

Abstract
We describe here a series of N-(quinolin-8-yl)benzenesulfonamides capable of suppressing the NFκB pathway identified from two high-throughput screens run at two centers of the NIH Molecular Libraries Initiative. These small molecules were confirmed in both primary and secondary assays of NFκB activation and expanded upon through analogue synthesis. The series exhibited potencies in the cell-based assays as low as 0.6 µM, and several indications suggest that the targeted activity lies within a common region of the NFκB pathway.
Nuclear factor-κB (NFκB) is the designation for a collection of transcription factors including homo- and heterodimers of the Rel family proteins (p50, p52, c-Rel, RelA/p65 and RelB).1,2 First described3 in 1986 as an enhancer of immunoglobulin-κ, NFκB is now understood to bind to the promoter region of more than 400 genes and play an important role in numerous physiological events, most prominently as a primary regulator of the immune and inflammatory responses.4 Several pathways leading to activation of NFκB are currently understood.5 Within the canonical pathway, the NFκB heterodimer exists predominantly in the cytoplasm as an inactive complex with the inhibitory IκB proteins. Phosphorylation of the IκB proteins by an IκB kinase (IKK) targets the IκBα protein for polyubiquitination by an E3 ligase and degradation by the proteasome. This permits the NFκB complex to translocate to the nucleus and commence gene expression. The exploration of this pathway has given the biomedical community several potential targets for manipulation of NFκB activity including IKK inhibition, proteasome regulation, and transcriptional inhibitors of NFκB.6,7 Therefore, NFκB has emerged as a highly studied therapeutic target and small molecule regulation of signaling cascades associated with NFκB may provide novel approaches to alleviate numerous disease states.6,7
Several high-throughput campaigns in search of novel inhibitors of the NFκB signal transduction pathway have been undertaken by the Molecular Libraries Initiative (MLI) of the NIH Roadmap for Medical Research.8 In one effort, a cell-based assay designed to identify IκBα stabilization was undertaken by the NIH Chemical Genomics Center in collaboration with the Metabolism Branch of the NCI Center for Cancer Research.9 A second effort was initiated by another center of the MLI located at Columbia University, wherein a cell-based assay was designed to identify inhibitors of TNFα-induced translocation of NFκB. Importantly, both assays were performed utilizing the same compound collection provided to each center by the MLI. Despite the differing assay design, cell type and signal readout a N-(quinolin-8-yl)benzenesulfonamide core scaffold was noted as a primary lead in both screens.
The IκBα stabilization screen was performed using a dual luciferase reporter system in the cell line OCI-Ly3, an excellent model for primary tumors of the activated B-cell subtype (ABC) of diffuse large B-cell lymphoma (DLBCL) (Figure 1a; PubChem AID: 445).9–11 Abnormally high constitutive NFκB activity levels have been noted in ABC-DLBCL, as in several types of cancer, due to high levels of IKKβ activity leading to elevated expression of NFκB targeted genes.11 In this setting, IKK and proteasome activity produce high degradation of IκBα, liberating p50/65 and/or p50/c-Rel heterodimers to translocate to the nucleus. Because lines of ABC-DLBCL and other cancer types are dependent on constitutive NFκB activity, the NFκB pathway is a therapeutic target. This is particularly true for those processes governing IκBα degradation, and the therapeutic potential of small molecules for this purpose has been shown by studies in ABC-DLBCL lines using a specific IKKβ inhibitor. Furthermore, the use of an ABC-DLBCL line for a small-molecule screen of IκBα stabilization provides a context that is especially close to that of the targeted disease, with the potential that inhibitors may be found that affect specific upstream points in IKK activation.
Figure 1.

a.) The two-color dual luciferase based assay. IκBα is fused to a luciferase emitting green light (CBG68), while a red light emitting luciferase (CBR) is present in its native state. Both luciferases are regulated by binding of the tet repressor to the tet operator (TO) binding site (TO), allowing simultaneous induction of the luciferases by the CMV promoter upon addition of doxycycline and test compounds. The CBR protein serves to normalize for nonspecific effects, while the levels of the IκBα-CBG68 protein increase uniquely with inhibition of the NFκB pathway leading to IκBα degradation. b.) The nuclear translocation assay. TNFα stimulation results in nuclear translocation of NFκB, which is detected by immunofluorescence using an anti-p65 antibody and a secondary antibody labeled with an Alexa 647 fluorophore (A647).
