


Abstract
Adenovirus VA RNAs are short non-coding transcripts that assist in maintaining viral protein expression in infected cells. Six sets of mismatch and compensatory base pair mutants of VA RNAI were examined by gel mobility and RNA UV melting to assess the contribution of each structural domain to its overall structure and stability. Each domain of VA RNAI was first assigned to one of two apparent unfolding transitions in the wild-type melting profile. The Terminal Stem and Central Domain unfold in a single cooperative apparent transition with an apparent Tm of ∼60°C. In contrast, the Apical Stem unfolds independently and with much higher apparent Tm of ∼83°C. Remarkably, this domain appears to behave as an almost entirely autonomous unit within the RNA, mirroring the functional division within the RNA between PKR binding and inhibition. The effects of mismatch and compensatory mutations at five of the six sites on the RNA melting profile are consistent with proposed base pairing and provide further validation of the current secondary structure model. Mutations in the Central Domain were tested in PKR inhibition assays and a component of the VA RNAI Central Domain structure essential for PKR inhibitory activity was identified.
INTRODUCTION
Adenoviruses devote a significant portion of their genome to ‘immune evasion’ genes that allow them to maintain persistent infections (1). Among these are the genes encoding the ‘Virus-associated’ (VA) RNAs, short non-coding RNA polymerase III transcripts that accumulate to extremely high levels in late stages of infection (2,3). All adenoviruses encode at least one such RNA and sometimes two, named VA RNAI and VA RNAII, respectively. Where two are produced, VA RNAI is the most active species and accumulates to higher levels. This may reflect a level of redundancy or it is possible that other undefined roles of VA RNAII may also exist (4). The best characterized role of VA RNAI is to inhibit the double stranded RNA-activated protein kinase PKR that would otherwise phosphorylate the eukaryotic initiation factor 2 (eIF2) to halt protein synthesis in the infected cell (3,5–8).
VA RNAI sequences from different adenovirus serotypes vary considerably (9), but each can be drawn in a similar extended secondary structure containing three major domains (10,11): a Terminal Stem (including the paired 5′- and 3′-ends), a Central Domain and an Apical Stem (Figure 1). Specific functions have been delineated for each domain. The Terminal Stem contains internal promoter sequences, may fulfil a role in protecting the RNA against exonuclease degradation (9), and has been implicated in suppression of RNAi mechanisms (12). The Apical Stem is the primary site of interaction for the two dsRNA binding domain (dsRBD) motifs of PKR (13–17) while the Central Domain contributes to PKR binding and is responsible for its inhibitory activity (10,13–15,18–20).
Figure 1.

Adenovirus type 2 (Ad2) VA RNAI secondary structure and compensatory base pair mutations. Secondary structure model proposed following identification of two highly conserved complementary tetranucleotides (shown in outline typeface) and redrawing the Central Domain to base pair these sequences (9,14,20). Compensatory base pair modifications were made at six sites (1–6) distributed through the Terminal Stem, Central Domain and Apical Stem (see Table 1).
Initial secondary structure models for VA RNAI (10,11,21) were refined by extensive mutagenesis and structure probing studies (14,22,23) and by comparative sequence analysis (4,9). The most recently proposed secondary structure (Figure 1) incorporates the pairing of two highly conserved tetranucleotides (GGGU/ACCC, nucleotides 37–40/119–122) within the Central Domain (9,14,20). Mutation of either strand reduced RNA activity significantly and although compensatory changes partially restored function, PKR inhibition did not reach wild-type levels (20). Other investigations into VA RNAI structure by mutagenesis have typically used either large deletions or alterations of the RNA sequence (10,14,18,22). From these it was broadly concluded that the length and position of the Apical Stem are essential for VA RNAI function but the precise sequence is less important (22). However, one study using a series of point mutations within the Central Domain showed that approximately 40% of the changes reduced RNA function to some extent suggesting a degree of sequence specificity (24).
