


Abstract
The hepatitis C virus (HCV) NS5B is essential for viral RNA replication and is therefore a prime target for development of HCV replication inhibitors. Here, we report the identification of a new class of HCV NS5B inhibitors belonging to the coumestan family of phytoestrogens. Based on the in vitro NS5B RNA-dependent RNA polymerase (RdRp) inhibition in the low micromolar range by wedelolactone, a naturally occurring coumestan, we evaluated the anti-NS5B activity of four synthetic coumestan analogues bearing different patterns of substitutions in their A and D rings, and observed a good structure-activity correlation. Kinetic characterization of coumestans revealed a noncompetitive mode of inhibition with respect to nucleoside triphosphate (rNTP) substrate and a mixed mode of inhibition towards the nucleic acid template, with a major competitive component. The modified order of addition experiments with coumestans and nucleic acid substrates affected the potencies of the coumestan inhibitors. Coumestan interference at the step of NS5B–RNA binary complex formation was confirmed by cross-linking experiments. Molecular docking of coumestans within the allosteric site of NS5B yielded significant correlation between their calculated binding energies and IC50 values. Coumestans thus add to the diversifying pool of anti-NS5B agents and provide a novel scaffold for structural refinement and development of potent NS5B inhibitors.
INTRODUCTION
Hepatitis C virus (HCV), a major human pathogen and causative agent of parenteral non-A non-B hepatitis, is often associated with the development of malignant chronic disease, including steatosis, liver cirrhosis and hepatocellular carcinoma (1–3). Hepatitis C Virus infection is estimated to be four to five times more prevalent than HIV-1, with over 200 million cases globally, of which ∼4.1 million infections exist in the United States alone (4,5). There is no vaccine against HCV at present, or any effective therapy broadly targeting all genotypes of HCV. Treatment options against HCV include pegylated interferon α (PEG-IFN-α) alone or in combination with ribavirin, a broad spectrum antiviral agent (6–8). Sustained virological response (SVR) rates for the combination therapy are below 50% for genotype 1 and up to 80% for genotypes 2 and 3, and are associated with severe side effects resulting in limited patient compliance of these drugs (9–12). Moreover, HCV undergoes rapid genetic evolution during replication, resulting in a vast mix of variants, thereby presenting additional challenge towards eradication of the virus in infected patients (13–15). Thus, there is an urgent need to develop improved therapeutic options to combat HCV infections.
HCV is an enveloped, positive-stranded RNA virus with a ∼9.6 kb genome which encodes a single large polyprotein of 3010 amino acids. Host and viral proteases process this polyprotein into four structural (Core, E1, E2 and p7) and six nonstructural proteins (NS2, -3, -4A, -4B, -5A and -5B) (16). Most efforts in the development of novel HCV antiviral agents have focused on two viral targets: the NS3 serine protease that cleaves HCV proteins from the polyprotein precursor and the NS5B RNA-dependent RNA polymerase (RdRp), a crucial and unique component of the viral replication machinery (17).
HCV NS5B is a 66 kDa phosphoprotein with predominant perinuclear localization. Purification of full-length NS5B has been challenging due to its hydrophobic C-terminal membrane anchorage domain. Consequently, recombinant NS5B with 21 to 55 amino acid C-terminal truncations has been purified from Escherichia coli or baculovirus-infected insect cells, thereby facilitating its structure-function investigations (18–24). Similar to other polymerases, crystal structures of NS5B have revealed a classical ‘right hand’ shape, with the characteristic fingers, palm and thumb subdomains (25–27). In vitro, NS5B nonspecifically utilizes a wide range of homo- or heteropolymeric RNA templates in a primer-dependent or independent (de novo synthesis) fashion (18,20,22,24,28,29), and its activity is stimulated by GTP under specified conditions (30). A number of recent studies have contributed towards establishing some insight into the mechanism of NS5B inhibition (31–35). This provides a platform for developing new inhibitors belonging either to the nucleoside analogue class (NI) that function as rNTP substrate mimics and block the elongation of new viral RNA strands or the nonnucleoside class of inhibitors (NNI), which inhibit the RdRp activity by an alternative mechanism (36,37).
Coumestans belong to the flavonoids category of phytoestrogens. Members of this family have been reported to possess diverse pharmacological properties such as antihemorrhagic, antiproteolytic and antiphospholipase activities (38). For a long time, traditional and herbal folk medicines have relied on naturally occurring coumestans from plants of the family Fabaceae against a variety of ailments. Wedelolactone, the naturally occurring active ingredient of herbal medicine derived from Eclipta prostrata and Wedelia calendulacea, has been extensively used in South American native medicine as snake antivenom (38). In traditional Chinese medicine, coumestans are used to treat septic shock and in Indian Ayurvedic medicine as a treatment for liver diseases, skin disorders and viral infections (39). Coumestans have been shown to reduce cancer risk (40), potentially due to their structural similarity to phytoestrogens. A series of coumestan derivatives were recently reported to inhibit the rat Na+, K+-ATPase activity and to bind to the GABAA receptors from the rat brain (41). In another study, wedelolactone and six coumestan analogues were reported to possess antihepatotoxic activity (42). More recently, wedelolactone has been shown to inhibit the NF-κB-mediated gene transcription in cells by blocking the phosphorylation and degradation of IκBα (43).
To date, the effect of coumestan class of compounds on any RdRp family of enzymes including HCV NS5B has not been investigated. In this study, we describe the identification and characterization of coumestans as a novel class of anti-NS5B agents. Based on structure-activity relationship (SAR) investigations on wedelolactone and four synthetic coumestan derivatives and computer modeling analysis to comprehend the SAR, we propose that this class of compounds may provide a new scaffold for developing potent anti-NS5B inhibitors.
MATERIALS AND METHODS
Materials
Reagents were purchased from the following sources: nickel-nitrilotriacetic acid (Ni-NTA) agarose and NAP-10 columns were from GE Health care; radiolabeled [α-32P] rNTPs were purchased from Perkin Elmer; HPLC-grade nucleoside triphosphate (NTP), RNase Out and glycogen were from Roche; GF-B filters from Whatman; anti-His antibody from Santa Cruz Biotechnology, Inc. and T4 polynucleotide kinase was obtained from Invitrogen. All other chemicals were of the highest available molecular biology grade and purchased from Fisher, Sigma or Bio-Rad.
Synthesis of inhibitors
Wedelolactone (7-methoxy-5,11,12-trihydroxy-coumestan) was purchased from EMD Chemicals Inc. and stored as 50 mM stock in 100% dimethylsulfoxide (DMSO). The coumestan analogues LQB16, LQB34 (PCALC36), LQB93 and LQB96 were synthesized as described previously (41,44). The purity of the synthesized coumestan derivatives was >95% as assessed by 1H NMR and 13C NMR spectroscopy. These analogues were dissolved in 100% DMSO as a 30 mM stock. All compounds were stored at −20°C for not more than 2 weeks. Serial dilutions of the inhibitors were made in DMSO immediately prior to the assay such that the final concentration of DMSO in all reactions was constant at 10%.
Purification of recombinant HCV NS5BCΔ21
Plasmid pThNS5BCΔ21 was transformed in E. coli DH5α and used for purification of HCV NS5BCΔ21 (45,46). This plasmid carries a hexahistidine tag (His-Tag) at the N-terminus of NS5BNIH1b strain and lacks the C-terminal 21 amino acid membrane-spanning domain. Purification was carried out following the method of Oh et al. with minor modifications (45). Briefly, the protein was induced at 25°C for 16 h by addition of 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The sonicated cell lysates were clarified by centrifugation at 30 000g for 45 min at 4°C, chromatographed on a Ni-NTA column (GE Health care) and washed in succession with three different buffers: NWB1 (50 mM sodium phosphate [pH 8.0], 10 mM beta mercaptoethanol, 20 mM imidazole, 2 M NaCl, 2% Nonidet NP-40 and 10% glycerol), NWB2 (similar to buffer NWB1 except that it contains 1 M NaCl and 1% Nonidet NP-40), and NWB3 (similar to buffer NWB1 except that it contains 100 mM NaCl and no NP-40) as described by Vo et al. (46). The bound protein was eluted in 1 mL fractions with NEB buffer (50 mM sodium phosphate [pH 8.0], 10 mM beta mercaptoethanol, 100 mM NaCl, 10% glycerol and 300 mM imidazole) and monitored by the Bradford colorimetric assay. The purity of NS5BCΔ21 was determined by Coomassie-stained SDS-PAGE analysis. Fractions enriched in NS5BCΔ21 (>95% purity) were pooled and dialysed against buffer A (50 mM Tris–HCl [pH 8.0], 1 mM dithiothreitol (DTT), 100 mM NaCl, 5 mM MgC12 and 50% glycerol), divided into aliquots and stored at −80°C.
