


Abstract
A facile, sensitive method for detecting specific sequences of oligonucleotides was developed. Detection of DNA sequences with single nucleotide discrimination is achieved by combining the selectivity of hybridization with an efficient cross-linking reaction. Readily synthesized bifunctional oligonucleotide probes containing a modified pyrimidine that is capable of forming interstrand cross-links under mild oxidative conditions internally, and biotin at their 5′-termini were used to discriminate between 16-nt long sites in plasmid DNA that differ by a single nucleotide. The target sequence was detected via fluorescence spectroscopy by utilizing conjugates of avidin and horseradish peroxidase in a microtiter plate assay. The method is able to detect as little as 250 fmol of target without using PCR and exhibits single nucleotide discrimination that approaches 200:1. In principle, this method is capable of probing any target sequence containing a 2′-deoxyadenosine.
INTRODUCTION
Detecting single nucleotide polymorphisms (SNPs) is an important activity in the current post-genomic era. SNP analysis is potentially useful for predicting the likelihood of genetic disease by using nucleic acid sequence as a biomarker, and for predicting response to therapeutic agents. Given the importance of this information, a number of methods are being developed to detect SNPs (1–6). However, there are still opportunities for the development of facile, inexpensive, sensitive methods for SNP detection. Methods that report on and amplify a binding event are attractive because they can eliminate the need to PCR amplify the target (7–10). PCR free detection is also necessary for any method that is carried out in cells (11,12). We have developed a method for detecting DNA sequences that is capable of single nucleotide discrimination. The method utilizes efficient cross-linking of a modified thymidine (1) in a biotinylated oligonucleotide probe to an opposing dA in the target strand (Scheme 1) (13,14). The ‘tagged’ target is detected using a common fluorescence reporter assay that takes advantage of tight binding between biotin and avidin that is conjugated to horseradish peroxidase (HRP; Scheme 2)(15).
Scheme 1.

Formation of interstrand cross-links via oxidation of a modified thymidine (1).
Scheme 2.

Sequence-selective detection of DNA via fluorescence using cross-linked DNA.
MATERIALS AND METHODS
General methods
Unless otherwise specified, chemicals were purchased from Aldrich or Fisher Scientific and enzymes was obtained from New England Biolabs. Oligonucleotides were synthesized via standard automated DNA synthesis techniques using an Applied Biosystems model 394 instrument. The Pac-dA and iPr-Pac-dG phosphoramidites were employed for the synthesis of oligonucleotides containing 1. Pivaloyl anhydride/2,6-lutidine/THF (8:1:1) was used as capping reagent and 1M t-butyl-hydroperoxide in toluene was used as oxidizing reagent. The oxidation time is 40 s and the capping time is 25 s. Deprotection of the nucleobases and phosphate moieties as well as cleavage of the linker were carried out under mild deprotection conditions (28% aq. NH3, room temperature, 2 h). Oligonucleotides were purified by 20% denaturing polyacrylamide gel electrophoresis and characterized by ESI-MS. Oligonucleotides 3 and 4 were subjected to additional purification by reversed-phase HPLC on a RP-C18 column (Varian, Microsorb-MV 100-5 C18 250 × 4.6 mm). Monitoring was carried out at 260 nm. The peak of interest (retention time: 38–39 min for 3 and 42–43 min for 4) was collected using the following gradient conditions: 0–5 min 0–2% B in A, 5–15 min 2–12% B in A, 15–50 min 12–25% B in A, 50–60 min 25–80% B in A, 60–65 min 80–100% B in A, 65–80 min 100% A, at a flow rate 1.0 ml/min) [A: 0.05 M (Et3NH)OAc (pH 7.0)/MeCN 95:5; B: 0.05 M (Et3NH)OAc (pH 7.0)/MeCN 50:50]. Radiolabeling was carried out according to the standard protocols (16). [γ-32P]ATP was purchased from Amersham Pharmacia Biotech. Quantification of radiolabeled oligonucleotides was carried out using a Molecular Dynamics Phosphorimager equipped with ImageQuant Version 5.1 software. Fluorescence spectra were collected on a Varian Cary Eclipse fluorescence spectrophotometer equipped with a microplate reader. Ninety-six-well flat-bottom assay plates (black) were purchased from Corning (Fisher, Cat. No. 07200509). Protamine sulfate and Tween-20 were purchased from Sigma (Cat. No. P 4020 and P 9416). SuperBlocking buffer in PBS and BlockBSA in PBS (10×) were purchased from Pierce (Cat. No. 37515 and 37525). ABC Elite ultra kit was purchased from Vector Laboratories (Cat. No. PK-6100). Amplex Red and Amplex Red Stop reagents were purchased from Invitrogen (Cat. No. A36006 and A33855). Cross-linking reactions were carried out in an Eppendorf Mastercycler Thermal Cycler in order to achieve maximal temperature control.