While considerable insight into VA RNAI structure and function has been gleaned from this large body of work, it is clear that alternative approaches are required for further understanding. Adenovirus VA RNAs are highly G/C rich (∼65% for Adenovirus type 2; Figure 1) and have high thermal stability. We designed a strategy to exploit these properties to probe the RNA stability and structure of Adenovirus type 2 (Ad2) VA RNAI using site-directed mutagenesis and UV melting analysis. Mismatch and compensatory mutations could be introduced in continuous helical regions without resulting in complete destabilisation of the RNA structure and changes in UV melting profiles could be readily detected upon switching tandem G–C base pairs to A–U. The sites of modification were distributed throughout the VA RNAI structure in order to assess the contribution of each domain to the global RNA architecture and stability. These studies support the current VA RNAI secondary structure model (Figure 1) but uncover a remarkable division between the two functional domains of VA RNAI, responsible for binding or inhibition of PKR, which appear to exist independently within the RNA structure.
MATERIALS AND METHODS
Mutagenesis and preparation of VA RNAI in vitro transcripts
A plasmid for in vitro transcription of Ad2 VA RNAI with a 3′-hepatitis delta virus (HDV) ribozyme was created as described previously (26). Mutant sequences (Table 1 and Figure 1) were generated in this plasmid using the QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by automated DNA sequencing. Templates for in vitro transcription reactions with T7 RNA polymerase (T7 RNAP) were prepared by DraI digestion of CsCl gradient purified plasmid DNA. Transcription reactions (0.5–1.0 ml) were performed using 100 μg/ml template DNA under optimal conditions for VA RNAI (27). RNA transcripts were purified by preparative polyacrylamide gel electrophoresis with gels containing 50% urea and 8% acrylamide and eluted from excised bands using a Biotrap device (Schleicher and Schuell). The integrity of the recovered RNA was assessed by urea denaturing PAGE. This process uncovered the electrophoretic mobility differences for one set of mutants (Figure 2).
Table 1.
Mismatch and compensatory base pair mutations in VA RNAI
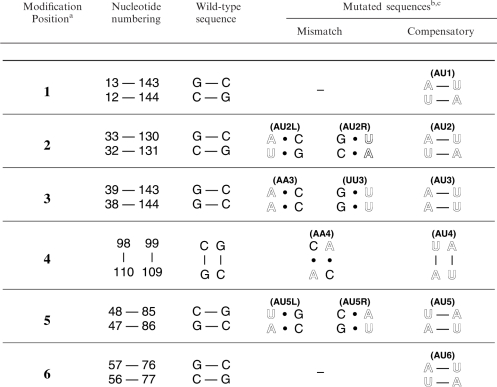
|
aCorrespond to boxed sites 1–6 on the VA RNAI secondary structure (Figure 1).
bMutated nucleotides are shown in outline typeface.
cRNA names in parenthesis are those used in the main text and Figures 2, 4 and 5 to denote the RNA containing the shown mutations.
Figure 2.

Denaturing polyacrylamide gel mobility assay. Wild-type and mutant VA RNAIs were resolved on an 8% polyacrylamide gel containing 50% urea. Size markers (M) correspond to RNAs of 170 and 220 nt.
UV melting analysis of wild-type and mutant VA RNAI
Typically, samples contained ∼20–25 μg RNA in a buffer solution containing 10 mM MOPS buffer pH 7.0 and 50 mM KCl. UV melting curves were measured on a Varian Cary 400 UV/Vis spectrophotometer, equipped with a 6-cell multichanger and in-sample temperature probe. Up to five melting curves were collected in a single run with a heating rate of 0.5°C/min. First derivatives (‘melting profiles’) of the melting curves (Figure 3) were calculated using a Savitsky–Golay algorithm as implemented in the program ‘OD Deriv’ (D.E. Draper, Johns Hopkins University; http://www.jhu.edu/~chem/draper/). The melting profile of wild-type VA RNAI was essentially unaffected by choice of monovalent salt. Higher concentrations of salt or addition of Mg2+ produced a general stabilization of both peaks in the melting profile with no evidence of specific stabilization of one component of the structure. However, under conditions of higher ionic strength than those used here, the second apparent transition rapidly increased in stability to beyond the measurable range (>98°C; data not shown). To simplify comparison of the relative hypochromicities at 260 and 280 nm for one set of mutants (site 2; Figure 5A and inset), their melting profiles were normalized at both wavelengths to the maximum value of the peak centred on 60°C. This corresponds to the portion of the Terminal Stem/Central Domain structure unaffected by the mutation and is identical in Tm and hypochromicity for each mutant at the same wavelength. The large increase in relative hypochromicity at 260 compared to 280 nm for the compensatory mutant AU2 is indicative of base pairing between these nucleotides.
