyacrylamide gels. G-rich oligonucleotides—LSPas, LSPas18 and TG6T; control oligonucleotides—LSPs, dsLSP, LSPs18, dsLSP18, an 18-mer ssDNA and 18-mer dsDNA. Arrows indicate the higher-ordered structures of LSPas and LSPas18. A 20-bp ladder DNA marker was loaded but not shown here under UV shadowing (stained with SYBR Gold).
Figure 2.

Formation of G-quartets in LSPas and TG6T demonstrated by circular dichroism spectroscopy. CD spectra of LSPas (A and B) and TG6T (C and D) were monitored from 10°C to 90°C in the presence or absence of urea. The buffer used in A and C contains 10 mM sodium cacodylate (pH 7.5), 100 mM NaCl, 5 mM MgCl2 and 0.1 mM EDTA. The buffer used in B and D consists of 10 mM sodium cacodylate (pH 7.5) and 1 M urea.
CD was employed to identify the possible structural differences in these G-rich ssDNA oligonucleotides. The CD spectra of LSPas in a native buffer containing 100 mM NaCl exhibited a strong positive peak at 265 nm and a negative one at 245 nm, reflecting the presence of a parallel G-quartet (Figure 2A). Typically, parallel G-quartets are characterized by a positive CD band at ∼260 nm and a negative one at ∼240 nm, whereas antiparallel G-quartets have a positive band at ∼295 nm and a negative one at ∼260 nm (45,46). The structure of LSPas remained mostly intact between 10°C and 30°C, the range of which was thus selected for monitoring its association with hLon in the ITC and DSC experiments (discussed later). LSPas became relatively unstructured at above 50°C (Figure 2A). The CD profile of LSPas with respect to temperature in a Na+-containing denaturing buffer (the native buffer with 1-M urea; spectra not shown) was compared to that in the native buffer. However, the G-quartet signature of LSPas was dramatically weakened in the denaturing buffer without NaCl; the ellipticity not only decreased but also red shifted with the temperature increment (Figure 2B). This result suggested the importance of monovalent cations in stabilizing G-quartet LSPas DNA. It has been reported that K+, Na+, NH4+ and several other monovalent cations stack between adjacent G-tetrads in G-quartet DNA to coordinate the eight carbonyl groups in every two tetrads, thereby making the G-quartet rigid (47–49). In particular, Na+ and NH4+ have been shown to neutralize the peripheral phosphate groups of G-quartets (50). In contrast to LSPas, TG6T exhibited a comparatively rigid G-quartet as revealed in the CD results; it was only mildly denatured by high temperature or by denaturing buffer (Figure 2, compare C with D; Figure S1, compare lane 5 with 6). TG6T showed a strong positive peak at 262 nm and a negative one at 242 nm (Figure 2C). The sequences of both LSPas and TG6T contain no contiguous guanines other than a 6-nt G cluster; the contexts hardly allow the formation of hairpins or intramolecular G-quartets. Based on the electrophoretic and CD data, LSPas and TG6T form intermolecular parallel G-quartet structures in Na+ solutions. LSPas was more vulnerable than TG6T to thermal and chemical denaturation because of the flanking sequence effect: the repulsion caused by the unpaired bases hinders the formation of LSPas quadruplex (see the schematic structures in Figure 2).
Human Lon selectively binds to G-quartets
The number of consecutive guanine bases in an intermolecular G-quartet is the main determinant of its stability. When we replaced the third and fourth guanines within LSPas with adenines, the resulting ssDNA oligonucleotide referred to as LSPasGA did not form a G-quartet (Figure S1, lane 4). The EMSA was used to examine the binding of hLon to fluorescein-conjugated LSPas or LSPasGA (Figure 3A). The results showed that hLon selectively interacted with the G-quartet form of LSPas but not with the monomer. When 7-deaza-guanines (7-deaza-Gs) were substituted for the 6-nt G cluster of LSPas, the sequence failed to form a G-quartet because the C-7 cannot be an electron donor for Hoogsteen hydrogen bonding. The 7-deaza-G modified LSPas was not bound by hLon (data not shown). The binding of hLon to fluoresceinated LSPas was competed by the addition of 50-fold excess of unlabeled LSPas but not LSPasGA. Moreover, hLon interacted only weakly with fluorescein-conjugated LSPasGA, which lacked G-quartet-forming ability.
Figure 3.

hLon binding to G-quartet DNA. (A) hLon binding to fluorescently labeled LSPas (lanes 1–4) and LSPasGA (lanes 5–8) probes was analyzed by electrophoresis mobility shift assay using 6% polyacrylamide gels. The unlabeled DNA competitors were added at a 50-fold excess the amount of the fluorescent probes. (B) A fixed amount of LSPas or TG6T (1 µM) was incubated with increasing concentration of hLon (0, 1.1, 5.5, 11, 16.5 and 22 µM). The reaction mixtures were analyzed on 6% polyacrylamide gels.
It is noteworthy that the heavy strand of human mtDNA exhibits a high frequency of 4–12 contiguous guanine residues every ∼100 bases. The binding of hLon to G-rich sequences in vivo is supported by a combination of cell-based, in vitro and bioinformatics analyses (25,28). In vitro experiments examining hLon binding to ssDNA oligonucleotides containing several of these G-rich sites present within the mitochondrial genome, show that hLon binds with higher affinity to LSPas than to the other G-rich sequences (25,28). However, previous studies did not directly determine the extent to which any of these G-rich ssDNAs form G-quartets. For this purpose, we analyzed the G-quartet-forming capabilities of these G-rich oligonucleotides by native gel electrophoresis (Figure S2). Some of these G-rich ssDNAs formed extensive high order structures. Although spectroscopic analysis was not performed because of the structural complexities, the high order forms likely represented G-quartets resulting from 2–3 discrete G-clusters in their sequence contexts. Results showed that the sequences capable of forming high order structures were better able to compete with LSPas for hLon binding (Figure S3). Dose-dependent binding of LSPas as well as TG6T to hLon is shown in Figure 3B.
The binding thermodynamics of hLon to DNA measured by ITC
Knowing that hLon binds to G-quartet DNA, we used ITC to investigate the energetics of hLon complexed with LSPas and TG6T. The binding reaction of hLon to LSPas is exothermic over the temperature range inspected (Figure 4A). The amount of heat release was integrated and fitted nonlinearly (Figure 4B). The enthalpy of association of hLon with LSPas decreased from −1.48 to −13.72 kcal/mol over a temperature range from 10°C to 30°C (Table 2). In contrast to the downward trend in the enthalpy change, the entropy penalty (−TΔS) increased from −5.49 to 6.21 kcal/mol with increasing temperature. The association constant (Ka) was ∼105 M−1 and was essentially temperature independent. In addition, the free energy change corresponding to the Ka was not susceptible to temperature changes, as a result of strong enthalpy–entropy compensation (Figure 4C).