Interstrand cross-link reaction performed with short oligonucleotides 5a–d and the ICL growth kinetics
The appropriate 32P-labeled oligonucleotide and its complementary strand (1.5 eq.) were hybridized in 100 mM NaCl. NaIO4 (5 mM) reactions of DNA duplexes (30 nM) were carried out in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl. For determination of the rate constant for interstrand cross-link formation (kICL), samples were incubated at 37°C. Aliquots were taken at the prescribed times and immediately quenched with equal volumes of 95% formamide loading buffer, heated to 80°C for 2 min and stored at –80 °C until subjecting to 20% denaturing PAGE analysis.
Construction of site-specifically modified M13 genome (plasmid 12a or 12b containing target 5a or 5b)
M13mp7 (7240 bp, circular single-stranded plasmid, 10 pmol, 10 μl) was digested with EcoRI to produce a linearized single-stranded plasmid (7200 nt) (17). The EcoRI digest was performed at 37°C for 4 h in the presence of EcoRI (1.0 μl, 20 U), EcoRI buffer (100 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2 and 0.025% Triton X-100, pH 7.5, 2.3 μl) and H2O (9.2 μl). To the digestion mixture, two scaffolds (15 pmol for each) were added, which are annealed to the newly formed ends of a linearized M13mp7 by heating at 90°C for 5 min, then cooling to 25°C over 2 h. In this step, the scaffolds 5′-AAA ACG ACG GCC AGT GAA TTG ATG GGC CTC-3′ and 5′-GGT TCA TGC CGC ACT GAA TCA TGG TCA TAG C-3′ anneal to 20 bases of the M13mp7 vector, with 10- or 11-base overhangs to house a modified 22mer 5′-CGG CAT GAA CCA GAG GCC CAT C-3′ (5a) or 5′-CGGCAT GAA CCG GAGGCC CAT C-3′ (5b), leaving a 1-base gap at the site of the variable nucleotide. Meanwhile, 15 pmol of 5a or 5b is 5′-phosphorylated in 30 μl containing T4-PNK buffer (10×, 3.0 μl), ATP (100 mM, 0.3 μl), 5 mM DTT and T4 PNK (0.4 μl, 12 U). After incubating the plasmid, scaffold and probe at 37°C for 1 h, the ligation was performed by adding T4 ligase (0.5 μl, 200 U), T4 ligase buffer (0.5 μl), DTT (1%, 2.25 μl), ATP (100 mM, 0.25 μl) and H2O (2.5 μl) and incubation at 16°C for 12 h. T4 DNA polymerase (0.4 μl, 3.9 U), EcoRI buffer (1.0 μl) and H2O (8.6 μl) were added and incubated at 37°C for 4 h to remove the excess scaffolds. The mixture volume was increased to 100 μl by adding water, extracted once with phenol/chloroform/isoamyl alcohol (25:24:1, 100 μl), and the aqueous phase passed through a home-made Sephadex-G25 Fine (Sigma-Aldrich) spin column to remove traces of phenol and salt. The M13 construct (12a or 12b) was obtained with a final concentration of 18 nM (150 μl).
