

Abstract
Our understanding of human phase II metabolic pathways which facilitate detoxification and excretion of warfarin (Coumadin) is limited. The goal of this study was to test the hypothesis that there are specific human hepatic and extrahepatic UDP-glucuronosyltransferase (UGT) isozymes, which are responsible for conjugating warfarin and hydroxylated metabolites of warfarin. Glucuronidation activity of human liver microsomes (HLMs) and eight human recombinant UGTs toward (R)- and (S)-warfarin, racemic warfarin, and major cytochrome P450 metabolites of warfarin (4′-, 6-, 7-, 8-, and 10-hydroxywarfarin) has been assessed. HLMs, UGT1A1, 1A8, 1A9, and 1A10 showed glucuronidation activity toward 4′-, 6-, 7-, and/or 8-hydroxywarfarin with Km values ranging from 59 to 480 μM and Vmax values ranging from 0.03 to 0.78 μM/min/mg protein. Tandem mass spectrometry studies and structure comparisons suggested glucuronidation was occurring at the C4′-,C6-, C7-, and C8-positions. Of the hepatic UGT isozymes tested, UGT1A9 exclusively metabolized 8-hydroxywarfarin, whereas UGT1A1 metabolized 6-, 7-, and 8-hydroxywarfarin. Studies with extrahepatic UGT isoforms showed that UGT1A8 metabolized 7- and 8-hydroxywarfarin and that UGT1A10 glucuronidated 4′-, 6-, 7-, and 8-hydroxywarfarin. UGT1A4, 1A6, 1A7, and 2B7 did not have activity with any substrate, and none of the UGT isozymes evaluated catalyzed reactions with (R)- and (S)-warfarin, racemic warfarin, or 10-hydroxywarfarin. This is the first study identifying and characterizing specific human UGT isozymes, which glucuronidate major cytochrome P450 metabolites of warfarin with similar metabolic rates known to be associated with warfarin metabolism. Continued characterization of these pathways may enhance our ability to reduce life-threatening and costly complications associated with warfarin therapy.
Warfarin (Coumadin) is a coumarin anticoagulant drug that is used worldwide to manage thromboembolic disease. Despite difficulties in effective patient management, which can be attributed to a very narrow therapeutic range, considerable genetic variations, slow onset of action, and many known drug-drug interactions, warfarin remains one of the most commonly prescribed cardiovascular medications (Bauer, 2006; Rettie and Tai, 2006). Warfarin is primarily administered as an oral medication consisting of a racemic mixture of enantiomers. The (S) enantiomer exhibits approximately 2 to 5 times more anticoagulant activity than the (R) enantiomer in humans (Breckenridge, 1977; Chan et al., 1994; Pitsiu et al., 2003). Both enantiomers inhibit the regeneration of reduced vitamin K by blocking vitamin K epoxide reductase complex, thereby reducing the formation of active clotting factors II, VII, IX, and X (Hirsh et al., 2001; Holbrook et al., 2005; Wajih et al., 2005; Rettie and Tai, 2006). It is readily absorbed, and peak systemic concentrations occur within 60 to 90 min (O’Reilly et al., 1963; Holford, 1986; Chan et al., 1994). Approximately 99% of warfarin is systemically bound to serum albumin. The bound state is thought to be pharmacologically inactive and protected from biotransformation and excretion, but formation of the bound state is reversible (Yacobi and Levy, 1977; Chan et al., 1994).
Variations in hepatic metabolism are thought to be a major determinant for warfarin response variations. Warfarin is readily oxidized via hepatic cytochrome P450s to produce 4′-, 6-, 7-, 8-, and 10-hydroxywarfarin. The monohydroxylated derivatives have little anticoagulation activity and are excreted through urine. (S)-Warfarin is predominantly metabolized via CYP2C9 to produce 6- and 7-hydroxywarfarin (Rettie and Tai, 2006). (R)-Warfarin is thought to be predominantly metabolized via CYP1A2, CYP3A4, CYP2C9, CYP2C18, and CYP2C19. CYP1A2 and/or CYP2C19 primarily produce 6-, 7-, and 8-hydroxywarfarin, whereas CYP3A4, CYP2C9, and CYP2C18 produce 4′- and 10-hydroxywarfarin (Rettie and Tai, 2006) in the presence of (R)-warfarin. Despite having a vast understanding of warfarin metabolism via cytochrome P450 oxidation, little progress has been made in developing more efficacious anticoagulant drugs or therapeutic strategies that reduce adverse side effects associated with warfarin administration.
Glucuronidation is one of the main phase II metabolic pathways whereby xenobiotics, such as drugs and natural compounds present in the diet, are biotransformed into polar conjugates. These conjugates are more water-soluble than the parent compounds and are easily excreted in bile or urine. The reaction is mediated by a family of enzymes, UDP-glucuronosyltransferases (UGTs), that catalyze the transfer of glucuronic acid (GlcUA) from UDP-GlcUA, to a wide range of structurally unrelated molecules bearing hydroxyl, carboxyl, amine, or thiol groups (Mackenzie et al., 2005). In humans, up to 20 different UGT isoforms belonging to the subfamilies 1 and 2 have been characterized following expression of their corresponding cDNA in heterologous cells (Ritter, 2000). Studies have shown that these isoforms present distinct, but frequently overlapping, substrate specificities (Radominska-Pandya et al., 1999). It is known that warfarin, as well as the monohydroxylated derivatives of warfarin, can be potential substrates for phase II conjugating enzymes (Jansing et al., 1992; Guo et al., 2006). However, human data are very limited with regard to warfarin and hydroxywarfarin conjugation.
The primary goal of this study is to elucidate which human hepatic and extrahepatic UGTs have the potential to conjugate warfarin and/or the primary hydroxylated metabolites of warfarin. Data show that glucuronidation of hydroxylated warfarin derivatives is catalyzed by several UGT1A isoforms. These reactions are controlled by both hepatic and extrahepatic pathways, as well as specific substrate-enzyme interactions. Future studies continuing to describe these pathways may enhance our ability to reduce warfarin toxicity and develop new anticoagulant therapies.
Materials and Methods
Materials
All chemicals used for this study were of at least reagent grade. 4′-Hydroxywarfarin, 6-hydroxywarfarin, 7-hydroxywarfarin, 8-hydroxywarfarin, 10-hydroxywarfarin, (R)-(+)-warfarin, (S)-(−)-warfarin, racemic warfarin, and UDP-glucuronic acid were purchased from Sigma-Aldrich (St. Louis, MO). Ethyl alcohol (95%) was purchased from AAPER (Shelbyville, KY). Unless otherwise specified, all other chemicals and reagents were purchased from Sigma-Aldrich. Recombinant human UGT 1A1, 1A3, 1A4, and 1A6–1A10 were produced in baculovirus-infected insect cells as described previously (Kurkela et al., 2003; Kuuranne et al., 2003). These preparations were shown to contain similar amounts of protein by Western blot analysis using an anti-His antibody directed at the His-tag modification present on each of these recombinant isoforms. The human UGT2B7 isoform was expressed in HEK293 cells (Coffman et al., 1997). UGT2B4, -2B15, and -2B17, which primarily catalyze the glucuronidation of bile acids and steroid hormones, rarely glucuronidate drugs and were not investigated. Recombinant human CYP2C9 expressed in baculovirus-infected insect cells was purchased from BD Biosciences (San Jose, CA). HLMs were obtained from a liver donated from a 67-year-old female who died of a stroke on December 31, 1999. Each enzyme tested in this study is known to be active toward substrates specific for that isoform.