To identify modulators of IκBα stability in ABC-DLBCL lines, such as small molecules inhibiting IKK or proteasome activity, we measured changes in the level of an exogenous IκBα-luciferase fusion reporter expressed by an NFκB-insensitive promoter.12,13 The dual luciferase IκBα stabilization screen in OCI-Ly3 employed here was designed to be suitable for HTS, by using IκBα fused to a green light-emitting beetle luciferase, with a red light-emitting beetle luciferase expressed in a native form to monitor cell uniformity and nonspecific effects.9 Fold-responsiveness was further increased by having both reporters under the control of inducible promoters regulated by doxycycline. Upon doxycycline induction of both luciferase reporters, compounds that increased green luminescence with minimal effects on the red luminescence signal were scored as IκBα stabilizers.9
The translocation-based assay was a high-content screen performed in human umbilical vein endothelial cells (HUVEC) using TNFα to stimulate nuclear translocation of endogenous NFκB (Figure 1b; PubChem AID: 438). NFκB is sequestered in the cytoplasm due to its binding to IκBα, which blocks exposure of a nuclear localization sequence. Activation by cytokines such as TNFα results in proteasome degradation of IκBα and subsequent translocation of NFκB from the cytoplasm to the nucleus. In the assay, nuclear translocation of the endogenous p65 RelA subunit of NFκB, at 30 min post-stimulation, was monitored using fluorescent antibody detection and an automated imaging platform.14,15 NFκB inhibitors in this assay, such as IκBα stabilizers, were detected as compounds that interfered with p65 translocation to the nucleus.
There were several common aspects of the two screens in this study. Importantly, both screens were run as cell-based assays utilizing an identical set of small molecules. Additionally, neither assay monitored NFκB gene transcription, and therefore transcriptional inhibitors, a potentially large source of non-specific positives, were not likely to be detected by these assays. The assays were validated using known inhibitors of the NFκB pathway (Figure 2). The proteasome inhibitor MG-132 (1)16 served as a positive control for the IκBα stabilization assay. The translocation assay used BAY 11-7082 (2), an agent that inhibits the TNFα-induced phosphorylation of IκBα.17–19 Additionally, a substituted 2-(thiophen-2-yl)quinazoline 3 which acts as an inhibitor of NFκB and AP1 mediated transcriptional activation20 was used as a positive control in a secondary assay that used TNFα to stimulate NFκB dependent expression of a β-lactamase reporter (NFκB-bla in Table 1 and Table 2). This compound also served as a negative control for the dual luciferase IκBα stabilization assay.
Figure 2.

Structures of MG-132 (1), BAY 11-7082 (2), a quinazoline based inhibitor of AP1 and NFκB mediated transcription (3), N-(quinolin-8-yl)benzenesulfonamide; PubChem CID: 161167 (4) and C7-locked N-(quinolin-8-yl)benzenesulfonamide 5; PubChem CID: 659101.
Table 1.
