


Abstract
The cell-cycle inhibitor p21CDKN1A has been suggested to directly participate in DNA repair, thanks to the interaction with PCNA. Yet, its role has remained unclear. Among proteins interacting with both p21 and PCNA, the histone acetyltransferase (HAT) p300 has been shown to participate in DNA repair. Here we report evidence indicating that p21 protein localizes and interacts with both p300 and PCNA at UV-induced DNA damage sites. The interaction between p300 and PCNA is regulated in vivo by p21. Indeed, loss of p21, or its inability to bind PCNA, results in a prolonged binding to chromatin and an increased association of p300 with PCNA, in UV-irradiated cells. Concomitantly, HAT activity of p300 is reduced after DNA damage. In vitro experiments show that inhibition of p300 HAT activity induced by PCNA is relieved by p21, which disrupts the association between recombinant p300 and PCNA. These results indicate that p21 is required during DNA repair to regulate p300 HAT activity by disrupting its interaction with PCNA.
INTRODUCTION
The cyclin-dependent kinase (CDK) inhibitor p21CDKN1A plays multiple roles not only as a cell-cycle regulator, but also as a regulator of transcription and apoptosis (1,2). In addition, p21 interacts with proliferating cell nuclear antigen (PCNA), and inhibits DNA synthesis by displacing DNA polymerases, as well as other proteins involved in DNA replication, from PCNA (3–6). Due to this behavior, p21 has also been considered as a potential inhibitor of DNA repair (7,8). However, previous works suggested that p21 might be required for DNA repair (9,10), particularly for nucleotide excision repair (NER). Recently, several findings have supported such hypothesis: p21 does not interfere with PCNA recruitment to DNA repair sites (11,12), nor with PCNA-dependent NER in vitro (13). In addition, p21-null human fibroblasts are more sensitive to UV-induced DNA damage, and show defects in NER (14). p21 protein is immediately recruited to DNA damage sites, together with repair factors, such as DNA polymerase δ, XPG and CAF-1 (12). This event does not occur in fibroblasts from patients affected by xeroderma pigmentosum (XP) belonging to complementation group A (XP-A), thus indicating a NER-related process (12).
Other findings support an additional role for p21 in base excision repair (BER), since p21 has been shown to interact with poly(ADP-ribose) polymerase 1 (PARP-1), thereby regulating PCNA (15). Lastly, p21 may regulate translesion DNA synthesis, an error-prone DNA repair process, to keep the mutation load to low levels (16). All these lines of evidence indicate that p21 may play an active role in DNA repair, whose significance still requires further elucidation.
Among proteins known to interact with p21, the transcriptional co-activator p300 deserves a particular interest (17), given that this protein has been shown to interact with PCNA, and to participate in DNA repair (18). In particular, p300 might play an important role in the DNA repair of the non-transcribed regions of the genome, a NER sub-pathway known as global genome repair (GG-NER) (19). In fact, the histone acetyltransferase (HAT) activity of p300 has been suggested to provide chromatin accessibility to the GG-NER machinery, depending on p53 expression (20). However, the role of p300 in DNA repair is still unclear and further complicated by the evidence that PCNA has an inhibitory effect on the p300 HAT activity (21).
Here, we have analyzed whether p21 may have a role in NER by regulating p300 activity through modulation of PCNA binding. We show that p21 localizes and interacts with both p300 and PCNA at DNA damage sites, and that p21 may physically interact with p300 in vitro. In addition, by using human fibroblasts deficient in p21 expression (22), and through ectopic expression of p21 and p300, we show that p21 and PCNA may bind independently to p300, but their mutual interaction regulates binding to p300, and that persistent binding of p300 to PCNA inhibits p300 HAT activity during NER. Finally, by in vitro binding and enzymatic assays, we provide evidence of a role for p21 in dissociating the interaction of p300 with PCNA, thereby restoring p300 HAT activity.
MATERIALS AND METHODS
Plasmids, proteins and antibodies
p21wt was cloned in pEGFP-N1 as previously described (6). RFP-PCNA and Flag-p300 plasmids were kindly provided by M.C. Cardoso (M. Delbruck Center for Molecular Medicine, Berlin, Germany) and by P.L. Puri (Telethon Institute, Rome, Italy), respectively. p21wt-HA and p21PCNA–-HA (mutated in PCNA binding domain) expression plasmids were previously described (6). Human PCNA protein, GST-fused full-length p21, a p21 mutant (ASM19) deficient in PCNA binding (23), or a C-terminal fragment, were expressed in BL21 bacteria and purified as described (23,24). Histidine (6×)-tagged human p21 (p21-His, cloned in pET-28 vector, Novagen), was purified by HisTrapTM Kit (Amersham). Flag-p300 fusion protein was purchased from Active Motif (USA).
Rabbit polyclonal anti-p21 (C-19 and N-20), and anti-p300 (N-15) antibodies were from Santa Cruz. Mouse monoclonal antibodies to p21waf1 (CP74 and DCS-60 clones) and to XPG (clone 8H7) were from NeoMarkers. Mouse monoclonal antibodies to actin (AC40), FLAG (M2), HA (H7) epitopes and polyclonal anti-GST antibody, were from Sigma. Rabbit polyclonal antibodies to acetyl-histone H3 (K9–K14) and to histone H3, were from Cell Signaling and Upstate. Mouse monoclonal antibodies to PCNA (PC10), and to CPDs (TDM-2) were from DakoCytomation and MBL, respectively. Rabbit anti-GFP polyclonal antibody was from Molecular Probes.
Cell cultures and UV irradiation
LF1 human lung embryonic fibroblasts and h-TERT-immortalized human p21+/+ and p21−/− fibroblasts (gift from J. Sedivy, Brown University, Providence, RI), were grown in MEM, or in HAM-F10 medium (Gibco BRL), respectively, supplemented with 15% foetal bovine serum (FBS) (22). HEK 293 and HeLa S3 were grown in DMEM medium supplemented with 10% FBS. Cell transfection was performed with Effectene (Qiagen), or by calcium phosphate method.