Electroporation of M13 constructs (12a or 12b) into GW 5100 cells
GW 5100 cells were grown in Luria Broth (LB) (10 ml) overnight at 37°C and diluted (1.0 ml in 100 ml LB buffer). The diluted cells were incubated at 37°C until OD600 = 0.5 and then harvested by centrifugation at 5500 r.p.m. for 10 min (4°C). The cells were resuspended in 50 ml of ice-cold deionized water and centrifuged at 5500 r.p.m. for 10 min. The bacterial pellet was resuspended in glycerol/water (10%, 1.0 ml) and kept on ice until further use. For each transformation, an aliquot (100 μl) of the cell suspension was mixed with 1 pmol of M13 construct (12a or 12b) and transferred to the bottom of an ice-cold BTX disposable cuvette (2.0 mm electrode gap). Electroporation of cells was carried out in a BTX ECM 399 Generator at 36 μF and 2.5 kV. Immediately after electroporation, LB medium (10 ml) was added and the mixture was incubated at 37°C for 1 h. An aliquot (10 μl) of regrown cells was plated and M13 constructs (12a or 12b) with modified insert gave blue plaques in the presence of isopropyl-β-d-thiogalactopyranoside (IPTG) and 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal) after incubation overnight at 37°C. A blue plaque was isolated and mixed in LB medium (1 ml). An aliquot (50 μl) of overnight grown GW5100 cells were added and diluted to 50 ml with LB buffer. The mixture was incubated at 37°C for 7 h and centrifuged at 9500 r.p.m. for 15 min (4°C). The supernatant containing M13 progeny phage (12a or 12b) was stored at 4°C for the next step.
Preparative isolation of plasmids (12a or 12b) in GW 5100 cells
An overnight culture of GW5100 cells (∼0.2 μl) was grown in 2 × YT (10 ml) with orbital shaking (250 r.p.m.) at 37°C. An aliquot (1 ml) of this stock was added to 2 × YT (500 ml) and grown at 37°C for 2 h. The M13 progeny phage (12a or 12b) (2 ml) (from last step) was then added and grown for 8 h at 37°C. The cells were cooled on ice for 10 min and then centrifuged at 9500 r.p.m., 4°C, for 15 min. The supernatant (500 ml) was transferred to a beaker containing 125 ml of 20% PEG-800 and NaCl (2.5 M) and stirred at 25°C for 5 min. The mixture was transferred to centrifuge tubes, stored at 4°C for 24 h and spun at 9500 r.p.m., 4°C, for 18 min. The supernatant was decanted and the pellet was resuspended in TE buffer (pH 8.0, 8.0 ml), which was extracted four times with phenol/chloroform/isoamyl alcohol (25:24:1, 3 ml). The aqueous layer was loaded onto a hydroxyapatite column (2 g + 30 ml H2O), which was first washed with H2O (10 ml) and then TE buffer pH 8.0 (5.0 ml). DNA was eluted using 10 ml of elution buffer (0.07 M K2HPO4, 0.07 M KH2PO4) and further purified using a Centricon cartridge (100 000 NMWL) following the manufacturer's protocol. Isolated plasmids containing the appropriate inserts (12a or 12b) were verified by direct sequencing (see Supplementary Data).
Interstrand cross-linking using plasmid DNA target
Targets 12a and 12b (1.5 pmol) were hybridized with probe (3 or 4, 2 eq.) in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl by heating at 70°C for 5 min and then cooling to 4°C over 2 h. NaIO4 (5 mM) reactions of DNA duplexes (30 nM) were carried out in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl at 25°C for 6 h (direct detection) or 45°C for 5 h (indirect detection).