SAR surrounding the N-(quinolin-8-yl)benzenesulfonamides
| Control compound # | I kBa stabilization EC50 (µM) and efficacy* | IkBa stabilization EC50 (µM) ratio** | translocation of NF-κB IC50 (µM)*** | NF-κB bla IC50 (µM) | Cytotoxcity IC50 (µM) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.0, 100% | 5.2, 100% | ND | 0.6 | >10 | ||||
| 2 | NA | NA | 3.0 | ND | ND | ||||
| 3 | Inactive | Inactive | Inactive | Inactive | Inactive | ||||
| Analogue # | R | R′ | R″ | ||||||
 |
4 | phenyl | H | H | 17, 350% | 20 | >10 | 8.0 | Inactive |
| 6 | 4-nitrophenyl | H | H | 6.5, 560% | 17 | 1.5 | 5.0 | Inactive | |
| 7 | 2-nitrophenyl | H | H | 11, 620% | 12 | 3.2 | 10 | Inactive | |
| 8 | 2-nitrophenyl | H | Me | 6.8, 200% | 8.0 | 3.8 | 6.0 | Inactive | |
| 9 | 3-nitrophenyl | H | Me | 6.0, 110% | 13 | 0.9 | 1.0 | >57 | |
| 10 | 4-methyl-2-nitrophenyl | H | H | 6.8, 275% | 10 | 2.1 | 6.0 | >57 | |
| 11 | 2-methyl-5-nitrophenyl | H | H | 1.0, 18% | >10 | 1.6 | 1.8 | >57 | |
| 12 | 2-nitro-4-(trifluoromethyl)phenyl | H | H | 3.6, 200% | 20 | 3.7 | 1.3 | >57 | |
| 13 | 2-nitro-4-(trifluoromethyl)phenyl | H | Me | 10, 50% | 3.6 | 1.1 | 13 | >57 | |
| 14 | 4-methyl-2-nitrophenyl | OMe | H | 6.8, 60% | 7.6 | 2.1 | 4.0 | >57 | |
| 15 | 4-methoxy-2-nitrophenyl | H | Me | ND | ND | 1.0 | ND | ND | |
| 16 | 2-methyl-5-nitrophenyl | H | Me | >20 | >57 | Inactive | >57 | Inactive | |
| 17 | 4-methyl-2-nitrophenyl | OMe | Me | >10 | >57 | Inactive | Inactive | >57 | |
| 18 | 4-methylphenyl | OMe | Me | 20, 150% | 20 | >10 | >57 | >57 | |
| 19 | 2-aminophenyl | H | H | 11, 300% | 8.5 | 2.0 | 13.5 | >57 | |
| 20 | 2-aminophenyl | H | Me | 8.0, 250% | 8.0 | 2.8 | 5.5 | Inactive | |
| 21 | 2-amino-4-methylphenyl | OMe | H | 4.0, 90% | 4.0 | 1.0 | 2.0 | >57 | |
| 22 | 2-amino-4-methylphenyl | OMe | Me | 9.4, 23% | 3.0 | Inactive | >57 | Inactive | |
| 23 | thiophen-2-yl | H | H | 5.3, 250% | 9.0 | 3.6 | 7.0 | Inactive | |
| 24 | 5-chlorothiophen-2-yl | H | H | 2.7, 190% | 2.7 | 1.3 | 1.0 | >57 | |
| 25 | 5-bromothiophen-2-yl | H | H | 5.9, 170% | 6.8 | 1.1 | 3.4 | Inactive | |
| 26 | 5-chlorothiophen-2-yl | H | Me | 1.4, 60% | 2.1 | 1.0 | 1.3 | >10 uM | |
| 27 | 5-bromothiophen-2-yl | H | Me | 6.1, 22% | 6.4 | 1.4 | 5.0 | >57 | |
EC50 values from the IκB stabilization assay shown for the green luminescence reporter along with the %efficacy.
EC50 values from the ratio of the green and red luminescent values for the IκBα stabilization. Data are averages from two to three experiments where each experiment consisted of concentration-titration for each compound performed and duplicate and fitting concentration-response curves to the response after the bioassay. NA = not applicable, the compound only showed a strong inhibitory response in the original qHTS (IC50 = 2.5 µM, 95% inhibition) in the non-specific (red luminescence) dataset.
IC50 values are derived from curve-fitting to data from a single experiment performed in triplicate. ND = not determined. All compounds showed >90% efficacy in the translocation assay except compounds 15 (78%) and 27 (75%). The cytotoxicity assay was performed in OCI-Ly3 cells using a 4 hr endpoint.
Table 2.