Human Liver and Recombinant UGT Isoform Incubations
HLM (50 μg) or UGT recombinant membrane protein (5 μg) were incubated in 100 μM Tris-HCl, pH 7.4/5 mM MgCl2/5 mM saccharolactone with 100 to 2000 μM substrate, in a total volume of 30 μl. Substrates were added in dimethyl sulfoxide with a final concentration of 2%, and controls omitting substrates were run with each assay. No additional detergents or other activators used in the incubations. Reactions were started by the addition of UDP-GlcUA (4 mM) and incubated for up to 180 min at 37°C. The rate of glucuronidation of hydroxywarfarin was linear for up to 3 h (data not shown). The reactions were stopped by addition of 40 μl of ethanol. Each sample was centrifuged at 14,000 rpm for 8 min to spin down the protein, and 60 μl of the supernatant was transferred to an autosampler vial for analysis as described above. All incubations were performed in duplicate, and corresponding error was calculated.
Recombinant Cytochrome P450 Incubations
Recombinant CYP2C9 was expressed from human CYP2C9 (Arg144) cDNA using a baculovirus expression system. Microsomes also contained cDNA-expressed human cytochrome P450 reductase and human cytochrome b5. CYP2C9 (50 pmol) was incubated in 400 μl of a Tris-HCl buffer, pH 7.5, containing 1.3 mM NADP+, 3.3 mM MgCl2, 3.3 mM glucose 6-phosphate, and 0.4 U/ml glucose-6-phosphate dehydrogenase with 100 μM (S)-warfarin. Some incubations also contained 100 μM 8-hydroxywarfarin. All cytochrome P450 reactions were incubated for 60 min at 37°C. Reactions were terminated by the addition of 400 μl of ethanol. Protein was pelleted by centrifugation, and supernatant was transferred to autosampler vials and stored at −80°C before analysis.
High-Performance Liquid Chromatography-UV/Vis Analysis
High-performance liquid chromatography (HPLC) methods were elaborated for the initial separation and identification of warfarin glucuronides. Analyses were performed using an HP 1050 HPLC system equipped with a UV-Vis diode array detector. Instrument operation and data acquisition were controlled through the Agilent ChemStation software package (Agilent Technologies, Santa Clara, CA). Samples were separated using a Supelcosil LC-18 (25 cm × 4.6 mm, 5 mm) column warmed to 37°C. The solvent system consisted of 0.1% acetic acid in water (A) and methanol (B) at a flow rate of 1 ml/min. The separation of warfarin and metabolites was achieved using the following elution gradient: 100% A (5 min), linear gradient from 100% A to 100% B (5–25 min), and 100% B (25–30 min). The column was then re-equilibrated at initial conditions for 10 min between runs. The elution of each warfarin metabolite was monitored at 313 nm. Primary standards for the glucuronidated monohydroxylated warfarin metabolites were not available; therefore, product concentrations were semiquantified using the responses for external standards of each warfarin substrate. It has been shown previously that the addition of the glucuronic acid moiety does not alter the extinction coefficients from that of the unreacted substrate (Doerge et al., 2000). A minimum detection limit (0.3 nmol ± 9.6% relative standard deviation) was determined by measuring 3 times the S.D. of the 7-hydroxwarfarin low level standard (1 nmol). Detection limit studies were conducted with eight independent analyses conducted over 4 days.
Liquid Chromatography-Tandem Mass Spectrometry Analysis
Liquid chromatography (LC)-tandem mass spectrometry (MS/MS) analyses for product confirmation were performed using an Agilent 1100 HPLC system, which was interfaced with an API 4000 triple quadrupole (MS/MS) mass spectrometer (Applied Biosystems, Foster City, CA). Instrument operation and data acquisition were controlled through the Analyst software package (version 1.4.1; Applied Biosystems). The HPLC system consisted of an autosampler, a binary pump, and a column oven. Samples were loaded and resolved at a flow rate of 1 ml/min on a 4.6 × 150-mm C18 column (ZORBAX Eclipse 5μ XDB-C18; Agilent) maintained at 40°C. Mobile phases were 0.1% acetic acid in water (A) and 0.1% acetic acid in methanol (B). Compounds of interest were eluted using the following gradient: 50% B (0 min), linear gradient from 50 to 60% B (0–2 min), linear gradient from 60 to 90% B (2–3 min), linear gradient from 90 to 100% B (3–4.1 min), 100% B (4.1–7.9 min), linear gradient from 100 to 50% B (7.9–8 min), and 50% B (8.0 min and after). Total run time, including a 2-min column pre-equilibration period, was 12 min. Injection volume was 5 μl. All MS/MS analyses were performed in positive ion mode by electrospray ionization using a Turbo IonSpray source. Curtain, nebulizer, turbo, and collisionally activated dissociation gases were 40, 50, 65, and 6 pounds per square inch gauge, respectively. Turbo heater temperature was 510°C, and ion spray voltage was 5500 V. Specific MS/MS experimental conditions are noted in Table 1. 6-Hydroxywarfarin formation was quantitatively measured by comparing multiple reaction monitoring (MRM) responses associated with authentic 6-hydroxywarfarin standards (5–40 nM).
TABLE 1.
MS/MS experimental conditions for product ion, MRM, and neutral loss scans
| Analyte | Q1 | Q3 | CE | EP | DP | CXP | |
|---|---|---|---|---|---|---|---|
| m/z | V | ||||||
| Product ion | |||||||
| 1 | Hydroxywarfarin glucuronides | 501 | 50–550 | 25 | 10 | 52 | 5–20 (2-s cycle) |
| 2 | Warfarin Glucuronides | 485 | 50–550 | 25 | 10 | 52 | 5–20 (2-s cycle) |
| MRM | |||||||
| 1 | Warfarin | 309 | 163 | 45 | 10 | 52 | 10 |
| Warfarin Glucuronide | 485 | 163 | 45 | 10 | 52 | 12 | |
| 2 | 4′-, 6-, 7-, and 8-Hydroxywarfarin | 325 | 179 | 20 | 10 | 52 | 10 |
| 4′-, 6-, 7-, and 8-Hydroxywarfarin glucuronide | 501 | 179 | 45 | 10 | 52 | 12 | |
| 3 | 10-Hydroxywarfarin | 325 | 251 | 31 | 10 | 52 | 14 |
| 10-Hydroxywarfarin glucuronide | 501 | 251 | 31 | 10 | 52 | 14 | |
| Neutral loss | |||||||
| 1 | Loss of glucuronic acid | Loss of 176 (350–700 amu) | 30 | 10 | 52 | 5–20 (2-s cycle) | |
CE, collision energy; EP, entrance potential; DP, declustering potential; CXP, collision cell exit potential; Q1, quadrupole 1; Q3, quadrupole 3; amu, atomic mass unit.