Detection of mismatches via fluorescence spectroscopy
(i) The cross-linked samples were dissolved in 0.5 ml with PPF buffer (100 mM potassium phosphate pH 4.7 and 50% formamide). The sample was then heated at 80°C for 10 min and centrifuged in the Centricon cartridge (100 000 NMWL) for 10 min at 5000 r.c.f. The retentates were dissolved in 0.5 ml of the hot PPF buffer (preheated at 70°C) and centrifuged. This procedure was repeated. The retentates were dissolved in 0.5 ml of TE buffer (10 mM Tris, 1.0 mM EDTA, pH 7.5) and centrifuged (2×). The final retentate was dissolved in 200 μl of TE buffer and quantified by UV-measurement. (ii) A 96-well black flat-bottom plate was incubated with 0.1% protamine (200 μl) for 20 h at 4°C, followed by washing with water (3 × 250 μl). Plasmid DNA (300 fmol, 200 μl) in TE buffer (10 mM Tris, 1 mM EDTA, pH 7.5) was incubated in the pretreated assay plate in an incubator at 37°C for 2 h without shaking. After washing with TPBS buffer (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4 and 0.1% Tween-20, pH 7.5, 10 × 250 μl), the plate was incubated with BSA/SuperBlock solution (1 ml/10 ml) (2 × 250 μl) for 15 min at room temperature with shaking. The plate was then washed with TPBS buffer (5 × 250 μl), followed by incubation with ABC ultra solution (50 times dilution, Vector lab) for 30 min at room temperature. After washing with TPBS (10 × 250 μl), the plate was incubated with Amplex Ultra Red substrate solution (100 μl, 0.1 mM Amplex Ultra Red and 0.015% H2O2 in 50 mM sodium phosphate buffer, pH 7.5) for 30 min at room temperature with shaking, followed by addition of 20 μl Amplex Ultra Red Stop solution (Invitrogen). Sodium phosphate buffer (180 μl, 50 mM, pH 7.5) was added into each well to achieve the final volume (300 μl). The fluorescence of each well in the plate was then determined using a fluorescence plate reader [excitation: 568 nm, emission: 581 nm, PMT voltage 550 (when using indirect probe 4), 520 (when using direct probe 3), slit width 10 nm]).
RESULTS AND DISCUSSION
Experimental design
Hybridization methods distinguish between fully matched and single nucleotide mismatched sequences by taking advantage of higher melting temperatures of duplexes containing the former. The utilization of hybridization methods are complicated by conflicting properties of duplex DNA. Differences in melting temperatures of fully matched versus single nucleotide mismatched duplexes are inversely proportional to duplex length. However, the overall stability of duplexes increases with increasing length. Duplex stability is important because one needs to remove the mismatched probes, typically by washing, without disrupting the fully matched complexes. Alternative backbones that form more stable complexes with DNA targets, such as peptide nucleic acids (PNAs) can help overcome these conflicting properties (7). We anticipated that the recently discovered interstrand cross-linking reaction (Scheme 1) induced upon mild oxidation of the modified thymidine (1) would enable us to utilize short oligonucleotides to selectively target nucleic acid sequences.
Detection using HRP conjugated to avidin is attractive because it is sensitive, easy to carry out and large numbers of scientists are familiar with it. As described below, the ability to covalently link the probe and target is crucial for employing this detection method (Scheme 2). Sandwich assays that utilize biotin binding to an avidin-horseradish peroxidase (avidin-HRP) conjugate detect as little as 25 fmol of DNA (15,18). The detection limit of the assay is dependent upon the ability to achieve low background fluorescence levels by effectively separating unbound avidin-HRP from biotinylated target. We anticipated that a biotinylated oligonucleotide probe covalently linked to target DNA (Scheme 1) would withstand rigorous conditions for removing excess probe and avidin-HRP (Scheme 2).
The cross-linking reaction is useful in the presence of a variety of flanking sequences
The cross-linking reaction between 1 and an opposing dA upon mild oxidation by NaIO4 proceeds through the rearranged methide (2, Scheme 1). In order for this reaction to be useful as part of a probe to specifically detect a complementary sequence, it must proceed comparably regardless of flanking sequence. Cross-linking was examined in 4 of the 16 possible flanking sequences at 37°C (Table 1). The rate constants spanned <3-fold range. Furthermore, cross-linking proceeded more rapidly when 1 was flanked by pyrimidines than by larger purines. We propose that the greater size of the purines and Π-stacking energy increases the barrier to interconversion between the anti and syn forms of the methide like electrophile (2, Scheme 1). Overall, the kinetic experiments indicate that the cross-linking reaction is not strongly biased by flanking sequence and will be generally useful for detecting oligonucleotide sequences.
Table 1.