SAR surrounding the C7-locked N-(quinolin-8-yl)benzenesulfonamides
| Analogue # | R | R′ | R″ | R″′ | IkBa stabilization EC50 (µM) and efficacy* | IkBa stabilization EC50 (µM) ratio** | translocation of NF-κB IC50 (µM)*** | NF-κB bla IC50 (µM) | Cytotoxcity IC50 (µM) | |
|---|---|---|---|---|---|---|---|---|---|---|
 |
5 | H | H | H | H | 2.2, 120% | 2.6 | 1.0 | 3.4 | Inactive |
| 28 | H | H | H | OH | ND | ND | Inactive | ND | ND | |
| 29 | H | H | H | OMe | 1.3, 20% | 1.4 | 1.0 | 1.3 | Inactive | |
| 30 | H | H | H | Me | 0.5, 60% | 0.8 | 0.6 | 0.8 | >57 | |
| 31 | H | H | H | CF3 | 0.7, 32% | 1.0 | 1.0 | 1.2 | Inactive | |
| 32 | H | H | OMe | Me | 1.3, 34% | 1.6 | 1.2 | 2.0 | Inactive | |
| 33 | H | H | H | F | 4.1, 122% | 12 | 1.7 | 1.8 | Inactive | |
| 34 | H | H | H | Cl | 1.1, 66% | 11 | 1.1 | 1.9 | Inactive | |
| 35 | H | OMe | H | H | ND | ND | 2.5 | ND | ND | |
| 36 | H | Me | H | H | 7.0, 32% | 10 | ND | 8.1 | Inactive | |
| 37 | Me | H | OMe | Me | 1.4, 300% | 2.3 | 0.9 | 1.1 | >57 | |
| 38 | Me | H | H | OH | 8.0, 120% | 11 | 10 | 6.7 | >57 | |
| 39 | Me | H | H | OMe | 3.7, 200% | 3.4 | 2.5 | 3.2 | >57 | |
| 40 | Me | H | H | CF3 | 8.9, 700% | 7.2 | 1.2 | 3.6 | Inactive |
EC50 values from the IκB stabilization assay shown for the green luminescence reporter along with the %efficacy.
EC50 values from the ratio of the green and red luminescent values for the IκBα stabilization. Data are averages from two to three experiments where each experiment consisted of concentration-titration for each compound performed and duplicate and fitting concentration-response curves to the response after the bioassay.
IC50 values are derived from curve-fitting to data from a single experiment performed in triplicate. ND = not determined. All compounds showed >90% efficacy in the translocation assay. The cytotoxicity assay was performed in OCI-Ly3 cells using a 4 hr endpoint.
In addition to the required deposit of screening results within the PubChem database, the collaborative nature of the MLI allowed members of each research team to recognize N-(quinolin-8-yl)benzenesulfonamide (4) and the related C7-locked N-(quinolin-8-yl)benzenesulfonamide 5 as the two common lead structures that were found within both screens (Figure 2). To further confirm that these agents interfered with the NFκB activation in a genuine manner, a reporter assay obtained from Invitrogen where induction of β-lactamase occurred in an NFκB-dependent manner was also performed on the active compounds. This data confirmed the ability of these compounds to inhibit the NFκB pathway, and the compounds were then advanced for further study (Table 1 and Table 2).
A significant number of N-(quinolin-8-yl)benzenesulfonamides (both open form and C7-locked form) are commercially available, and the collection was expanded quickly through vendor purchase. Further, the synthesis of analogues of 4 was undertaken through a one-step process involving the addition of unsubstituted or 2-substituted, 6-substituted or 2,6-disubstituted quinolin-8-amines and a variety of sulfonyl chlorides (Scheme 1).21 Amino-substituted analogues (19–22)18 were obtained by reduction of the corresponding nitro compounds with stannous chloride in ethanol (Scheme 2). Over 40 novel analogues were prepared to establish preliminary structure-activity relationships.
Scheme 1.
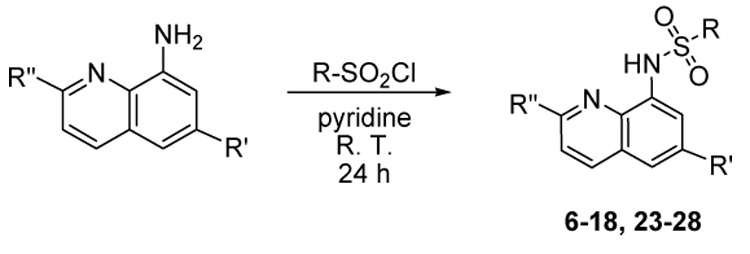
Scheme 2.