Enzyme Kinetics
Kinetic parameters and Vmax and Km values, were determined by incubating the UGT membrane protein (5 μg) in the presence of varying concentrations of substrate (100–2000 μM) at a fixed concentration of UDP-GlcUA (4 mM) for 90 min. All other conditions were identical to those of the screening experiments. Assuming Michaelis-Menten kinetics, the parameters were determined using Prism4 software (GraphPad Software, San Diego, CA).
Statistics
Cytochrome P450 inhibition studies were analyzed by analysis of variance and tested for significance using a p value of <0.05.
Results
Glucuronidation of Warfarin and Its Hydroxylated Derivatives by Human Hepatic Microsomes and Recombinant UGT Isoforms
Eight human recombinant UGTs expressed as His-tag proteins in baculovirus-infected Sf9 insect cells from the UGT1A family and UGT2B7 overexpressed in HEK293 cells were evaluated for their ability to glucuronidate warfarin and racemic mixtures of hydroxylated warfarin derivatives. Racemic incubations represented clinical dosing because warfarin is usually administered as a racemic mixture of its two enantiomeric forms. Both the (R)- and (S)-enantiomers of native warfarin were commercially available and tested as specific substrates. During preliminary assessments, a series of screening studies were purposely designed at a high substrate concentration (750 μM) to maximize the potential of forming and identifying any potential glucuronide conjugate. Screening data with (R)-, (S)-, and racemic warfarin, as well as five racemic monohydroxylated warfarin metabolites (4′-, 6-, 7-, 8-, and 10-hydroxy) are shown (Fig. 1).
Fig. 1.
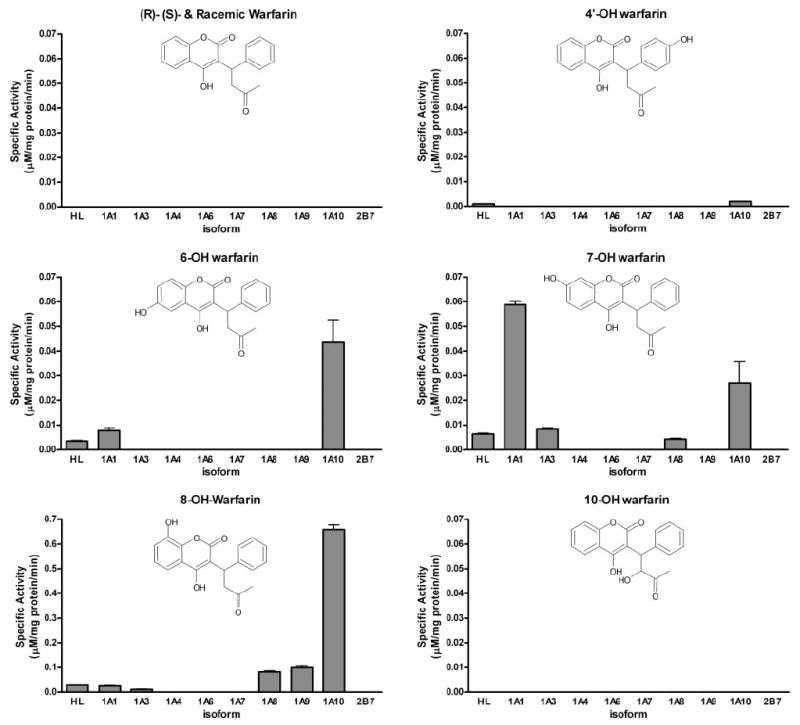
Glucuronidation activities of human liver microsomes and human recombinant UGTs toward native warfarin and its hydroxylated derivatives. Glucuronidation activities UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, and 1A10 were measured using membrane fractions of recombinant UGTs expressed as His-tag proteins in baculovirus-infected Sf9 insect cells (5 μg). The substrate and cosubstrate (UDP-GlcUA) concentrations were 750 μM and 4 mM, respectively. Specific activities are expressed in micromolar per milligram of protein per minute and shown with S.E.s of the mean based on two experiments.
Neither enantiomerically pure isoform of warfarin served as a direct substrate for human UGTs (Fig. 1). This is important new information indicating that native warfarin biotransformation is exclusively dependent on the oxidative metabolism catalyzed by P450s. Two other monohydroxylated warfarin derivatives, 4′- and 10-hydroxy, showed very little or no activity with any isoform under experimental conditions tested (Fig. 1). The two major products of CYP2C9 metabolism, 6- and 7-hydroxywarfarin, were glucuronidated by UGT1A1, -1A3, and -1A10 and to a lesser extent -1A8 (Fig. 1). UGT1A10 glucuronidated both 6- and 7-hydroxywarfarin with similar efficiency (~0.04 μM/mg protein/ min). 7-Hydroxywarfarin was a slightly better substrate for UGT1A1 (~0.06 μM/mg protein/min), but this isoform’s activity toward 6-hydroxywarfarin was lower (<0.01 μM/mg protein/min). UGT1A3 and -1A8 also glucuronidated 7-hydroxywarfarin with low efficiency (~0.005 μM/mg protein/min). 8-Hydroxywarfarin seemed to be the best substrate for glucuronidation (Fig. 1). UGT 1A1, -1A3, -1A8, 1A9, and -1A10 showed activity with 8-hydroxywarfarin, with UGT1A10 showing the highest activity (>0.6 μM/mg protein/min). This compound was also a very good substrate for UGT1A8 and -1A9 (~0.1 μM/mg protein/min). UGT1A1 and -1A3 had much lower activity toward 8-hydroxywarfarin (<0.05 μM/mg protein/min, respectively). Human recombinant UGT1A4, 1A6, 1A7, and 2B7 did not show any measurable activity toward any substrate tested (Fig. 1).
Kinetic Analysis
Kinetic analyses revealed UGT1A10 universally accepted the racemic 6-, 7-, and 8-hydroxywarfarins as substrates, whereas the remaining UGTs were more selective (Fig. 2; Table 2). Despite variations in the Vmax and Km values, the UGT1A10 catalytic efficiencies toward the 6- and 7-hydroxylated warfarins were similar, whereas the efficiency toward 8-hydroxywarfarin was approximately 10-fold higher (Table 2). Compared with UGT1A10, 7-hydroxywarfarin glucuronidation by UGT1A1 was 4-fold more efficient due to a higher Vmax and lower Km (Table 2). The turnover rates for 8-hydroxywarfarin by UGT1A8 and UGT1A9 were similar, although UGT1A9 displayed a 2-fold lower Km, resulting in a more efficient enzyme toward this substrate (Table 2).
