


Abstract
Minichromosome maintenance (MCM) helicases are the presumptive replicative helicases, thought to separate the two strands of chromosomal DNA during replication. In archaea, the catalytic activity resides within the C-terminal region of the MCM protein. In Methanothermobacter thermautotrophicus the N-terminal portion of the protein was shown to be involved in protein multimerization and binding to single and double stranded DNA. MCM homologues from many archaeal species have highly conserved predicted amino acid similarity in a loop located between β7 and β8 in the N-terminal part of the molecule. This high degree of conservation suggests a functional role for the loop. Mutational analysis and biochemical characterization of the conserved residues suggest that the loop participates in communication between the N-terminal portion of the helicase and the C-terminal catalytic domain. Since similar residues are also conserved in the eukaryotic MCM proteins, the data presented here suggest a similar coupling between the N-terminal and catalytic domain of the eukaryotic enzyme.
INTRODUCTION
The minichromosome maintenance (MCM) complex is thought to function as the replicative helicase of archaea and eukarya (1,2). In eukaryotes the MCM complex is a family of six essential polypeptides (Mcm2–7) with highly conserved amino acid sequences. Biochemical studies have shown that a dimeric complex of the Mcm4,6,7 heterotrimer possesses 3′-5′ DNA helicase activity, can translocate on single and double stranded DNA, can bind DNA and RNA, and has the ability to unwind DNA–RNA duplexes while translocating on the DNA strand (3,4). The interactions of Mcm4,6,7 with either Mcm2 or Mcm3,5 were shown to inhibit helicase activity and therefore were suggested to play regulatory roles (3,4).
Most archaeal species examined contain a single MCM homologue (1,2) with biochemical properties similar to the eukaryotic Mcm4,6,7 complex. The archaeal MCM proteins were shown to contain 3′-5′ DNA helicase activity, bind and translocate along ss and dsDNA, unwind DNA-RNA duplex substrate while translocating along the DNA, and to displace proteins from DNA [(5), and references therein].
The MCM helicases are divided into a C-terminal portion, which contains the helicase catalytic domain, and a N-terminal region (6–8). To date, a high-resolution structure has been determined only for the N-terminal portion of the MCM protein from the archaeon Methanothermobacter thermautotrophicus (6). That structure revealed a dumbbell-shaped double hexamer. Each monomer folds into three distinct domains. Domain A, at the N-terminus, is mostly α-helical. Domain B has three β-strands and contains a zinc-finger motif shown to be needed for DNA binding (9,10). Domain C contains five β-strands that form an oligonucleotide/oligosaccharide binding (OB) fold. This domain connects the N-terminal portion of the enzyme to the C-terminal catalytic region. Domain C contains a β-finger motif shown to be essential for ss and dsDNA binding (6,10). The domain was also shown to be necessary for MCM multimerization (7).
Sequence alignment of MCM proteins from many archaeal species has revealed highly conserved residues in a loop between β7 and β8 in domain C (Figure 1A, 100% identity in pink, 95% identity in blue and 90% identity in green). Based on the crystal structure of the N-terminal part of the molecule, the loop is located in the opposite side of the dimer interface between the two hexamers (Figure 1B and C). Electron micrograph (EM) reconstruction of the full-length M. thermautotrophicus MCM helicase (8,11) also suggest that the loop is in close proximity to the catalytic domain located at the C-terminal part of the molecule (Figure 1F). Loop regions are known to be less conserved than other secondary structures unless they play an important functional role. The high conservation suggests that the loop between β7 and β8 may play a role in MCM function. Biochemical characterization of proteins harboring mutations in this region suggest that the loop region is likely to be involved in coupling the N-terminal multimerization and DNA binding domains with the C-terminal catalytic domain. Thus the loop may function as a bridge, allowing a movement between the two domains to transmit a signal.
Figure 1.

A conserved loop at the N-terminal portion of the M. thermautotrophicus MCM protein is in close proximity to the catalytic domain. (A) An alignment of the amino acid sequences of the loop between β7 and β8 in 21 archaeal MCM proteins belonging to subgroup I. An alignment of human Mcm2–7 proteins is shown at the bottom. β7 and β8 are marked at the top of the alignment. Highlighted colors represent residues with 100% identity in pink, 95% identity in blue and 90% identity in green. The accession numbers used are: Methanothermobacter thermautotrophicus, NP_276876; Thermoplasma acidophilum, NP_394261; Thermoplasma volcanium, NP_111551; Picrophilus torridus, YP_023995; Ferroplasma acidarmanus, ZP_01708764; Haloquadratum walsbyi, YP_659323; Haloarcula marismortui, YP_137231; Natronomonas pharaonis, CAI50644; Methanocorpusculum labreanum, YP_001030268; Methanospirillum hungatei, YP_502454; Methanoculleus marisnigri, YP_001047160; Methanosarcina acetivorans, NP_618700; Methanosarcina barkeri, YP_305120; Methanosarcina acetivorans, NP_615641; Methanosarcina mazei, NP_633860; Methanococcoides burtonii, YP_567033; Methanosaeta thermophila, YP_842696; Methanosphaera stadtmanae, YP_447408; Sulfolobus tokodaii, NP_376352; Archaeoglobus fulgidus, NP_069353; Halobacterium, NP_280836. (B) and (C). Three dimensional structure of the dodecameric N-terminal portion of the M. thermautotrophicus MCM protein [PDB ID: 1LTL (6)] is shown with the loop between β7 and β8 highlighted in red. B, top view; C, side view. (D) and (E) The structure of a monomer of the N-terminal part of M. thermautotrophicus MCM with domain A shown in green, domain B in red and domain C in blue. The structure was constructed using PyMOL and PDB ID: 1LTL. The loop between β7 and β8 is highlighted in yellow and the conserved residues are also shown. The loop and the conserved residues are enlarged in panel E. (F). Electron micrograph (EM) reconstruction of the intact M. thermautotrophicus MCM was generated using PyMol and the coordinates that were previously reported (11). The N-terminal part of the molecule is shown in green, the C-terminal part is shown in blue and the loop between β7 and β8 is highlighted in red.