Figure 4.

Isothermal titration calorimetric analysis for the binding of hLon to LSPas and TG6T. (A) LSPas and (D) TG6T were titrated (350 µM) into solutions containing hLon (30 µM); heat responses were registered isothermally at the temperatures as indicated. Integrals of the heat responses were normalized and fitted with a nonlinear least-square method to obtain thermodynamic parameters (B and E). The parameters including −TΔS, ΔH and ΔG representing the association of hLon with LSPas (C) and TG6T (F) were plotted against temperature.
Table 2.
Thermodynamic parameters for the binding of hLon to LSPas and TG6T
| Target DNA | Temperature (°C) | ΔH (kcal/mol) | −TΔS (kcal/mol) | Ka (×105 M−1) | ΔG (kcal/mol) | ΔCp (cal/mol/K) |
|---|---|---|---|---|---|---|
| LSPas | 10 | −1.48 ± 0.12 | −5.49 | 2.45 ± 0.43 | −6.99 ± 0.09 | −607.82 |
| 15 | −5.04 ± 0.12 | −1.98 | 2.11 ± 0.11 | −7.03 ± 0.03 | ||
| 20 | −8.37 ± 3.31 | 1.01 | 3.03 ± 0.83 | −7.29 ± 0.17 | ||
| 30 | −13.72 ± 6.91 | 6.21 | 2.60 ± 0.72 | −7.52 ± 0.15 | ||
| TG6T | 5 | −2.04 ± 0.75 | −4.14 | 1.24 ± 0.58 | −6.49 ± 0.21 | 67.29 |
| 10 | −1.84 ± 0.88 | −4.71 | 1.35 ± 0.89 | −6.58 ± 0.36 | ||
| 20 | −1.30 ± 0.14 | −5.69 | 1.36 ± 0.22 | −6.80 ± 0.09 | ||
| 30 | −0.63 ± 0.02 | −7.05 | 3.48 ± 0.36 | −7.69 ± 0.06 |
ΔCp was obtained from linear regression of ΔH against temperature. The binding thermodynamic parameters of both LSPas and TG6T were calculated on the monomeric concentrations of hLon and DNA. The LSPas constants corrected for the G-quartet fraction of LSPas are aΔH, −T [ΔS + (a/T + Rlna)], alnK, ΔG-RTlna and aΔCp, respectively. a represents the reciprocal of the LSPas G-quartet fraction, which was estimated 1.15 as analyzed by ImageQuant ver. 5.2 (Molecular Dynamics) with Figure 3A lane 1 (G-quartet 0.87; monomer 0.13).
The binding of hLon to LSPas showed a characteristic thermodynamic switch at Ts = 18.86°C, where the reaction entropy was zero. The zero-enthalpy temperature TH was extrapolated to 6.98°C. This binding reaction was enthalpy-driven and entropy-unfavorable when the temperature was above 18.86°C, being entropy-driven and enthalpy-unfavorable when the temperature was below 6.98°C. At temperatures between 6.98°C and 18.86°C, the reaction was both enthalpy- and entropy-driven. This result demonstrates that the association of hLon with LSPas was dominated by a large negative enthalpy change and a reduction in entropy at physiologically relevant temperatures. The apparent heat capacity change evaluated by the temperature dependence of the enthalpy changes was −607.82 cal·K−1·mol−1.
As demonstrated by EMSA, hLon formed a complex with TG6T (Figure 3B), which competed for hLon binding to LSPas, albeit poorly (Figure S3A). We proceeded with a mechanistic study of hLon binding to TG6T under the same conditions used to analyze LSPas binding. The thermodynamic data revealed that similar to hLon binding to LSPas, binding to TG6T was exothermic, and exhibited moderate temperature dependence (Figure 4D and Table 2). However, in contrast to what was observed with LSPas, the heat capacity change for hLon binding to TG6T was signed positive and relatively small, giving a value of 67.29 cal·K−1·mol−1. The reaction entropy in the case of TG6T was positive over the temperature examined. The two thermodynamic switch temperatures, TH and TS, were 38.65°C and −30.86°C, respectively (Figure 4F). Within the broad temperature range between 38.65°C and −30.86°C, the binding of hLon to TG6T was enthalpy-and-entropy-driven, and was mostly dominated by the entropy term. This reaction was mainly entropy-driven both near and above physiologically relevant temperatures. It was entropy-unfavorable at the temperature below −30.86°C, and enthalpy-unfavorable at the temperature above 38.65°C.
The magnitude of heat capacity change is generally regarded as a thermodynamic signature of hydrophobic interactions. It has been reported that ligand binding involving specific interactions is generally accompanied by a significant decrease in heat capacity (51–53). The heat capacity change for the binding of hLon to LSPas was large in comparison with that for the unfolding of globular proteins reported to be 12 cal·K−1·mol−1 per residue, suggesting the possibility that the association of hLon with LSPas might be coupled with local folding of 51 amino acid residues (54,55). The large negative heat capacity change for their interactions reflects a significant decrease in the translational and rotational degrees of freedom, as a result of ensemble conformational restriction manifested by Equation (9).
| 9 |
The scenario that reduction in thermal fluctuations as hLon binds to LSPas is consistent with the finding of this complex in temperature-dependent CD spectra (Figure S4A and B). The result indicated that the association of hLon with LSPas provides a substantial enhancement of thermal stability, which was also observed for TG6T albeit to a lesser extent (Figure S4C). Moreover, the CD spectral analysis of these hLon–DNA complexes suggested that neither hLon nor the DNA molecules were subjected to global conformational transitions upon binding (Figure S5). The G-quartet structures recognized by hLon remain largely unperturbed as demonstrated by the CD spectra of DNA from 250 to 350 nm, within which DNA is sensitive to conformational changes whereas the protein has very weak signal (Figure S5). Although the CD intensity of DNA weakened in the presence of hLon, it underwent no structural transition (no chromatic shift was observed). The data suggest that the G-quartet structures bound by hLon are not disrupted; at least they are generally intact. However, it remains to be discovered whether the protein locally distorts or bends the G-quartet structures.