Purification of Klenow polymerase and HIV-1 RT
Recombinant Klenow fragment of E. coli DNA polymerase I was purified from an overproducing exonuclease deficient clone (pET-3a-K) expressed in E. coli BL-21 (DE-3) by ammonium sulfate fractionation and Biorex-70 column chromatography as described before (47). Enzyme stocks were stored in aliquots at −20°C in buffer I (50 mM Tris–HCl [pH 7.0], 1 mM DTT, 100 mM NaCl and 50% glycerol).
Two recombinant plasmids, pET-28a-RT66 and pET-28a-RT51 encoding p66 and p51 subunits of HIV-1 RT, respectively, with metal binding His-Tag sequences at their N-terminal region were used for isolating wild-type heterodimeric HIV-1 RT by Ni-NTA chromatography as described before (48). The purified enzyme preparation was found to be greater than 95% pure as judged by SDS-PAGE and was stable at −20°C for several months.
NS5B RdRp assay
The effect of coumestan on the RdRp activity of NS5B was evaluated by the standard primer-dependent elongation reactions employing synthetic homopolymeric template-primers (TP) according to previously described procedures with some modification (24,47). Unless otherwise specified, NS5B and RNA TP were incubated on ice for 5 min before the addition of NTPs. Enzymatic reaction mixtures containing 20 mM Tris–HCl (pH 7.0), 100 mM NaCl, 100 mM sodium glutamate, 0.5 mM DTT, 0.01% BSA, 0.01% Tween-20, 5% glycerol, 20 U/mL of RNase Out, 0.5 μM of poly rA/U12, 25 μM UTP, 2–5 μCi [α-32P]UTP, 300–500 ng of NS5BCΔ21 and 0.5 mM MnCl2 with or without inhibitors in a total volume of 25 μl were incubated for 1 h at 30°C. The concentration of DMSO in all reactions was maintained constant at 10%. Reactions were terminated by the addition of ice-cold 5% (v/v) trichloroacetic acid (TCA) containing 0.5 mM pyrophosphate. The quenched reaction mixtures were incubated at −20°C for 1 h to precipitate out denatured polymeric RNA products, transferred to GF-B filters, washed twice with 5% (v/v) TCA containing 0.5 mM pyrophosphate to remove unincorporated UTP, and rinsed three times with water and once with ethanol before vacuum drying (47). The amount of radioactive UMP incorporated into RNA products was quantified on a liquid scintillation counter (Packard). Activity of NS5B in the absence of the inhibitor but containing an equivalent amount of DMSO (control reaction) was set at 100% and that in the presence of the inhibitor was quantified relative to this control. The concentration of coumestans inhibiting 50% of NS5B RdRp activity (IC50) were calculated from the inhibition curves as a function of inhibitor concentration and values obtained represent an average of at least two independent measurements. N,N-disubstituted phenylalanine derivative # 14, a documented NS5B inhibitor (33), was included as an internal standard in each set of experiments.
Reactions involving pre-incubation conditions were designed as follows: in one set, coumestans were added to the preformed NS5B-TP complex (45 min at 4°C) and incubated further on ice for 5 min. In a second set, NS5B-compound complex (45 min at 4°C) was preformed prior to the addition of the TP. In a third set, TP-coumestan complex (45 min at 4°C) was preformed prior to the addition of NS5B. In all cases, reactions were initiated by the addition of a mixture of rNTPs, incubated at 30°C for 1 h and quenched by the addition of 5% (v/v) TCA containing 0.5 mM pyrophosphate. Product formation was determined by GF-B filter binding assay as described above. For each set, the efficacy of the inhibitor under modified order of reactions components was evaluated (49,50).
HIV-1 RT and Klenow enzymatic assay
The specificity of the coumestans as inhibitors of HCV NS5B was evaluated through HIV-1 RT and Klenow polymerase counter screen employing standard methods as described before (47,48) with slight modification. In brief, assays were carried out in a final volume of 25 μl in a reaction mixture containing 50 mM Tris–HCl [pH 7.8], 1 mM DTT, 0.01% BSA, 50 mM KCl, 200 nM poly rA/dT18, 25 μM dTTP, 1 μCi [α-32P]dTTP, 10 mM MgCl2 and 10 nM enzyme in the absence or presence of the inhibitor. The concentration of DMSO in all reactions was kept constant at 10%. Incubations were carried out at 37°C for 20 min, and reactions were terminated by the addition of ice-cold TCA (5%) containing 0.5 mM pyrophosphate. The reaction mixtures were filtered through GF-B filters, and radioactivity in the acid insoluble fraction was determined by scintillation counting.
Gel-based incorporation assay
The incorporation of UMP on poly rA/U12 TP by NS5BCΔ21 at varying compound concentrations was evaluated by gel analysis of products essentially as described above for the HCV RdRp assay except that the elongation time was reduced to 20 min. Reactions were stopped by the addition of 25 mM EDTA and 0.5% SDS. The RNA products recovered by phenol-chloroform extraction and ethanol precipitation were dissolved in formamide gel loading buffer (51). The samples were heated at 90°C for 5 min and approximately one-fifth of the RNA products were resolved on a denaturing 6% polyacrylamide gel containing 7 M urea. The extent and pattern of synthesis were visualized by phosphorimager analysis and quantified using ImageQuant software (Molecular Dynamics).
Cross-linking of NS5B to TP
For cross-linking of NS5B to RNA, we engaged a synthetic rA20/U12 TP. The U12-primer was 5′-end-labeled with [γ-32P] ATP using T4 polynucleotide kinase, purified on a NAP-10 column as suggested by the manufacturer, adjusted to the required specific radioactivity with unlabeled primer and annealed with equimolar concentrations of unlabeled rA20 template. Cross-linking reactions as a function of inhibitor concentration were carried out as described earlier (47). The reaction mixture, in a final volume of 50 μl contained 20 mM Hepes (pH 7.0), 50 mM NaCl, 0.5 mM DTT, 0.01% BSA, 5% (v/v) glycerol, 20 U/ml of RNase Out, 200 nM of 32P-labeled U12/rA20 (15 000 c.p.m./pmol), 1.5 μg of NS5BCΔ21 and 0.5 mM MnCl2. Reactions were incubated on ice for 10 min in the absence or presence of the coumestans and were exposed to 254 nm UV irradiation at a dose of 300 mJ/cm2 (Spectronic Corporation). The cross-linked species were resolved by SDS-PAGE (8%). The dried gel was analysed on a phosphorimager and the extent of cross-linking was quantified using ImageQuant software.
Determination of the constant(s) of inhibition (Ki) and mode of inhibition
The mode of inhibition by the coumestan derivatives was evaluated essentially in two series of experiments as described by McKercher et al. (52). The first series of experiments was performed by varying the TP (poly rA/U12) and inhibitor concentrations. The concentration of poly rA/U12 ranged from 0.2 to 2.5 μM with a fixed concentration of UTP at 25 μM. In the second set of experiments, reaction velocities were determined at a fixed concentration of poly rA/U12 (0.25 μM) and varying concentrations of inhibitor and UTP (ranging from 2.5 to 80 μM). In both series of experiments, the concentration of inhibitors ranged 0.25–4 times their IC50 value. Assays were carried out in the standard RdRp buffer described above containing 300–500 ng of NS5BCΔ21 and 3–6 μCi [α-32 P] UTP. Aliquots were withdrawn at defined times and terminated by quenching with 5% TCA containing 0.5 mM pyrophosphate. The extent of incorporation in each set was determined by GF-B filter binding assay. Kinetic results were then plotted according to the methods of Dixon and Cornish-Bowden (53) in order to determine the mode of inhibition and the Ki. Kinetic experiments were performed at least twice and values are averages of at least triplicate samples. Standard deviations for all Ki were <10%.