To follow UV-induced histone H3 acetylation, cells were pre-treated for 1 h with 2 mM hydroxyurea, as described (20).
The siRNA transfections were performed in LF1 cells using Interferin reagent (Polyplus) and p21-siRNA or GFP-siRNA (as a control) pools (Dharmacon), for 48 h at the final concentration of 10 nM.
Cells were either subjected to global (10 J/m2), or local UV-irradiation (20 J/m2) with a UV-C lamp (254 nm), as described (12).
Cell fractionation, co-immunoprecipitation and Western blot analysis
Whole cell extracts were prepared in lysis buffer containing 50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet NP-40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.2 mM Na3VO4 and a mixture of protease and phosphatase inhibitor cocktails (Sigma). To obtain the chromatin-bound fraction, cells were lysed in hypotonic buffer (10 mM Tris–HCl, pH 7.4, 2.5 mM MgCl2, 0.5% Nonidet NP-40) containing 1 mM PMSF, 0.2 mM Na3VO4 and protease and phosphatase inhibitor cocktails. Lysed cells were washed in hypotonic buffer and chromatin-bound proteins were released with DNase I, as described (25). Whole or fractionated cell extracts were incubated with antibodies pre-bound to protein A-sepharose CLB4 (Amersham), or protein G-magnetic beads (Dynal, Invitrogen), for 3 h at 4°C under constant agitation, washed and eluted from beads by SDS-denaturing buffer, as described (12). The immunoprecipitated proteins were resolved on NuPage 4–12% gradient gels (Invitrogen), transferred to nitrocellulose membranes, probed with specific antibodies and revealed using enhanced chemiluminescence (Amersham).
Immunofluorescence and confocal microscopy analysis
Cells were seeded on coverslips, transfected and locally irradiated as described earlier and lysed in situ with hypotonic lysis buffer before fixation to analyze chromatin-bound proteins, as described (25). Immunofluorescence staining with primary antibodies was followed by appropriate Alexa 488 or 594-conjugated secondary antibodies (Molecular Probes). Confocal microscopy analysis, image capture and processing was performed with a Leica TCS SP2 confocal microscope, as previously described (12).
Pull-down and competitive binding assays
Equimolar concentrations (1 μM each) of full-length (GST-p21), GST-p21 (ASM19) mutant that does not bind PCNA (23), a C-terminal peptide (GST-p21C), or GST alone were pre-incubated with 250 ng of recombinant Flag-p300 protein for 1 h at 4°C in 50 mM Tris–HCl buffer (pH 8.0), containing 2.5 mM MgCl2, 75 mM KCl, 10% glycerol, 1 mM dithiothreitol (DTT) and 0.2 mM PMSF. Fifty microliters of GSH-agarose beads were added and further incubated for 1 h under agitation, washed and eluted by SDS-denaturing buffer, as described (24). For competitive binding assay, 250 ng of recombinant Flag-p300 were pre-incubated with 500 ng PCNA, or with 800 ng GST-p21 in 100 μl of HAT buffer (50 mM Tris–HCl, pH 8.0, 10% glycerol, 1 mM DTT, 0.1 mM EDTA, 0.1 mM PMSF, 10 mM sodium butyrate, 0.5 μM acetylCoA) for 30 min at 4°C. Increasing concentrations (molar excess 2.5×, 10×, 20×) of GST-p21, or PCNA (1.5–10×), were added, and further incubated for 30 min before addition of 50 μl of anti-Flag antibody pre-bound to protein G-magnetic beads. Samples were kept under agitation for 2 h, then washed and eluted by SDS-denaturing buffer.
HAT assay
HAT assay (26) was performed with purified recombinant Flag-p300 protein (100 ng). Reaction was performed at 30°C in 50 μl of HAT buffer containing 1 μg each of purified histones H3 and H4, and 1 μCi of 3H-acetyl-Coenzyme A (Amersham Biosciences; specific activity 159 GBq/mmol). Recombinant PCNA, p21-His or GST-p21 fusion proteins were added to the reaction, as required. To follow acetylation kinetics, 200 μl reaction volumes were used, and aliquots of 50 μl were taken at 5, 10, 15, 20 min and spotted onto GF-C glass fiber filters (Whatman). Incorporated radioactivity was precipitated and counted with a Beckman LS 6000 liquid scintillation instrument. Data were expressed as mean values of counts, subtracted from background values and normalized to HAT activity measured in samples containing only p300 and substrates. Statistical analysis was performed with the Student's t-test.
RESULTS
p21 localizes and interacts with p300 at DNA damage sites
To study the role of p21 during DNA repair, we initially investigated the possibility that p21 protein could localize with p300 at DNA damage sites. For this purpose, the localization of p300 in relation to the main lesion induced by UV, i.e. cyclobutane pyrimidine dimers (CPDs), was first determined after local UV-irradiation, by immunofluorescence and confocal microscopy analysis. Figure 1A shows that, although p300 was distributed throughout the whole nucleus, it partially co-localized with CPDs (merged yellow spots), as further verified by analysis of pixel intensity profiles of the two fluorescence signals (Figure S1a). Co-localization specificity was checked by using an antibody against ribonucleoproteins, which did not co-localize with CPDs (Figure S1b). Then, we expressed p21 fused to GFP (p21-GFP) in HeLa cells and performed local UV-irradiation experiments to evaluate chromatin-binding of p21-GFP and p300 at DNA damage sites. Figure 1B shows that p300 protein was again spread throughout the whole nucleus, but p21-GFP was recruited to sites of DNA damage, spatially close to endogenous p300. To investigate whether p300 was present close to DNA damage sites where PCNA is also recruited (12), PCNA fused to Red Fluorescent Protein (RFP-PCNA) and p300 fused to FLAG epitope (Flag-p300) were co-expressed in HeLa cells. Similarly to the endogenous protein, Flag-p300 was distributed throughout the whole nucleus, but showed also an accumulation at irradiated spots, where clearly co-localized with RFP-PCNA, as indicated by the yellow spots in the merged image (Figure 1C). To assess whether co-localization was accompanied by physical interaction between p21 and p300, HeLa cells expressing p21-GFP (or empty vector pEGFP) were UV-irradiated and then cells extracts were immunoprecipitated with anti-GFP antibody. As seen in Figure 2A, p21-GFP, but not pEGFP, interacts specifically with p300 and PCNA, both before and after UV irradiation. The presence of XPG in the immunoprecipitates confirmed the previously observed co-localization of XPG and p21-GFP at UV-irradiated sites (12).