DSC analysis of the sequence-dependent heat capacity change
DSC was used to further investigate whether the significant decrease in heat capacity in the binding of hLon to LSPas was sequence-specific. The heat capacity of hLon as a function of temperature showed an accumulation of energy in the pretransition region (Figure 5A). The transition temperature of hLon was ∼46°C, and was followed by an abrupt decrease in heat capacity that indicated a step of dramatic exothermic reaction. The onset of this exothermic reaction occurred at ∼50°C, which most likely resulted from the aggregation of hLon immediately after or partly during the thermal denaturation. When significant interactions between hLon and DNA occur, the complex can be populated with multiple microstates in the pretransition region [Equation (8)], which means an extra energy accumulation attributed to this binding. The effect of DNA binding on the overall heat capacity in the pretransition region from 10°C to 30°C was qualitatively assessed by Equation (8). Since the transition temperatures of the DNA oligonucleotides were all found beyond 80°C, at which the heat capacity of buffer began to deviate from the baseline, we therefore undertook the evaluation by assuming that the differences caused by DNA were negligible in these cases.
Figure 5.

Heat capacity thermograms of hLon in the presence and absence of DNA. (A) The partial molar heat capacity of hLon is shown between 10°C and 70°C. The heat capacity of hLon in complex with LSPas (B) or TG6T (C) at a 1:1.5 molar ratio after buffer correction was normalized by hLon molarity.
The difference in the pretransition slope (∂Cp/∂T) between hLon and the hLon/LSPas complex was +1.844 kcal/mol/K2 (Figure 5B and Δ(∂Cp/∂T) in Table 3). The positive value signified that more heat was absorbed to weaken the interactions between hLon and LSPas. In addition, the endothermic dissociation of the hLon-LSPas complex appeared to partly compensate the drastic decrease of heat capacity that arose from hLon aggregation. However, the gain of extra ∂Cp/∂T for DNA binding was not observed in other G-rich sequences. The Δ(∂Cp/∂T) of the hLon–TG6T complex was −0.271 kcal/mol/K2, signed negative and relatively small, and the hLon in complex with LS1as, LS2as and LS3as also showed little significant change in ∂Cp/∂T (Figure 5C and Table 3). The data of LS4as and LS5as are likely to be invalid because the hLon aggregated when either sequence was added in the solution prior to scan. In brief, these results revealed that hLon binding to LSPas was significantly involved in weak interactions as indicated by the relatively large positive change in the pretransition heat capacity relative to temperature, but it was not the case in other G-rich sequences although they formed G-quartets. The DSC data are fairly consistent with the ITC results: hLon binding to LSPas was accompanied by a highly negative heat capacity change, which was absent from the binding to TG6T. The combination of ITC and DSC analyses sheds new light on the DNA-binding mechanism of hLon; whereby the specific binding of the protease to LSPas is coupled with structural adaptation as demonstrated by the large negative heat capacity change of the association. By contrast, the heat capacity measurements on hLon in complex with other G-rich sequences showed little or no significant change in heat capacity. Moreover, the dissociation constants for hLon binding to LSPas and TG6T at 20°C were 3.30 and 7.35 µM, respectively. This result suggests that hLon has similar affinity for both sequences, but the binding to TG6T has a relatively negligible heat capacity change and a lesser extent of structural stabilization, which can be referred to as rigid-body association.
Table 3.
Determination of the pretransition slopes of heat capacity in hLon and hLon/DNA complexes
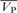 |
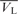 |
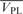 |
〈 ∂ Cp /∂ T〉 (kcal/mol/K2) | Δ 〈 ∂ Cp /∂ T〉 (kcal/mol/K2) | |
|---|---|---|---|---|---|
| hLon | 0.745 | 0.871 | |||
| hLon/LSPas | 0.572 | 0.727 | 2.715 | +1.844 | |
| hLon/TG6T | 0.596 | 0.739 | 0.600 | −0.271 | |
| hLon/LS1as | 0.581 | 0.730 | 0.711 | −0.160 | |
| hLon/LS2as | 0.581 | 0.729 | 0.914 | +0.043 | |
| hLon/LS3as | 0.577 | 0.729 | 0.804 | −0.067 | |
| hLon/LS4as | 0.589 | 0.730 | 0.199 | −0.672a | |
| hLon/LS5as | 0.587 | 0.730 | -0.556 | −1.427a |
Calculation of the partial-specific volume of hLon () was performed with the program SEDNTERP (available at http://www.jphilo.mailway.com/download.htm). The V-bars of DNA molecules () were estimated from the individual V-bars of bases, the values of which have been resolved by Dr Olwyn Byron (http://www.bbri.org/RASMB/rasmb.html). The V-bars of the hLon/DNA mixtures () were calculated with the equation, (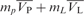 )/(mp + mL) (41).
)/(mp + mL) (41).
aThe Δ of hLon/LS4as and hLon/LS5as are likely to be invalid because hLon aggregated upon binding.
DISCUSSION
Heat capacity change arising from a ligand-binding reaction can be attributed to polyelectrolyte and nonpolyelectrolyte effects. Polyelectrolyte effects are a consequence of dehydration upon complex formation, and the relocation of water molecules from the solute hydration shell to the bulk solvent is entropy oriented. Nonpolyelectrolyte effects resulting from hydrophobic and other weak interactions are directed by enthalpy changes. The influence of these effects on the binding of hLon to DNA is determined by thermodynamic-driving forces: the enthalpy and entropy changes. The polyelectrolyte effect is noticeably temperature dependent compared to the nonpolyelectrolyte effect; apolar hydration heat capacity decreases with temperature increment, and thus the favorable gain of dehydration entropy for a complex formation is expected to increase with temperature (56–58). Meanwhile, dehydration is likely to occur with a mild positive change in enthalpy. This inherent dependence of entropy and enthalpy on temperature gives rise to a partial positive heat capacity change. Nevertheless, hLon binding to LSPas was associated with a dramatic decrease in heat capacity; the ensemble enthalpy change was negative over the temperature examined, and the entropy change became quite negative at higher temperatures. The compensation temperature TC (where the ΔH and the −TΔS converged) of hLon binding to LSPas was 12.79°C, above which the enthalpy term predominated. By contrast, hLon binding to TG6T was accompanied by a slight increase in heat capacity; its TC was −5.15°C, above which the reaction was primarily driven by entropy. Taken together, these results suggested that hLon binding to LSPas revolves around nonpolyelectrolyte effects, whereas the binding to TG6T is primarily influenced by polyelectrolyte effects.