The synthesis of various analogues of 5 was more detailed and the method has been recently reported.22 While several synthetic strategies had been attempted, the construction of the sultam scaffold via a diazotization-induced cyclization of accessible N-8-quinolinyl-2-aminobenzenesulfonamides proved to be the most efficient way of synthesizing this unique type of C7-locked sulfonamide (Scheme 2).
Over 300 analogues of 4 and 5 were analyzed within both screens. Results for selected analogues of 4 are shown in Table 1, while the results for derivatives of 5 are shown in Table 2. In general, there was good agreement in the relative potencies of selected analogues tested in both screening assays, as well as the NFκB β-lactamase reporter assay (although exceptions do exist). We take this as an indication that the same cellular target is being manipulated.
The results enabled a number of structure-activity relationships to be observed. In general, the presence of electron withdrawing groups upon the sulfonyl phenyl ring produced a favorable potency response for analogues of 4. Interestingly, the presence of a 2-methyl substituent on the quinoline ring often reduced the potency of the response that is best illustrated by compounds 11 and 16. Heterocyclic rings in place of the sulfonyl phenyl ring were well tolerated as indicated by potency values associated with thiophene analogues 23–27.
A more rigid structure was associated with compound 5, because a bridge between the quinoline carbon 7 and the ortho-position of the sulfonyl phenyl ring locks the core scaffold in a rigid conformation. Two compounds (30 and 31) with the best overall potency values were identified here, although the efficacy in the IκBα stabilization assay was sometimes lower in this series than the unfused series. Substituents at the 9 position (para to the sulfonylamide moiety designated R”’ in Table 2) are generally well tolerated as are 6-methoxy and 2-methyl substitutions (analogues 32 and 37–40, respectively). Unfortunately, despite numerous modifications made to both core structures of 4 and 5, we were unsuccessful in identifying compounds with potencies equivalent to or better than the proteasome inhibitor MG-132 in these assays.
Several points should be made regarding the behavior of these compounds. First, the concentration-response curves associated with these compounds were generally steep (Hill slope showed an average = 4 in the β-lactamase reporter and IκBα stabilization assays, data not shown). Shoichet has recently provided an evaluation of such phenomena and provides rationales for why steep curves emerge, pointing to the formation of colloidal aggregation as the most deleterious reason.23 However, these N-(quinolin-8-yl)benzenesulfonamides were evaluated in a recent profile of the MLSCN compound collection for persistent, small molecule colloidal aggregators and were not noted as being ‘aggregation-like.’24 Further, the colloidal aggregation phenomenon is most significant in purified protein assays and is likely to be less prevalent in cell-based assays. Additionally, for cell-based assays measuring signal transduction pathways, Hill slopes often differ from unity and values of 1,000 are theoretically possible.25 Also, while the potencies of these compounds were generally similar, the efficacies varied in the IκBα stabilization assay. Interestingly, some analogues showed efficacies more than three fold higher than that noted for MG-132 while others showed efficacies of less than 50% of the MG-132 control levels (all potency values reported here for the IκBα stabilization are relative to the observed efficacy of the particular compound). For example, compound 11 showed a low efficacy in both the IκBα stabilization assay (18%) and the NFκB β-lactamase assay (30% inhibition) with similar potency, although this compound showed an efficacy of 97% in the NFκB translocation. Finally, the current series shows lower potencies when serum concentrations are increased to levels between 10% and 20% (data not shown). We attempted to alter the water solubility profile of these compounds to alleviate this issue with no affect.