Figure 1.
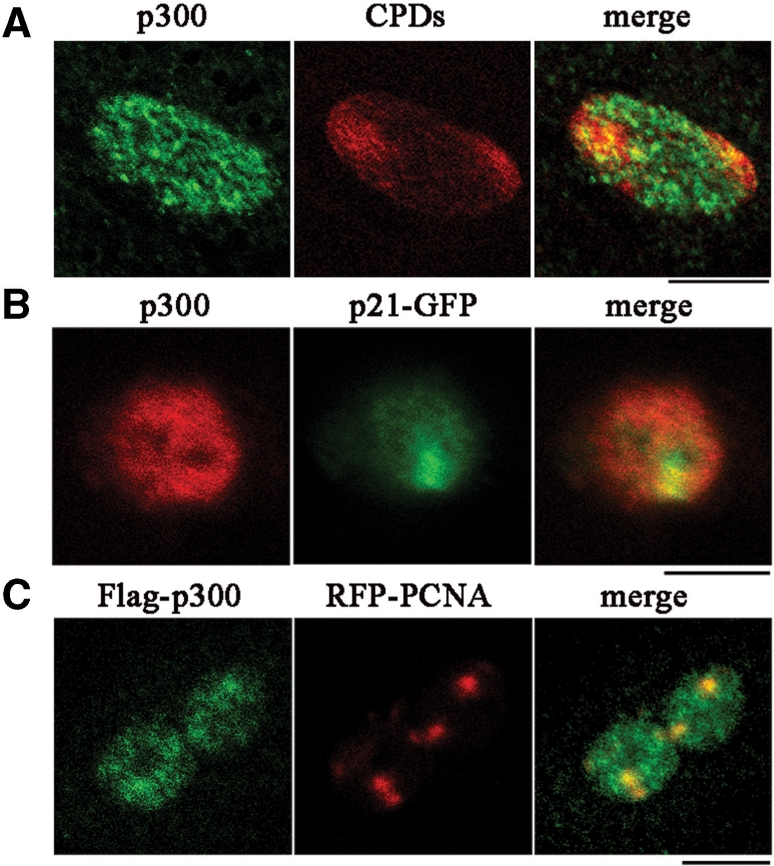
p300 co-localizes with p21 at sites of DNA damage and DNA repair. (A) Immunofluorescence confocal microscopy analysis of chromatin-bound p300 (green fluorescence) at sites of CPDs (red fluorescence). LF1 fibroblasts grown on coverslips were lysed in situ and fixed 30 min after local UV-irradiation, as described in ‘Materials and Methods’ section. Scale bar, 10 μm. (B) Confocal microscopy analysis of chromatin-bound p300 immunofluorescence (red) and p21-GFP autofluorescence (green) detected in HeLa cells at sites of local UV-irradiation. Transfected cells were UV-irradiated and fixed as described in ‘Materials and Methods’ section. Scale bar, 10 μm. (C) Confocal microscopy analysis of chromatin-bound Flag-p300 (green immunofluorescence), and RFP-PCNA autofluorescence (red), detected in HeLa cells after local UV-irradiation. Transfected cells were UV-irradiated and fixed as described in ‘Materials and Methods’ section. Scale bar, 10 μm.
Figure 2.

p21 interacts and physically associates with p300 in vivo and in vitro. (A) Immunoprecipitation (Ip) with anti-GFP antibody on extracts from HeLa cells expressing pEGFP, or p21-GFP, before and 30 min after UV-C irradiation, as described in ‘Materials and Methods’ section. Input load: 1/30 of cell extracts. (B) Immunoprecipitation (Ip) with anti-p21 antibodies of soluble (S) and chromatin-bound (C-b) fractions from control and 30 min after UV-irradiation (10 J/m2) of LF1 fibroblasts. Immunoprecipitation with irrelevant antibody (Ig) is shown together with bands of immunoglobulin heavy chains (Ig hc) of anti-p21 antibodies. Input load: 1/30 and 1/15 of soluble or chromatin-bound fraction, respectively. (C) Direct association between recombinant p300 and p21. Equimolar concentrations (1 μM) of full-length GST-p21 (GST-p21wt), a p21 mutant that does not bind PCNA (GST-p21ASM19), a p21 C-terminal peptide (GST-p21C) or GST alone were pre-incubated with recombinant Flag-p300 protein (250 ng), and then pulled-down with GSH-agarose beads, as described in ‘Materials and Methods’ section. The lower portion of the gel shown was stained with Coomassie, while the upper part was analysed by Western blot (WB) with anti-Flag antibody. M: molecular weight markers (kDa).
To further characterize the interaction between p21 and p300 in DNA repair process, immunoprecipitation of endogenous p21 was performed in human fibroblasts, after isolation of the soluble and the chromatin-bound fractions. As shown in Figure 2B, anti-p21 antibodies could immunoprecipitate PCNA, from the soluble fraction of both non-irradiated and UV-irradiated cells. No significant levels of PCNA were immunoprecipitated from the chromatin-bound fraction of non-irradiated cells, due to the absence of p21. Interestingly, in the immunoprecipitates from both fractions of UV-damaged cells, significant levels of p300 and XPG were detected together with p21 and PCNA. To provide evidence of a direct physical interaction between p21 and p300, recombinant GST-tagged p21wt was compared with a mutant form (p21ASM19) that does not bind PCNA (23), or with a C-terminal fragment of p21, in a pull-down assay with recombinant p300. Both GST-p21wt and GST-p21ASM19 mutant protein, but not the GST-p21 C-terminal fragment, nor GST alone, were able to physically interact with recombinant p300 (Figure 2C). These results indicate that p21 may directly associate with p300, in addition to DNA repair factors, and that this binding is not dependent on the C-terminal region required for interaction with PCNA (1,3).