Despite striking differences in their heat capacity changes and major driving forces, hLon binding to LSPas and TG6T showed similar association constants and free energy changes (Table 2). An underestimation of the LSPas parameters unavoidably occurs for hLon binding to the G-quartet form because because LSPas DNA was present in reactions as both monomeric and G-quartet forms. The thermodynamic parameters including the association constants and free energy changes are of the right order of magnitude; however, they should be corrected for the fraction of LSPas G-quartets present in order to compare more accurately the binding of LSPas and TG6T to hLon (see Table 2). In energetic terms, the DNA-binding affinity of a protein is reflected by the free energy change, which is relatively insensitive to subtle changes in either binding component. It has been shown in recent studies that only a few crucial regions within a macromolecule complex determines the free energy of binding (55,59). The remaining regions are more relevant to binding specificity (stereochemistry), and contribute little to the overall ΔG. The deletion or mutation of specificity-determining groups that do not damage the structural integrity of the complex would not likely affect the binding free energy; thus thermodynamic homeostasis could be maintained. Nonetheless, the ΔH and the ΔS would be susceptible to minor structural changes, even though these two terms may compensate in a similar fashion resulting in an invariant free energy change.
The thermodynamic ‘neutral’ was observed in the binding of hLon to TG6T, whose free energy change was comparable to that of LSPas. This phenomenon revealed that TG6T, the core region of G-quartet formation residing in LSPas, is essential for the stabilization of hLon–DNA complex by conferring most of the binding energy. As indicated by EMSA results, disrupting the G-quartet in LSPas by replacing two guanines with adenines interfered with binding to hLon. This result suggested that the DNA substrate determines the binding preference of hLon at free energy level. In other words, hLon binding to LSPas is significantly weakened if LSPas loses its ability to form G-quartet. However, the binding specificity should be addressed by its thermodynamic properties and heat capacity change. The weak dependence of entropy and enthalpy on temperature for hLon binding to TG6T parallels the association with LSPas at low temperatures. But at high temperatures, weak bond formation apparently dominates the interactions between hLon and LSPas, rendering more negative the enthalpy as well as the entropy of reaction; while in the case of TG6T, dehydration appears to dominate the thermodynamics of this complex formation. In addition, the drastic difference in heat capacity change between these two cases signifies their fundamental discrepancy in the degree of temperature dependence for the individual thermodynamic parameters including the enthalpy and entropy changes. For a ligand-binding reaction, heat capacity change is primarily concerned with hydrophobic interactions and dehydration of apolar groups. Summing up the thermodynamic results, we infer that the small change in heat capacity for the binding of hLon to TG6T mainly accounts for the extent of dehydration, whereas the binding to LSPas associated with the large negative heat capacity change reflects a considerable involvement of hydrophobic interactions. Supported further by the DSC results, these findings suggest that hLon binds to most G-quartet-forming sequences present in mtDNA principally through electrostatic interactions which are thermodynamically inadequate for specific binding. By contrast, hLon recognizes LSPas as a specific target by enhancing the structural stability of the complex. In addition, these results raise the possibility that a sequence-specific recognition step might be preceded by nonspecific binding to G-quartet structures (60).
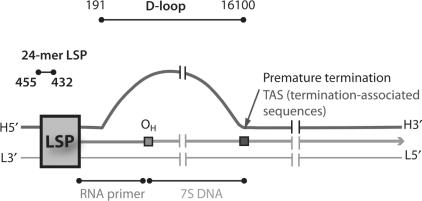
The human mitochondrial genome (mtDNA) consists of 16,569 bp. Although the base composition of mammalian mtDNA is biased with a high frequency of contiguous guanines on the heavy (H) strand as compared with the complementary light (L) strand, only a few studies have addressed the physiological implications of G-quartets in this closed circular genome (61,62). Analysis of human mtDNA with two G-quartet motif-searching programs, Quadfinder and QGRS Mapper, revealed 20 intramolecular G-quartet-forming hits on the H strand, while none were found on the L strand (Table S1) (63,64). Fourteen out of these 20 sequences are located between LSP and OL on the heavy strand. LSPas is contained within one of these hits positioned between 438 and 471.
One model of mtDNA replication as proposed by Clayton and the colleagues (65,66) holds that replication proceeds in an asymmetric manner. Synthesis is initiated at the origin of the H strand (OH) that is downstream from the LSP. The single-stranded parental H strand is displaced during replication, and expands until approximately two-thirds the genome is replicated reaching the origin of the L strand (OL), at which point L-strand replication ensues in the opposite direction. Studies have found that replication of the leading H strand terminates prematurely at TAS (termination-associated sequences), resulting in a 7S DNA; this three-stranded structure referred to as D-loop (displacement-loop), consists of the parental L-strand template, the nascent prematurely terminated H strand and the displaced single-stranded parental H strand (Table 1) (67,68). It has been proposed that the rate of mtDNA replication is regulated by the turnover of the D-loop, which is likely mediated by the formation of secondary structures at certain conserved sequences within the D-loop (69,70) and some trans-acting factors binding to these regions (71–73). We suggest that the single-stranded parental H strand exposed to solution might allow the formation of G-quartets at various G-rich clusters between LSP and OL as identified by Quadfinder (Table S1). Recent findings by Maizels and the colleagues (74) on the G-quartet-containing structures called G-loops may strengthen the validity of our suggestion. However, it is unclear whether hLon interacts with intramolecular LSPas quadruplex, and if the binding is accompanied by similar heat capacity effect: it remains to be clarified whether the specific recognition of the LSPas sequence requires the flanking bases to present quadruply. Although it has been debated on the biological relevance of intramolecular parallel G-quartets arising from topological constraints, a FRET-based investigation reveals that parallel and antiparallel G-quartet conformations can coexist and interconvert under physiological relevant conditions (75). In our study, the combination of electrophoretic, spectroscopic and thermodynamic data indicates that hLon preferentially binds to intermolecular parallel G-quartet structures. Given that multiple copies of mtDNA molecules frequently concentrate to form heritable units of mtDNA or to facilitate intermitochondrial recombination (76–78), formation of G-quartets between separate mtDNA molecules could be possible in vivo (see the schematic structures in Figure 2). We have found that the G-quartet-forming motifs on mtDNA are thermodynamically favorable to form intermolecular G-quartets while kinetically prone to form intramolecular G-quartets in vitro (our unpublished data). However, whether mtDNA forms G-quartets intra- and/or intergenomically remains to be determined.