Figure 3.

UV melting analysis of wild-type Adenovirus type 2 (Ad2) VA RNAI. Normalized UV melting curves (grey) collected at 260 nm (solid line) and 280 nm (dashed line) shown scaled to the 280 nm data. The corresponding ‘melting profiles’, the first derivative of the melting curve, at each wavelength are shown in black.
Figure 5.

Uncoupling of apparent transitions in VA RNAI with mismatch and compensatory mutations at sites 2–5. Melting profiles for single strand and partial mutations (denoted AA, UU or R/L, see Table 1), and compensatory mutations at (A) site 2, (B) site 3, (C) site 4 and (D) site 5. The melting profile of wild-type VA RNAI at 280 nm is shown as a shadow in each plot for comparison. In (A), the inset plot shows the shoulder region with the profiles at 260 nm and 280 nm normalized to the height of the apparent transition centred on 60°C; this indicates a significant increase in relative hypochromicity at 260 nm for the compensatory mutant (marked with double-headed arrow).
PKR autophosphorylation inhibition assays
PKR was expressed in a non-phosphorylated form in E. coli and purified as described previously (28). PKR was dialysed into 2× reaction buffer (100 mM Tris pH 7.8, 100 mM KCl, 10% glycerol, 5 mM DTT) and purified VA RNAI diluted into the same buffer to prepare 5× stocks for each concentration tested. PKR (∼0.1 μg) was pre-incubated with each VA RNAI concentration at room temperature for 15 min. An equal volume of the activating solution (0.02 μg/ml poly(I).poly(C) activator RNA, 0.2 mCi/ml 32P ATP (6000 Ci/mmole; 10 mCi/ml in buffered solution; MP Biomedicals), 40 μM ATP, 4 μM MgCl2) was added and the mixture incubated at room temperature for a further 10 min. The reaction was stopped by the addition of 0.5 volume of 3× SDS PAGE loading dye. Samples were heated at 90°C for 2 min and fractionated on a 7.5% acrylamide SDS PAGE gel. Gels were fixed, dried and exposed to an imaging plate (Molecular Dynamics) for 30 min and viewed using a Typhoon 8600 variable mode phosphorimager. Quantitation of autophosphorylation was performed using ImageJ software.
RESULTS
Site-directed mutagenesis was used to incorporate compensatory base pair mutations into each structural domain of VA RNAI, at six sites in total (Figure 1 and Table 1). Each modification involved the exchange of two Watson–Crick G–C for A–U base pairs within a continuous helical segment of secondary structure of not less than four base pairs. In three cases both flanking base pairs were G–C (sites 4, 5 and 6), and for the others by one G–C and one A–U base pair (sites 1, 2 and 3). The modifications were located to probe each of the helical regions of VA RNAI, specifically: the lower (1) and upper (2) regions of the Terminal Stem, the Central Domain (3 and 4), and the lower (5) and upper (6) regions of the Apical Stem (Figure 1). Seven further mutations that introduced A • C and G • U mismatched pairs at four sites were also created (Table 1).
Denaturing PAGE analysis of wild-type and mutant VA RNAI
VA RNAI exhibits an aberrant mobility on denaturing polyacrylamide gels, migrating with an apparent size of ∼220 nt (14). None of the compensatory or mismatch modifications altered the length of the RNA and the effect of each on the global RNA structure was expected to be small, particularly for the compensatory base pair changes. No significant effect on the VA RNAI gel mobility was therefore anticipated and this was found to be the case for all mutants generated except those at site 5, at the base of the Apical Stem. Here, each mutation dramatically altered the RNA's electrophoresis mobility (see Figure 2; compare wild-type VA RNAI in Lane 1 with Lanes 6, 13 and 14). The effect is greatest for the 5′-strand partial mutant (AU5L; Lane 13), which migrates as expected for an RNA of 155 nucleotides. However, the reduction in gel retardation appears largely independent of which strand is altered and is substantial even for the compensatory base-paired mutant (Lane 6).