Molecular modeling
All computations were carried out on a Dell Precision 470n workstation with the RHEL 4.0 operating system using Glide 4.5 (Schrodinger LLC). For docking experiments all the compounds were constructed using the fragment dictionary of Maestro 8.0 and geometry optimized using the Optimized Potentials for Liquid Simulations-all atom (OPLS-AA) force field (54) with the steepest descent followed by truncated Newton conjugate gradient protocol as implemented in Macromodel 9.5. Water molecules of crystallization were removed from the complex, and the protein was optimized for docking using the protein preparation wizard provided by Schrodinger LLC and the Impact program (First Discovery v4.5). Partial atomic charges for compounds as well as protein were assigned according to the OPLS-AA force field. The extra precision (XP) Glide docking method as described elsewhere (55–58), was then applied to dock the coumestans into the HCV NS5B NNI binding sites for tetracyclic indole (PDB ID: 2DXS) (59), N,N-disubstituted phenylalanine (PDB ID: 1NHU) (35) and benzothiadiazine (PDB ID: 2FVC) (60) inhibitors. Molecular dynamic (MD) simulations were carried out on the NS5B-LQB34 complex (obtained by docking LQB34 at the NS5B NNI binding site for tetracyclic indole) using the OPLS-AA force field and a generalized Born/solvent-accessible surface area (GB/SA) implicit water solvent model with a dielectric constant of 78. The complex was subjected to 1000 ps MD simulations at 300 K with an integration step of 1 fs. System coordinates were saved every 2 ps for further analysis. 3D structures and trajectories were visually inspected using the Maestro graphical interface. Root-mean-square (r.m.s) deviations from the initial structures were calculated using superposition option in Maestro. An average structure obtained from the last 500 ps of MD simulations was refined by means of 1000 steps of steepest descent followed by conjugate gradient energy minimization. Conjugate gradient energy minimizations were performed four times using the positional restraints to all heavy atoms with 1000, 500, 100 and 0 kJ/mol Å2 force constants in sequence. The maximum number of cycles of minimization was 5000 and the convergence criterion for the energy gradient was 0.001 kJ/mol Å.
RESULTS
Expression and purification of recombinant HCV NS5BCΔ21
To investigate the influence of selected coumestan family of compounds on the RdRp activity of NS5B, we employed the N-terminal His-tagged HCV NS5B polymerase (genotype 1b), lacking the C-terminal 21-amino acid membrane-spanning domain (NS5BCΔ21). Deletion of the last 21 hydrophobic residues of NS5B has been reported to enhance protein solubility (20), without compromising its kinetic properties or its ability to perform both de novo and primer-initiated RNA synthesis (51,61). We expressed the recombinant NS5BCΔ21 in bacteria (Figure 1, panel A), and purified it by Ni-NTA affinity chromatography. In the final step, the protein was batch eluted in 1 ml fractions with 300 mM imidazole buffer. To ascertain the purity and homogeneity of the eluted NS5BCΔ21, aliquots of eluted fractions were analysed by Coomassie blue staining of the SDS-PAGE prior to pooling the fractions. Eluates with the highest purity (>95%) were pooled, dialysed and stored in aliquots at −80°C. The purified NS5BCΔ21 corresponding to a single band of 64 kDa as visualized on a Coomassie blue-stained SDS-PAGE (Figure 1, panel B), and analysed by western blot with anti-His antibody (Figure 1, panel C) was used for all the experiments described in this study. The RdRp activity and integrity of the stored aliquots of NS5BCΔ21 was found to remain intact over a 2-year period.
Figure 1.

Purifications of recombinant NS5B. Recombinant NS5BCΔ21 with N-terminal 6X Histidine-epitope tag was purified from lysates of E. coli DH5α harboring the NS5BCΔ21 expression plasmid pThNS5BCΔ21. The cells grown at 25°C were induced with IPTG (200 μM). An aliquot of the induced culture was analysed by SDS-PAGE and visualized by Coomassie blue staining. Panel A depicts the induction profile of NS5BCΔ21 (indicated by asterisk). Clarified lysates of the induced culture were purified by nickel affinity column chromatography, eluted with 300 mM imidazole containing NEB buffer and dialysed overnight against buffer A (50 mM Tris–HCl [pH 8.0], 1 mM DTT, 100 mM NaCl, 5 mM MgC12 and 50% glycerol). The purified protein corresponding to a single band of 64 kDa is shown in panel B. Panel C shows the immunoblot of purified NS5BCΔ21 employing anti-His probe H-3 antibody (Santa Cruz Biotechnology).
Identification of coumestans as inhibitors of HCV NS5B RdRp activity
In a search for novel HCV NS5B inhibitors, we screened the coumestan class of phytoestrogens for their inhibitory potency against HCV NS5BCΔ21. The assay consisted of an in vitro nucleotide incorporation assay on a homopolymeric poly rA/U12 TP by a functionally active recombinant NS5BCΔ21 enzyme. This culminated in the identification of wedelolactone, a naturally occurring coumestan with a prototype coumestan scaffold (42), as an inhibitor of HCV NS5B with an IC50 of 36.1 μM (Table 1). A positive control consisted of NS5B in the absence of compound but containing equivalent amount of DMSO (10%). N,N-disubstituted phenylalanine derivative # 14, included as an internal reference standard in this investigation yielded an IC50 value of 0.3 μM versus the reported IC50 = 0.7 μM under our experimental condition. This variance in IC50 value may be attributed to the full-length recombinant NS5B expressed from baculovirus-infected Sf9 insect cells and a Flash-Plate scintillation proximity assay employed by Chan et al. (33). The specificity of wedelolactone as inhibitor of HCV NS5B polymerase was confirmed by HIV-1 RT (IC50 > 250 μM) and Klenow polymerase (IC50 > 700 μM) counter screen.
Table 1.
Structure–activity relationship of the Coumestan analogues
| Compounds | Structure | NS5B inhibition (IC50 μM) | Log P | Gscorea | Gscoreb |
|---|---|---|---|---|---|
| Wedelolactone | 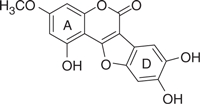 |
36.1 | 0.87 | −7.34 | −6.71 |
| LQB16 | 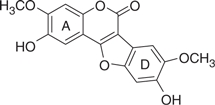 |
311 | 1.70 | −4.61 | −4.72 |
| LQB34 | 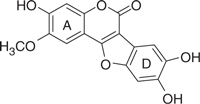 |
18.5 | 0.82 | −7.53 | −6.91 |
| LQB93 | 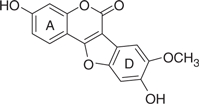 |
174 | 1.52 | −6.63 | −6.32 |
| LQB96 | 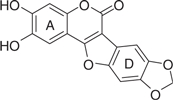 |
63.8 | 0.98 | −6.91 | −6.28 |
The concentration of DMSO in all reactions was kept constant at 10%. The IC50 values of the coumestan analogues were determined from dose–response curves using 8–12 concentrations for each compound in duplicate. Curves were fitted to data points and IC50 values were interpolated from the resulting curves using SigmaPlot 8.0 software. The values represent an average from at least two independent experiments. N,N-disubstituted phenylalanine derivative 14 (IC50 = 0.3 μM) was included as an internal reference standard. The predicted hydrophobicity of the coumestans was determined from their log P values employing QikProp 3.0 program (Schrodinger software package). Gscorea and Gscoreb values were determined by docking of coumestans into NS5B NNI binding site for tetracyclic indole (PDB ID: 2DXS) and N,N-disubstituted phenylalanine (PDB ID: 1NHU), respectively. A more negative Gscore indicates a better fit at the binding site.
Based on this preliminary investigation, we carried out SAR analysis and biological characterization of wedelolactone and four synthetic coumestan derivatives bearing different patterns of substitution on their rings A and D (Table 1). All the five coumestans tested inhibited the RdRp activity of NS5B, but with significant differences in their potency, with IC50 values that differed by up to more than one order of magnitude (Table 1). Of these, LQB34 and wedelolactone, bearing catechol group in their D-ring were found to be the most potent, inhibiting NS5B activity with IC50 values of 18.5 μM and 36.1 μM, respectively. This result suggests that change in the positions of the hydroxyl and methoxy groups in the A-ring of these coumestans did not substantially affect their anti-NS5B activity. Similarly, LQB96 bearing a less polar cyclic methylenedioxy group in the D-ring and a different pattern of oxygenation at the A-ring exhibited a slight decrease in its potency (2–3.5-fold) when compared to wedelolactone and LQB34. LQB16, on the other hand, carrying methylation of the phenol group in D-ring, in addition to inversion in the position of the hydroxyl and methoxy groups in A-ring as compared to LQB34, was the least potent of the five coumestans with a near 20-fold increase in its IC50 value. Interestingly, this dramatically decreased potency was improved by nearly two-fold with LQB93, bearing identical D-ring to LQB16, but carrying only the hydroxyl group in its A-ring. Together, these data suggest that the presence of the hydroxyl substituent in A-ring along with the catechol group in the D-ring may be important for the anti-NS5B efficacy of the coumestans.