MATERIALS AND METHODS
Materials
ATP, [γ-32P]ATP and [α-32P]ATP were obtained from GE Healthcare, and oligonucleotides were synthesized by the CARB DNA synthesis facility. All proteins used in the study were purified as previously described (7) and are derived from the full-length MCM gene.
Methods
Multiple alignment
The M. thermautotrophicus MCM protein sequence (MTH1770) was aligned using BLAST against 41 archaeal genomes (National Center for Biotechnology Information, NCBI). Full length MCM sequence relatives with expectation scores <10−5 from the 41 archaeal genomes were pooled and aligned using the MUSCLE program and default parameters. Aligned sequences were loaded onto Jalview 2.2.1, and the N-terminal portion (MTH1770 residues 1–244) was kept for the following analysis. PHYLIP promlk (version 3.6) was used to build the maximum likelihood phylogenetic tree, which resulted in four subgroups (Group I–IV). From the tree, a total of 21 MCM proteins from subgroup I that contain the M. thermautotrophicus MCM sequence (MTH1770) were selected to view as an alignment (Figure 1A).
Expression and purification of MCM mutant proteins
All M. thermautotrophicus MCM mutant proteins used in this study are derivatives of the full-length enzyme and were generated using PCR-based site-directed mutagenesis as previously described (7). All constructs contain a C-terminal His6-tag and were cloned into the NdeI and XhoI sites of the pET-21a vector (Novagene). The oligonucleotides used for the mutagenesis are shown in Supplementary Table 1. The wild-type and mutant proteins were overexpressed in E. coli codon plus cells (Stratagene) at 37°C and purified on a Ni-column as previously described (12).
ATPase assay
ATPase activity was measured in reaction mixtures (15 μl) containing 25 mM Hepes-NaOH (pH 7.5), 1 mM dithiothreitol, 5 mM MgCl2, 100 μg/ml bovine serum albumin (BSA), and 1500 pmol of [γ-32P]ATP (3000 Ci/mmol; GE healthcare) and 10 or 30 nM MCM protein (as monomer) in the presence or absence of 50 ng ssDNA (5′-GGCAGATAACAGTTGTCCTGGAGAACGACCTGGTTGACACCCTCACACCC -3′). After incubation at 60°C for 60 min, samples were placed on ice, then an aliquot (1 μl) was spotted onto a polyethyleneimine cellulose thin layer plate, and ATP and Pi were separated by chromatography in 1 M formic acid and 0.5 M lithium chloride. The extent of ATP hydrolysis was quantitated by phosphorimager analysis. All ATPase assays were repeated three times, and their averages with SD are shown in Figure 6A.
Figure 6.

Mutations in the loop between β7 and β8 alter the ATPase activity of MCM. (A) ATPase activity of wild-type and mutant MCM proteins in the absence and presence of 50 ng ssDNA was determined as described in ‘Materials and Methods’ using 10 and 30 nM of MCM (as monomer). (B–D) Km and kcat ATP hydrolysis by the wild-type and mutant MCM proteins were determined using 50 nM of proteins in the absence (B) or presence (C) of 50 ng ssM13 and various concentrations of ATP (0.01, 0.05, 0.1, 0.25, 0.5, 0.75 1 and 1.4 mM) as described in ‘Materials and Methods’. Rate of reaction and kcat were calculated as described in ‘Materials and Methods’ (D). NI, not informative due to high standard error.
For the kinetics measurement time-based ATPase assays were performed. ATPase activities were measured in reaction mixtures (50 μl) containing 25 mM Hepes-NaOH (pH 7.5), 1 mM dithiothreitol, 5 mM MgCl2, 100 μg/ml BSA, and 50 nM of MCM protein (as monomer) in the presence or absence of 50 ng ssM13 (New England Biolabs) with various ATP concentrations (0.01, 0.05, 0.1, 0.25, 0.5, 0.75, 1 or 1.4 mM) containing 50 nM of [γ-32P]ATP (3000 Ci/mmol; GE healthcare) at 60°C.
At 0, 20, 40, and 80 minute intervals, a 3 μl aliquot was taken out and quenched on ice. At all ATP concentrations, the reactions were determined to be in steady state at about 40 min (data not shown). Thus, a single time point measurement at 30 min was performed to determine the Km and Vmax with various ATP concentrations (0.01, 0.05, 0.1, 0.25, 0.5, 0.75, 1 and 1.4 mM). The kinetic parameters were calculated using GraFit version 5.0.1. (Erithacus software) and the Michaelis-Menton equation, where graphs were fit to v = (Vmax · [S])/(Km + [S]); where v is the rate of reaction in μM min−1, S is the substrate concentration in μM, Vmax is the maximum reaction rate of the enzyme, and Km is the Michaelis-Menton constant. The turnover rate, kcat, was determined by dividing the Vmax by the total amount of MCM protein in the reaction (50 nM). Several data resulted in non-informative values for Km and Vmax due to the fact that the standard errors were >90% of the values calculated. All experiments repeated three times and the averages are shown in Figure 6B–D.
Nitrocellulose filter DNA binding assay
Single stranded and dsDNA substrates for filter binding assays were prepared by labeling the oligonucleotide using [γ-32P]ATP and T4 polynucleotide kinase. Unincorporated [γ-32P]ATP was removed from the DNA substrate by extraction from polyacrylamide gel as previously described (13). For ssDNA binding a 49-mer oligonucleotide with the sequence 5′-GCAGATAACAGTTGTCCTGGAGAACGACCTGGTTGACACCCTCACACCC-3′ was used. For dsDNA the same oligonucleotide was hybridized to its complementary sequence.
Filter binding assays were carried out in a reaction mixture (20 μl) containing 25 mM Hepes-NaOH (pH 7.5), 2 mM dithiothreitol, 10 mM MgCl2, 100 μg/ml BSA, 50 fmol of 32P-labeled ss or dsDNA substrate and 10, 30 or 90 nM of MCM protein (as monomer).
After incubation at 60°C for 10 min the mixture was filtered through an alkaline-washed nitrocellulose filter (Millipore, HA 0.45 μm) (14) which was subsequently washed with 20 mM Tris-HCl (pH 7.5). The radioactivity adsorbed to the filter was measured by liquid scintillation counting. All DNA binding experiments were repeated three times, and their averages with SD are shown in Figure 5A and B.