Fig. 2.
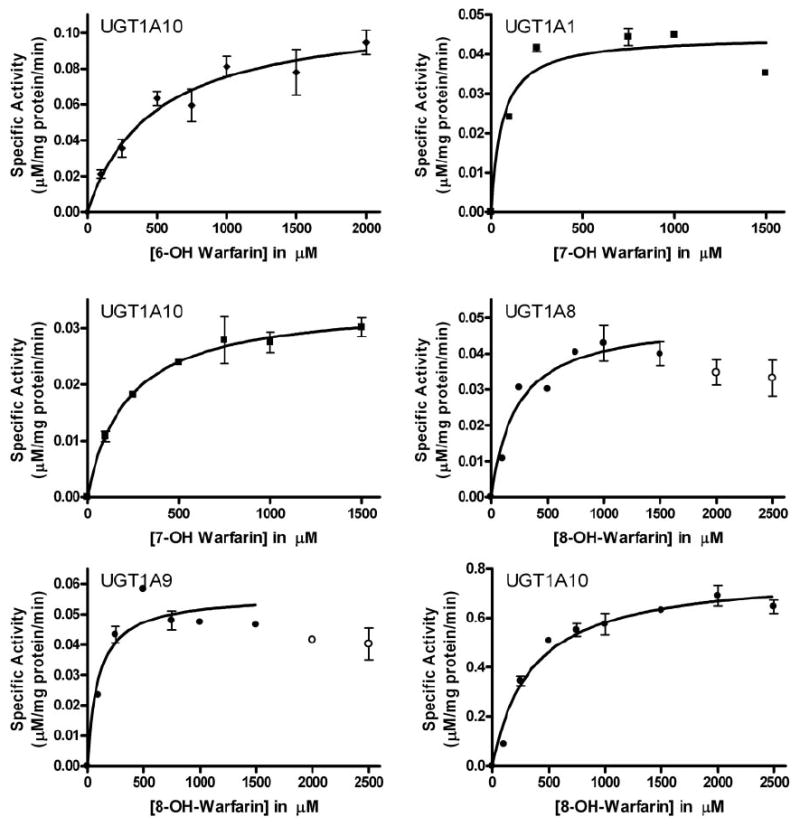
Steady-state glucuronidation of hydroxywarfarins for selected recombinant UGT isoforms. Glucuronidation activities of recombinant proteins were measured by incubating membrane fractions containing recombinant UGT1A10 (5 μg) with increasing concentrations (shown in the figure) of substrate at a constant concentration of UDP-GlcUA (4 mM). Diamonds, 6-hydroxywarfarin incubations; squares, 7-hydroxywarfarin incubations; circles, 8-hydroxywarfarin incubations. Curve fits and kinetic constants were determined using GraphPad Prism 4 software, and the resulting parameters are included in Table 2.
TABLE 2.
Steady-state parameters for glucuronidation of warfarin metabolites by UGT isoformsa
| Substrate | UGT Isoform | Vmax | Km | Vmax/Km |
|---|---|---|---|---|
| μM/min/mg protein | μM | ×10−3 min−1 mg−1 protein) | ||
| 6-OH-warfarin | UGT1A10 | 0.11 ± 0.01 | 480 ± 130 | 0.23 ± 0.06 |
| 7-OH-warfarin | UGT1A1 | 0.045 ± 0.003 | 59 ± 24 | 0.76 ± 0.31 |
| UGT1A10 | 0.030 ± 0.003 | 165 ± 41 | 0.18 ± 0.05 | |
| 8-OH-warfarin | UGT1A8 | 0.050 ± 0.003 | 225 ± 65 | 0.22 ± 0.06 |
| UGT1A9 | 0.057 ± 0.003 | 97 ± 36 | 0.59 ± 0.22 | |
| UGT1A10 | 0.78 ± 0.03 | 346 ± 57 | 2.3 ± 0.4 |
Parameters determined from the fit of initial velocities to a Michaelis-Menten kinetic scheme using the software GraphPad Prism.
Product Confirmations/MS Spectral Interpretation
Metabolite identification by LC-MS/MS confirmed that 4′-, 6-, 7-, and 8-hydroxywarfarin served as substrates for human UGTs, but not 10-hydroxywarfarin, racemic warfarin, (S)-(−)-warfarin, or (R)-(+)-warfarin (Figs. 3 and 4). The presence of m/z 325 in product ion analyses suggested the loss of glucuronic acid (Fig. 4), and identification of other fragment ions has been proposed (Fig. 5).
Fig. 3.
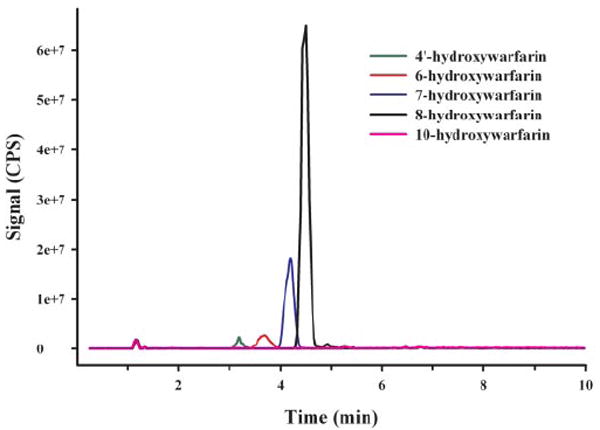
Reverse phase-HPLC chromatograph of product ion (m/z 501) experiments. Tracings represent organic-soluble metabolites generated during incubation of human liver microsomes with UDP-glucuronic acid (4 mM) and 750 μM of each substrate (green, 4′-hydroxywarfarin; red, 6-hydroxywarfarin; blue, 7-hydroxywarfarin; black, 8-hydroxywarfarin; pink, 10-hydroxywarfarin). No specific metabolites were generated when 10-hydroxywarfarin (750 μM), racemic warfarin (750 μM), (S)-warfarin (750 μM), or (R)-warfarin (750 μM) were used as substrates (data not shown). Each substrate was incubated individually for 180 min. All other incubation conditions are noted under Materials and Methods.
Fig. 4.