Gel-based incorporation assay
To determine the effect of the coumestan compounds on NS5BCΔ21-mediated incorporation of rNTP substrates into a growing RNA strand, we carried out gel-based analyses of the reaction products. These assays are useful in analysing the nature and pattern of the product synthesized by polymerases family of enzymes and have been reported by several groups in context of NS5B-inhibition mechanism on a variety of TP (34,49,52). In our studies, we employed the poly rA/U12 TP, and monitored incorporation of radiolabeled [α-32P] UTP substrate by NS5BCΔ21 in the presence of increasing concentrations of the coumestans. The products were resolved on a 6% denaturing urea gel and visualized by phosphorimaging. As seen in Figure 2, reactions reconstituted with increasing amounts of wedelolactone or LQB34 inhibited full-length product formation as a function of inhibitor concentration (Figure 2, lanes 2–5). The reduction in product formation was higher in case of LQB34 compared to wedelolactone at similar concentrations of the inhibitor and is in agreement with their IC50 values. The reactions containing inhibitors did not appear to produce shorter products as a result of abortive initiation or premature termination. As expected, no detectable extension product was visible at the highest concentration of wedelolactone (Figure 2, lane 5).
Figure 2.

Effect of coumestan derivatives on the RNA polymerase activity of NS5B. The RdRp activity of NS5BCΔ21 at increasing concentrations of the indicated compounds was determined in a standard RdRp assay containing poly rA/U12 as the TP and Mn2+ as the divalent cation at 30°C for 20 min. The RNA product synthesized was resolved on a 6% denaturing urea gel and visualized by phosphorimaging. Lane 1 in each set represents the control reaction carried out in the absence of the coumestan compound. Panel A: lanes 2–5 represent wedelolactone concentrations of 10, 25, 50 and 100 μM. Panel B: lanes 2–5 indicate LQB34 concentrations of 1, 5, 10, 25 and 50 μM, respectively. The position of the U12-primer is indicated by an arrow to the left of the gel.
Mode of inhibition
In order to gain insight into the mechanism of NS5B inhibition by the three most potent coumestans, we conducted two different series of kinetic analyses of enzyme inhibition. In the first series, reaction velocities were determined under conditions of varying concentrations of TP and inhibitor in the presence of a constant amount of UTP (Figure 3). In the second series, the concentrations of UTP and inhibitor were varied in the presence of a constant amount of TP (Figure 4). The data obtained were analysed by Dixon and Cornish-Bowden plots to determine the mode of inhibition and evaluate the Ki values. It is clear from Figure 3 that the three coumestan compounds displayed a mixed mode of inhibition towards the TP, with a significant competitive component (intercept on the Dixon plot above the x-axis) and a minor uncompetitive component (intercept extrapolated from the Cornish-Bowden plot below the x-axis) which corresponded to Ki competitive values of 11.2, 4.5 and 42.6 μM and Ki uncompetitive values of 43, 37.5 and 342.3 μM for wedelolactone, LQB34 and LQB96, respectively. In contrast, the mode of inhibition by all three coumestans emerged to be noncompetitive with respect to UTP as seen in Figure 4, with distinct location of the intercept on the x-axis in both the Dixon and the Cornish-Bowden plots. The kinetic constants determined from these plots corresponded to Ki values of 8.4, 4.2 and 42 μM for wedelolactone, LQB34 and LQB96, respectively. In all cases, the Ki constants correlated with the IC50 values of the respective coumestans.
Figure 3.

Ki determination of coumestan compounds and mode of inhibition with regard to template/primer substrate. Reaction velocities were measured at varying template/primer concentrations (0.2, 0.5, 0.7, 1.0, 1.5 and 2.3 μM plotted as closed circles, open circles, closed triangles, open triangles, closed squares and open squares, respectively) with fixed concentration of UTP (25 μM). Data were analysed by the Dixon and Cornish-Bowden plots of the reciprocal velocity at indicated concentrations of the individual coumestan derivatives. All three coumestan derivatives displayed a mixed mode of inhibition towards template/primer with a major competitive component (intercept on the Dixon plot above the x-axis) and a minor uncompetitive component (intercept extrapolated from the Cornish-Bowden plot below the x-axis). The kinetic parameters corresponded to Ki competitive values of 11.2, 4.5 and 42.6 μM and Ki uncompetitive values of 43, 37.5 and 342.3 μM for wedelolactone, LQB34 and LQB96, respectively.
Figure 4.

Evaluation of inhibition kinetic parameters of coumestan compounds with regard to UTP substrate. Reaction velocities were measured at increasing concentrations of UTP substrate (2.5–80 μM) on poly rA-U12 template/primer (0.25 μM) in the absence or presence of increasing concentrations (5–140 μM) of the indicated inhibitor. Panels A, B and C depict the Dixon and Cornish-Bowden plots for wedelolactone, LQB34 and LQB96, respectively. Both plots display similar intercept profiles on the x-axis indicating a noncompetitive mode of inhibition with regard to UTP. The Ki values corresponded to 8.4, 4.2 and 42 μM for wedelolactone, LQB34 and LQB96, respectively.
Modified order of addition influences potency of coumestans
Given the mixed mechanistic mode towards the RNA template, we employed the modified order of reagent addition experiments to identify the mechanistic step at which this inhibition may be mediated. NS5B was pre-incubated with either inhibitor or RNA before adding the remaining components to initiate RNA synthesis. To exclude the possibility that inhibition may be mediated as a consequence of direct inhibitor binding to the RNA and precluding it from the RdRp reaction, we set up another series of reactions wherein the inhibitor was pre-incubated with the RNA prior to the addition of the other components. As seen in Figure 5, both wedelolactone and LQB34 exhibited a downward shift in their IC50 values upon pre-incubation with NS5B. Moreover, under these conditions, complete inhibition was gradually reached at higher inhibitor concentrations. Further, separation of wedelolactone from the NS5B-inhibitor pre-incubation complex on a Sephadex G25 column also decreased the RdRp activity of NS5B (data not shown), suggesting the direct binding of the inhibitor to NS5B. Conversely, NS5B-RNA pre-incubation prior to the addition of the inhibitor resulted in an upward shift of the IC50 values. This increase was approximately four-fold in case of LQB34 (from 18.5 to 70 μM) against a modest near two-fold increase seen with wedelolactone (36 to 80 μM). In this scenario, the inhibition curves for LQB34 and wedelolactone depicted an altered trend since a significant fraction of NS5B activity was not inhibited even at very high coumestan concentrations, this effect being more pronounced with LQB34. In addition, the observation that the IC50 values did not change upon pre-incubation of coumestans with the RNA suggests that the inhibitor did not bind the RNA. Cumulatively, the pre-incubation data is consistent with the kinetic experiments and mode of inhibition and suggests that the coumestans bind to NS5B directly and exert their inhibitory effect by interfering with an early step in the RdRp reaction such as productive RNA binding to the enzyme.
Figure 5.

NS5B inhibition by wedelolactone (A) and LQB34 (B). Dependence of inhibition curves on NS5B-RNA (TP + E), NS5B-Inhibitor (E + I) and RNA-Inhibitor (TP + I) complex formation was examined by pre-incubating the indicated components for 30 min at 4°C followed by evaluation of the NS5B RdRp activity as described in the text. UMP incorporation into product was expressed as percent of control (no inhibitor) and plotted against increasing concentrations of the indicated coumestan compounds.
RNA competes with coumestan for NS5B binding
Similar to other polymerases, NS5B requires multiple substrates, including a TP or a template-initiation nucleotide complex in a sequential fashion, in which the first step is formation of the binary complex involving binding of NS5B to the TP (62). We have earlier demonstrated the formation of this binary complex by polymerase-TP cross-linking experiments for the Klenow fragment of E. coli DNA polymerase I (47), MuLV-RT (63) and HIV-1 RT (48). The observation that pre-incubation of NS5B-RNA protected the enzyme from being inhibited by the coumestans, prompted us to ascertain whether the coumestans adversely affect the binding of the TP to NS5B. A direct photochemical cross-linking of 5′-32P-labeled U12/rA20 to NS5B was performed by UV irradiation of the NS5B-TP complexes in the presence of increasing amounts of coumestans under standard RdRp assay conditions. The extent of NS5B-TP covalent complex formed as a function of inhibitor concentration was analysed by SDS-PAGE. Results shown in Figure 6 indicate a reduction in the E-TP binary complex formation in the presence of the inhibitor. This reduction directly correlated with the inhibitor concentration (Figure 6, lanes 2–9) and is in agreement with the competitive mode of TP binding as well as the protective effect of RNA TP against coumestan-mediated inhibition of NS5B. Further, the reduction of NS5B-RNA binary complex species was higher in case of LQB34 compared to wedelolactone and correlated well with their respective IC50 values. The binary complex formation data substantiates our hypothesis that inhibition may be mediated at the RNA binding step among others.