Interaction of p21 with PCNA regulates mutual association with p300
In order to understand whether p21 affects the interaction between p300 and PCNA, cell extracts obtained from hTERT-immortalized human p21-null (p21−/−), and parental (p21+/+) fibroblasts were used for immunoprecipitation of endogenous p300 with anti-p300 antibody. As previously reported for primary p21-null fibroblasts (14), immortalized p21−/− fibroblasts exhibited a higher sensitivity to UVC-induced DNA damage, and showed deficiency in NER (supplementary Figure S2A and B). Figure 3A shows that in p21+/+ fibroblasts, the interaction between p300 and PCNA occurred to a higher extent after UV-irradiation, as compared with untreated control samples. In contrast, the interaction was already detectable in non-irradiated control samples of p21−/− fibroblasts, and it was higher in UV-irradiated samples. To clarify this aspect, a HA-tagged wild-type p21 protein (p21wt), or a mutant form (p21PCNA–) unable to interact with PCNA (27), were expressed in HeLa cells. After immunoprecipitation with anti-HA antibody (Figure 3B), it was found that PCNA interacted with p21wt-HA both before and after UV-irradiation, while p300 was present in the immunoprecipitate obtained from UV-treated cells, being almost undetectable in non-irradiated control samples. In the samples transfected with the mutant p21 construct (p21PCNA–-HA), no significant interaction with PCNA was detectable, as expected, while p300 protein was present in the immunoprecipitates from both irradiated and non-irradiated samples. In parallel aliquots of cell extracts, immunoprecipitation was performed with an antibody to p300 protein. Remarkably, the amount of PCNA interacting with p300 in the samples expressing the p21PCNA–-HA protein was significantly higher than those expressing p21wt-HA. Similarly, the p21 mutant was immunoprecipitated with p300 to a higher extent than the p21wt protein.
Figure 3.

p21 modulates the interaction between p300 and PCNA. (A) Immunoprecipitation (Ip) with anti-p300 antibody, or irrelevant IgG (Ig) of whole cell extracts from p21+/+ and p21−/− fibroblasts, obtained before or 30 min after UV-irradiation, as described in ‘Materials and Methods’ section. Input load: 1/30 of cell extracts. (B) Immunoprecipitation (Ip) with irrelevant antibody (Ig) with anti-HA or with anti-p300 antibodies, of whole cells extracts from HeLa cells non-transfected (nt) or transfected with plasmid for expression of p21wt-HA (p21wt), or p21PCNA−-HA (p21PCNA−). Input load: 1/30 of cell extracts. (C) Immunoprecipitation (Ip) with anti-Flag antibody of whole cell extracts from 293 cells transfected (tr.) or not according to the scheme reported: no plasmid (lanes 1,5), Flag-p300 plasmid (lanes 2 and 6), Flag-p300 in combination with p21wt-HA (lanes 3 and 7), or with p21PCNA−-HA plasmid (lanes 4 and 8). Input load: 1/30 of cell extracts.
To further characterize this interaction, HA-tagged p21wt and mutant proteins were co-expressed with Flag-tagged p300 in HEK 293 cells. Figure 3C shows that after immunoprecipitation with anti-Flag antibody, PCNA was associated with exogenous p300 in cells expressing Flag-p300 alone, or in combination with mutant p21PCNA–-HA protein, but not in samples co-expressing Flag-p300 and p21wt-HA. As observed above in HeLa cells, association of p300 with mutant p21PCNA–-HA occurred to higher levels than with p21wt-HA. These results indicate that, as well as in vitro, p21 and PCNA may independently bind to p300 in vivo. However, interaction of p21 with PCNA regulates mutual association with p300.
Loss of p21 induces accumulation and persistent binding of p300 to PCNA in concomitance with reduction in HAT activity
To understand whether the expression of p21 could influence the recruitment of p300 and PCNA to DNA damage sites, this process was investigated in p21+/+ and p21−/− fibroblasts. Time course analysis of chromatin-bound proteins (Figure 4A) in p21+/+ fibroblasts showed that PCNA was recruited to chromatin after UV-irradiation and its levels remained relatively constant with time. Levels of p300 did not change initially (as also shown in Figure 1A, B and C), but a decline was observed at late repair times (11). In contrast, both PCNA and p300 accumulated with time in the chromatin-bound fraction of p21−/− cells, as also shown by immunofluorescence (Figure S3). In order to confirm by a different approach that prolonged binding to chromatin of p300 and PCNA after DNA damage was related to the absence of p21, expression of this protein was silenced by RNA interference (RNAi) in primary LF1 fibroblasts. Western blot analysis (Figure 4B) showed that at 24 h after UV-irradiation, the levels of chromatin-bound PCNA and p300 in fibroblasts transfected with siRNA to p21 were remarkably higher than those detected in cells not exposed to p21 siRNA, and in cells transfected with control siRNA (GFP). The persistent binding to chromatin of both p300 and PCNA after UV irradiation suggested that they were associated. Immunoprecipitation with anti-p300 antibody (Figure 4C) confirmed that chromatin-bound PCNA and p300 interacted early after DNA damage (0.5 h) in p21+/+ cells, while their association was undetectable at late repair time (24 h), due to the decrease in p300 levels. In contrast, the interaction between p300 and PCNA persisted even 24 h after DNA damage, in p21−/− fibroblasts.
Figure 4.