Interstrand cross-link (ICL) yields from 1 as a function of flanking sequence
| 5′-d(AGA TGG AN1 NAG GTA C) | |||
|---|---|---|---|
| 3′-d(TCT ACC TN′A N′TC CAT G) | |||
| N:N′ | k (s−1)a | t1/2 (min) | Yield ICL (%) |
| T:A | 1.2 ± 0.3 × 10−3 | 9.4 ± 0.6 | 33.8 ± 1.4 |
| A:T | 5.1 ± 0.2 × 10−4 | 22.6 ± 0.3 | 24.9 ± 0.5 |
| C:G | 1.5 ± 0.2 × 10−3 | 7.7 ± 0.6 | 34.8 ± 0.5 |
| G:C | 6.4 ± 0.4 × 10−4 | 18.0 ± 0.6 | 18.6 ± 1.2 |
aAverage of three independent experiments each carried out in triplicate.
Analysis of single nucleotide discrimination by gel electrophoresis
With this strategy in mind, bifunctional oligonucleotide probes (3, 4) equipped with biotin at their 5′-termini and 1 at an internal site were synthesized using commercially available reagents and the previously reported requisite phosphoramidite for introducing 1 (13,14,19). The oligonucleotides were prepared using standard solid-phase oligonucleotide synthesis methods. The probes were chosen to be 16-nt long in order to target unique sequences in the human genome. The specific sequences were designed to target codon 248 in exon 7 of the p53 gene. The p53 gene is often mutated in human cancer, and there is a high frequency of G → A transitions at codon 248 (the following website catalogues p53 mutations: http://p53.free.fr.). The ‘direct’ probe (3) was designed to utilize 1 to cross-link (‘tag’) and to discriminate between matched (dA) and mismatched sequences. Denaturing polyacrylamide gel electrophoresis (PAGE) was used to test the ability of the biotinylated cross-linking probes to discriminate against single nucleotide mismatches. The chemoselectivity of the cross-linking reaction was exploited when using the direct probe (3) to distinguish between a target having an opposing dA (5a) or dG (5b). The methide intermediate produced upon oxidation of 1 reacts with the N1-position of dA (Scheme 1), which is more nucleophilic than the corresponding nitrogen in dG (14). Indeed, cross-linked product was observed in 47.2 ± 0.8% when 3 was hybridized with the matched (5a) sequence (6) and treated with NaIO4 at 25°C. No product was detected when dG was opposite 1, despite the fact that the Tm (7, Table 2) was ∼20°C higher than the reaction temperature. Carrying out the incubation at 40°C decreased the yield of cross-link in the matched duplex (6) to 34.7%, but still no product was observed when 1 was opposite dG (7). The lower cross-link yield may be due to decreased residence time of the duplex, but we also cannot eliminate other effects, such as changes in the relative reaction rates of the methide with dA and water.
Table 2.
Duplex melting temperatures
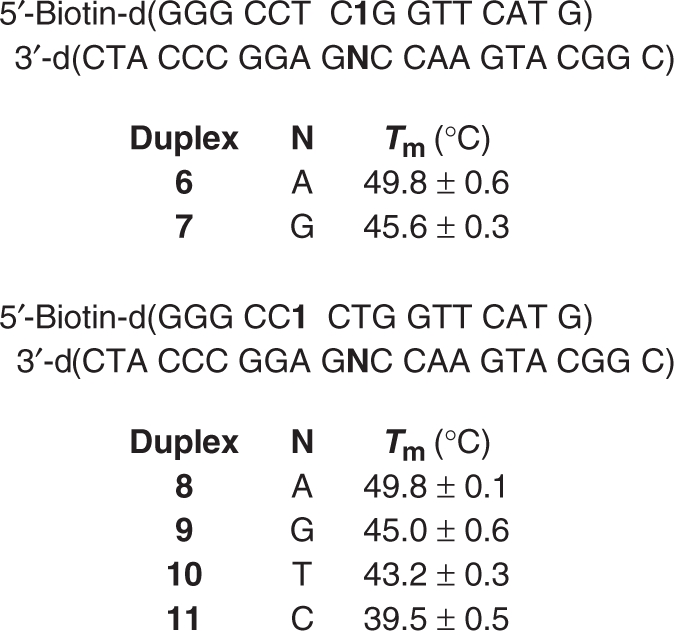 |