Figure 6.

Coumestans inhibit NS5B-RNA binary complex formation. Binding of NS5B to RNA as a function of individual coumestan derivative was evaluated by UV-mediated cross-linking. NS5BCΔ21 (1.5 μg) was incubated with 30 nM [5′-32P] labeled rA20/U12 template/primer (200K Cerenkov c.p.m.) in a standard irradiation mixture in the absence or presence of increasing concentrations of the indicated coumestan compound and exposed to UV radiation. The cross-linked species were resolved by SDS-PAGE and visualized on a phosphorimager. The extent of NS5B-RNA cross-linked species formed was quantified using ImageQuant software (Molecular Dynamics). Lane 1 in each set represents the control reaction carried out in the absence of the inhibitor. Lanes 2–9 represent indicated compounds at concentrations of 1, 5, 10, 15, 30, 50, 100 and 200 μM, respectively.
Hydrophobicity of coumestans
Hydrophobicity assessment is a valuable index utilized during drug development since it serves as an excellent predictor of drug absorption, bioavailability and drug-receptor interactions. In order to examine the relationship between the coumestan-mediated inhibition of NS5B and the chemical properties of this group of compounds, we assessed the relationship between the calculated logP and IC50 values. The logP value represents the partition coefficient, which is a measure of differential solubility of a compound in octanol/water mixture. The logP values of the coumestan compounds were calculated using QikProp 3.0 program available in Schrodinger software package (Table 1) and plotted against their corresponding IC50 values (Figure 7). The derived logP values were least for LQB34 and wedelolactone and highest for LQB16. A plot of logP versus IC50 values of coumestans shows an inverse correlation between hydrophobicity and inhibitory potency of this class of compounds (Figure 7; P < 0.02, Spearman test). This observation may be predictive of a trend for less hydrophobic coumestans to be better inhibitors of NS5B and may assist in efforts to design better coumestan-based inhibitors.
Figure 7.

Correlation between hydrophobicity and potency of coumestans. The predicted hydrophobicity of the coumestans was determined from their logP values employing QikProp 3.0 program (Schrodinger software package) and plotted against their IC50 values to establish a correlation between the potency and hydrophobicity of the coumestans (P < 0.02, Spearman test).
Binding mode of the most potent coumestan (LQB34)
To further understand SAR and investigate the potential binding mode of the coumestan derivatives to HCV NS5B, we performed molecular modeling studies employing Glide docking software. To rule out any bias, each of the three reported HCV NS5B NNI binding site represented by tetracyclic indole (59), N,N-disubstituted phenylalanine (35) and benzothiadiazine (60) inhibitors was examined for coumestan binding. As a preliminary step, we validated the accuracy of our docking approach by determining how closely ‘the lowest energy pose (binding conformation)’ predicted by the object scoring function, Glidescore (Gscore) in our case, resembles the experimental binding mode as determined by X-ray crystallography. Towards this end, we removed the aforementioned crystallographic bound inhibitors from their binding sites and then re-docked them into their respective binding site on HCV NS5B polymerase. We found a good agreement between the localization of the inhibitor upon docking and from the crystal structure as is evident from the 0.827, 0.748 and 0.297 Å root mean square (rms) deviations for tetracyclic indole, N,N-disubstituted phenylalanine and benzothiadiazine inhibitors, respectively. This confirms the reliability of the Glide docking procedure in reproducing the experimentally observed binding mode for NS5B inhibitors. Therefore, the parameters set for Glide docking appear reasonable to provide meaningful insight into the predicted binding mode for coumestans.
Employing Glide molecular docking, we investigated the interactions of the coumestan analogues at the HCV NS5B NNI binding site and analysed the relationship between their calculated binding energies (Gscore) and IC50 values. As seen in Table 1, docking of coumestans into NS5B NNI binding site for tetracyclic indole (allosteric pocket 1) yielded an excellent correlation between their Gscores and IC50 values. LQB34, the best inhibitor of this series exhibited the most negative Gscore in comparison to the other analogues, indicating a better fit at the binding site, whereas LQB16, the least potent coumestan displayed the least negative Gscore, with other analogues displaying binding scores consistent with their IC50 values. A somewhat similar trend of overall agreement between the binding energies and IC50 values was also observed upon docking the coumestans at the NS5B NNI binding site for N,N-disubstituted phenylalanine (allosteric pocket 2), with the exception that LQB93 and LQB96 which exhibited a three-fold difference in their IC50 values showed near similar Gscores. Further comparison revealed that the coumestans exhibited a better fit in allosteric pocket 1 versus allosteric pocket 2, as deduced from the relatively more negative Gscores in the former pocket. In contrast, binding of coumestans into the NS5B benzothiadiazine-binding site (allosteric pocket 3) located adjacent to the active site was ruled out based on the poor Gscore values derived from the scoring function at this site (data not shown). Together this analysis suggests that allosteric pocket 1, represented by the NS5B NNI binding site for tetracyclic indole may be the potential site of coumestan binding.
To gain insight into the mechanism of inhibition by the coumestans, we analysed the interactions of the docked conformation of LQB34 within allosteric pocket 1 (Figure 8A and 8B). The benzofuran moiety of LQB34 was located in a deep hydrophobic pocket formed by residues Leu392, Ala395, Ala396, Ile424, Leu425 and His428 whereas its coumarin moiety interacts with Val494, Pro495 and Trp500 and was fairly solvent-exposed. Further, LQB34 was engaged in a series of electrostatic interactions across the entire shallow-binding pocket of NS5B. Notably, its 8-OH group forms hydrogen bond with the backbone of Leu392 whereas its 9-OH group was located at hydrogen bonding distances (3.37 Å) from the backbone of Ile424. Its benzofuran ring oxygen exhibited electrostatic contact with the His428 ring –NH function whereas the oxygen atom of its 2-OCH3 group was closely located (2.34 Å) to the guanidine function of Arg503 and possibly interacts through hydrogen bond. The predicted binding mode agrees well with the observed SAR. For instance, conversion of the 8-OH into 8-OCH3 group results in the loss of hydrogen bond interaction between the backbone of Leu392 of NS5B and the inhibitor, in addition to causing steric hindrance with the side chain of Ala395. This may account for the dramatically reduced inhibitory potency of LQB16 and LQB93 (Table 1). Removal of this steric hindrance by modification of 8- and 9-OH function into a methylenedioxy derivative such as in LQB96 reasonably improved the potency of the inhibitor. Thus, the predicted pose of LQB34 suggests that extension of the 8- and 9-OH function may be detrimental to the potency of the inhibitor.
Figure 8.

(A) Docked conformation of the most potent compound LQB34 in the HCV NS5B NNI binding site for tetracyclic indole (PDB ID: 2DXS) (54). Hydrogen bonds are shown as dotted yellow lines. Distances are shown as dotted pink lines. Active site amino acid residues are represented as sticks while the inhibitor is shown as ball and stick model. (B) Docked (ball and stick model) conformation of LQB34 overlaid within the Macromodel surface of the allosteric-binding site of NS5B. (C) Hydrophilic (green color) and hydrophobic (white color) surface area of the allosteric-binding site of NS5B (PDB ID: 2DXS) in the presence of the most potent compound LQB34.
DISCUSSION
Identification and characterization of several structural classes of NNIs of NS5B has previously been reported. In this study, we have identified the coumestan family of isoflavones as novel inhibitors of HCV NS5B RdRp activity in vitro. Coumestans occur naturally in a balanced diet, are readily bioavailable, and are not associated with any type of toxicity (64,65), thus making them potentially attractive candidates for development as clinically relevant agents.
The basic structure of the coumestan scaffold including its coumarin moiety can be modified chemically to enhance SAR, an aspect corroborated in the present investigation where LQB34 was the most potent (∼2 to 20-fold lower IC50 value) of the five coumestans tested. The variable potency of these compounds may be attributed to the different pattern of oxygenation on their A- and D-ring structures as is substantiated by our data, wherein LQB16 carrying methoxy groups on both rings A and D exhibited reduced anti-NS5B activity in contrast to wedelolactone and LQB34, the two most potent coumestans of this group, both of which harbored hydroxyl and catechol groups on their rings A and D, respectively. Not surprisingly, the anti-NS5B efficacy of these compounds inversely correlated with their hydrophobicity, thus providing an important physicochemical parameter to be considered for future optimization of coumestan structures.