Figure 5.

Mutations in the loop between β7 and β8 do not affect MCM DNA binding. (A) and (B) Filter binding assays were performed as described under ‘Materials and Methods’ using 50 fmol of 32P-labled single strand (A) or double strand (B) DNA in the presence of 10, 30 and 90 nM of proteins (as monomer). The average result of three experiments is shown. (C–E) The Kd values of the interactions between the MCM mutant proteins and DNA were measured using Fluorescence polarization anisotropy (FPA) analysis as described under ‘Materials and Methods’ in the absence (C) and presence (D) of 1 mM ATP and 10 nM Cy3-labeled ssDNA. The anisotropy change was measured as the proteins were titrated into the reaction mixture. Data were analyzed as described in ‘Materials and Methods’ and the Kd values are shown in panel E.
Fluorescence polarization anisotropy (FPA) measurement
Fluorescence anisotropy measurements were performed at 25°C using a Fluoromax-3 spectrofluorimeter equipped with an autopolarizer (Jobin Yvon Inc.), using a 3 mm path length cuvette with a starting volume of 150 μl. A 50-mer ssDNA (5′-CGCAGATAACAGTTGTCCTGGAGAACGACCTGGTTGACACCCTCACACCC -3′) was 5′-labeled with Cy3 and purified with a HPLC C18 column. The concentration of DNA was calculated using an absorbance of 260 nm with extinction coefficient 477 300 M−1·cm−1 and absorbance at 546 nm using extinction coefficient 136 000 M−1cm−1 for Cy3 dye. DNA concentrations calculated by both measurements differed by <10%. The measurements using absorbances at 260 nm were used to calculate the concentrations for the experiments. The initial reaction mixture contained 25 mM Hepes-NaOH (pH 7.5), 2 mM DTT, 5 mM MgCl2 and 10 nM DNA, with or without 1 mM ATP. Following the addition of proteins the reaction mixture was incubated for 10 min and then measured with a setting of 5 sec integration and with three averaged measurements. The DNA was excited at 545 nm and emission spectra was set at 570 nm. Anisotropy values were directly tabulated in the analysis and with measured G factor and dark correction acquired at each blank for each experiment. The binding constant (Kd) was determined using GraFit version 5.0.1 (Erithacus software), using the following quadratic equation for fluorescent polarization anisotropy experiments (15,16)  ; where ΔA is the change in anisotropy, ΔAT is the total anisotropy change, ET is the enzyme concentration at each titration point, DT is the total concentration of DNA (assuming it is constant at 10 nM) and Kd is the dissociation constant for the binding isotherm. The experiments were repeated twice and their averages with SD are shown in Figure 5C–E.
; where ΔA is the change in anisotropy, ΔAT is the total anisotropy change, ET is the enzyme concentration at each titration point, DT is the total concentration of DNA (assuming it is constant at 10 nM) and Kd is the dissociation constant for the binding isotherm. The experiments were repeated twice and their averages with SD are shown in Figure 5C–E.
DNA helicase assay
Substrates for helicase assays were made as previously described (17) using the following oligonucleotides. For forked-like DNA substrate the two oligonucleotides were 5′-GGGACGCGTCGGCCTGGCACGTCGGCCGCTGCGGCCAGGCACCCGATGGCGTT TGTTTGTTTGTTTGTTTGTTT-3′ and 5′-TTTGTTTGTTTGTTTGTTTGTTTGTTTGTTTGTTTGCCGACGTGCCAGGCCGACGCGTCCC-3′ and for the substrate with only a 3′-overhang region (flat substrate) the oligonucleotides used were 5′-CCGACGTGCCAGGCCGACGCGTCCC-3′ and 5′-GGGACGCGTCGGCCTGGCACGTCGGCCGCTGCGGCCAGGCACCCGATGGC-3′
DNA helicase activity assays were measured in reaction mixtures (15 μl) containing 20 mM Tris-HCl (pH 8.5), 2 mM DTT, 10 mM MgCl2, 100 μg/ml BSA, 3.33 mM ATP, 10 fmol (0.66 nM) of 32P-labeled substrate and 10, 30 or 90 nM MCM protein (as monomer) for flat substrate or 3, 9 or 27 nM MCM protein (as monomer) for forked substrate. Following incubation at 60°C for 1 hr the reactions were stopped by adding 5 μl of loading buffer containing 1% SDS, 100 mM EDTA, 0.1% xylene cyanol, 0.1% bromophenol blue and 50% glycerol and chilling on ice. Aliquots (10 μl) were loaded onto an 8% native polyacrylamide gel in 0.5X TBE and electrophoresed for 40 min at 180V. Gels were visualized and quantitated by phosphorimaging. All helicase experiments were repeated three times, and their averages with SD are shown in Figure 2.
Figure 2.

Conserved residues in the loop between β7 and β8 affect MCM helicase activity. Helicase assays of wild-type and mutant MCM proteins were performed as described in ‘Materials and Methods’ with 10 fmol of flat (A and B) or forked (C and D) substrates. In panels A and B the protein concentrations used were 10, 30 and 90 nM (as monomer) while in panels C and D they were 3, 9 and 27 nM (as monomer). The average result of three experiments are shown in panels B and D. Panels A and C are representative gels. Panel A lane 1, substrate only; lane 2, boiled substrate; lanes 3, 6, 9, 12, 15, 18 and 21 are 10 nM; lanes 4, 7, 10, 13, 16, 19 and 22 are 30 nM; lanes 5, 8, 11, 14, 17, 20 and 23 are 90 nM of MCM proteins. Panel C lane 1, substrate only; lane 2, boiled substrate; lanes 3, 6, 9, 12, 15, 18, and 21 are 3 nM; lanes 4, 7, 10, 13, 16, 19 and 22 are 9 nM; lanes 5, 8, 11, 14, 17, 20, and 23 are 27 nM of MCM proteins.