This study reveals that hLon has a general preference for G-quartet DNA, and selectively binds to LSPas along with structural adaptations for the overall structural tightening. Our findings are consistent with recent work identifying a G-rich consensus sequence for hLon binding that matches well with a sequence contained within LSPas (28). Additional consensus sequences showing the best fit for hLon binding sites are primarily located downstream of OH and before OL. In vitro, purified hLon binds with higher affinity to ssDNA oligonucleotides that conform to this G-rich consensus. Consistent with these findings, hLon binds preferentially in cultured human cells to the CR/D-loop region as well as to sites between OH and OL. Our data support the notion that hLon binding to mtDNA sequences in vitro and in vivo is determined by the propensity of ssDNA to form G-quartets. We demonstrate that hLon is able to recognize its binding target with a notable reduction in thermal fluctuations. In addition, the high proportion of G-rich clusters in the H strand raises the possibility that these sites serve as hot spots of binding for hLon. We propose that hLon associates with the G-quartet-forming regions during the expansion of D-loop, and selectively binds to target sequence(s) on the basis of different binding heat capacities. Thus, the finding of specific target(s) by hLon to mtDNA might be facilitated by initial binding to these hot spots rather than random diffusion or one-dimensional sliding (79,80).
Potential G-quartet-forming regions that have been characterized in eukaryotic nuclear genome are principally found in telomeres, immunoglobulin heavy-chain switch regions, ribosomal DNA, G-rich microsatellites and certain promoter regions (81,82). Telomeres are guanine-rich tandem repeats at the ends of linear chromosomes, which tend to form G-quartet structures to prevent the 3′ single-stranded overhang from nonhomologous end joining. Telomere integrity is regulated by some G-quartet destabilizing factors as well as telomerase. Telomere-binding proteins such as Pot1 and G-quartet-specific helicases like BLM and WRN syndrome proteins render the telomeric DNA accessible to telomerase for end extension (83). Deregulations of telomerase activity and the helicase-mediated unwinding of G-quartets at telomeres are potentially associated with many types of cancer. On the other hand, many putative G-quadruplexes have been identified in nontelomeric regions, of which promoter sequences are intensely concerned with the correlation between the G-quartet stability and gene expression. Stabilization and destabilization of the G-quartet structures at the promoter regions of c-myc and Ki-ras proto-oncogenes have been revealed to be important to the gene regulation (84,85). It is of great interest to discover how gene expression is regulated by some G-quartet-binding proteins that are involved in the turnover of G-quartet structures at the control regions.
The specialized nucleic acid structures found in nuclear genome broaches an intriguing issue of whether they exist in organelle genomes, and if they are under similar or shared regulation. Unlike nuclear DNA, typical mitochondrial genomes are circular and thus do not require telomere/telomerase system for an end-replication problem. However, linear mtDNA molecules are found in certain yeast strains, which have mitochondrial telomere-binding proteins for maintaining their genome stability (86,87). Our data raise the possibility that some regions of mtDNA may form G-quadruplexes. Although this conjecture awaits further experimental validation, recent findings on localization of potential G-quartet-binding proteins at mitochondria suggest they might interact with G-quartet structures on mtDNA. Telomere reverse transcriptase (TERT) that normally guards the end integrity of chromosomes in nuclei carries a mitochondrial targeting sequence and retains telomerase activity in mitochondria (88,89). Translocation of TERT from nuclei to mitochondria is triggered by oxidative stress and exacerbates mtDNA damage, in contrast to its protective role in nuclei. Topoisomerase I (Top1) has also been identified in mitochondria with yet unclear functions (90,91). The existence of mitochondrial Top1 might be relevant to the topologic problems encountered during mtDNA replication/transcription. Its nuclear counterpart interacts with and promotes the formation of G-quartet structures in vitro (92). However, whether the mitochondrial Top1 as well as TERT associates with G-quartet structures on mtDNA is unknown.
The dissociation constants of hLon binding to LSPas and TG6T were found at micromolar range by using ITC. The binding strength seems to be weak, but reasonable because hLon most likely functions as a regulatory protein. It has been reported that hLon binding to DNA is inhibited by nucleotides whereas is stimulated by protein substrates (25). The physiological functions of mtDNA bound by hLon are still unclear. As hLon functions as an ATP-dependent protease and is a likely sensor of environmental stress in mitochondria, it is possible that hLon selectively degrades components of the mtDNA replication, repair and transcription machinery in response to changes in cellular metabolism. A variety of mitochondrial intermediate enzymes seemingly unrelated to mtDNA transactions such as aconitase, acetohydroxyacid reductoisomerase and serine/threonine deaminase have been identified in mitochondrial nucleoids (34,93–95), suggesting that they may be important in coordinating metabolic regulation with mtDNA maintenance. Further investigation is required to determine whether these respective enzymatic activities are functionally linked to their association with mtDNA binding, and whether they are selectively degraded and thereby regulated by hLon during cellular stress such as the increased generation of reactive oxygen species and/or hypoxia.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank Dr Ming-Chya Wu for access to the differential scanning calorimeter. We are also grateful to Dr Lou-Sing Kan, Dr Chia-Ching Chang, Dr Jiunly Chir and Dr Kamal Singh for helpful comments, and to Drs Hui-Chuan Chang and Chris S.-C. Jao for technical support in circular dichronism and isothermal titration calorimeter. This study was funded by National Science Council, Taiwan (96-2311-B-001-010 to S.-H.W.). Funding to pay the Open Access publication charges for this article was provided by Academia Sinica, Taiwan.
Conflict of interest statement. None declared.