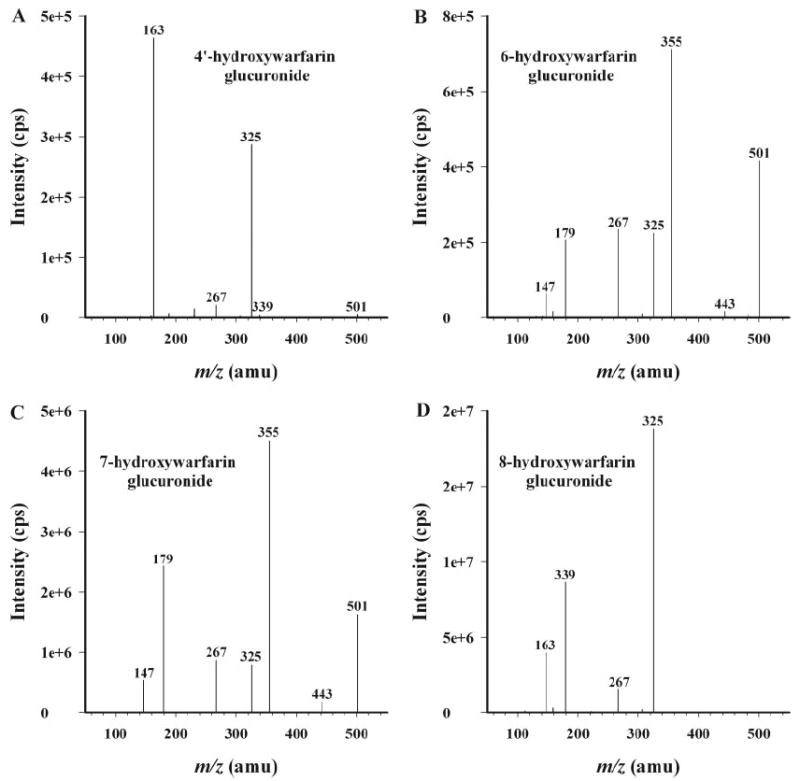
Product ion (m/z 501) spectra of the organic-soluble metabolites generated during incubation of human liver microsomes with UDP-glucuronic acid (4 mM) and 750 μM 4′-hydroxywarfarin (A), 6-hydroxywarfarin (B), 7-hydroxywarfarin (C), and 8-hydroxywarfarin (D). Each substrate was incubated individually for 180 min. All other incubation conditions are noted in Materials and Methods. MS/MS spectra are representative of the major products resolved in Fig. 3. Minor products were also observed in negative controls.
Fig. 5.
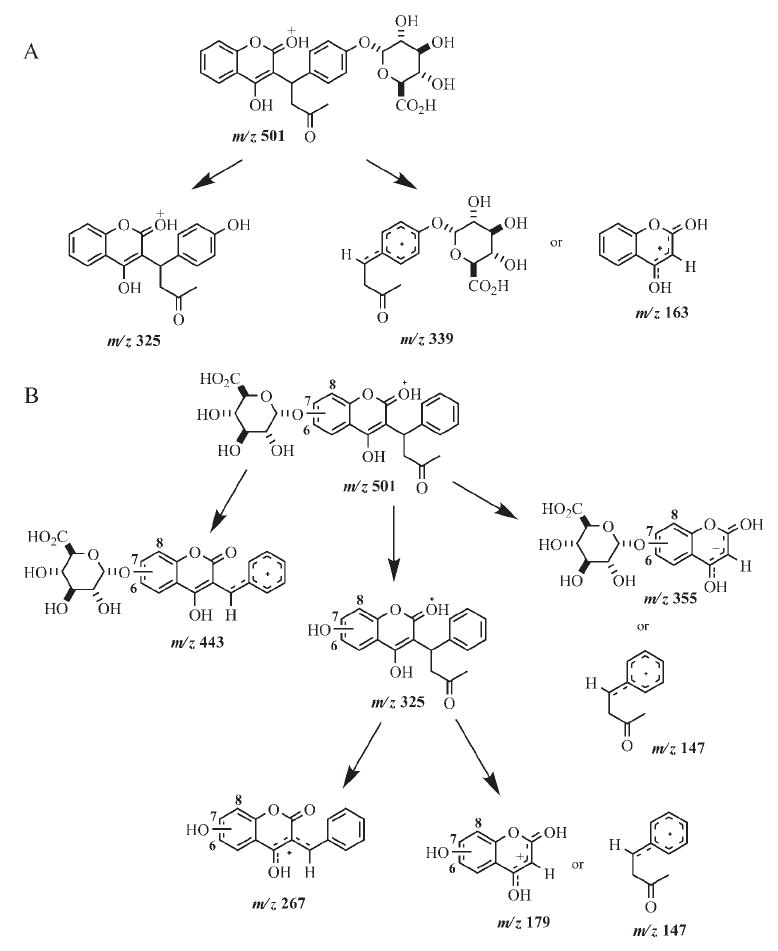
Proposed MS/MS fragmentation pathways for 4′-hydroxywarfarin glucuronide (A) and 6-, 7-, and 8-hydroxywarfarin glucuronides (B).
For each of the hydroxywarfarins, formation of two possible regioisomeric products is possible. Glucuronidation can occur at either the 4-hydroxyl group of the original warfarin skeleton or at the P450 oxidation site (C4′-, C6-, C7-, and C8-positions). Examination of the MS/MS spectrum for 4′-hydroxywarfarin glucuronide (Fig. 4A) revealed a peak at m/z 339, which corresponds to the mass of the side chain plus glucuronic acid (Fig. 5A). This suggests that glucuronidation occurred at the C4′-position. The MS/MS spectra for 6-hydroxywarfarin glucuronide (Fig. 4B) and 7-hydroxywarfarin glucuronide (Fig. 4C) are virtually identical with regard to the masses of the fragment ions, although there are slight differences in relative abundance of the fragments. The base peak in the MS/MS spectrum appears at m/z 355, which corresponds to loss of the side chain from the glucuronidated hydroxycoumarin skeleton (Fig. 4, B and C). The MS/MS spectrum of 8-hydroxywarfarin glucuronide (Fig. 4D) was different from the spectra of 6- and 7-hydroxywarfarin glucuronide. The base peak of the 8-hydroxywarfarin glucuronide was m/z 325 rather than 355 (Fig. 4D). Although these data allow for conclusive identification of the hydroxywarfarin glucuronides, they do not allow for specific assignments of product regiochemistry.
Hydroxywarfarin glucuronides demonstrated a propensity to undergo in-source fragmentation during MS/MS analysis. MRM experiments (data not shown) and neutral loss studies (data not shown) were designed to assess whether additional metabolites were formed but not identified during product ion scans (Fig. 3). Neither study identified major glucuronidated metabolites other than those observed in product ion scans (Fig. 3). MRM studies showed a small degree of in-source fragmentation.
Cytochrome P450 Warfarin Product Inhibition Study
To begin assessing the physiological significance of hydroxywarfarin glucuronidation, 6-hydroxylase activity of CYP2C9 was measured with (S)-warfarin (100 μM) in the presence and absence of 8-hydroxywarfarin (100 μM). Reaction rates were approximately 6.0 ± 4.7 pmol product/nmol P450/min (mean ± S.D.) in the absence of 8-hydroxywarfarin. The addition of 8-hydroxywarfarin (100 μM) significantly inhibited this reaction by approximately 85% (Fig. 6, A and B).