The relative rates of unwinding were measured in reaction mixtures (140 μl) containing 20 mM Tris-HCl (pH 8.5), 2 mM DTT, 10 mM MgCl2, 100 μg/ml BSA, 3.33 mM ATP, 93.33 fmol (0.66 nM) of 32P-labeled substrate and 30 nM proteins for flat substrate and 10 nM proteins for forked substrate. Following incubation at 60°C samples (10 μl) were removed at 0, 5, 10, 15, 30, 45 and 90 min, quenched with 5 μl of loading buffer containing 1% SDS, 100 mM EDTA, 0.1% xylene cyanol, 0.1% bromophenol blue, and 50% glycerol, and put on ice. Aliquots (10 μl) were loaded onto an 8% native polyacrylamide gel in 0.5X TBE and electrophoresed for 40 min at 180 V. Gels were visualized and quantitated by phosphorimaging. All helicase experiments were repeated three times, and their averages with SD are shown in Figure 3. The slope of initial (linear) rate was used to calculate the relative rate of unwinding and it is presented as μmoles of DNA unwound per moles of protein per minute (μmol·mol−1·min−1).
Figure 3.

Mutations in the loop between β7 and β8 alter the rate of DNA unwinding. Helicase assays were performed as described in ‘Materials and Methods’ with flat (A) or forked (B) DNA substrates and 10 nM MCM (A) or 30 nM MCM (B). Following incubation at 60°C aliquots were analyzed at 0, 5, 10, 15, 30, 45 and 90 min. The rate of unwinding was calculated as described in ‘Materials and Methods’ and shown in panel C.
Gel filtration analysis
One hundred micrograms of wild-type and mutant MCM proteins were applied to a superose-6 gel filtration column (HR10/30, GE Healthcare) pre-equilibrated with buffer containing 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, and 10% glycerol. The column was run at a flow rate of 0.2 ml/min at room temperature and 250 μl fractions were collected. The presence of protein was determined by ultraviolet absorbance at 280 nm.
Differential scanning calorimetry (DSC) measurements
The purified proteins were dialyzed at room temperature against buffer containing 20 mM potassium phosphate (pH 7.5), 100 mM NaCl and 10% glycerol. The protein concentrations were determined using an absorbance at 280 nm and a calculated extinction coefficient of ε280 of 28 730 cm−1·M−1. The solution outside the dialysis bag was retained and used as reference for the experiments.
DSC measurements were performed using a VP-DSC Microcalorimeter from Microcal Inc. (Northampton, MA). The volume of the solution and the reference vessels were 0.511 ml and the scan was either at slow rate (15 K hr−1) or medium rate (60 K hr−1), with the temperature ranging from 25–85°C. Since the scans of protein samples were irreversible, the second scan for each sample was used as the baseline. After subtraction of the baseline from the protein scan, the resulting raw data of differential powers as a function of time were divided by the scan rate to convert into a heat capacity as a function of temperature. Utilizing the EXAM program (18), a two state, A ⇔ B, transition model was then fitted to the heat capacity as a function of temperature scan to determine the van't Hoff enthalpy (ΔHVH) for the scan from the shape of the transition peak; a transition temperature (Tm), and a calorimetric transition enthalpy (ΔHcal) was calculated from the area under the transition peak (mJ) divided by the moles of protein in the sample cell (concentration of protein × 0.511 ml). DSC scans on the samples were repeated twice.
Circular dichroism
Circular dichroism (CD) measurements were performed with a Model J-720 Jasco Spectropolarimeter using quartz cells with a 0.005 cm path length at room temperature. The proteins used for CD were prepared as described above for the DSC experiments, with concentrations of proteins ranging from 10–30 μM. Far-UV wavelength scans were recorded at room temperature from 250–190 nm. Averages for three CD spectra were presented. Ellipticity results were expressed as mean residue ellipticity [θ] = degrees × centimeter2 × decimole−1.
UV crosslinking
The UV crosslinking experiments were performed using 250 ng of wild-type, K325A and the various loop mutant proteins as previously described (9) in a 20 μl reaction mixture containing 3.3 pmol of [α-32P]ATP, 20 mM Hepes-NaOH (pH 7.5), 5 mM MgCl2, 2 mM DTT, and 250 ng BSA. The samples were incubated for 10 min at 60°C followed by exposure to 2.5 J/cm2 UV irradiation in a model 2400 Stratalinker (Stratagene) and analyzed by SDS-PAGE followed by Coomassie Blue staining and autoradiography. The percent of ATP cross linked was calculated using the following equation: 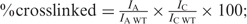 where IA and IAWT are the intensity of radiolabeled ATP cross linked to the mutant or wild-type proteins, respectively, detected by autoradiography and Ic and IcWT are the intensity of Coomassie stained mutant and wild-type MCM proteins, respectively.
where IA and IAWT are the intensity of radiolabeled ATP cross linked to the mutant or wild-type proteins, respectively, detected by autoradiography and Ic and IcWT are the intensity of Coomassie stained mutant and wild-type MCM proteins, respectively.
RESULTS
The archaeal MCM helicase contains highly conserved residues in the N-terminal portion
Comparison of the primary amino acid sequences of the MCM helicases from a number of archaea identified highly conserved residues in the N-terminal region of all species (data not shown). Not only have most of the highly conserved residues been identified in domain C of the N-terminal portion of the molecule, but when the conserved residues are mapped on the three dimensional structure of the M. thermautotrophicus MCM many of them are located at a loop located between β7 and β8 in domain C (Figure 1).
Mutations in the loop region between β7 and β8 affect MCM helicase activity
As shown in Figure 1A there are several highly conserved amino acids in the loop between β7 and β8 in the N-terminal part of MCM. As the loop is on the surface of the N-terminal part of the molecule (Figure 1B and C) and loops are usually not highly conserved unless they have a functional role, one may hypothesize that the conserved residues play an important role for MCM function. To evaluate whether the hypothesis is correct, conserved residues were mutated in the background of the wild-type MCM from the archaeon M. thermautotrophicus. This MCM was chosen as, to date, this is the only MCM homologue with high-resolution structural information.