REFERENCES
- 1.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 2.Fukui T, Eguchi T, Atomi H, Imanaka T. A membrane-bound archaeal Lon protease displays ATP-independent proteolytic activity towards unfolded proteins and ATP-dependent activity for folded proteins. J. Bacteriol. 2002;184:3689–3698. doi: 10.1128/JB.184.13.3689-3698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Besche H, Tamura N, Tamura T, Zwickl P. Mutational analysis of conserved AAA+ residues in the archaeal Lon protease from Thermoplasma acidophilum. FEBS Lett. 2004;574:161–166. doi: 10.1016/j.febslet.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 4.Chung CH, Goldberg AL. The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La. Proc. Natl Acad. Sci. USA. 1981;78:4931–4935. doi: 10.1073/pnas.78.8.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin DT, Goff SA, Webster T, Smith T, Goldberg AL. Sequence of the lon gene in Escherichia coli. A heat-shock gene which encodes the ATP-dependent protease La. J. Biol. Chem. 1988;263:11718–11728. [PubMed] [Google Scholar]
- 6.Van Dyck L, Pearce DA, Sherman F. PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1994;269:238–242. [PubMed] [Google Scholar]
- 7.Lee AY, Tsay SS, Chen MY, Wu SH. Identification of a gene encoding Lon protease from Brevibacillus thermoruber WR-249 and biochemical characterization of its thermostable recombinant enzyme. Eur. J. Biochem. 2004;271:834–844. doi: 10.1111/j.1432-1033.2004.03988.x. [DOI] [PubMed] [Google Scholar]
- 8.Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T, Taniguchi H. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem. 2004;279:421–428. doi: 10.1074/jbc.M305623200. [DOI] [PubMed] [Google Scholar]
- 9.Ostersetzer O, Kato Y, Adam Z, Sakamoto W. Multiple intracellular locations of Lon protease in Arabidopsis: evidence for the localization of AtLon4 to chloroplasts. Plant Cell Physiol. 2007;48:881–885. doi: 10.1093/pcp/pcm052. [DOI] [PubMed] [Google Scholar]
- 10.Charette MF, Henderson GW, Markovitz A. ATP hydrolysis-dependent protease activity of the lon (capR) protein of Escherichia coli K-12. Proc. Natl Acad. Sci. USA. 1981;78:4728–4732. doi: 10.1073/pnas.78.8.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waxman L, Goldberg AL. Selectivity of intracellular proteolysis: protein substrates activate the ATP-dependent protease (La) Science. 1986;232:500–503. doi: 10.1126/science.2938257. [DOI] [PubMed] [Google Scholar]
- 12.Mizusawa S, Gottesman S. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc. Natl Acad. Sci. USA. 1983;80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurizi MR. Degradation in vitro of bacteriophage lambda N protein by Lon protease from Escherichia coli. J. Biol. Chem. 1987;262:2696–2703. [PubMed] [Google Scholar]
- 14.Bonnefoy E, Almeida A, Rouviere-Yaniv J. Lon-dependent regulation of the DNA binding protein HU in Escherichia coli. Proc. Natl Acad. Sci. USA. 1989;86:7691–7695. doi: 10.1073/pnas.86.20.7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright R, Stephens C, Zweiger G, Shapiro L, Alley MR. Caulobacter Lon protease has a critical role in cell-cycle control of DNA methylation. Genes Dev. 1996;10:1532–1542. doi: 10.1101/gad.10.12.1532. [DOI] [PubMed] [Google Scholar]
- 16.Torres-Cabassa AS, Gottesman S. Capsule synthesis in Escherichia coli K-12 is regulated by proteolysis. J. Bacteriol. 1987;169:981–989. doi: 10.1128/jb.169.3.981-989.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stout V, Torres-Cabassa A, Maurizi MR, Gutnick D, Gottesman S. RcsA, an unstable positive regulator of capsular polysaccharide synthesis. J. Bacteriol. 1991;173:1738–1747. doi: 10.1128/jb.173.5.1738-1747.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whistler CA, Stockwell VO, Loper JE. Lon protease influences antibiotic production and UV tolerance of Pseudomonas fluorescens Pf-5. Appl. Environ. Microbiol. 2000;66:2718–2725. doi: 10.1128/aem.66.7.2718-2725.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002;4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 20.Rep M, van Dijl JM, Suda K, Schatz G, Grivell LA, Suzuki CK. Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science. 1996;274:103–106. doi: 10.1126/science.274.5284.103. [DOI] [PubMed] [Google Scholar]
- 21.Hori O, Ichinoda F, Tamatani T, Yamaguchi A, Sato N, Ozawa K, Kitao Y, Miyazaki M, Harding HP, et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J. Cell Biol. 2002;157:1151–1160. doi: 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu GK, Smith MJ, Markovitz DM. Bacterial protease Lon is a site-specific DNA-binding protein. J. Biol. Chem. 1997;272:534–538. [PubMed] [Google Scholar]
- 23.Fu GK, Markovitz DM. The human LON protease binds to mitochondrial promoters in a single-stranded, site-specific, strand-specific manner. Biochemistry. 1998;37:1905–1909. doi: 10.1021/bi970928c. [DOI] [PubMed] [Google Scholar]
- 24.Lu B, Liu T, Crosby JA, Thomas-Wohlever J, Lee I, Suzuki CK. The ATP-dependent Lon protease of Mus musculus is a DNA-binding protein that is functionally conserved between yeast and mammals. Gene. 2003;306:45–55. doi: 10.1016/s0378-1119(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Lu B, Lee I, Ondrovicova G, Kutejova E, Suzuki CK. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J. Biol. Chem. 2004;279:13902–13910. doi: 10.1074/jbc.M309642200. [DOI] [PubMed] [Google Scholar]
- 26.Nomura K, Kato J, Takiguchi N, Ohtake H, Kuroda A. Effects of inorganic polyphosphate on the proteolytic and DNA-binding activities of Lon in Escherichia coli. J. Biol. Chem. 2004;279:34406–34410. doi: 10.1074/jbc.M404725200. [DOI] [PubMed] [Google Scholar]
- 27.Lee AY, Hsu CH, Wu SH. Functional domains of Brevibacillus thermoruber lon protease for oligomerization and DNA binding: role of N-terminal and sensor and substrate discrimination domains. J. Biol. Chem. 2004;279:34903–34912. doi: 10.1074/jbc.M403562200. [DOI] [PubMed] [Google Scholar]
- 28.Lu B, Yadav S, Shah PG, Liu T, Tian B, Pukszta S, Villaluna N, Kutejova E, Newlon CS, et al. Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J. Biol. Chem. 2007;282:17363–17374. doi: 10.1074/jbc.M611540200. [DOI] [PubMed] [Google Scholar]
- 29.Shah IM, Wolf R.E., Jr. Inhibition of Lon-dependent degradation of the Escherichia coli transcription activator SoxS by interaction with ‘soxbox’ DNA or RNA polymerase. Mol. Microbiol. 2006;60:199–208. doi: 10.1111/j.1365-2958.2006.05086.x. [DOI] [PubMed] [Google Scholar]