UV melting analysis of wild-type and base pair compensatory mutants of VA RNAI
UV melting analysis can be used in simple cases to derive a range of thermodynamic parameters for nucleic acids. For large structured RNAs, however, unfolding typically occurs in multiple overlapping transitions and the resulting unfolding ‘profiles’ are often complex (25). The peaks in the melting profiles presented here are therefore referred to as ‘apparent’ transitions, and the analysis of thermodynamics of RNA unfolding is limited to an estimation of the apparent Tm associated with each apparent transition in the profile.
Wild-type Ad2 VA RNAI unfolds with two major apparent transitions, with Tm values of approximately 60°C and 83°C (Figure 3) under the solution conditions used. All UV melting profiles were collected under identical conditions of 10 mM MOPS buffer at neutral pH with 50 mM KCl (see ‘Methods’ section), which was found to be optimal for measuring the apparent transition Tm values. KCl could be substituted for other monovalent salts with little effect. However, addition of higher concentrations of salt or addition of Mg2+ dramatically increased both apparent Tms but with little effect on the shape of the melting profile for wild-type VA RNAI (data not shown). Addition of as little as 0.5 mM MgCl2 increased the Tm of the first apparent transition by 12°C to ∼72°C and increased the Tm of the second apparent transition beyond the measurable range (>98°C). The stabilising effect of Mg2+ on the first apparent transition reached a maximum at ∼5 mM with an apparent Tm of 81°C (data not shown).
The compensatory base pair changes at each site in VA RNAI were first used to assign each structural domain of VA RNAI to these apparent transitions (Figure 4). Compensatory changes in the Terminal Stem (AU1 and AU2 RNAs) and Central Domain (AU3 and AU4 RNAs) affect only the first apparent transition. In contrast, the Apical Stem compensatory changes (AU5 and AU6 RNAs) affect only the second, higher temperature apparent transition. These mutations clearly assign unfolding of the Terminal Stem and Central Domain to the first, and the unfolding of the Apical Stem to the second apparent transition. For two of the compensatory changes, AU1 and AU6 RNAs, the shape of the melting profile was unaltered but a shift in hypochromicity (increased at 260 nm compared to 280 nm) and reduction in apparent Tm (decreases of approximately 4°C and 8°C, respectively) were observed. These are the changes that might be predicted upon exchange of G–C for A–U base pairs within an otherwise unaltered RNA structure (see ‘Discussion’ section). Compensatory mutations at the remaining four sites, AU2-5 RNAs, produce more complex effects on the melting profile. This indicates that each apparent transition is composed of at least two or more cooperative unfolding transitions in the wild-type RNA and that these transitions can be uncoupled by changes to the stability of specific regions of the RNA. These were investigated further using partial, mismatch mutants at sites 2–5 (Table 1). In all these additional mutant RNAs, the effect of each mutation remained almost exclusively restricted to the same apparent transition as for the complete compensatory change.
Figure 4.

UV melting profiles for wild-type VA RNAI and compensatory base pair mutants. (A) Melting profiles collected at 280 nm for wild-type (black) and compensatory base pair mutant VA RNAIs: AU1 (red), AU2 (orange), AU3 (light green), AU4 (dark green), AU5 (cyan), and AU6 (blue). (B) Melting profiles collected at 260 nm coloured as in (A). Note that the hypochromicity is significantly greater at 280 nm than 260 nm; however, compensatory base pair changes from G–C to A–U may increase the relative hypochromicity at 260 nm for the apparent transitions affected. Mutations in the Terminal Stem and Central Domain affect only the first apparent transition (Tm ∼60°C) while mutations in the Apical Stem affect only the second (Tm ∼83°C).
Uncoupling of connected unfolding transitions using mismatch mutations
All mutations at site 2 cause an uncoupling of unfolding transitions within the first apparent transition with no effect on the second transition (Figures 4 and 5A). Mismatch mutations on either side of the helix that introduce adjacent A • C and G • U base pairs, in AU2L and AU2R RNAs (Table 1), both produce a shoulder on the low temperature side of the first apparent transition. The compensatory base pairing does not restore a wild-type like profile as shown by the AU2 RNA. All three RNAs exhibit an equal lowered hypochromicity in the apparent transition that remains centred at ∼60°C. However, when the 260 nm and 280 nm data for each RNA are normalized to the absorbance of the invariant apparent transition at Tm ∼60°C, it is clear that the difference in hypochromicity at 260 nm versus 280 nm is significantly greater in the compensatory base paired RNA than either mismatch mutant (Figure 5A, inset), consistent with base pairing of these nucleotides.