Loss of p21 enhances chromatin-binding and association between p300 and PCNA after DNA damage. (A) Western blot analysis of recruitment of p300, PCNA and p21 proteins in the chromatin-bound fraction. Human h-TERT–immortalized p21+/+ and p21−/− fibroblasts were irradiated with UV-C (10 J/m2), collected at the indicated time points (h) after UV irradiation, and fractionated as described in ‘Materials and Methods’ section. Actin was also determined as a loading control. (B) Western blot analysis of proteins remaining chromatin-bound 24 h after UV-irradiation in LF1 fibroblasts not transfected, or transfected with p21-specific or control (GFP) siRNA pools. (C) Immunoprecipitation (Ip) with anti-p300 or irrelevant antibody (Ig) was performed on extracts of chromatin-bound proteins from p21+/+ and p21−/− fibroblasts collected 0.5 or 24 h after UV irradiation. Input load: 1/15 of nuclear extracts.
It was previously reported that p300 is an accessibility factor for NER proteins, and that its HAT activity towards histones H3 and H4 was reduced in UV-irradiated p53-null human fibroblasts (20). Thus, we investigated whether this could occur also in p21-null fibroblasts. Western blot analysis of histone H3 acetylation (K9 and K14 residues) after UV-irradiation of p21+/+ and p21−/− human fibroblasts revealed a significant reduction in histone H3 acetylation at 3 h after DNA damage (Figure 5A). Cells treated with trichostatin A (TSA) were added as a control of acetylation. Densitometric analysis of immunoreactive bands (Figure 5B) showed that there was a clear reduction in the ratio acetylated form/total levels of histone H3 (by about 65% at 3 h, P = 0.002) in p21−/− fibroblasts, as compared with p21+/+ cells. At 24 h after UV irradiation, the decrease was less evident (about 30%), yet still significant (P = 0.01).
Figure 5.

Loss of p21 results in a reduced HAT activity after UV irradiation. (A) Western blot analysis of acetylated forms (K9-K14) and total content of histone H3 performed in p21+/+ (lanes 1–4) and p21−/− (lanes 5–8) cell extracts, before (lanes 1 and 5) and 1 h (lanes 2 and 6), 3 h (lanes 3 and 7) or 6 h (lanes 4 and 8) after UV irradiation. In lane 9, extract from cells treated with trichostatin A (TSA) to enhance H3 acetylated forms, is shown as positive control of acetylation. (B) Densitometric analysis of the ratio acetylated form (K9–K14)/total levels of histone H3 in p21+/+ (empty bars) and p21−/− (solid bars) fibroblasts collected 3 or 24 h after UV irradiation. Normalized mean values ± SD of three independent experiments are reported (3 h, P = 0.002; 24 h, P = 0.01). (C) Western blot analysis of acetylated form (K9–K14) and of the total levels of histone H3, and p21. LF1 fibroblasts not transfected, or transfected with p21-specific or control (GFP) siRNA pools were collected 24 h after UV irradiation. (D) Densitometric analysis of the ratio acetylated form (K9–K14)/total levels of histone H3, in UV-irradiated LF1 fibroblasts not transfected, or transfected with p21-specific or control (GFP) siRNA pools. Normalized mean values ± SD of three independent experiments, are reported (P = 0.03, p21-siRNA versus untreated control; P = 0.04, p21-siRNA versus GFP-siRNA).
Given that PCNA inhibits p300 HAT activity (21), and having shown that in p21−/− cells their interaction persists after DNA damage, we investigated the effect of the reduction in p21 levels by RNAi on p300 HAT activity. Western blot analysis of acetylated forms (K9 and K14) and total levels of histone H3 showed a significant reduction in acetylated forms (Figure 5C). Densitometric evaluation of the ratio acetylated form/total level of histone H3 (Figure 5D), confirmed that reduction in p21 protein levels by RNAi in normal fibroblasts resulted in a concomitant decrease in HAT activity after UV irradiation, similar to that observed in p21−/− fibroblasts.
p21 dissociates the interaction between p300 and PCNA restoring p300 HAT activity
To provide a functional link between HAT activity and modulation of p300/PCNA interaction by p21, recombinant Flag-p300 and PCNA were pre-incubated before addition of increasing amounts of GST-p21, or GST alone. Immunoprecipitation with anti-Flag antibody (Figure 6A) showed that PCNA was able to bind directly to p300 in the absence of GST-p21. Addition of low molar excess (2.5×) amounts of GST-p21 resulted in concomitant binding of GST-p21 and PCNA to p300. In contrast, higher excess induced the dissociation of PCNA, but also a reduction in the levels of GST-p21 associated with p300. In reverse experiments, binding of GST-p21 to p300 was enhanced by low PCNA concentration (about equimolar to GST-p21), while a 10-fold excess of PCNA, reduced the amount of GST-p21 associated to p300 to the levels observed in the absence of PCNA. The amount of PCNA still bound to p300 was also reduced at the higher concentration, again suggesting turn-over of this interaction.
Figure 6.

p21 dissociates the binding of PCNA to p300, restoring HAT activity. Competition binding assays: (A) recombinant PCNA (500 ng) was incubated with Flag-p300, in the absence (lane 2), or in the presence of increasing molar excess of GST-p21 protein (2.5×, 10× and 20×, lanes 3–5), or with 10× GST (lane 6). (B) GST-p21 (800 ng) was incubated with Flag-p300 in the absence (lane 2), or in the presence of increasing molar excess of PCNA (1.5×, 10×, lanes 3 and 4). Proteins bound to Flag-p300 were immunoprecipitated (Ip) with anti-Flag antibody and analyzed by Western blot. Input load (lane 1): 1/2 of Flag-p300, and 1/20 of either PCNA or GST-p21 proteins. (C) HAT assay with recombinant p300 (100 ng) in the absence or in the presence of 0.8 μM PCNA, 0.8 μM PCNA + 8 μM p21-His or GST-p21 (p21wt), 0.8 μM PCNA + 8 μM GST-p21ASM19 (p21ASM19) or p21wt alone (8 μM). The reaction was performed for 10 min (i.e. within the linear part of the reaction). Normalized mean values (±SD) of radioactivity counts from three to five independent experiments are reported (P = 0.007, PCNA+p21wt versus PCNA; P = 0.001, p21wt versus PCNA).