A number of basic similarities were revealed in the mechanism of inhibition of coumestans with other characterized NNIs of HCV polymerase. Kinetic experiments yielded a noncompetitive mode of inhibition of the coumestan analogues towards nucleotide substrates, akin to representative compounds of the six chemically distinct scaffolds of NNIs (i) benzothiadiazine (31,60), (ii) benzylidene (49,66), (iii) benzimidazole (50,67), (iv) N,N-disubstituted phenylalanine (33), (v) dihydropyranone (68) and (vi) pyrano indole (69), all of which are noncompetitive inhibitors of NTP incorporation and reported to bind directly to NS5B polymerase. In the present study, we have no direct evidence linking coumestan binding to NS5B. However, the somewhat increased potency of these compounds upon pre-incubation with NS5B, in addition to the observation that the resolved NS5B-inhibitor complex demonstrated reduced RdRp activity, argues in favor of direct coumestan binding to NS5B.
In the modified order of addition experiments, the coumestans, like the benzylidenes (49) but contrary to the benzothiadiazines (70), exhibited decreased efficacy under conditions of NS5B-RNA pre-incubation. Hence, access to the coumestan-binding site may be partially occluded either directly by the presence of nucleic acid, or by conformational changes that follow RNA binding. Evidence for RNA-mediated protection of NS5B inactivation is also indicated by the steady state kinetic data wherein these analogues exhibited a mixed mode of inhibition towards the template/primer with a significant competitive component similar to that reported for the benzimidiazoles (52). We believe that the coumestans, similar to the benzylidene and benzimidazole series, inhibit NS5B at an early step during initiation of RNA synthesis, possibly concurrent with NS5B-RNA complex formation step. This speculation is consistent with our observation that no abortive or prematurely terminated products were observed in the primer-elongation reactions containing the coumestans. Given that NS5B like other polymerases follows an ordered mechanism of RNA synthesis, wherein nucleic acid substrate must first bind, followed by nucleotide substrate in order to form a productive polymerase complex, evidence that the two most active compounds, wedelolactone and LQB34, compete with the RNA/RNA TP in the cross-linking experiments, is certainly indicative of inhibition of the formation of a prepolymerase complex. Importantly, the decrease in formation of binary complex correlates well with the inhibition of polymerization activity, suggesting that these compounds inhibit NS5B by binding mostly the free enzyme rather than a preformed E-RNA/RNA complex. Hence, the coumestans are likely to bind at an allosteric site and decrease binding of RNA through an allosteric mechanism.
Crystallographic studies on NS5B in complex with allosteric inhibitors have suggested the presence of a relatively shallow-binding site (59). A shallow-binding pocket facilitates access to the binding partner (in our case coumestans) and enhances the on-rate. However, a shallow interface will also increase access of free water molecules, which would undermine the stability of the partnering complex unless the complex is stabilized by other means. In order to design tight-binding inhibitors for this site of NS5B, it is crucial to evaluate the advantages and disadvantages of the shallow pocket. For example, NS5B allosteric-binding site is predominantly solvent-exposed, thus for an inhibitor to approach and bind to this site requires significant displacement of solvent molecules while the inhibitor itself can simultaneously undergo predominant desolvation upon moving from bulk to the shallow-binding pocket. In case of coumestans, the coumarin moiety at the binding site remains solvent-exposed thereby reducing desolvation penalty to a minimum. Displacement of water molecules from the binding site usually leads to increase in entropy, provided lost protein-water contacts are reestablished by the inhibitor. We propose that the two hydroxyl groups present in the D-ring form hydrogen bonding network with the binding site residues. Such interactions can compensate for the lost water-protein interactions. Our docking results have shown that free coumestans do not undergo conformational change upon binding to NS5B due to the lack of rotatable bonds (except –OCH3 groups) and therefore do not loose entropy thereby reflecting a strong correlation between their inhibitory activities and calculated binding energies (Gscore).
Considering protein flexibility in solution, the behavior of the predicted complex of the most potent inhibitor LQB34 was studied in a dynamic context. MD simulations yielded a stable complex reinforcing the validity of the Gscore. The superposition of the coordinates of an energy-minimized average structure obtained from the last 250 trajectories, onto the starting complex yielded r.m.s.d values of 1.430 and 1.210 Å for all atom-based and ligand atom-based superposition, respectively. The r.m.s.d value of 1.430 Å for all atoms of the NS5B-LBQ34 complex suggests that NS5B undergo conformational changes which might affect the RNA binding to the enzyme upon coumestan binding. The stability of the hydrogen bonding network predicted by Glide XP docking method was examined by monitoring the percentage occurrence of predicted hydrogen bonds during simulation time. Analysis of the MD trajectory suggested the occurrence of Glide-predicted hydrogen bond in 85% of the complexes. The new hydrogen bond (9-OH- - - -O Ile424) was predicted in 27% of the complexes. A detailed investigation on the binding modes of coumestan inhibitors to NS5B followed by MD simulations is likely to provide useful guideline to design future inhibitors with enhanced activity.
The binding mode of LQB34 suggests ways to further improve upon potency of coumestan derivatives. For example, the C-7 position of the benzofuran moiety can be substituted to fit the narrower hydrophobic pocket formed by Val37, Leu392, Ala393, Leu492 and Val494 (Figure 8B). Structural studies have shown that Arg503 stabilizes the NS5B NNI such as benzimidazole/indole-based inhibitors (59). Hence, a –COOH modification of the C-2 methoxy group located in proximity of Arg503 would allow a favorable charge–charge interaction for binding. In addition, the 3-hydroxy group of LQB34, which is not in direct contact with NS5B, can be modified to attain desired pharmacokinetic profile. Also, modification of coumestans’ benzofuran and/or coumarin core structure and peripheral hydroxyl and/or methoxy groups to better accommodate the hydrophobic/hydrophilic surface map of NS5B binding pocket (Figure 8C) may prove instrumental for further SAR studies on coumestan derivatives.
In conclusion, we have identified and described the mechanism of inhibition of a new series of NNIs belonging to the coumestan family. Coumestans represent novel structural templates of anti-NS5B inhibitors which can be further modified into very potent drugs against HCV through the application of molecular modeling and SAR analyses. Studies are underway to develop and screen other coumestan derivatives and evaluate their efficacy in HCV replicon system.
ACKNOWLEDGEMENTS
We thank Dr Spencer Knapp for providing N,N-disubstituted phenylalanine derivative # 14 and Dr Ye Chen for help in preparing the figures and the reference library. This research was supported by National Institute of Health (NIH) research grant R01-DK066837 to NKB. TT was supported by start-up funds and resources provided by the College of Pharmacy and the Department of Pharmaceutical Sciences, St John's University, NY. Funding to pay the Open Access publication charges for this article was provided by National Institute of Health research grant R01-DK066837.
Conflict of interest statement. None declared.