If this was the only manner in which 1 could be used to probe DNA sequence, the method would be limited to detecting sequences in which dA was the target. Consequently, ‘indirect’ probe 4 was prepared in order to demonstrate that 1 could in principle be used to detect any DNA sequence containing a dA. The incubation temperature was critical when using the indirect probe (4) where duplex thermal stability (Table 2) was expected to be the primary determinant for achieving selectivity. The matched duplex (8) containing the indirect probe (4) melts more than 4°C higher than that of any mismatch, indicating that the presence of the nonnative nucleotide (1) does not eliminate the differences in thermal stabilities of matched and mismatched duplexes. Using the Tm's as a guide we carried out the hybridization/cross-linking reactions using the indirect probe at the approximate Tm of the most stable mismatch (9, T:G, 45°C, Table 2). Higher cross-linking yields for the matched sequence (8) were obtained at temperatures lower than the Tm of the T:G mismatch (9), but at a cost in selectivity (Table 3). Increasing the reaction temperature above the Tm of 9 improved selectivity, but also resulted in a significant decrease in cross-linking yield for the matched (8, T:A) sequence (Table 3). The most satisfactory combination of cross-link yield (Table 3) and selectivity was obtained by carrying out the reaction at the Tm of the most stable mismatch (Table 2). Consequently, the selectivity of the cross-linking reaction involving 1 opposite dA was screened (8–11) at 45°C (Table 4). As expected, there was a correlation between the ICL yield and Tm. The matched cross-link yield was lower than that observed for the direct probe (3) which was measured at lower temperatures. The decreased yield using 4 may affect the selectivity observed in the fluorescence experiments (see below).
Table 3.
Interstrand cross-link (ICL) yields from 1 as a function of temperature
| Temp. (°C) | % Yield ICL | ||
|---|---|---|---|
| 8a | 9a | Selectivityb | |
| 25 | 47.2 ± 0.8 | 44.5 ± 0.4 | 1.1 |
| 40 | 34.7 ± 0.9 | 11.0 ± 0.3 | 3.2 |
| 43 | 26.3 ± 1.7 | 2.3 ± 0.2 | 11.4 |
| 44 | 23.7 ± 1.0 | 1.8 ± 0.3 | 13.2 |
| 45 | 21.4 ± 0.3 | 0.9 ± 0.1 | 23.8 |
| 46 | 8.9 ± 0.5 | 0.1 ± 0.1 | 89.0 |
aAverage of three independent samples.
bRatio of average ICL yield in 8 : 9.
Table 4.
Interstrand cross-link (ICL) yields from 4 as a function of opposing nucleotide at 45°C
| Duplex | N | Yield ICLa (%) | Selectivityb |
|---|---|---|---|
| 8 | A | 21.4 ± 0.3 | – |
| 9 | G | 0.9 ± 0.1 | 23.8 |
| 10 | T | 0.3 ± 0.1 | 71.3 |
| 11 | C | 0.1 ± 0.1 | 214.0 |
aAverage of three independent samples.
bRatio of ICL yield in 8 : respective duplex.
Fluorescence detection of single nucleotide discrimination in plasmid DNA
Having established the sequence selectivity of the tagging reaction, the utility of the SNP method was demonstrated using a fluorescence assay in microtiter plates (Scheme 2). Single-stranded plasmids (12a, 12b) containing 5a or 5b were prepared from M13mp7. The plasmid was linearized by cleaving a hairpin site with EcoR1 and then recircularized with the appropriate insert. Cells were transfected with the circular plasmids via electroporation and recovered by standard methods following overnight growth.