Selected analogues were submitted to numerous additional assays in an attempt to isolate the target region in the NFκB pathway. The compounds were inactive in biochemical assays of IKKβ using recombinant kinase. Additionally, a full screen for proteasome inhibition (PubChem AID: 526) against the library that included the leads described here did not yield any actives other than MG-132. As well, selected compounds were screened versus a panel of kinases including IKKα, IRAK1, IRAK4, JNK1α1, JNK2α2, JNK3, MAPK1, MAPK2, PKBα, RIPK2, SAPK2a, TAK1, and TBK1 without any inhibition. An E3 ligase target was ruled out by treating HUVEC with compound 5 and staining for phosphorylated IκBα. Treatment of HUVEC with Ro106-9920, a known E3 ligase inhibitor26, but not compound 5, blocked IκBα ubiquitination and subsequent proteasome degradation, resulting in detectable staining of IκBα (data not shown). Thus, the target of this class of small molecule NFκB pathway inhibitors has not been identified at present.
In summary, we have identified a series of N-(quinolin-8-yl)benzenesulfonamides that inhibit NFκB activation with good potency, and selected derivatives maintained high efficacy levels. Lead compounds 4 and 5 provided templates for optimization where novel analogues were obtained with improved potencies (< 1 uM). This series provides a starting point for more detailed selectivity and mechanistic studies.
Acknowledgments
The authors would like to thank Jeremy Smith, Adam Yasgar and Paul Shinn for assistance in compound management at the NIH Chemical Genomics Center, and Darryl Leja of the National Human Genome Research Institute for graphical artwork. We thank Ms. Allison Peck for critical reading of this manuscript. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Gilmore TD. Oncogene. 2006;25:6680. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 2.Perkins ND. Nature Rev. Mol. Cell Biol. 2007;8:49. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 3.Sen R, Baltimore D. Cell. 1986;47:921. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal BB. Expert Opin. Ther. Targets. 2007;11:109. doi: 10.1517/14728222.11.2.109. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann A, Natoli G, Ghosh G. Oncogene. 2006;25:6706. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 6.D’Acquisto FD, Ianaro A. Curr. Opin. Pharmacol. 2006;6:387. doi: 10.1016/j.coph.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Gilmore TD, Herscovitch M. Oncogene. 2006;25:6887. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 8.Austin CP, Brady LS, Insel TR, Collins FS. Science. 2004;306:1138. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 9.Davis RE, Zhang Y-Q, Southall N, Staudt LM, Austin CP, Inglese J, Auld DS. Assay Drug Dev. Technol. 2007;5:85. doi: 10.1089/adt.2006.048. [DOI] [PubMed] [Google Scholar]
- 10.Davis RE, Brown KD, Siebenlist U, Staudt LM. J. Exp. Med. 2001;184:1861. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okamoto T, Sanda T, Asamitsu K. Curr. Pharm. Des. 2007;13:447. doi: 10.2174/138161207780162944. [DOI] [PubMed] [Google Scholar]
- 12.Lam LT, Davis RE, Pierce J, Hepperle M, Xu Y, Hottelet M, Nong Y, Wen D, Adams J, Dang L, Staudt LM. Clin. Cancer Res. 2005;11:28. [PubMed] [Google Scholar]
- 13.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. Nature. 2006;441:106. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 14.Mayer T, Jagla B, Wyler M, Kelly P, Aulner N, Beard M, Barger G, Toebben U, Smith DH, Branden L, Rothman J. Methods in Enz. 2006;414:266. doi: 10.1016/S0076-6879(06)14015-X. [DOI] [PubMed] [Google Scholar]