REFERENCES
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Seeff LB. Natural history of hepatitis C. Am. J. Med. 1999;107:10S–15S. doi: 10.1016/s0002-9343(99)00374-5. [DOI] [PubMed] [Google Scholar]
- 3.Di Bisceglie AM, Order SE, Klein JL, Waggoner JG, Sjogren MH, Kuo G, Houghton M, Choo QL, Hoofnagle JH. The role of chronic viral hepatitis in hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 1991;86:335–338. [PubMed] [Google Scholar]
- 4.WHO. Global surveillance and control of hepatitis C. Report of a WHO consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J. Viral Hepat. 1999;6:35–47. [PubMed] [Google Scholar]
- 5.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann. Intern. Med. 2006;144:705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 6.Myles DC. Recent advances in the discovery of small molecule therapies for HCV. Curr. Opin. Drug Discov. Devel. 2001;4:411–416. [PubMed] [Google Scholar]
- 7.Dillon JF. Hepatitis C: what is the best treatment? J. Viral Hepat. 2004;11(Suppl. 1):23–27. doi: 10.1111/j.1365-2893.2004.00573.x. [DOI] [PubMed] [Google Scholar]
- 8.Ni ZJ, Wagman AS. Progress and development of small molecule HCV antivirals. Curr. Opin. Drug Discov. Devel. 2004;7:446–459. [PubMed] [Google Scholar]
- 9.Lauer GM, Walker BD. Hepatitis C virus infection. N. Engl. J. Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 10.Cornberg M, Huppe D, Wiegand J, Felten G, Wedemeyer H, Manns MP. Treatment of chronic hepatitis C with PEG-interferon alpha-2b and ribavirin: 24 weeks of therapy are sufficient for HCV genotype 2 and 3. Z. Gastroenterol. 2003;41:517–522. doi: 10.1055/s-2003-39649. [DOI] [PubMed] [Google Scholar]
- 11.Ideo G, Bellobuono A. New therapies for the treatment of chronic hepatitis C. Curr. Pharm. Des. 2002;8:959–966. doi: 10.2174/1381612024607009. [DOI] [PubMed] [Google Scholar]
- 12.Reichard O, Norkrans G, Fryden A, Braconier JH, Sonnerborg A, Weiland O. Randomised, double-blind, placebo-controlled trial of interferon alpha-2b with and without ribavirin for chronic hepatitis C. The Swedish study group. Lancet. 1998;351:83–87. doi: 10.1016/s0140-6736(97)06088-1. [DOI] [PubMed] [Google Scholar]
- 13.Arataki K, Kumada H, Toyota K, Ohishi W, Takahashi S, Tazuma S, Chayama K. Evolution of hepatitis C virus quasispecies during ribavirin and interferon-alpha-2b combination therapy and interferon-alpha-2b monotherapy. Intervirology. 2006;49:352–361. doi: 10.1159/000095155. [DOI] [PubMed] [Google Scholar]
- 14.Farci P, Quinti I, Farci S, Alter HJ, Strazzera R, Palomba E, Coiana A, Cao D, Casadei AM, Ledda R, et al. Evolution of hepatitis C viral quasispecies and hepatic injury in perinatally infected children followed prospectively. Proc. Natl Acad. Sci. USA. 2006;103:8475–8480. doi: 10.1073/pnas.0602546103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pawlotsky JM. Hepatitis C virus population dynamics during infection. Curr. Top. Microbiol. Immunol. 2006;299:261–284. doi: 10.1007/3-540-26397-7_9. [DOI] [PubMed] [Google Scholar]
- 16.Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 2000;242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 17.De Francesco R, Tomei L, Altamura S, Summa V, Migliaccio G. Approaching a new era for hepatitis C virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral Res. 2003;58:1–16. doi: 10.1016/s0166-3542(03)00028-7. [DOI] [PubMed] [Google Scholar]
- 18.Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 19.Adachi T, Ago H, Habuka N, Okuda K, Komatsu M, Ikeda S, Yatsunami K. The essential role of C-terminal residues in regulating the activity of hepatitis C virus RNA-dependent RNA polymerase. Biochim. Biophys. Acta. 2002;1601:38–48. doi: 10.1016/s1570-9639(02)00433-8. [DOI] [PubMed] [Google Scholar]
- 20.Ferrari E, Wright-Minogue J, Fang JW, Baroudy BM, Lau JY, Hong Z. Characterization of soluble hepatitis C virus RNA-dependent RNA polymerase expressed in Escherichia coli. J. Virol. 1999;73:1649–1654. doi: 10.1128/jvi.73.2.1649-1654.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita T, Kaneko S, Shirota Y, Qin W, Nomura T, Kobayashi K, Murakami S. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J. Biol. Chem. 1998;273:15479–15486. doi: 10.1074/jbc.273.25.15479. [DOI] [PubMed] [Google Scholar]
- 22.Yuan ZH, Kumar U, Thomas HC, Wen YM, Monjardino J. Expression, purification, and partial characterization of HCV RNA polymerase. Biochem. Biophys. Res. Commun. 1997;232:231–235. doi: 10.1006/bbrc.1997.6249. [DOI] [PubMed] [Google Scholar]
- 23.Ishii K, Tanaka Y, Yap CC, Aizaki H, Matsuura Y, Miyamura T. Expression of hepatitis C virus NS5B protein: characterization of its RNA polymerase activity and RNA binding. Hepatology. 1999;29:1227–1235. doi: 10.1002/hep.510290448. [DOI] [PubMed] [Google Scholar]
- 24.Tomei L, Vitale RL, Incitti I, Serafini S, Altamura S, Vitelli A, De Francesco R. Biochemical characterization of a hepatitis C virus RNA-dependent RNA polymerase mutant lacking the C-terminal hydrophobic sequence. J. Gen. Virol. 2000;81:759–767. doi: 10.1099/0022-1317-81-3-759. [DOI] [PubMed] [Google Scholar]
- 25.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure. 1999;7:1417–1426. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 26.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl Acad. Sci. USA. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 28.Luo G, Hamatake RK, Mathis DM, Racela J, Rigat KL, Lemm J, Colonno RJ. De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus. J. Virol. 2000;74:851–863. doi: 10.1128/jvi.74.2.851-863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong W, Ferrari E, Lesburg CA, Maag D, Ghosh SK, Cameron CE, Lau JY, Hong Z. Template/primer requirements and single nucleotide incorporation by hepatitis C virus nonstructural protein 5B polymerase. J. Virol. 2000;74:9134–9143. doi: 10.1128/jvi.74.19.9134-9143.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohmann V, Overton H, Bartenschlager R. Selective stimulation of hepatitis C virus and pestivirus NS5B RNA polymerase activity by GTP. J. Biol. Chem. 1999;274:10807–10815. doi: 10.1074/jbc.274.16.10807. [DOI] [PubMed] [Google Scholar]
- 31.Dhanak D, Duffy KJ, Johnston VK, Lin-Goerke J, Darcy M, Shaw AN, Gu B, Silverman C, Gates AT, Nonnemacher MR, et al. Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 2002;277:38322–38327. doi: 10.1074/jbc.M205566200. [DOI] [PubMed] [Google Scholar]
- 32.Gu B, Johnston VK, Gutshall LL, Nguyen TT, Gontarek RR, Darcy MG, Tedesco R, Dhanak D, Duffy KJ, Kao CC, et al. Arresting initiation of hepatitis C virus RNA synthesis using heterocyclic derivatives. J. Biol. Chem. 2003;278:16602–16607. doi: 10.1074/jbc.M210891200. [DOI] [PubMed] [Google Scholar]
- 33.Chan L, Reddy TJ, Proulx M, Das SK, Pereira O, Wang W, Siddiqui A, Yannopoulos CG, Poisson C, Turcotte N, et al. Identification of N,N-disubstituted phenylalanines as a novel class of inhibitors of hepatitis C NS5B polymerase. J. Med. Chem. 2003;46:1283–1285. doi: 10.1021/jm0340400. [DOI] [PubMed] [Google Scholar]
- 34.Carroll SS, Tomassini JE, Bosserman M, Getty K, Stahlhut MW, Eldrup AB, Bhat B, Hall D, Simcoe AL, LaFemina R, et al. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J. Biol. Chem. 2003;278:11979–11984. doi: 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Ng KK, Cherney MM, Chan L, Yannopoulos CG, Bedard J, Morin N, Nguyen-Ba N, Alaoui-Ismaili MH, Bethell RC, et al. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 2003;278:9489–9495. doi: 10.1074/jbc.M209397200. [DOI] [PubMed] [Google Scholar]
- 36.Beaulieu PL, Tsantrizos YS. Inhibitors of the HCV NS5B polymerase: new hope for the treatment of hepatitis C infections. Curr. Opin. Investig. Drugs. 2004;5:838–850. [PubMed] [Google Scholar]
- 37.Walker MP, Yao N, Hong Z. Promising candidates for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs. 2003;12:1269–1280. doi: 10.1517/13543784.12.8.1269. [DOI] [PubMed] [Google Scholar]