- 30.Shadel GS. Mitochondrial DNA, aconitase ‘wraps’ it up. Trends Biochem. Sci. 2005;30:294–296. doi: 10.1016/j.tibs.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005;6:815–825. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- 32.Chen XJ, Wang X, Kaufman BA, Butow RA. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307:714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 33.Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl Acad. Sci. USA. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 2006;281:25791–25802. doi: 10.1074/jbc.M604501200. [DOI] [PubMed] [Google Scholar]
- 35.Bota DA, Ngo JK, Davies KJ. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 2005;38:665–677. doi: 10.1016/j.freeradbiomed.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Chung CH, Goldberg AL. DNA stimulates ATP-dependent proteolysis and protein-dependent ATPase activity of protease La from Escherichia coli. Proc. Natl Acad. Sci. USA. 1982;79:795–799. doi: 10.1073/pnas.79.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 38.Fasman GD and Chemical Rubber Company. Handbook of Biochemistry and Molecular Biology: Nucleic Acids. 3rd. Cleveland, p.589: CRC Press; 1975. [Google Scholar]
- 39.Hassur SM, Whitlock H.W., Jr. UV shadowing – a new and convenient method for the location of ultraviolet-absorbing species in polyacrylamide gels. Anal. Biochem. 1974;59:162–164. doi: 10.1016/0003-2697(74)90020-7. [DOI] [PubMed] [Google Scholar]
- 40.Jelesarov I, Bosshard HR. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 1999;12:3–18. doi: 10.1002/(SICI)1099-1352(199901/02)12:1<3::AID-JMR441>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 41.Hinz HJ. Thermodynamic parameters for protein-protein and protein-ligand interaction by differential scanning microcalorimetry. Methods Enzymol. 1986;130:59–79. doi: 10.1016/0076-6879(86)30008-9. [DOI] [PubMed] [Google Scholar]
- 42.Ladbury JE, Chowdhry BZ. Biocalorimetry: Applications of Calorimetry in the Biological Sciences. Chichester, New York, pp.183–205: J. Wiley; 1998. [Google Scholar]
- 43.Privalov PL, Potekhin SA. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 1986;131:4–51. doi: 10.1016/0076-6879(86)31033-4. [DOI] [PubMed] [Google Scholar]
- 44.Kasamatsu H, Robberson DL, Vinograd J. A novel closed-circular mitochondrial DNA with properties of a replicating intermediate. Proc. Natl Acad. Sci. USA. 1971;68:2252–2257. doi: 10.1073/pnas.68.9.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williamson JR. G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- 46.Keniry MA. Quadruplex structures in nucleic acids. Biopolymers. 2000;56:123–146. doi: 10.1002/1097-0282(2000/2001)56:3<123::AID-BIP10010>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 47.Kang C, Zhang X, Ratliff R, Moyzis R, Rich A. Crystal structure of four-stranded Oxytricha telomeric DNA. Nature. 1992;356:126–131. doi: 10.1038/356126a0. [DOI] [PubMed] [Google Scholar]
- 48.Laughlan G, Murchie AI, Norman DG, Moore MH, Moody PC, Lilley DM, Luisi B. The high-resolution crystal structure of a parallel-stranded guanine tetraplex. Science. 1994;265:520–524. doi: 10.1126/science.8036494. [DOI] [PubMed] [Google Scholar]
- 49.Hud NV, Schultze P, Sklenar V, Feigon J. Binding sites and dynamics of ammonium ions in a telomere repeat DNA quadruplex. J. Mol. Biol. 1999;285:233–243. doi: 10.1006/jmbi.1998.2327. [DOI] [PubMed] [Google Scholar]
- 50.Wong A, Wu G. Selective binding of monovalent cations to the stacking G-quartet structure formed by guanosine 5′-monophosphate: a solid-state NMR study. J. Am. Chem. Soc. 2003;125:13895–13905. doi: 10.1021/ja0302174. [DOI] [PubMed] [Google Scholar]
- 51.Ha JH, Spolar RS, Record M.T., Jr. Role of the hydrophobic effect in stability of site-specific protein-DNA complexes. J. Mol. Biol. 1989;209:801–816. doi: 10.1016/0022-2836(89)90608-6. [DOI] [PubMed] [Google Scholar]
- 52.Spolar RS, Record M.T., Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 53.Ladbury JE, Wright JG, Sturtevant JM, Sigler PB. A thermodynamic study of the trp repressor-operator interaction. J. Mol. Biol. 1994;238:669–681. doi: 10.1006/jmbi.1994.1328. [DOI] [PubMed] [Google Scholar]
- 54.Cooper A. Heat capacity of hydrogen-bonded networks: an alternative view of protein folding thermodynamics. Biophys. Chem. 2000;85:25–39. doi: 10.1016/s0301-4622(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 55.Cooper A, Johnson CM, Lakey JH, Nollmann M. Heat does not come in different colours: entropy-enthalpy compensation, free energy windows, quantum confinement, pressure perturbation calorimetry, solvation and the multiple causes of heat capacity effects in biomolecular interactions. Biophys. Chem. 2001;93:215–230. doi: 10.1016/s0301-4622(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 56.Baldwin RL. Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl Acad. Sci. USA. 1986;83:8069–8072. doi: 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Privalov PL, Makhatadze GI. Heat capacity of proteins. II. Partial molar heat capacity of the unfolded polypeptide chain of proteins: protein unfolding effects. J. Mol. Biol. 1990;213:385–391. doi: 10.1016/S0022-2836(05)80198-6. [DOI] [PubMed] [Google Scholar]
- 58.Prabhu NV, Sharp KA. Heat capacity in proteins. Annu. Rev. Phys. Chem. 2005;56:521–548. doi: 10.1146/annurev.physchem.56.092503.141202. [DOI] [PubMed] [Google Scholar]
- 59.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 60.Kalodimos CG, Biris N, Bonvin AM, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004;305:386–389. doi: 10.1126/science.1097064. [DOI] [PubMed] [Google Scholar]
- 61.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 62.Nonin-Lecomte S, Dardel F, Lestienne P. Self-organisation of an oligodeoxynucleotide containing the G- and C-rich stretches of the direct repeats of the human mitochondrial DNA. Biochimie. 2005;87:725–735. doi: 10.1016/j.biochi.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 63.Scaria V, Hariharan M, Arora A, Maiti S. Quadfinder: server for identification and analysis of quadruplex-forming motifs in nucleotide sequences. Nucleic Acids Res. 2006;34:W683–W685. doi: 10.1093/nar/gkl299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kikin O, D’Antonio L, Bagga PS. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34:W676–W682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown TA, Cecconi C, Tkachuk AN, Bustamante C, Clayton DA. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 2005;19:2466–2476. doi: 10.1101/gad.1352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clayton DA. Mitochondrial DNA replication: what we know. IUBMB Life. 2003;55:213–217. doi: 10.1080/1521654031000134824. [DOI] [PubMed] [Google Scholar]