Given that PCNA inhibits HAT activity of p300 (21), we performed a HAT assay under conditions of saturating p21 concentration toward PCNA, and using either a GST- or histidine-tagged recombinant p21. The results (Figure 6B) showed that PCNA significantly inhibited p300 HAT activity toward histones H3 and H4, and that this inhibition was relieved by the addition of p21wt, but not by the p21ASM19 mutant protein. Addition of either p21wt or mutant form alone in the absence of PCNA, had no significant effect on p300 HAT activity. Similarly, addition of GST alone was uneffective (not shown). Neither PCNA, nor p21 appeared to be good substrates for p300, as compared with histones, in our experimental conditions (Figure S4). Thus, these results indicate that p21 dissociates the inhibitory binding of PCNA to p300, thereby restoring p300 HAT activity.
DISCUSSION
The present study suggests that p21 is required to regulate the HAT activity of p300, which has been indicated to provide accessibility of NER machinery to DNA damage sites (20). It was previously shown that p21 could regulate the transcriptional activity of p300 by modulating the CRD1 repression domain (28), although no direct binding was observed within this region of p300 (17). Interestingly, a low binding level of p21 was observed in the C-terminal region of p300, which is also involved in the interaction with PCNA (18). Here, we show that p21 co-localizes at DNA damage sites and interacts directly with p300, and that for this activity the full-length protein is probably required because a C-terminal peptide (capable of interaction with PCNA), did not bind to recombinant p300. In addition, binding of p21 to p300 is not dependent on interaction with PCNA, given that a p21 mutant unable to bind PCNA (23) still retained its ability to associate with p300.
Considering that both p21 and PCNA may independently bind to p300, p21 appears to have a role in the regulation of mutual interaction between p300 and PCNA. In fact, our in vivo experiments showed that their association was enhanced, or persisted longer, in the absence of p21, or in the presence of a mutant protein unable to bind PCNA. In vitro, it appears that p21 and PCNA at or near equimolar concentrations, cooperate to bind p300. In contrast, an excess of one over the other, induces dissociation from p300. The ability of p21 to displace PCNA-interacting proteins (PIP) is well known, at least for those proteins that bind PCNA through the conserved sequence named PIP box (5). However, p300 does not seem to have a PIP box (18), and its interaction with PCNA might be provided by alternative sequences (29,30).
It has been recently observed that p21 binds to the myc and cdc25a promoters upon DNA damage, thereby functioning as a transcriptional inhibitor (31). However, in that study the presence of PCNA was not investigated, and no conclusions can be drawn on the eventual contribution of PCNA to inhibit both transcriptional, as well as HAT activity of p300 (21).
In this study, we have found that loss of p21 results in a reduced acetylation of histone H3 in vivo, concomitantly with a persistent association between p300 and PCNA. In addition, we have confirmed that PCNA inhibits HAT activity of p300 in vitro. Remarkably, p21 was able to relieve the inhibitory effect of PCNA by dissociating the binding with p300. Thus, our results indicate that p21 regulates the HAT activity of p300 required for NER (18,20) by disrupting its interaction with PCNA. Interestingly, it was previously reported that also p33ING1b protein was able to dissociate the complex between p300 and PCNA in a DNA damage-dependent manner, and that ING1b overexpression enhanced histone acetylation (32). However, a direct relation between p33ING1b and p300 activity was not investigated in that study.
It is important to note that the role of p300 in NER could not be limited to the acetylation of histones. In fact, it is known that p300 acetylates some proteins participating in BER, like the endonuclease FEN1 (33), DNA polymerase β (34), and the DNA glycosylases TDG, NEIL2, OGG1 and MPG (35–38). In contrast, there is no substantial information concerning acetylation of NER factors, and knowledge is limited to the interaction of p300 with both DDB1 and DDB2 proteins (39,40), XPA protein (18) and to the evidence that during transcription-coupled repair, CSB protein recruits p300 onto chromatin, together with other repair factors (41). In the case of BER, it is interesting to note that many of the proteins listed above are known to interact with PCNA (29). Acetylation may influence the activity and/or recruitment of such BER factors to DNA repair sites, suggesting that NER proteins could be similarly regulated.
Remarkably, p300 appears to contribute to NER by inducing, in the presence of transcriptional activators (TA), a transcription-independent chromatin remodeling process (42). Some of these TAs interact with repair factors, and are acetylated by p300, thereby modulating transcription and DNA repair (38,43). Moreover, both p21 and PCNA may also interact with some of these TAs (29,44–46), which in turn may also bind to NER factors (47,48). Based on these lines of evidence and on our observations, we propose the model illustrated in Figure 7. Both p21 and PCNA may bind to p300 (and possibly to TAs) at basal levels. After DNA damage, PCNA is first recruited to sites where p300 is located, thereby sequestering and inhibiting p300 HAT activity, blocking TA acetylation and promoting the shift from transcription to DNA repair (35,41,42). Immediately after PCNA, p21 is recruited to the same sites (12) where it may dissociate p300/PCNA interaction. The released p300 HAT activity may contribute to chromatin remodeling of surrounding sequences (41), thereby promoting PCNA-dependent DNA repair of chromatin regions not readily accessible to NER machinery. In this scenario, another crucial role for p21 in maintaining genome stability might be played through the regulation of p300 HAT activity required at the chromatin level during GG-NER (49).
Figure 7.

Schematic representation of the hypothetical role of p21 in global genome repair (GG-NER), through destabilization of the association of p300 to PCNA. Under normal conditions, both p21 and PCNA may bind to p300 and/or transcriptional activator (TA). After DNA damage, PCNA is first recruited to sites of damaged DNA where p300 is present, thereby inhibiting HAT activity. Immediately afterward, p21 is also recruited, until its local concentration is high enough to bind both PCNA and p300, thereby disrupting their association. The release of p300 restores its HAT activity, thus promoting accessibility of PCNA-dependent NER machinery in surrounding chromatin regions. For simplification, NER factors (e.g. XPG) found in the p21–p300 complex have not been drawn here.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work was supported in part by a CNR grant (DG.RSTL.040.002), and by MIUR grant (PRIN 2006/069475). P.P. was supported by a PRIN scholarship from Pavia University. B.D. is supported by La Ligue contre le Cancer. We gratefully thank J.M. Sedivy (Providence, RI), M.C. Cardoso (Berlin, Germany), M. Crescenzi, P. Pichierri, P.L. Puri (Rome, Italy), M. Malanga (Naples, Italy), for their generous gift of cells, plasmids and proteins. We thank also P. Vaghi (Centro Grandi Strumenti, Università di Pavia) for help in confocal microscopy analysis, and M. Parks for English revision. Funding to pay the Open Access publication charges for this article was provided by MIUR-PRIN (2006/069475).