The assay conditions were optimized using the direct probe (3). Plasmids were hybridized with a 2-fold molar excess of the direct or indirect probes. Following addition of NaIO4 (5 mM) to initiate the cross-linking reaction, the samples were incubated with the direct probe at 25°C for 6 h. Removal of the excess biotinylated probe (3), crucial for achieving sufficiently low background fluorescence levels needed for sensitive detection, was enabled by the cross-linking reaction. The dehybridized (80°C) mixture was filtered through a membrane, which was then washed with warm buffered formamide solution (2×), followed by pH 7.5 buffer (2×) (20). The biotinylated plasmid was then transferred to the well of an activated microtiter plate, conjugated to avidin-HRP and reacted with Amplex Red and H2O2. The emission (581 nm) was measured for the matched (opposing dA, 12a) and mismatched (opposing dG, 12b) samples (Figure 1). The fluorescence was measured as a function of plasmid loading in the microtiter plate well and reached a maximum at 250 fmol (Figure 1). Subsequent experiments were carried out using 300 fmol of plasmid. Background measurements (in triplicate) were made for samples in which plasmid was hybridized with probe but not oxidized. The reported fluorescence signals (Figure 2) were determined by subtracting the average background signal from the average signal obtained from samples in the presence of NaIO4. Most importantly, the discrimination for the matched versus mismatched sequence was 193:1 (Figure 2A, Table 5).
Figure 1.

Fluorescence intensity versus amount of plasmid loaded (plasmid 12a, black square; plasmid 12b, red circle).
Figure 2.

Selective DNA detection in plasmids (12a, 12b) via fluorescence using bifunctional oligonucleotides. (A) Using direct probe 3. (B) Using indirect probe 4. Signal for matched pairs are in black; mismatched are in red.
Table 5.
Fluorescence detection of DNA in single stranded plasmids
| Plasmid | Probe | Fluor. Int.a | Selectivityb |
|---|---|---|---|
| 12a | 3 | 964 ± 4 | – |
| 12b | 3 | 5 ± 3 | 193 |
| 12a | 4 | 849 ± 9 | – |
| 12b | 4 | 19 ± 4 | 45 |
aAverage of three independent samples.
bRatio of average fluorescence intensity in matched versus mismatched plasmid.
The ability of the indirect probe (4) to discriminate between plasmids containing a matching (dA, 12a) or mismatching (dG, 12b) target sequences was determined using the procedure optimized for the direct probe with the exception that the reaction was carried out at the previously optimized temperature (Table 3, 45°C). The fluorescence intensities (Figure 2B, Table 5) correlated well with the cross-linking yields measured by denaturing PAGE (Table 4). For instance, the overall intensity for the matched sequence was lower than that observed using the direct probe (3). In addition, the signal from the mismatched target was slightly greater than when the direct probe was used. Of the two effects the signal from the mismatched indirect probe (4 and 12b) accounted for most of the reduction in selectivity when using the indirect probe. Nonetheless, the indirect probe (4) showed 45-fold selectivity for the matched versus mismatched target sequence.
CONCLUSION
We have developed a facile method for detecting DNA sequences capable of single nucleotide discrimination that detects target DNA at nanomolar concentration. By relying upon DNA interstrand cross-linking to dA by a modified thymidine, oligonucleotides (16 nt) whose complexes would otherwise be too unstable can be used (7). In addition, cross-linking to the target enables us to detect and amplify a specific sequence using biotin-avidin binding. The bifunctional probes are, in principle, capable of probing any sequence that contains a dA and show excellent selectivity of plasmid targets using short oligonucleotides, which along with eliminating PCR as a step in this process, makes it a rapid and inexpensive DNA detection method.
SUPPLEMENTARY DATA
Supplementary Data are available are NAR Online.
ACKNOWLEDGEMENTS
We are grateful for support of this research from the National Institute of General Medical Sciences (GM-054996). Funding to pay the Open Access publication charges for this article was provided by GM-054996.
Conflict of interest statement. None declared.