First, the effect of the mutation on helicase activity was determined. It was previously shown that helicase activity on forked substrate is higher than on flat substrate (12). Therefore, both substrates were used in the study but higher protein concentrations were used with the flat substrate. As shown in Figure 2A and B, three mutant forms of the protein have lost the ability to unwind flat DNA substrate (E182R, E185R and P193G). When forked DNA substrate was tested (Figure 2C and D) the P193G mutant protein remained inactive while E182R showed limited activity. However, on forked DNA the E185R mutant protein showed activity that is as good if not better than that of wild-type MCM at low enzyme concentrations (3 nM). Additionally, the E185R mutant protein cannot unwind more than ∼70% of the substrate even at higher protein concentrations (270 nM, data not shown).
The experiments described above were performed using a single time point and thus cannot provide information regarding the differences in unwinding rates between the various mutant proteins. To determine the effect of the mutations on the rate of DNA unwinding, a time course experiment was performed (Figure 3). As was the case with the experiments described in Figure 2, no appreciable activity could be detected with the E182R, E185R and P193G mutant proteins when flat substrate was used. On forked substrate the E185R mutant protein exhibits helicase activity which is similar to the wild-type MCM (Figure 3). The L184A mutant protein has a slightly higher rate of unwinding on flat substrate compared to forked substrate (Figure 3C).
Why some mutant enzymes are not active on a flat substrate is not yet known. One possibility is that the central hole created by the N-terminal part and the catalytic domain is larger and thus more of the enzyme moves along the duplex when provided with only a 3′-overhang region, as has previously been reported for the archaeal and eukaryal MCM helicases (17,19).
Mutation of conserved loop residues does not affect MCM structural integrity
It was previously shown that the N-terminal portion of MCM plays a major role in MCM multimerization and that domain C, in particular, is needed for MCM hexamerization (7). It was also shown that protein hexamerization is needed for helicase activity (J.-H. Shin and ZK, unpublished observation) and the deletion of domain C results in an inactive enzyme (7). Thus, it is possible that the substitutions in the loop, which is located in domain C, alter the structure of the MCM molecule, leading to the reduced helicase activity observed in the mutant proteins. Therefore, the oligomeric state, structural integrity and thermostability of the mutant proteins were determined using gel filtration, differential scanning calorimetry (DSC) and circular dichroism (CD).
As shown in Figure 4A the wild-type and all mutant proteins form dodecamers in solution as was previously reported for the wild-type enzyme (20–22). Next, the overall structural integrity of the mutant proteins was evaluated using CD spectra. The results indicate that all mutant proteins are folded, retaining a secondary structure similar to the wild-type enzyme (Figure 4B).
Figure 4.

Mutations in the loop between β7 and β8 do not change the overall structure of MCM. (A) Gel-filtration analysis of the wild-type and mutant MCM proteins. One hundred micrograms of protein were loaded onto a superose-6 gel filtration column and analyzed as described in ‘Materials and Methods’. The peak elutions of several molecular weight standards are shown at the top of the figure. Thyroglobulin (Thy, 670 kDa), ferritin (Fer, 440 kDa), albumin (Alb, 67 kDa) and ovalbumin (Ova, 45 kDa). (B) Circular dichroism (CD) spectra of wild-type and mutant MCM proteins. Each spectra was normalized to mean residue ellipticity [θ] = degrees × centimeter2 × decimole−1 according to the concentrations of protein determined as described in ‘Materials and Methods’. The spectra were taken at 23°C.
However, it is possible that the mutations do not affect the oligomeric state of the helicase but do affect its conformational stability. Because the helicase assays are performed at high temperature (60°C), it is important to determine whether the mutations affect the stability of the enzyme at high temperature. The DSC study showed that all mutant proteins, except for P193G, have melting temperatures similar to the wild-type MCM (Table 1). For P193G melting is ∼7°C higher than the wild-type protein suggesting a more stable structure. DSC scans of several of the mutants were performed at the slower scan rate of 15 K hr−1 and it was observed that the transition temperature and enthalpies were within experimental uncertainty of the results at 60 K hr−1. Manly et al. (23) have justified that those proteins that exhibit transition properties independent of scan rate could be analyzed in terms of a thermodynamic two-state transition model although the transitions do not re-appear upon a re-scan of the solutions.
Table 1.
Differential Scanning Calorimetry (DSC) analysis of wild-type and mutant MCM proteins
| Protein | Tm (°C) | ΔHVH (kJ/mole) | ΔHcal (kJ/mole) |
|---|---|---|---|
| Wild-type | 66.2 ± 1.7a | 1247 ± 159 | 531 ± 3 |
| Q181A | 62.5 ± 2.4a | 834 ± 56 | 297 ± 96 |
| E182R | 67.7 ± 3.9a | 1023 ± 334 | 384 ± 197 |
| L184A | 66.0 ± 0.2 | 1366 ± 343 | 189 ± 98 |
| E185R | 68.7 ± 0.2 | 694 ± 178 | 336 ± 208 |
| G190A | 65.5 ± 1.9a | 1378 ± 127 | 122 ± 20 |
| P193G | 73.0 ± 1.2a | 952 ± 337 | 332 ± 62 |
aAverage of medium (60 K hr−1) and slow (15 K hr−1) scan
Thus, all mutant proteins are dodecameric in solution and appear to form intact secondary structures with melting temperatures in the range of the wild-type enzyme. Thus, gross structural alterations are not likely to be the cause for the decrease in helicase activity observed with the E182R and P193G mutant MCM proteins.
Mutations in the loop do not affect DNA binding by MCM
The N-terminal portion of the M. thermautotrophicus MCM protein was shown to contain two motifs required for DNA binding. Domain B contains a zinc-finger motif shown to participate in DNA binding, as substitution of one of the Cys residues in the motif to Ser (C158S) substantially reduced ss and dsDNA binding (9,10), and deletion of domain B also resulted in reduced DNA binding by MCM (7). Domain C contains a β-finger motif shown to be essential for DNA binding because substitution of key Arg and Lys residues in the motif with Ala (R227A and K229A) completely abolished both ss and dsDNA binding (6,10). Although the mutations in the loop between β7 and β8 are not in close proximity to either of the DNA binding motifs, it is possible that the changes slightly alter the structure of the molecule and thus affect DNA binding. As proper DNA binding is needed for helicase activity, loss of DNA binding may result in the effects observed in the mutant forms of the protein. Therefore, filter binding assays were performed to evaluate the ability of the various mutants to bind ss and dsDNA (Figure 5A and B). As shown in Figure 5, all mutant proteins bind ssDNA (Figure 5A) and dsDNA (Figure 5B).