Fig. 6.
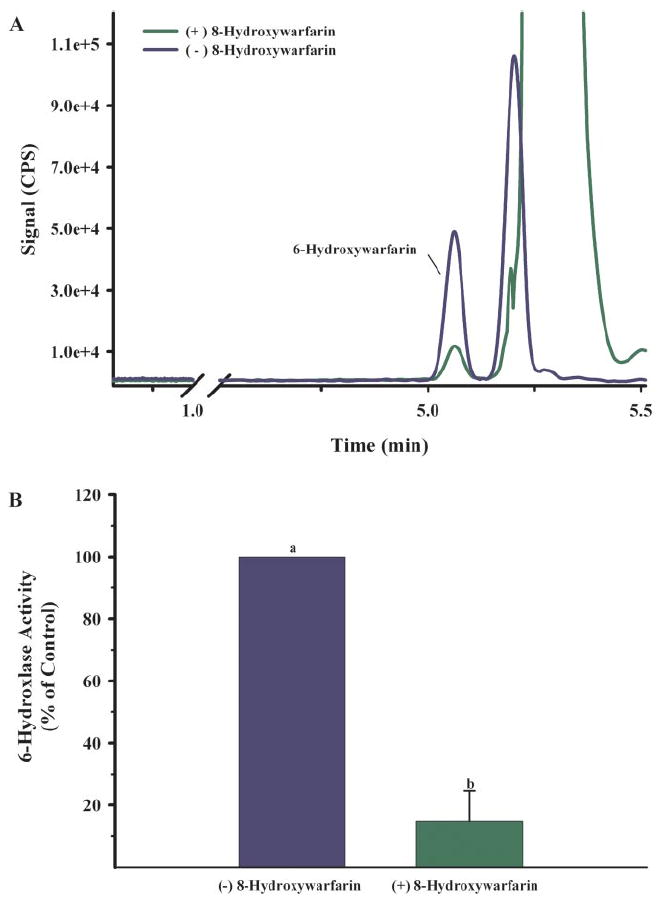
A, reverse phase-HPLC chromatographs of the organic-soluble metabolites generated during the incubation of Sf9 microsomes containing recombinant CYP2C9 and (S)-warfarin (100 μM). Incubations were conducted in the presence (green) and absence (blue) of 8-hydroxywarfarin (100 μM). 6-Hydroxywarfarin was identified through the evaluation of MS/MS spectrum and retention times of authentic standards. B, relative 6-hydroxylase activity of CYP2C9 in the presence and absence of 8-hydroxywarfarin (100 μM). Data represent three independent experiments. 6-Hydroxylase activity of incubations excluding 8-hydroxywarfarin was 6.0 ± 4.7 pmol product/nmol P450/min (mean ± S.D.). Means with different superscripts are statistically different (p < 0.05).
Discussion
Phase II metabolism of warfarin and warfarin metabolites is important for proper detoxification and excretion. However, the specific human isozymes involved in glucuronidation and excretion of warfarin and the corresponding cytochrome P450 metabolites have never been characterized. In this report, we evaluate the ability of specific human recombinant UGT isoforms to metabolize warfarin and several hydroxywarfarin derivatives. These studies show that warfarin itself is not a substrate for any of the assayed UGTs, despite a hydroxyl group at the C4-position that could be a potential target for conjugation with glucuronic acid. Rather, warfarin becomes a substrate for human UGTs after being processed via oxidative biotransformation. A possible explanation for the lack of activity at the C4-position may be related to the formation of a cyclic hemiketal isomer within the active site of the UGT. This is analogous to what has been observed with CYP2C9 interactions (Heimark and Trager, 1985; He et al., 1999).
Previous studies support the finding that biotransformation by human UGTs is exclusively dependent on oxidative metabolism catalyzed by P450s. Isolated rat hepatocytes as well as in vivo rodent models produce glucuronides of hydroxywarfarin metabolites (Jansing et al., 1992; Guo et al., 2006). There is also one report indicating that hydroxywarfarin glucuronides are excreted in human urine (Kaminsky and Zhang, 1997), which is consistent with our unpublished observations showing that hydroxywarfarin glucuronides are a major metabolite excreted in human urine.
Of all the specific UGTs examined in the current study, the extrahepatic isoform, UGT1A10, exhibits the highest glucuronidation activity toward the largest number of monohydroxylated warfarin derivatives tested. UGT1A10 glucuronidates 4′-, 6-, 7-, and 8-hydroxywarfarin at relatively high metabolic rates. UGT1A8, another extrahepatic isoform, has a similar metabolic capacity but exhibits more enzyme substrate specificity compared with the reactivity of UGT1A10. UGT1A8 only metabolizes 7- and 8-hydroxywarfarin. Of the hepatic UGTs tested, UGT1A1 and -1A9 are the only isoforms shown to be involved in the glucuronidation of hydroxywarfarins. It is noteworthy that UGT1A9 metabolizes 8-hydroxywarfarin with the highest efficiency as measured by Vmax/Km. UGT1A1 metabolized all three substrates, including the highly efficient glucuronidation of 7-hydroxywarfarin. Even though the extrapolation of Km and Vmax values derived in this study to in vivo processes is difficult, the reported metabolic parameters are comparable and/or much higher than values stated in previous reports measuring warfarin oxidation rates catalyzed by P450s (Rettie et al., 1992; Kaminsky et al., 1993; Sullivan-Klose et al., 1996; Zhang et al., 1999). This comparison as well as the fact that these products are known to be excreted in human urine (Kaminsky and Zhang, 1997) suggests that hydroxywarfarin conjugation via UGTs is important for human detoxification and excretion.
It is very interesting that UGT1A8 and 1A10 react readily with hydroxywarfarin metabolites. UGT1A8 and 1A10 are known to be primarily expressed in human stomach and intestine and are thought to be important for detoxifying xenobiotics (Strassburg et al., 1997; Cheng et al., 1999; Fisher et al., 2001). Data presented in this report are consistent with published reports studying extrahepatic P450 oxidation of warfarin. For example, CYP2C19 is known to be expressed in human intestine and is known to specifically produce 8-hydroxywarfarin in the presence of (R)-warfarin (Rettie and Tai, 2006). The current study shows that 8-hydroxywarfarin is one of the primary metabolites of warfarin glucuronidated by UGT1A8 and UGT1A10. This suggests that UGTs expressed in human intestine work in concert with CYP2C19 to preferentially detoxify and remove (R)-warfarin. Perhaps intestinal glucuronidation of (R)-warfarin oxidation products contributes to the lower efficacy of this enantiomer (Breckenridge, 1977; Chan et al., 1994; Pitsiu et al., 2003).