Conflict of interest statement. None declared.
REFERENCES
- 1.Dotto GP. p21WAF1/Cip1: more than a break to the cell cycle? Biochim. Biophys. Acta. 2000;1471:M43–M56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 2.Coqueret O. New roles for p21 and p27 cell-cycle inhibitor: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 3.Gulbis JM, Kelman Z, Hurtwitz J, O’Donnel M, Kuriyan J. Structure of the C-terminal region of p21waf1/cip1 complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 4.Waga S, Stillman B. Cyclin-dependent kinase inhibitor p21 modulates the DNA primer-template recognition complex. Mol. Cell. Biol. 1998;18:4177–4187. doi: 10.1128/mcb.18.7.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warbrick E. The puzzle of PCNA's many partners. BioEssays. 2000;22:997–1006. doi: 10.1002/1521-1878(200011)22:11<997::AID-BIES6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 6.Cazzalini O, Perucca P, Riva F, Stivala LA, Bianchi L, Vannini V, Ducommun B, Prosperi E. p21CDKN1A does not interfere with loading of PCNA at DNA replication sites, but inhibits subsequent binding of DNA polymerase delta at the G1/S phase transition. Cell Cycle. 2003;2:596–603. [PubMed] [Google Scholar]
- 7.Cooper MP, Balajee AS, Bohr VA. The C-terminal domain of p21 inhibits nucleotide excision repair in vitro and in vivo. Mol. Biol. Cell. 1999;10:2119–2129. doi: 10.1091/mbc.10.7.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21WAF1 to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 9.McDonald ER, III, Wu GS, Waldman T, El-Deiry WS. Repair defect of p21waf1/cip1−/− human cancer cells. Cancer Res. 1996;56:2250–2255. [PubMed] [Google Scholar]
- 10.Sheikh MS, Chen YQ, Smith ML, Fornace AJ., Jr Role of p21waf/cip1/sdi1 in cell death and DNA repair as studied using a tetracycline-inducible system in p53-deficient cells. Oncogene. 1997;14:1875–1882. doi: 10.1038/sj.onc.1201004. [DOI] [PubMed] [Google Scholar]
- 11.Savio M, Stivala LA, Scovassi AI, Bianchi L, Prosperi E. p21waf1/cip1 protein associates with the detergent-insoluble form of PCNA concomitantly with disassembly of PCNA at nucleotide excision repair sites. Oncogene. 1996;13:1591–1598. [PubMed] [Google Scholar]
- 12.Perucca P, Cazzalini O, Mortusewicz O, Necchi D, Savio M, Nardo T, Stivala LA, Leonhardt H, Cardoso MC, Prosperi E. Spatiotemporal dynamics of p21CDKN1A protein recruitment to DNA-damage sites and interaction with proliferating cell nuclear antigen. J. Cell Sci. 2006;119:1517–1527. doi: 10.1242/jcs.02868. [DOI] [PubMed] [Google Scholar]
- 13.Shivji MKK, Ferrari E, Ball K, Hübscher U, Wood RD. Resistance of human nucleotide excision repair synthesis in vitro to p21CDKN1. Oncogene. 1998;17:2827–2838. doi: 10.1038/sj.onc.1202352. [DOI] [PubMed] [Google Scholar]
- 14.Stivala LA, Riva F, Cazzalini O, Savio M, Prosperi E. p21waf1/cip1-null human fibroblasts are deficient in nucleotide excision repair downstream the recruitment of PCNA to DNA repair sites. Oncogene. 2001;20:563–570. doi: 10.1038/sj.onc.1204132. [DOI] [PubMed] [Google Scholar]
- 15.Frouin I, Maga G, Denegri M, Riva F, Savio M, Spadari S, Prosperi E, Scovassi AI. Human proliferating cell nuclear antigen, poly (ADP-ribose) polymerase-1, and p21waf1/cip1: a dynamic exchange of partners. J. Biol. Chem. 2003;278:39265–39268. doi: 10.1074/jbc.C300098200. [DOI] [PubMed] [Google Scholar]
- 16.Avkin S, Sevilya Z, Toube L, Geacintov N, Chaney SG, Oren M, Livneh Z. p53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Mol. Cell. 2006;22:407–413. doi: 10.1016/j.molcel.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 17.Gregory DJ, Garcia-Wilson E, Poole JC, Snowden AW, Roninson IB, Perkins ND. Induction of transcription through the p300 CRD1 Motif by p21WAF1/CIP1 is core promoter specific and cyclin dependent kinase independent. Cell Cycle. 2002;1:343–350. [PubMed] [Google Scholar]
- 18.Hasan S, Hassa PO, Imhof R, Hottiger MO. Transcriptional coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature. 2001;410:387–391. doi: 10.1038/35066610. [DOI] [PubMed] [Google Scholar]
- 19.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 20.Rubbi CP, Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003;22:975–986. doi: 10.1093/emboj/cdg082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong R, Chakravarti D. The human proliferating cell nuclear antigen regulates transcriptional coactivator p300 activity and promotes transcriptional repression. J. Biol. Chem. 2003;278:44505–44513. doi: 10.1074/jbc.M303138200. [DOI] [PubMed] [Google Scholar]
- 22.Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cell through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 23.Podust VN, Podust LM, Goubin F, Ducommun B, Hubscher U. Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry. 1995;34:8869–8875. doi: 10.1021/bi00027a039. [DOI] [PubMed] [Google Scholar]
- 24.Riva F, Savio M, Cazzalini O, Stivala LA, Scovassi IA, Cox LS, Ducommun B, Prosperi E. Distinct pools of proliferating cell nuclear antigen associated to DNA replication sites interact with the p125 subunit of DNA polymerase δ or DNA ligase I. Exp. Cell Res. 2004;293:357–367. doi: 10.1016/j.yexcr.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Scovassi AI, Prosperi E. Analysis of proliferating cell nuclear antigen (PCNA) associated with DNA excision repair sites in mammalian cells. Methods Mol. Biol. 2006;314:457–475. doi: 10.1385/1-59259-973-7:457. [DOI] [PubMed] [Google Scholar]