To obtain a more quantitative thermodynamic comparison on the binding of the various mutant proteins to DNA and to determine whether ATP has an effect on the DNA binding, a fluorescence polarization anisotropy (FPA) analysis was performed (Figure 5C–E). As a negative control a β-finger mutant protein that cannot bind DNA (6,10) was used. As was the case in previous studies, the mutant protein did not bind DNA in the FPA assay (Figure 5C and D). The results show that all mutant proteins bind ssDNA with similar affinity as the wild-type MCM (Figure 5E). The data also show that ATP does not affect DNA binding by MCM as the Kd values do not substantially change whether ATP is present or not (Figure 5E). These results suggest that the reduced helicase activity observed with two of the mutant MCMs proteins, E182R and P193G, is not due to their inability to bind DNA.
Mutations in the loop region of the N-terminal domain alter the ATPase activity of MCM
The data presented in Figure 5 show that mutations in the loop between β7 and β8 do not substantially affect the ability of the proteins to interact with DNA. This suggests that DNA binding is not the reason for the impaired helicase activity observed. ATP hydrolysis is required for helicase activity, as it fuels the unwinding reaction. A mutant protein in which a key Lys residue needed for ATPase activity is replaced by Ala (K325A) does not have ATPase activity (21,22) and is not active in helicase assays (21,22). It has also been shown that the MCM ATPase activity is stimulated in the presence of DNA (20,21). Thus, the effect of the mutations on the ATPase activity and the stimulation by DNA was determined (Figure 6). As a negative control the K325A mutant protein was used (Figure 6A). The data show that the two mutant proteins with reduced helicase activity, E182R and P193G, also have very limited, if any, ATPase activity with no stimulation by DNA (Figure 6A) and no activity was observed when three fold higher protein concentrations were used (data not shown). In addition, several mutant proteins (Q181A, L184A, E185R and G190A) showed higher ATPase activity than the wild-type enzyme (Figure 6A).
To get a more quantitative picture of the ATPase activity of the mutant MCMs in comparison to the wild-type enzyme the Km and kcat were determined in the presence and absence of ssDNA (Figure 6B–D). When the two mutant proteins with limited helicase activity, E182R and P193G, were analyzed, the Km and kcat values could not be determined regardless of whether ssDNA was present or not (Figure 6B–D). All other proteins show an increase of Kcat in the presence of ssDNA but not to the same extent. Even with those differences between the mutant proteins, the ability of ssDNA to stimulate ATPase activity suggests that they are able to translocate along ssDNA. These results are consistent with the ability of these mutant proteins to unwind DNA (Figure 2). The observation that several mutations at the N-terminal, non-catalytic, portion of the enzyme either stimulate or inhibit ATPase activity suggests that the N-terminal part regulates the ATPase and helicase activities of the catalytic domain.
However, the lack of ATPase activity by the E182R and P193G mutant proteins could be due to their inability to bind ATP. Therefore, as a control, and in order to demonstrate the ability of the mutant proteins to bind ATP a UV-cross linking experiment was performed (Figure 7). This method was previously used to demonstrate ATP binding to the wild-type enzyme (9,24). As shown in Figure 7 all mutant proteins, including E182R and P193G, can bind ATP. The only mutant protein that did not bind ATP is K325A, as has previously been reported (9,24).
Figure 7.

MCM proteins with mutations in the loop can bind ATP. UV crosslinking of ATP to the various MCM proteins was performed as described in ‘Materials and Methods’. The proteins were separated on 10% SDS-PAGE and visualized by staining with Coomassie Blue (A) and phosphorimaging (B). Lane 1, molecular weight marker; lane 2, no proteins added; lane 3, wild-type MCM lane 4, K325A; lane 5, Q181A; lane 6, E182R; lane 7, L184A; lane 8, E185R; lane 9, G190A; lane 10, P193G. The ATP cross-linked to the wild-type and mutant MCMs is indicated (%).
DISCUSSION
The archaeal MCM and probably the eukaryotic enzymes are composed of two main parts in which the C-terminal region contains the catalytic activity and the N-terminal portion participates in DNA binding. However, the mutational analysis presented here suggests that the loop between β7 and β8 in the N-terminal part is involved in coupling the DNA binding of the N-terminal portion of the molecule to the ATPase and helicase activity of the C-terminal AAA+ catalytic region. This is based on the observation that some of the mutant proteins have DNA binding and basal ATPase activity similar to the wild-type enzyme but do not show DNA stimulation of the ATPase activity and/or helicase activity.
This is not the first example of a N-terminal mutation in the M. thermautotrophicus MCM that affects catalytic activity. It was shown that a Pro to Leu substitution at position 62 (P62L) affects helicase activity but not DNA binding or basal ATPase activity (25). A similar mutation in the eukaryotic Mcm5 protein (P83L) from Saccharomyces cerevisiae, known as the BOB1 mutation, was shown to bypass the requirement for the activity of the cell-cycle regulator Dbf4-dependent kinase Cdc7 required for the initiation of DNA replication (26). It was suggested that this mutation causes a conformational change in the MCM structure (6).
Another residue in the N-terminal part of the M. thermautotrophicus MCM protein important for the biochemical properties of the enzyme is Arg at position 99. It was shown that although substitution of Arg to Ala at that position (R99A) has no effect on basal ATPase activity it reduces the stimulation of the ATPase activity by ssDNA (27).
The data suggest that the loop between β7 and β8 in the N-terminal region may interact with a region on the C-terminal part of the helicase and that these interactions mediate communication between the two regions of the enzyme. Such an interaction between the loop and the catalytic domain may also be postulated from the EM studies of the full-length M. thermautotrophicus MCM protein (Figure 1F) (8,11). The structure suggests that the loop between β7 and β8 is in close proximity to the C-terminal part of the molecule and seems to be the only interacting region between the N and C part of the molecule.