LC-MS/MS analysis of product mixtures confirm that glucuronide conjugation does indeed occur for 4′-, 6-, 7-, and 8-hydroxywarfarin but not for racemic warfarin, (R)-warfarin, (S)-warfarin, or 10-hydroxywarfarin. These data show that warfarin oxidation at the 4′-, 6-, 7-, and 8-position is required for glucuronidation. Product ion scans of the desired monoglucuronides provide MS/MS spectra for species with appropriate mass (m/z 501 for the hydroxywarfarins) and in the case of 4′-hydroxywarfarin glucuronide allow for a specific regiochemistry assignment at the C4′-position. This finding differs from results obtained with in vivo and in vitro rodent models that assigned glucuronidation at the C4-position (Jansing et al., 1992; Guo et al., 2006). The reason for this difference is unknown but is possibly due to differences between rodent and human metabolic systems.
Assignment of product regiochemistry is not as straightforward for the 6-, 7-, and 8-hydroxywarfarin glucuronides. The presence of two reactive hydroxyl groups on the coumarin skeleton (Fig. 5) limits MS/MS spectral information because potential fragment ions have the same mass regardless of which hydroxyl group (4- or 6-, 7-, or 8-hydroxy) is glucuronidated. The lack of UGT activity with warfarin, (R)-warfarin, (S)-warfarin, and 10-hydroxywarfarin (Fig. 1; Jansing et al., 1992) suggests that the hydroxyl group located at the C4-position is sterically hindered and less reactive with human UGTs. Information provided by these structure comparisons suggests that glucuronidation is occurring at the C6-, C7-, and C8-positions. Figure 7 shows a representative scheme indicating which UGTs act on specific hydroxylated warfarin metabolites and where it is thought that this action takes place in humans.
Fig. 7.
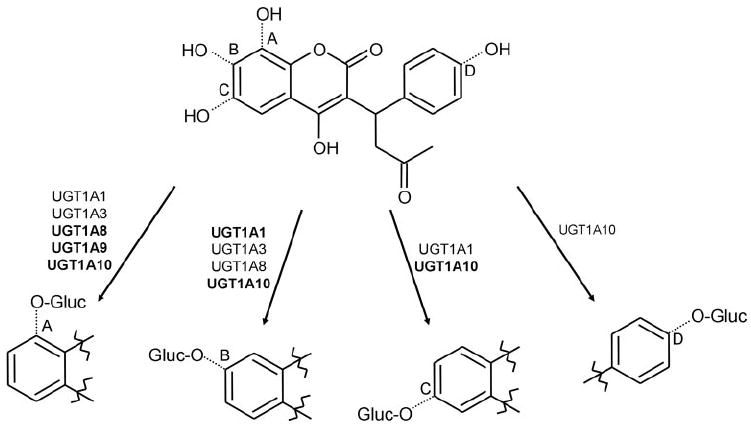
Sites of hydroxywarfarin glucuronidation catalyzed by human UGTs. The upper structure shows the combined structure of 8- (A), 7- (B), 6- (C), and 4′-hydroxywarfarin (D). The lower structures show specific glucuronide products formed by the indicated UGT isoforms. The size of the font used for each respective isoform represents the relative levels of activity of each enzyme.
Although it is widely recognized that oxidation by P450s inactivates warfarin, this is the first report suggesting that variations in UGT loci and metabolism need to be considered as important variables while adjusting warfarin dosing algorithms. Our in vitro data showing significant inhibition (approximately 85%) of CYP2C9 catalytic activity toward (S)-warfarin in the presence of 8-hydroxywarfarin highlight the potential importance of this pathway. Thus, CYP2C9 inhibition would be avoided by removing the P450 products through UGT-mediated conjugation with glucuronic acid. Alternatively, it is possible that hydroxywarfarin glucuronides may inhibit P450 activity. Glucuronides inhibiting P450 activity is a relatively new concept and is considered an unexpected phenomenon (Ogilvie et al., 2006). One can also speculate that glucuronidation of warfarin derivatives produces biologically active glucuronides, like the 6-O-glucuronide of morphine (Ritter, 2000). Interactions between glucuronides and specific transporters are another potentially mitigating but unexplored factor in the clearance of these compounds.
This report characterizes human UGTs associated with warfarin metabolism and provides vital information that will be necessary to fully explore the importance of this metabolic pathway. UGT deficiency due to epigenetic factors and/or polymorphisms could significantly alter the glucuronidation of warfarin metabolites and result in a compromised capacity for warfarin metabolism. Overall, these in vitro studies provide insight for understanding the phase II metabolic pathways that glucuronidate cytochrome P450 warfarin metabolites and facilitate their excretion. This study shows that UGT activity is controlled through specific P450 oxidations as well as specific enzyme substrate mechanisms. Future studies assessing potential stereochemical effects, UGT polymorphisms, and/or epigenetic factors may provide insight in developing better anticoagulant treatment options.
Acknowledgments
We thank Richard Bonner, Benjamin Jefferson, Suzanne Owen, and Amanda Fincher (Arkansas Department of Health) as well as Bob Kobelski (Centers for Disease Control) for valuable contributions while preparing this manuscript and Grazyna Nowak (University of Arkansas for Medical Sciences) for insightful comments during the preparation of this manuscript.
This study was supported by National Institutes of Health Grants DK60109, DK56262, and GM075893 (to A.R.-P.), by Bioterrorism Cooperative Agreement U90/CCU616974-07 (to J.H.M.), by an Association of Public Health Laboratories-Fellowship (to N.C.M. and J.H.M.), by the Academy of Finland (Project 210933), and by the Sigrid Juselius Foundation (to M.F.)