REFERENCES
- 1.Tyagi S, Marras SAE, Kramer FR. Wavelength-shifting molecular beacons. Nat. Biotechnol. 2000;18:1191–1196. doi: 10.1038/81192. [DOI] [PubMed] [Google Scholar]
- 2.Frutos AG, Pal S, Quesada M, Lahiri J. Method for detection of single-base mismatches using bimolecular beacons. J. Am. Chem. Soc. 2002;124:2396–2397. doi: 10.1021/ja012374d. [DOI] [PubMed] [Google Scholar]
- 3.Nakatani K. Chemistry challenges in SNP typing. Chembiochem. 2004;5:1623–1633. doi: 10.1002/cbic.200400161. [DOI] [PubMed] [Google Scholar]
- 4.Rosi NL, Mirkin CA. Nanostructures in biodiagnostics. Chem. Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 5.Grossmann TN, Roglin L, Seitz O. Triplex molecular beacons as modular probes for DNA detection. Angew. Chem. Int. Ed. 2007;46:5223–5225. doi: 10.1002/anie.200700289. [DOI] [PubMed] [Google Scholar]
- 6.Ogasawara S, Fujimoto K. SNP Genotyping by using photochemical ligation. Angew. Chem. Int. Ed. 2006;45:4512–4515. doi: 10.1002/anie.200600790. [DOI] [PubMed] [Google Scholar]
- 7.Zhang N, Appella DH. Colorimetric detection of anthrax DNA with a peptide nucleic acid sandwich - hybridization assay. J. Am. Chem. Soc. 2007;129:8424–8425. doi: 10.1021/ja072744j. [DOI] [PubMed] [Google Scholar]
- 8.Sando S, Sasaki T, Kanatani K, Aoyama Y. Amplified nucleic acid sensing using programmed self-cleaving DNAzyme. J. Am. Chem. Soc. 2003;125:15720–15721. doi: 10.1021/ja0386492. [DOI] [PubMed] [Google Scholar]
- 9.Takada T, Fujitsuka M, Majima T. Single-molecule observation of DNA charge transfer. Proc. Natl Acad. Sci. USA. 2007;104:11179–11183. doi: 10.1073/pnas.0700795104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nam JM, Stoeva SI, Mirkin CA. Bio-bar-code-based DNA detection with PCR-like sensitivity. J. Am. Chem. Soc. 2004;126:5932–5933. doi: 10.1021/ja049384+. [DOI] [PubMed] [Google Scholar]
- 11.Sando S, Kool ET. Imaging of RNA in bacteria with self-ligating quenched probes. J. Am. Chem. Soc. 2002;124:9686–9687. doi: 10.1021/ja026649g. [DOI] [PubMed] [Google Scholar]
- 12.Silverman AP, Baron EJ, Kool ET. RNA-templated chemistry in cells: discrimination of Escherichia, Shigella and Salmonella bacterial strains with a new two-color FRET strategy. Chembiochem. 2006;7:1890–1894. doi: 10.1002/cbic.200600278. [DOI] [PubMed] [Google Scholar]
- 13.Hong IS, Greenberg MM. DNA interstrand cross-link formation initiated by reaction between singlet oxygen and a modified nucleotide. J. Am. Chem. Soc. 2005;127:10510–10511. doi: 10.1021/ja053493m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong IS, Ding H, Greenberg MM. Oxygen independent DNA interstrand cross-link formation by a nucleotide radical. J. Am. Chem. Soc. 2006;128:485–491. doi: 10.1021/ja0563657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue L, Greenberg MM. Facile quantification of lesions derived from 2'-deoxyguanosine in DNA. J. Am. Chem. Soc. 2007;129:7010–7011. doi: 10.1021/ja072174n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- 17.Delaney JC, Essigmann JM. Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Methods Enzymol. 2006;408:1–15. doi: 10.1016/S0076-6879(06)08001-3. [DOI] [PubMed] [Google Scholar]
- 18.Dhar S, Kodama T, Greenberg MM. Selective detection and quantification of oxidized abasic lesions in DNA. J. Am. Chem. Soc. 2007;129:8702–8703. doi: 10.1021/ja073014e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong IS, Greenberg MM. Efficient DNA interstrand cross-link formation from a nucleotide radical. J. Am. Chem. Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 20.Hearst JE. A photochemical investigation of the dynamics of oligonucleotide hybridization. Ann. Rev. Phys. Chem. 1988;39:291–315. doi: 10.1146/annurev.pc.39.100188.001451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.