- 15.The NFκB translocation assay was performed essentially as described.14 HUVEC grown for fewer than 8 passages were seeded at 3,000 cells/well in 384-well view plates. For the concentration-response curves, one of two alternate protocols was used: compounds were added to a final concentration of 10 µM in media, 0.33% DMSO and serially diluted by 1:3, or compounds were added to a final concentration of 8 µM and serially diluted by 1:2. In either case, this produced eight data points. After a 3.5 h incubation with compound, cells were stimulated using 10 µM TNFα for 30 min. Fixed and permeabilized cells were stained with Hoechst to visualize the nuclei, and with mouse anti-p65 (Invitrogen) followed by a secondary antibody conjugated to Alexa Fluor 647 (Invitrogen). Images were scanned using the In Cell Analyzer 3000 (GE Healthcare) and analyzed using the Nuclear Trafficking module
- 16.Lee DH, Goldberg AL. J. Biol. Chem. 1996;271:27280. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- 17.Kamthong PJ, Wu M-C. Biochem. J. 2001;356:525. doi: 10.1042/0264-6021:3560525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izban KF, Ergin M, Qin J-Z, Martinez RL, Pooley RJ, Saeed S, Alkan S. Hum. Pathol. 2000;31:1482. doi: 10.1053/hupa.2000.20370. [DOI] [PubMed] [Google Scholar]
- 19.Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. J. Biol. Chem. 1997;272:21096. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 20.Palanki MSS, Erdman PE, Ren M, Suto M, Bennett BL, Manning A, Ransone L, Spooner C, Desai S, Ow A, Totsuka R, Tsao P, Toriumi W. Bioorg. Med. Chem. Lett. 2003;13:4077. doi: 10.1016/j.bmcl.2003.08.047. [DOI] [PubMed] [Google Scholar]
- 21.General procedure for analogs of 4: (Procedure for compound 27): To a solution of 2-methylquinolin-8-amine (0.16 g, 1.0 mmol) in pyridine (5 mL) was added 5-bromothiophene-2-sulfonyl chloride (0.26 g, 1.0 mmol). The mixture was stirred at room temperature overnight and was precipitated with water. The crude product was filtered and recrystallized from ethanol to afford 27 (0.33 g, 87%) as white crystals. ¹H NMR (300 MHz, CDCl3) δ 9.35 (br, 1H), 8.00 (d, 1H), 7.82 (d, 1H), 7.55-7.39 (m, 2H), 7.35-7.21 (m, 2H), 6.94 (d, 1H), 2.69 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 158.3, 140.7, 138.2, 136.5, 132.9, 132.6, 130.3, 126.6, 123.3, 122.9, 120.3, 115.7, 25.5; ESI-MS (M+ +H): 384.0. General procedure for analogs of 4 (amino-substituted analogs): (Procedure for compound 21): To a suspension of N-(6-methoxyquinolin-8-yl)-4-methyl-2-nitrobenzenesulfonamide (0.37 g, 1.0 mmol) in ethanol (5 mL) stannous chloride (0.38 g, 2.0 mmol) was added slowly. The mixture was refluxed for 2h. After removal of ethanol, the residue was treated with 1M sodium hydroxide. The aqueous solution was extracted with methylene chloride. The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a brown solid which was recrystallized from ethanol to yield 22 (0.29 g, 85 %) as a tan solid. ¹H NMR (300 MHz, CDCl3) δ 9.40 (br, 1H), 8.54 (s, 1H), 7.90 (d, 1H), 7.67 (d, 1H), 7.37-7.26 (m, 2H), 6.63 (s, 1H), 6.48 (d, 1H), 6.38 (s, 1H), 4.95 (br, 2H), 3.84 (s, 3H), 2.13 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 158.1, 146.1, 145.7, 145.6, 135.1, 135.0, 134.9, 130.2, 129.3, 122.5, 118.8, 118.1, 118.0, 106.8, 99.6, 55.9, 21.8; ESI-MS (M+ +H): 344.1. For general procedure for analogs of 5 see reference 22.
- 22.Xie Y, Gong G, Liu Y, Deng S, Rinderspacher KA, Branden L, Landry DW. Tet. Lett. submitted. [Google Scholar]
- 23.Shoichet BK. J. Med. Chem. 2006;49:7274. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 24.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. J. Med. Chem. 2007;50:2385. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 25.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. Nature Chem. Biol. 2007;3(8):466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 26.Swinney DC, Xu Y-Z, Scarafia L, Lee I, Mak A, Gan Q-F, Ramesha C, Mulkins M, Dunn J, So O-Y, Biegel T, Dinh M, Volkel P, Barnett J, Dalrymple S, Lee S, Huber M. J. Biol. Chem. 2002;277:23573. doi: 10.1074/jbc.M200842200. [DOI] [PubMed] [Google Scholar]