ABBREVIATIONS
- UGT
UDP-glucuronosyltransferase
- GlcUA
glucuronic acid
- HLM
human liver microsome
- HPLC
high-performance liquid chromatography
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- MRM
multiple reaction monitoring
- P450
cytochrome P450
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
References
- Bauer KA. New anticoagulants. Hematology Am Soc Hematol Educ Program. 2006:450–456. doi: 10.1182/asheducation-2006.1.450. [DOI] [PubMed] [Google Scholar]
- Breckenridge AM. Interindividual differences in the response to oral anticoagulants. Drugs. 1977;14:367–375. doi: 10.2165/00003495-197714050-00003. [DOI] [PubMed] [Google Scholar]
- Chan E, McLachlan A, O’Reilly R, Rowland M. Stereochemical aspects of warfarin drug interactions: use of a combined pharmacokinetic-pharmacodynamic model. Clin Pharmacol Ther. 1994;56:286–294. doi: 10.1038/clpt.1994.139. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Radominska-Pandya A, Tephly TR. Studies on the substrate specificity of human intestinal UDP-glucuronosyltransferases 1A8 and 1A10. Drug Metab Dispos. 1999;27:1165–1170. [PubMed] [Google Scholar]
- Coffman BL, Rios GR, King CD, Tephly TR. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos. 1997;25:1–4. [PubMed] [Google Scholar]
- Doerge DR, Chang HC, Churchwell MI, Holder CL. Analysis of soy isoflavone conjugation in vitro and in human blood using liquid chromatography-mass spectrometry. Drug Metab Dispos. 2000;28:298–307. [PubMed] [Google Scholar]
- Fisher MB, Paine MF, Strelevitz TJ, Wrighton SA. The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev. 2001;33:273–297. doi: 10.1081/dmr-120000653. [DOI] [PubMed] [Google Scholar]
- Guo Y, Weller P, Farrell E, Cheung P, Fitch B, Clark D, Wu SY, Wang J, Liao G, Zhang Z, et al. In silico pharmacogenetics of warfarin metabolism. Nat Biotechnol. 2006;24:531–536. doi: 10.1038/nbt1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Korzekwa KR, Jones JP, Rettie AE, Trager WF. Structural forms of phenprocoumon and warfarin that are metabolized at the active site of CYP2C9. Arch Biochem Biophys. 1999;372:16–28. doi: 10.1006/abbi.1999.1468. [DOI] [PubMed] [Google Scholar]
- Heimark LD, Trager WF. Stereoselective metabolism of conformational analogues of warfarin by beta-naphthoflavone-inducible cytochrome P-450. J Med Chem. 1985;28:503–506. doi: 10.1021/jm00382a021. [DOI] [PubMed] [Google Scholar]
- Hirsh J, Dalen J, Anderson DR, Poller L, Bussey H, Ansell J, Deykin D. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119:8S–21S. doi: 10.1378/chest.119.1_suppl.8s. [DOI] [PubMed] [Google Scholar]
- Holbrook AM, Pereira JA, Labiris R, McDonald H, Douketis JD, Crowther M, Wells PS. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005;165:1095–1106. doi: 10.1001/archinte.165.10.1095. [DOI] [PubMed] [Google Scholar]
- Holford NH. Clinical pharmacokinetics and pharmacodynamics of warfarin: understanding the dose-effect relationship. Clin Pharmacokinet. 1986;11:483–504. doi: 10.2165/00003088-198611060-00005. [DOI] [PubMed] [Google Scholar]
- Jansing RL, Chao ES, Kaminsky LS. Phase II metabolism of warfarin in primary culture of adult rat hepatocytes. Mol Pharmacol. 1992;41:209–215. [PubMed] [Google Scholar]
- Kaminsky LS, de Morais SM, Faletto MB, Dunbar DA, Goldstein JA. Correlation of human cytochrome P4502C substrate specificities with primary structure: warfarin as a probe. Mol Pharmacol. 1993;43:234–239. [PubMed] [Google Scholar]
- Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73:67–74. doi: 10.1016/s0163-7258(96)00140-4. [DOI] [PubMed] [Google Scholar]
- Kurkela M, Garcia-Horsman JA, Luukkanen L, Morsky S, Taskinen J, Baumann M, Kostiainen R, Hirvonen J, Finel M. Expression and characterization of recombinant human UDP-glucuronosyltransferases (UGTs). UGT1A9 is more resistant to detergent inhibition than other UGTs and was purified as an active dimeric enzyme. J Biol Chem. 2003;278:3536–3544. doi: 10.1074/jbc.M206136200. [DOI] [PubMed] [Google Scholar]
- Kuuranne T, Kurkela M, Thevis M, Schanzer W, Finel M, Kostiainen R. Glucuronidation of anabolic androgenic steroids by recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos. 2003;31:1117–1124. doi: 10.1124/dmd.31.9.1117. [DOI] [PubMed] [Google Scholar]
- Mackenzie PI, Walter Bock K, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, Miners JO, Owens IS, Nebert DW. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 2005;15:677–685. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–197. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- O’Reilly RA, Aggeler PM, Leong LS. Studies on the coumarin anticoagulant drugs: the pharmacodynamics of warfarin in man. J Clin Invest. 1963;42:1542–1551. doi: 10.1172/JCI104839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsiu M, Parker EM, Aarons L, Rowland M. A Bayesian method based on clotting factor activity for the prediction of maintenance warfarin dosage regimens. Ther Drug Monit. 2003;25:36–40. doi: 10.1097/00007691-200302000-00005. [DOI] [PubMed] [Google Scholar]
- Radominska-Pandya A, Czernik PJ, Little JM, Battaglia E, Mackenzie PI. Structural and functional studies of UDP-glucuronosyltransferases. Drug Metab Rev. 1999;31:817–899. doi: 10.1081/dmr-100101944. [DOI] [PubMed] [Google Scholar]
- Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol. 1992;5:54–59. doi: 10.1021/tx00025a009. [DOI] [PubMed] [Google Scholar]
- Rettie AE, Tai G. The pharmocogenomics of warfarin: closing in on personalized medicine. Mol Interv. 2006;6:223–227. doi: 10.1124/mi.6.4.8. [DOI] [PubMed] [Google Scholar]
- Ritter JK. Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chem Biol Interact. 2000;129:171–193. doi: 10.1016/s0009-2797(00)00198-8. [DOI] [PubMed] [Google Scholar]
- Strassburg CP, Oldhafer K, Manns MP, Tukey RH. Differential expression of the UGT1A locus in human liver, biliary, and gastric tissue: identification of UGT1A7 and UGT1A10 transcripts in extrahepatic tissue. Mol Pharmacol. 1997;52:212–220. doi: 10.1124/mol.52.2.212. [DOI] [PubMed] [Google Scholar]
- Sullivan-Klose TH, Ghanayem BI, Bell DA, Zhang ZY, Kaminsky LS, Shenfield GM, Miners JO, Birkett DJ, Goldstein JA. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics. 1996;6:341–349. doi: 10.1097/00008571-199608000-00007. [DOI] [PubMed] [Google Scholar]
- Wajih N, Sane DC, Hutson SM, Wallin R. Engineering of a recombinant vitamin K-dependent gamma-carboxylation system with enhanced gamma-carboxyglutamic acid forming capacity: evidence for a functional CXXC redox center in the system. J Biol Chem. 2005;280:10540–10547. doi: 10.1074/jbc.M413982200. [DOI] [PubMed] [Google Scholar]
- Yacobi A, Levy G. Protein binding of warfarin enantiomers in serum of humans and rats. J Pharmacokinet Biopharm. 1977;5:123–131. doi: 10.1007/BF01066216. [DOI] [PubMed] [Google Scholar]
- Zhang H, LeCulyse E, Liu L, Hu M, Matoney L, Zhu W, Yan B. Rat pregnane X receptor: molecular cloning, tissue distribution, and xenobiotic regulation. Arch Biochem Biophys. 1999;368:14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]