Mismatch and compensatory mutations at site 3 also affect only the first apparent transition (Figure 5B). The 5′-strand was modified to create tandem destabilising A • C mismatches (AA3 RNA; Table 1) and, accordingly, this was expected to have the most deleterious effect of all changes at site 3. However, the changes in the melting profile for AA3 RNA compared to wild-type VA RNAI are relatively modest. A shoulder is observed on the low temperature side of the 60°C apparent transition, which is still present in the melting profile with only a small reduction in hypochromicity. The melting profile is most similar to that of AU2L RNA, the mismatch mutation on the opposite strand about half a helical turn below. The melting profile for the compensatory base pair mutant has a broadened shoulder with reduced apparent Tm. The most dramatic effect at site 3, however, is caused by mutation of the 3′-strand to create tandem G • U mismatches at this site (UU3 RNA). Hypochromicity of Tm ∼60°C peak is reduced further as more apparent unfolding transition(s) are uncoupled. There are now at least three apparent transitions visible: one broad transition at low temperature, a sharper shoulder at ∼53°C, and the same apparent transition, but with further reduced hypochromicity, centred at ∼60°C. Interpretation of the effects of mutations at this site may be complicated by possible involvement of these nucleotides in a higher order structure of the Central Domain (see ‘Discussion’ section).
The compensatory mutation at site 4 (AU4 RNA) also produces a new lower temperature apparent transition that appears as a shoulder on the apparent transition at Tm ∼60°C (Figure 5C). As before, this is still present in the melting profile but with reduced hypochromicity, while the high temperature apparent transition is completely unaffected. One additional mutation was made at site 4 that introduces tandem A • C mismatches (AA4 RNA; Table 1). The shoulder observed for AU4 RNA is no longer present in AA4 RNA although the first apparent transition still exhibits significantly lower hypochromicity than wild-type (Figure 5C) indicating that this RNA lacks some element(s) of the wild-type RNA fold, potentially the Central Domain tertiary structure. To assess the importance of this lost structure for VA RNAI activity, PKR kinase inhibition assays were performed with wild-type, AU4 and AA4 RNAs (see below).
Compensatory mutations at sites 5 and 6 affected only the second apparent unfolding transition with a more dramatic effect at site 5 (Figure 4). Mismatch mutations creating adjacent A • C and G • U mismatches at this position (AU5L and AU5R) affect the high temperature transition similarly. Although the RNAs appear to retain their structure, the apparent transition is uncoupled into at least two separate apparent transitions. All three RNAs with mutations at site 5 retain some structure that unfolds in a broad transition centred around 83°C. AU5 and AU5R RNAs have an apparent transition in their melting profiles at ∼70°C that is most pronounced in the compensatory mutant consistent with restoration of base pairing. For the two mismatch mutants, the melting profiles suggest there may be some additional effect on the first apparent transition that corresponds to Terminal Stem and Central Domain unfolding. A slight shoulder is observed on the low temperature side of the transition and the hypochromicity for the first transition is significantly reduced for one single strand mutant, AU5R RNA. It is likely that the first observation corresponds to an uncoupled unfolding transition of the Apical Stem that is destabilised to such an extent that it unfolds in a broad transition overlapping with the first apparent transition at 60°C. The more significant reduction in hypochromicity in the first apparent transition for AU5R RNA, suggests that the stability of the base of the Apical Stem may have some, albeit limited, interaction with the Central Domain, presumably through stacking of bases at the interface of the two domains. The largest effect of this mismatch mutation in the UV melting analysis parallels its effect on the gel mobility of VA RNAI (Figure 2).
Inhibition of PKR autophosphorylation by wild-type VA RNAI and site 4 mutants
Finally, we wished to assess whether any observations made in the UV melting analysis might provide further insight into the structural determinants of VA RNAI function. While several mutations caused significant changes to the melting profile of VA RNAI, in only one instance (at site 4) did this appear to indicate a partial loss of structure. The effects of mutations at site 3 may be complicated by potential involvement of RNA tertiary structure (see Discussion). Mutations at sites outside the Central Domain are unlikely to affect PKR inhibition as the full structure appears to be retained, albeit destabilized in one specific domain. Furthermore, other mutations we have made that alter the bases making up sites 1, 2 and 6 but maintain base pairing and helical structure do not affect PKR inhibitory activity significantly (not shown).