- 26.Chakravarti D, Ogryzko V, Kao HY, Nash A, Chen H, Nakatani Y, Evans RM. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell. 1999;96:393–403. doi: 10.1016/s0092-8674(00)80552-8. [DOI] [PubMed] [Google Scholar]
- 27.Cayrol C, Knibiehelr M, Ducommun B. p21 binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient cells. Oncogene. 1998;16:311–320. doi: 10.1038/sj.onc.1201543. [DOI] [PubMed] [Google Scholar]
- 28.Snowden AW, Anderson LA, Webster GA, Perkins ND. A novel transcriptional repression domain mediates p21WAF1/CIP1 induction of transactivation. Mol. Cell. Biol. 2000;20:2676–2686. doi: 10.1128/mcb.20.8.2676-2686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prosperi E. The fellowship of the rings: distinct pools of proliferating cell nuclear antigen (PCNA) trimer at work. FASEB J. 2006;20:833–837. doi: 10.1096/fj.05-5469hyp. [DOI] [PubMed] [Google Scholar]
- 30.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Vigneron A, Cherier J, Barré B, Gamelin E, Coqueret O. The cell cycle inhibitor p21waf1 binds to the myc and cdc25A promoters upon DNA damage and induces transcriptional repression. J. Biol. Chem. 2006;281:34742–34750. doi: 10.1074/jbc.M602492200. [DOI] [PubMed] [Google Scholar]
- 32.Vieyra D, Loewith R, Scott M, Bonnefin P, Boisvert FM, Cheema P, Pastyryeva S, Meijer M, Johnston RN, Bazett-Jones DP, et al. Human ING1 proteins differentially regulate histone acetylation. J. Biol. Chem. 2002;277:29832–29839. doi: 10.1074/jbc.M200197200. [DOI] [PubMed] [Google Scholar]
- 33.Hasan S, Stucki M, Hassa PO, Imhof R, Gehrig P, Hunziker P, Hübscher U, Hottiger MO. Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol. Cell. 2001;7:1221–1231. doi: 10.1016/s1097-2765(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 34.Hasan S, El-Andaloussi N, Hardeland U, Hassa PO, Bürki C, Imhof R, Schär P, Hottiger MO. Acetylation regulates the DNA end-trimming activity of DNA polymerase β. Mol. Cell. 2002;10:1213–1222. doi: 10.1016/s1097-2765(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 35.Tini M, Benecke A, Um S-J, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 36.Bhakat KK, Hazra TK, Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006;26:1654–1665. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Likhite VS, Cass EI, Anderson SD, Yates JR, Nardulli AM. Interaction of estrogen receptor α with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. J. Biol. Chem. 2004;279:16875–16882. doi: 10.1074/jbc.M313155200. [DOI] [PubMed] [Google Scholar]
- 39.Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T, Raychaudhuri P. The p48 subunit of the damaged-DNA binding protein DDB associates with CBP/p300 family of histone acetyltransferase. Mutat. Res. 2001;486:89–97. doi: 10.1016/s0921-8777(01)00082-9. [DOI] [PubMed] [Google Scholar]
- 40.Rapic-Otrin V, McLenigan MP, Bisi DC, Gonsalez M, Levine AS. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 2002;30:2588–2598. doi: 10.1093/nar/30.11.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LHF. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodelling and repair factors of stalled RNA polymerase II in vivo. Mol. Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 42.Frit P, Kwon K, Coin F, Auriol J, Dubaele S, Salles B, Egly JM. Transcriptional activators stimulate DNA repair. Mol. Cell. 2002;10:1391–1401. doi: 10.1016/s1097-2765(02)00732-3. [DOI] [PubMed] [Google Scholar]
- 43.Schultz-Norton JR, Walt KA, Ziegler YS, McLeod IX, Yates JR, Raetzman LT, Nardulli AM. The deoxyribonucleic acid repair protein flap endonuclease-1 modulates estrogen-responsive gene expression. Mol. Endocrinol. 2007;21:1569–1580. doi: 10.1210/me.2006-0519. [DOI] [PubMed] [Google Scholar]
- 44.Fritah A, Saucier C, Mester J, Redeuilh G, Sabbah M. p21WAF1/CIP1 selectively controls the transcription of estrogen receptor α. Mol. Cell. Biol. 2005;25:2419–2430. doi: 10.1128/MCB.25.6.2419-2430.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin P, Lardeaux V, Lefebvre P. The proliferating cell nuclear antigen regulates retinoic acid receptor transcriptional activity through direct protein-protein interaction. Nucleic Acids Res. 2005;13:4311–4321. doi: 10.1093/nar/gki745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schultz-Norton JR, Gabisi VA, Ziegler YS, McLeod IX, Yates JR, Nardulli AM. Interaction of estrogen receptor α with proliferating cell nuclear antigen. Nucleic Acids Res. 2007;35:5028–5038. doi: 10.1093/nar/gkm533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keriel A, Stary A, Sarasin A, Rochette-Egly C, Egly JM. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARalpha. Cell. 2002;109:125–135. doi: 10.1016/s0092-8674(02)00692-x. [DOI] [PubMed] [Google Scholar]
- 48.Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockaine syndrome in XP-G/CS patients. Mol. Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 49.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.