- 67.Doda JN, Wright CT, Clayton DA. Elongation of displacement-loop strands in human and mouse mitochondrial DNA is arrested near specific template sequences. Proc. Natl Acad. Sci. USA. 1981;78:6116–6120. doi: 10.1073/pnas.78.10.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clayton DA. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- 69.Brown GG, Gadaleta G, Pepe G, Saccone C, Sbisa E. Structural conservation and variation in the D-loop-containing region of vertebrate mitochondrial DNA. J. Mol. Biol. 1986;192:503–511. doi: 10.1016/0022-2836(86)90272-x. [DOI] [PubMed] [Google Scholar]
- 70.Nass MM. Precise sequence assignment of replication origin in the control region of chick mitochondrial DNA relative to 5′ and 3′ D-loop ends, secondary structure, DNA synthesis, and protein binding. Curr. Genet. 1995;28:401–409. doi: 10.1007/BF00310807. [DOI] [PubMed] [Google Scholar]
- 71.Madsen CS, Ghivizzani SC, Hauswirth WW. Protein binding to a single termination-associated sequence in the mitochondrial DNA D-loop region. Mol. Cell. Biol. 1993;13:2162–2171. doi: 10.1128/mcb.13.4.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 73.Roberti M, Musicco C, Polosa PL, Milella F, Gadaleta MN, Cantatore P. Multiple protein-binding sites in the TAS-region of human and rat mitochondrial DNA. Biochem. Biophys. Res. Commun. 1998;243:36–40. doi: 10.1006/bbrc.1997.8052. [DOI] [PubMed] [Google Scholar]
- 74.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ying L, Green JJ, Li H, Klenerman D, Balasubramanian S. Studies on the structure and dynamics of the human telomeric G quadruplex by single-molecule fluorescence resonance energy transfer. Proc. Natl Acad. Sci. USA. 2003;100:14629–14634. doi: 10.1073/pnas.2433350100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lockshon D, Zweifel SG, Freeman-Cook LL, Lorimer HE, Brewer BJ, Fangman WL. A role for recombination junctions in the segregation of mitochondrial DNA in yeast. Cell. 1995;81:947–955. doi: 10.1016/0092-8674(95)90014-4. [DOI] [PubMed] [Google Scholar]
- 77.MacAlpine DM, Perlman PS, Butow RA. The numbers of individual mitochondrial DNA molecules and mitochondrial DNA nucleoids in yeast are co-regulated by the general amino acid control pathway. EMBO J. 2000;19:767–775. doi: 10.1093/emboj/19.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gibson T, Blok VC, Phillips MS, Hong G, Kumarasinghe D, Riley IT, Dowton M. The mitochondrial subgenomes of the nematode Globodera pallida are mosaics: evidence of recombination in an animal mitochondrial genome. J. Mol. Evol. 2007;64:463–471. doi: 10.1007/s00239-006-0187-7. [DOI] [PubMed] [Google Scholar]
- 79.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Flyvbjerg H, Keatch SA, Dryden DT. Strong physical constraints on sequence-specific target location by proteins on DNA molecules. Nucleic Acids Res. 2006;34:2550–2557. doi: 10.1093/nar/gkl271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat. Struct. Mol. Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 82.Oganesian L, Bryan TM. Physiological relevance of telomeric G-quadruplex formation: a potential drug target. Bioessays. 2007;29:155–165. doi: 10.1002/bies.20523. [DOI] [PubMed] [Google Scholar]
- 83.Opresko PL, Mason PA, Podell ER, Lei M, Hickson ID, Cech TR, Bohr VA. POT1 Stimulates recQ helicases WRN and BLM to unwind telomeric DNA substrates. J. Biol. Chem. 2005;280:32069–32080. doi: 10.1074/jbc.M505211200. [DOI] [PubMed] [Google Scholar]
- 84.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl Acad. Sci. USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cogoi S, Quadrifoglio F, Xodo LE. G-rich oligonucleotide inhibits the binding of a nuclear protein to the Ki-ras promoter and strongly reduces cell growth in human carcinoma pancreatic cells. Biochemistry. 2004;43:2512–2523. doi: 10.1021/bi035754f. [DOI] [PubMed] [Google Scholar]
- 86.Nosek J, Tomaska L, Pagacova B, Fukuhara H. Mitochondrial telomere-binding protein from Candida parapsilosis suggests an evolutionary adaptation of a nonspecific single-stranded DNA-binding protein. J. Biol. Chem. 1999;274:8850–8857. doi: 10.1074/jbc.274.13.8850. [DOI] [PubMed] [Google Scholar]
- 87.Tomaska L, Makhov AM, Nosek J, Kucejova B, Griffith JD. Electron microscopic analysis supports a dual role for the mitochondrial telomere-binding protein of Candida parapsilosis. J. Mol. Biol. 2001;305:61–69. doi: 10.1006/jmbi.2000.4254. [DOI] [PubMed] [Google Scholar]
- 88.Santos JH, Meyer JN, Skorvaga M, Annab LA, Van Houten B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell. 2004;3:399–411. doi: 10.1111/j.1474-9728.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 89.Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum. Mol. Genet. 2006;15:1757–1768. doi: 10.1093/hmg/ddl098. [DOI] [PubMed] [Google Scholar]
- 90.Tua A, Wang J, Kulpa V, Wernette CM. Mitochondrial DNA topoisomerase I of Saccharomyces cerevisiae. Biochimie. 1997;79:341–350. doi: 10.1016/s0300-9084(97)80028-4. [DOI] [PubMed] [Google Scholar]
- 91.Zhang H, Meng LH, Pommier Y. Mitochondrial topoisomerases and alternative splicing of the human TOP1mt gene. Biochimie. 2007;89:474–481. doi: 10.1016/j.biochi.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 92.Arimondo PB, Riou JF, Mergny JL, Tazi J, Sun JS, Garestier T, Helene C. Interaction of human DNA topoisomerase I with G-quartet structures. Nucleic Acids Res. 2000;28:4832–4838. doi: 10.1093/nar/28.24.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaufman BA, Newman SM, Hallberg RL, Slaughter CA, Perlman PS, Butow RA. In organello formaldehyde crosslinking of proteins to mtDNA: identification of bifunctional proteins. Proc. Natl Acad. Sci. USA. 2000;97:7772–7777. doi: 10.1073/pnas.140063197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bogenhagen DF, Wang Y, Shen EL, Kobayashi R. Protein components of mitochondrial DNA nucleoids in higher eukaryotes. Mol. Cell Proteomics. 2003;2:1205–1216. doi: 10.1074/mcp.M300035-MCP200. [DOI] [PubMed] [Google Scholar]
- 95.Malka F, Lombes A, Rojo M. Organization, dynamics and transmission of mitochondrial DNA: focus on vertebrate nucleoids. Biochim. Biophys. Acta. 2006;1763:463–472. doi: 10.1016/j.bbamcr.2006.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.