Comparison of the UV melting data for the compensatory (AU4) and mismatch (AA4) mutants at site 4 suggested that AA4 RNA may lack a component of the wild-type VA RNAI structure, visible as a low temperature shoulder on the first apparent transition in the profile. To test whether this component of RNA structure might be important for activity, mutants and wild-type VA RNAI were tested in assays of PKR autophosphorylation inhibition. AA4 RNA is a significantly poorer inhibitor of PKR autophosphorylation compared to wild-type VA RNAI and requires higher concentrations to reach complete inhibition (Figure 6). The AU4 RNA compensatory mutant, in contrast, is as active as wild-type VA RNAI.
Figure 6.

PKR kinase activity inhibition assays with wild-type, AA4 and AU4 VA RNAIs. Levels of PKR autophosphorylation were quantified after preincubation with various concentrations of each inhibitor VA RNA (0–10 μg/ml) and subsequent activation with poly(I)–poly(C) dsRNA. The partial mutant AA4, that lacks an unfolding transition (Figure 5C) that may correspond to Central Domain tertiary structure, shows reduced inhibitory activity while the compensatory mutant AU4 is as active as wild-type VA RNAI.
DISCUSSION
In order to assign the secondary structure domains of Adenovirus type 2 VA RNAI to one of the two apparent transitions in the wild-type RNA melting profile, six sets of tandem compensatory base pair changes were introduced. Exchanging G–C for A–U base pairs might be expected to have one or more of three major effects on the RNA UV melting profile: (1) the relative hypochromicity of the unfolding transition(s) associated with the modified region should be altered, with a relative increase at 260 nm, compared to 280 nm; (2) the associated Tm(s) for the transition(s) may decrease; (3) individual unfolding transitions that are coupled as a single cooperative unfolding event (i.e. apparent transition) in the wild-type RNA may be uncoupled and become visible as distinct peaks or shoulders. Each VA RNAI with compensatory base pair modifications was analysed by UV melting and all these effects were observed. The domains of VA RNAI could be immediately assigned to one of the two apparent unfolding transitions: the Terminal Stem and Central Domain unfold as a single cooperative apparent transition at ∼60°C while the Apical Stem unfolds at higher temperature with an apparent transition centred around 83°C.
Most strikingly, this division of the domains is maintained for all mutants examined suggesting that the two halves of the RNA behave as independent structural domains. In wild-type VA RNAI, the structure and stability of the Terminal Stem and Central Domain are tightly coupled and they unfold in a single cooperative apparent transition. The Apical Stem in contrast appears to be a highly stable and independently folded domain within the global architecture of the RNA. This structural division thus mirrors the functional division of VA RNAI, with the Apical Stem as the primary site of binding for PKR distinct from the Central Domain that inhibits the activity of PKR.
VA RNAI migrates on denaturing gels with an apparent size of ∼220 nt and this phenomenon has been used as a simple assay for the structural integrity of mutant sequences (14). The results presented here suggest some caution may also be required in interpreting this phenomenon as a proxy for global VA RNAI folding. The analysis of compensatory base pair changes here has confirmed that the structure at the base of the Apical Stem as the source of this effect. The aberrant gel mobility is almost certainly due to incomplete denaturation of this extraordinarily stable RNA domain structure even under the strongly denaturing conditions used. In support of this, in UV melting studies in buffer containing 2–6 M urea the Apical Stem structure of wild-type VA RNAI retains an apparent Tm of >70°C (data not shown). Since even compensatory mutations at the site analysed in the base of the Apical Stem (site 5) do not recover the wild-type gel mobility or apparent Tm, clearly both the sequence and structure in this region are critical to its stability and resistance to denaturation. The four nucleotides corresponding to site 5 are almost completely conserved across all VA RNAI and VA RNAII sequences from all Adenovirus serotypes (9). While this may simply reflect the evolutionary closeness of these sequences, there is obvious sequence variation in other base-paired regions of the VA RNAs suggesting that the conservation of these base pairs and the unusual stability they impart could arise from functional benefits to the virus. For example, the extreme stability of the Apical Stem may make this critical PKR-binding domain more resistant to unwinding or degradation by cellular RNases that form part of the host cell defences. It does not appear from the current data, however, that the Apical Stem structure/stability directly stabilises the Central Domain, as previously proposed (22), since it appears to be an essentially independent domain within the overall VA RNAI structure.