The mechanism by which the loop in the N-terminal part of MCM regulates the activity of the catalytic domain is not yet clear. However, a suggested model is shown in Figure 8. In this model the loop mediates communication between the two parts of the enzyme and transmits signals from the N-terminal part to the AAA+ catalytic region in the C-terminal part. These signals may include DNA binding by the N-terminal region and nucleotide binding and hydrolysis by the catalytic domain. Since a high resolution 3D structure of the full length MCM protein is not yet available one must rely on the structure of other helicases to suggest conformational changes occurring upon ATP binding and hydrolysis. Even with those similarities one cannot clearly know at which states, apo, ATP-, and ADP-bound state, the enzyme binds to DNA, releases and moves along it. Thus, in the model the three different states are shown without commitment to a specific nucleotide binding state (the nucleotide is therefore not shown, Figure 8A, states I-III). The data presented here suggest that the mutations in the loop disrupt the proper interactions between the N- and C-terminal parts and that these interactions are needed for helicase activity. The mutation(s) in the loop may have one of two effects (each mutation may have a different one). They may either stabilize the interaction between the two parts of the molecule and thus prevent the movement needed for activity (Figure 8B, stable interactions) or the mutations may have the opposite effect by preventing interactions between the two regions of the molecule, again resulting in impaired activity (Figure 8B, no interactions).
Figure 8.

A model for the roles played by the N-terminal loop in MCM function. In the model the C-terminal catalytic region is shown in blue, the N-terminal region in yellow and the loop between β7 and β8 in red. (A) A proposed role for the loop region in the activity of the helicase. The loop connects the N- and C-parts of the molecule and serves as a pivot to allow regulated movements of the domains needed for activity. See text for details. (B) Mutations in the loop may stabilize the interaction between the N- and C-parts of the helicase (top) or weaken the interaction (bottom), thus preventing the coupling (coordinate movement) between the two parts of the molecule. See text for details.
Interactions between the N-terminal part and the C-terminal AAA+ catalytic domains and the regulation of the catalytic domain by the N-terminal part have also been suggested by a study with the MCM homologue from the archaeon Solfolobus solfataricus (28). This study used purified N-terminal and C-terminal portions of MCM to determine their effect on the biochemical properties of the enzyme. The study demonstrated an interaction between the two parts of the molecule and suggested that the N-terminal portion functions to tether the catalytic domain to the DNA. Similar experiments, however, cannot be performed with the M. thermautotrophicus enzyme, as we and others have failed to obtain soluble and properly folded intact catalytic domain. However, as both S. solfataricus and M. thermautotrophicus MCM proteins are similar in primary amino acid sequence and in their biochemical properties one would expect that in M. thermautotrophicus similar direct interactions between the C- and N-terminal regions do exist. The data presented in this paper suggest that the loop between β7 and β8 of the N-terminal part may participate in interactions and regulation of the C-terminal catalytic region. As similar residues are also conserved in the S. solfataricus MCM one may expect that the same loop is involved in interactions between the N- and C-terminal regions of the S. solfataricus protein.
The N-terminal part of MCM was shown to interact with the two Cdc6 proteins of M. thermautotrophicus (Cdc6-1 and -2) (12,29). It was also shown that the interactions between MCM and Cdc6 proteins regulate MCM activity (29). Thus, it is possible that the loop between β7 and β8 of the N-terminal part of MCM also transmits the signal of Cdc6 binding, and perhaps other MCM interacting proteins [e.g. GINS (30)] to the catalytic part of the molecule.
To date, no high-resolution structure of the eukaryotic MCM is available. However, the similarity in primary amino acid sequence of the N-terminal region between the archaeal and eukaryal MCM proteins [in particular in domain C (7)] suggests that the eukaryotic MCM proteins will fold like the archaeal enzyme (1,6). As shown in Figure 1A the eukaryotic Mcm2-7 proteins contain similar conserved residues in the same location as the loop in the archaeal enzyme. As the structure of the eukaryotic enzyme is predicted to be the same as the archaeal, one can speculate that the loop will play a similar role in the eukaryotic MCM proteins. The loop may participate in interacting with the catalytic domain and, like the archaeal enzyme, couple the N-terminal region and the catalytic domain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Drs Phil Bryan, Vince LiCata and John Marino for help with data analysis, Drs Silvia Onesti and Alessandro Costa for the coordinates of the EM structure and Dr Lori Kelman for her comments on the manuscript. This work was supported by a Research Scholar Grant from the American Cancer Society (RSG-04-050-01-GMC) and a grant from the National Science Foundation (MCB-0450695) awarded to Z.K. Certain commercial materials, instruments and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments or equipment identified is necessarily the best available for the purpose. Funding to pay the Open Access publication charges for this article was provided by the grant from the American Cancer Society.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kelman Z, Hurwitz J. Structural lessons in DNA replication from the third domain of life. Nat. Struct. Biol. 2003;10:148–150. doi: 10.1038/nsb0303-148. [DOI] [PubMed] [Google Scholar]
- 2.Kelman Z, White MF. Archaeal DNA replication and repair. Curr. Opin. Microbiol. 2005;8:669–676. doi: 10.1016/j.mib.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Tye BK, Sawyer SL. The Hexameric Eukaryotic MCM Helicase: Building Symmetry from Nonidentical Parts. J. Biol. Chem. 2000;275:34833–34836. doi: 10.1074/jbc.R000018200. [DOI] [PubMed] [Google Scholar]