- 38.Mors WB, do Nascimento MC, Parente JP, da Silva MH, Melo PA, Suarez-Kurtz G. Neutralization of lethal and myotoxic activities of South American rattlesnake venom by extracts and constituents of the plant Eclipta prostrata (Asteraceae) Toxicon. 1989;27:1003–1009. doi: 10.1016/0041-0101(89)90151-7. [DOI] [PubMed] [Google Scholar]
- 39.Singh B, Saxena AK, Chandan BK, Agarwal SG, Anand KK. In vivo hepatoprotective activity of active fraction from ethanolic extract of Eclipta alba leaves. Indian J. Physiol. Pharmacol. 2001;45:435–441. [PubMed] [Google Scholar]
- 40.Horn-Ross PL, Barnes S, Lee M, Coward L, Mandel JE, Koo J, John EM, Smith M. Assessing phytoestrogen exposure in epidemiologic studies: development of a database (United States) Cancer Causes Control. 2000;11:289–298. doi: 10.1023/a:1008995606699. [DOI] [PubMed] [Google Scholar]
- 41.da Silva AJM, Melo PA, Silva NM, Brito FV, Buarque CD, de Souza DV, Rodrigues VP, Pocas ES, Noel F, Albuquerque EX, et al. Synthesis and preliminary pharmacological evaluation of coumestans with different patterns of oxygenation. Bioorg. Med. Chem. Lett. 2001;11:283–286. doi: 10.1016/s0960-894x(00)00621-1. [DOI] [PubMed] [Google Scholar]
- 42.Wagner H, Geyer B, Kiso Y, Hikino H, Rao GS. Coumestans as the main active principles of the liver drugs Eclipta alba and Wedelia calendulacea1. Planta Med. 1986;52:370–374. [PubMed] [Google Scholar]
- 43.Kobori M, Yang Z, Gong D, Heissmeyer V, Zhu H, Jung YK, Gakidis MA, Rao A, Sekine T, Ikegami F, et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 2004;11:123–130. doi: 10.1038/sj.cdd.4401325. [DOI] [PubMed] [Google Scholar]
- 44.Pocas ES, Costa PR, da Silva AJ, Noel F. 2-Methoxy-3,8,9-trihydroxy coumestan: a new synthetic inhibitor of Na+,K+-ATPase with an original mechanism of action. Biochem. Pharmacol. 2003;66:2169–2176. doi: 10.1016/j.bcp.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 45.Oh JW, Ito T, Lai MM. A recombinant hepatitis C virus RNA-dependent RNA polymerase capable of copying the full-length viral RNA. J. Virol. 1999;73:7694–7702. doi: 10.1128/jvi.73.9.7694-7702.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vo NV, Oh JW, Lai MM. Identification of RNA ligands that bind hepatitis C virus polymerase selectively and inhibit its RNA synthesis from the natural viral RNA templates. Virology. 2003;307:301–316. doi: 10.1016/s0042-6822(02)00095-8. [DOI] [PubMed] [Google Scholar]
- 47.Kaushik N, Pandey VN, Modak MJ. Significance of the O-helix residues of Escherichia coli DNA polymerase I in DNA synthesis: dynamics of the dNTP binding pocket. Biochemistry. 1996;35:7256–7266. doi: 10.1021/bi960537i. [DOI] [PubMed] [Google Scholar]
- 48.Kaushik N, Rege N, Yadav PN, Sarafianos SG, Modak MJ, Pandey VN. Biochemical analysis of catalytically crucial aspartate mutants of human immunodeficiency virus type 1 reverse transcriptase. Biochemistry. 1996;35:11536–11546. doi: 10.1021/bi960364x. [DOI] [PubMed] [Google Scholar]
- 49.Lee G, Piper DE, Wang Z, Anzola J, Powers J, Walker N, Li Y. Novel inhibitors of hepatitis C virus RNA-dependent RNA polymerases. J. Mol. Biol. 2006;357:1051–1057. doi: 10.1016/j.jmb.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 50.Tomei L, Altamura S, Bartholomew L, Biroccio A, Ceccacci A, Pacini L, Narjes F, Gennari N, Bisbocci M, Incitti I, et al. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003;77:13225–13231. doi: 10.1128/JVI.77.24.13225-13231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vo NV, Tuler JR, Lai MM. Enzymatic characterization of the full-length and C-terminally truncated hepatitis C virus RNA polymerases: function of the last 21 amino acids of the C terminus in template binding and RNA synthesis. Biochemistry. 2004;43:10579–10591. doi: 10.1021/bi049773g. [DOI] [PubMed] [Google Scholar]
- 52.McKercher G, Beaulieu PL, Lamarre D, LaPlante S, Lefebvre S, Pellerin C, Thauvette L, Kukolj G. Specific inhibitors of HCV polymerase identified using an NS5B with lower affinity for template/primer substrate. Nucleic Acids Res. 2004;32:422–431. doi: 10.1093/nar/gkh160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974;137:143–144. doi: 10.1042/bj1370143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 55.Bytheway I, Cochran S. Validation of molecular docking calculations involving FGF-1 and FGF-2. J. Med. Chem. 2004;47:1683–1693. doi: 10.1021/jm030447t. [DOI] [PubMed] [Google Scholar]
- 56.Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J. Chem. Inf. Model. 2006;46:401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- 57.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 58.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 59.Ikegashira K, Oka T, Hirashima S, Noji S, Yamanaka H, Hara Y, Adachi T, Tsuruha J, Doi S, Hase Y, et al. Discovery of conformationally constrained tetracyclic compounds as potent hepatitis C virus NS5B RNA polymerase inhibitors. J. Med. Chem. 2006;49:6950–6953. doi: 10.1021/jm0610245. [DOI] [PubMed] [Google Scholar]
- 60.Tedesco R, Shaw AN, Bambal R, Chai D, Concha NO, Darcy MG, Dhanak D, Fitch DM, Gates A, Gerhardt WG, et al. 3-(1,1-dioxo-2H-(1,2,4)-benzothiadiazin-3-yl) -4-hydroxy-2(1H)-quinolinones, potent inhibitors of hepatitis C virus RNA-dependent RNA polymerase. J. Med. Chem. 2006;49:971–983. doi: 10.1021/jm050855s. [DOI] [PubMed] [Google Scholar]
- 61.Kashiwagi T, Hara K, Kohara M, Kohara K, Iwahashi J, Hamada N, Yoshino H, Toyoda T. Kinetic analysis of C-terminally truncated RNA-dependent RNA polymerase of hepatitis C virus. Biochem. Biophys. Res. Commun. 2002;290:1188–1194. doi: 10.1006/bbrc.2001.6333. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y, Jiang WW, Pratt J, Rockway T, Harris K, Vasavanonda S, Tripathi R, Pithawalla R, Kati WM. Discovery of pyrano[3,4-b]indoles as potent and selective HCV NS5B polymerase inhibitors. Biochemistry. 2006;45:11312–11323. doi: 10.1021/bi060511j. [DOI] [PubMed] [Google Scholar]
- 63.Shi Q, Singh K, Srivastava A, Kaushik N, Modak MJ. Lysine 152 of MuLV reverse transcriptase is required for the integrity of the active site. Biochemistry. 2002;41:14831–14842. doi: 10.1021/bi0258389. [DOI] [PubMed] [Google Scholar]
- 64.Branca F, Lorenzetti S. Health effects of phytoestrogens. Forum Nutr. 2005;57:100–111. doi: 10.1159/000083773. [DOI] [PubMed] [Google Scholar]
- 65.Humfrey CD. Phytoestrogens and human health effects: weighing up the current evidence. Nat. Toxins. 1998;6:51–59. doi: 10.1002/(sici)1522-7189(199804)6:2<51::aid-nt11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 66.Powers JP, Piper DE, Li Y, Mayorga V, Anzola J, Chen JM, Jaen JC, Lee G, Liu J, Peterson MG, et al. SAR and mode of action of novel non-nucleoside inhibitors of hepatitis C NS5b RNA polymerase. J. Med. Chem. 2006;49:1034–1046. doi: 10.1021/jm050859x. [DOI] [PubMed] [Google Scholar]
- 67.Beaulieu PL, Bos M, Bousquet Y, DeRoy P, Fazal G, Gauthier J, Gillard J, Goulet S, McKercher G, Poupart MA, et al. Non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase: discovery of benzimidazole 5-carboxylic amide derivatives with low-nanomolar potency. Bioorg. Med. Chem. Lett. 2004;14:967–971. doi: 10.1016/j.bmcl.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 68.Love RA, Parge HE, Yu X, Hickey MJ, Diehl W, Gao J, Wriggers H, Ekker A, Wang L, Thomson JA, et al. Crystallographic identification of a noncompetitive inhibitor binding site on the hepatitis C virus NS5B RNA polymerase enzyme. J. Virol. 2003;77:7575–7581. doi: 10.1128/JVI.77.13.7575-7581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gopalsamy A, Lim K, Ciszewski G, Park K, Ellingboe JW, Bloom J, Insaf S, Upeslacis J, Mansour TS, Krishnamurthy G, et al. Mechanistic study of HCV polymerase inhibitors at individual steps of the polymerization reaction. J. Med. Chem. 2004;47:6603–6608. doi: 10.1021/jm0401255. [DOI] [PubMed] [Google Scholar]
- 70.Tomei L, Altamura S, Bartholomew L, Bisbocci M, Bailey C, Bosserman M, Cellucci A, Forte E, Incitti I, Orsatti L, et al. Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 2004;78:938–946. doi: 10.1128/JVI.78.2.938-946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]