Compensatory mutations in the outermost helical regions of the RNA, AU1 in the Terminal Stem and AU6 in the Apical Stem, produced the simplest changes in the melting profiles: a reduction in Tm and shift in relative hypochromicity at the two wavelengths measured but with no other alteration in the shape of the profile. At the other four sites of modification, more complex changes were observed and these were investigated further by analysis of several mismatch mutations (Table 1). Mutations at each of these sites uncoupled the unfolding of different regions within the Terminal Stem/Central Domain (sites 2, 3 and 4) or Apical Stem (site 5). The Terminal Stem apparent transition is split into at least two apparent unfolding transitions by all mutations at sites 2 and 3. A component of the structure retains wild-type stability and still unfolds in an apparent transition at the wild-type Tm (∼60°C) but with reduced hypochromicity. New apparent transitions are observed as lower temperature peaks or shoulders in the profile of varying Tm and broadness. The stability of the Central Domain appears to be dependent upon that of the Terminal Stem as they unfold cooperatively in the wild-type RNA and this is retained upon introducing compensatory mutations in the lower Terminal Stem (site 1). However, the reverse is not true; sequence changes in the Central Domain that reduce its stability do not necessarily affect the Terminal Stem structure/stability. Mutations within in the Central Domain therefore appear to uncouple unfolding of the Central Domain secondary and tertiary structure from the Terminal Stem. A similar dependency is observed in the Apical Stem where alterations at the top of the stem (site 6) reduce the stability of the entire stem, while those made at the base of the stem uncouple unfolding transitions leaving some part of the structure at the wild-type Tm.
For five of six sets of mutations the comparison of the melting profiles for mismatch and compensatory mutations shows changes consistent with base pairing of the altered nucleotides. At the one exception (site 3), a series of similar mutations were made in a previous study and assessed in functional and structure probing assays (20). Though direct evidence of base pairing was not observed in the structure probing experiments, both the restoration of partial inhibitory function and concerted changes in adjacent nucleotides upon compensatory mutation indicated pairing of these bases, possibly within a higher order tertiary structure of the Central Domain. For all other compensatory changes, an increase in relative hypochromicity at 260 nm or recovery of a transition in the melting profile is observed for compensatory mutants. In an alternative secondary structure model, proposed prior to comparative sequence analysis (11) and used in the interpretation of some mutational studies (10,24), the nucleotides at sites 3 and 4 are not base paired (Figure S1). While interpretation of the effects of mutations at site 3 remains unclear, the melting profiles show clear evidence of base pairing at site 4. A component of the Central Domain secondary or possibly tertiary structure that is lost upon introduction of destabilising A • C mismatches (AA4 RNA) is returned by the compensatory mutation (AU4 RNA) and is visible as a low temperature shoulder on the 60°C apparent transition. The mismatch mutant AA4 RNA lacking this structure was a less active inhibitor of PKR, while the compensatory mutant showed wild-type activity. Confirmation that these nucleotides are paired provides further experimental validation of the secondary structure proposed from sequence analysis (Figure 1) and confirms that the ‘shoulder’ in the UV melting profile corresponds to a critical element of VA RNAI structure, potentially the Central Domain tertiary structure. This opens the possibility of a focussed analysis of point mutations within this domain and their effect on this critical functional component of VA RNAI structure.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to C. Holtby for assistance with site-directed mutagenesis, and Dr C. M. Dunham for useful discussions during the preparation of the manuscript. This work was supported by a Wellcome Trust Research Career Development Fellowship (ref. 061444) to G.L.C. and Medical Research Council PhD studentship to V.K.C. Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mahr JA, Gooding LR. Immune evasion by adenoviruses. Immunol. Rev. 1999;168:121–130. doi: 10.1111/j.1600-065x.1999.tb01287.x. [DOI] [PubMed] [Google Scholar]