- 4.Forsburg SL. Eukaryotic MCM proteins: beyond replication initiation. Microbiol. Mol. Biol. Rev. 2004;68:109–131. doi: 10.1128/MMBR.68.1.109-131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shin JH, Santangelo TJ, Xie Y, Reeve JN, Kelman Z. Archaeal minichromosome maintenance (MCM) helicase can unwind DNA bound by archaeal histones and transcription factors. J. Biol. Chem. 2007;282:4908–4915. doi: 10.1074/jbc.M606847200. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher RJ, Bishop BE, Leon RP, Sclafani RA, Ogata CM, Chen XS. The structure and Function of MCM from archaeal M. thermoautotrophicum. Nature Struct. Biol. 2003;10:160–167. doi: 10.1038/nsb893. [DOI] [PubMed] [Google Scholar]
- 7.Kasiviswanathan R, Shin JH, Melamud E, Kelman Z. Biochemical characterization of the Methanothermobacter thermautotrophicus minichromosome maintenance (MCM) helicase N-terminal domains. J. Biol. Chem. 2004;279:28358–28366. doi: 10.1074/jbc.M403202200. [DOI] [PubMed] [Google Scholar]
- 8.Pape T, Meka H, Chen S, Vicentini G, van Heel M, Onesti S. Hexameric ring structure of the full-length archaeal MCM protein complex. EMBO Rep. 2003;4:1079–1083. doi: 10.1038/sj.embor.7400010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poplawski A, Grabowski B, Long SE, Kelman Z. The zinc finger domain of the archaeal minichromosome maintenance protein is required for helicase activity. J. Biol. Chem. 2001;276:49371–49377. doi: 10.1074/jbc.M108519200. [DOI] [PubMed] [Google Scholar]
- 10.Kasiviswanathan R, Shin JH, Kelman Z. DNA Binding by the Methanothermobacter thermautotrophicus Cdc6 Protein Is Inhibited by the Minichromosome Maintenance Helicase. J. Bacteriol. 2006;188:4577–4580. doi: 10.1128/JB.00168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa A, Pape T, van Heel M, Brick P, Patwardhan A, Onesti S. Structural basis of the Methanothermobacter thermautotrophicus MCM helicase activity. Nucleic Acids Res. 2006;34:5829–5838. doi: 10.1093/nar/gkl708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin JH, Grabowski B, Kasiviswanathan R, Bell SD, Kelman Z. Regulation of minichromosome maintenance helicase activity by Cdc6. J. Biol. Chem. 2003;278:38059–38067. doi: 10.1074/jbc.M305477200. [DOI] [PubMed] [Google Scholar]
- 13.Shin JH, Kelman Z. The replicative helicases of bacteria, archaea and eukarya can unwind RNA-DNA hybrid substrates. J. Biol. Chem. 2006;281:26914–26921. doi: 10.1074/jbc.M605518200. [DOI] [PubMed] [Google Scholar]
- 14.McEntee K, Weinstock GM, Lehman IR. recA protein-catalyzed strand assimilation: stimulation by Escherichia coli single-stranded DNA-binding protein. Proc. Natl Acad. Sci. USA. 1980;77:857–861. doi: 10.1073/pnas.77.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heyduk T, Lee JC. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl Acad. Sci. USA. 1990;87:1744–1748. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Licata VJ, Wowor AJ. Applications of Fluorescence Anisotropy to the Study of Protein-DNA Interactions. Methods Cell Biol. 2008;84:243–262. doi: 10.1016/S0091-679X(07)84009-X. [DOI] [PubMed] [Google Scholar]
- 17.Shin JH, Jiang Y, Grabowski B, Hurwitz J, Kelman Z. Substrate requirements for duplex DNA translocation by the eukaryal and archaeal minichromosome maintenance helicases. J. Biol. Chem. 2003;278:49053–49062. doi: 10.1074/jbc.M308599200. [DOI] [PubMed] [Google Scholar]
- 18.Kirchhoff WH. Exam: A two-state thermodynamic analysis program. NIST Tech. Note. 1993;1401:1–103. [Google Scholar]
- 19.Kaplan DL, Davey MJ, O'Donnell M. Mcm4,6,7 uses a “Pump in Ring” mechanism to unwind DNA by steric exclusion and actively translocate along a duplex. J. Biol. Chem. 2003;278:49171–49182. doi: 10.1074/jbc.M308074200. [DOI] [PubMed] [Google Scholar]
- 20.Kelman Z, Lee JK, Hurwitz J. The single minichromosome maintenance protein of Methanobacterium thermoautotrophicum DH contains DNA helicase activity. Proc. Natl Acad. Sci. USA. 1999;96:14783–14788. doi: 10.1073/pnas.96.26.14783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong JP, Hayashi MK, Simon MN, Xu RM, Stillman B. A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicase. Proc. Natl Acad. Sci. USA. 2000;97:1530–1535. doi: 10.1073/pnas.030539597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shechter DF, Ying CY, Gautier J. The intrinsic DNA helicase activity of Methanobacterium thermoautotrophicum DH minichromosome maintenance protein. J. Biol. Chem. 2000;275:15049–15059. doi: 10.1074/jbc.M000398200. [DOI] [PubMed] [Google Scholar]
- 23.Manly SP, Matthews KS, Sturtevant JM. Thermal denaturation of the core protein of lac repressor. Biochemistry. 1985;24:3842–3846. doi: 10.1021/bi00336a004. [DOI] [PubMed] [Google Scholar]
- 24.Grabowski B, Kelman Z. Autophosphorylation of the archaeal Cdc6 homologues is regulated by DNA. J. Bacteriol. 2001;183:5459–5464. doi: 10.1128/JB.183.18.5459-5464.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher RJ, Chen XS. Biochemical activities of the BOB1 mutant in Methanobacterium thermoautotrophicum MCM. Biochemistry. 2006;45:462–467. doi: 10.1021/bi051754z. [DOI] [PubMed] [Google Scholar]
- 26.Hardy CF, Dryga O, Seematter S, Pahl PM, Sclafani RA. mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc. Natl Acad. Sci. USA. 1997;94:3151–3155. doi: 10.1073/pnas.94.7.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkinson ER, Chong JP. Minichromosome maintenance helicase activity is controlled by N- and C-terminal motifs and requires the ATPase domain helix-2 insert. Proc. Natl Acad. Sci. USA. 2006;103:7613–7618. doi: 10.1073/pnas.0509297103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barry ER, McGeoch AT, Kelman Z, Bell SD. Archaeal MCM has separable processivity, substrate choice and helicase domains. Nucleic Acids Res. 2007;35:988–998. doi: 10.1093/nar/gkl1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasiviswanathan R, Shin JH, Kelman Z. Interactions between the archaeal Cdc6 and MCM proteins modulate their biochemical properties. Nucleic Acids Res. 2005;33:4940–4950. doi: 10.1093/nar/gki807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marinsek N, Barry ER, Makarova KS, Dionne I, Koonin EV, Bell SD. GINS, a central nexus in the archaeal DNA replication fork. EMBO Rep. 2006;7:539–545. doi: 10.1038/sj.embor.7400649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.