- 2.O’Malley RP, Mariano TM, Siekierka J, Mathews MB. A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell. 1986;44:391–400. doi: 10.1016/0092-8674(86)90460-5. [DOI] [PubMed] [Google Scholar]
- 3.Soderlund H, Pettersson U, Vennstrom B, Philipson L, Mathews MB. New species of virus-coded low-molecular weight RNA from cells infected with Adenovirus type 2. Cell. 1976;7:585–593. doi: 10.1016/0092-8674(76)90209-9. [DOI] [PubMed] [Google Scholar]
- 4.Ma YL, Mathews MB. Comparative analysis of the structure and function of Adenovirus virus associated RNAs. J. Virol. 1993;67:6605–6617. doi: 10.1128/jvi.67.11.6605-6617.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2-alpha kinase. Cell. 1986;45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 6.Kostura M, Mathews MB. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol. Cell Biol. 1989;9:1576–1586. doi: 10.1128/mcb.9.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathews MB, Shenk T. Adenovirus virus-associated RNA and translation control. J.Virol. 1991;65:5657–5662. doi: 10.1128/jvi.65.11.5657-5662.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma YL, Mathews MB. Structure, function, and evolution of adenovirus-associated RNA: A phylogenetic approach. J. Virol. 1996;70:5083–5099. doi: 10.1128/jvi.70.8.5083-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furtado MR, Subramanian S, Bhat RA, Fowlkes DM, Safer B, Thimmappaya B. Functional dissection of Adenovirus VAI RNA. J. Virol. 1989;63:3423–3434. doi: 10.1128/jvi.63.8.3423-3434.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mellits KH, Mathews MB. Effects of mutations in stem and loop regions on the structure and function of Adenovirus VA RNA1. EMBO J. 1988;7:2849–2859. doi: 10.1002/j.1460-2075.1988.tb03141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson MG, Haasnoot PCJ, Xu N, Berenjian S, Berkhout B, Akusjarvi G. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke PA, Mathews MB. Interactions between the double stranded RNA binding motif and RNA - definition of the binding site for the interferon-induced protein kinase DAI (PKR) on Adenovirus VA RNA. RNA. 1995;1:7–20. [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke PA, Pe’ery T, Ma YL, Mathews MB. Structural features of adenovirus 2 virus-associated RNA required for binding to the protein kinase DAI. Nucleic Acids Res. 1994;22:4364–4374. doi: 10.1093/nar/22.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellits KH, Kostura M, Mathews MB. Interaction of Adenovirus VA RNAI with the Protein Kinase DAI – Nonequivalence of Binding and Function. Cell. 1990;61:843–852. doi: 10.1016/0092-8674(90)90194-j. [DOI] [PubMed] [Google Scholar]
- 16.Spanggord RJ, Vuyisich M, Beal PA. Identification of binding sites for both dsRBMs of PKR on kinase-activating and kinase-inhibiting RNA ligands. Biochemistry. 2002;41:4511–4520. doi: 10.1021/bi0120594. [DOI] [PubMed] [Google Scholar]
- 17.Spanggord RJ, Beal PA. Selective binding by the RNA binding domain of PKR revealed by affinity cleavage. Biochemistry. 2001;40:4272–4280. doi: 10.1021/bi002512w. [DOI] [PubMed] [Google Scholar]
- 18.Ghadge GD, Malhotra P, Furtado MR, Dhar R, Thimmapaya B. In vitro analysis of Virus Associated RNA-I (VAI RNA) - Inhibition of the double stranded RNA activated protein kinase PKR by VAI RNA mutants correlates with the in vivo phenotype and the structural integrity of the central domain. J. Virol. 1994;68:4137–4151. doi: 10.1128/jvi.68.7.4137-4151.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghadge GD, Swaminathan S, Katze MG, Thimmapaya B. Binding of the adenovirus VAI RNA to the interferon-induced 68 kDa protein kinase correlates with function. PNAS. 1991;88:7140–7144. doi: 10.1073/pnas.88.16.7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma YL, Mathews MB. Secondary and tertiary structure in the central domain of adenovirus type 2 VA RNA(I) RNA. 1996;2:937–951. [PMC free article] [PubMed] [Google Scholar]
- 21.Monstein H-J, Philipson L. The conformation of adenovirus VAI-RNA in solution. Nucleic Acids Res. 1981;9:4239–4250. doi: 10.1093/nar/9.17.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mellits KH, Pe'ery T, Mathews MB. Role of the apical stem in maintaining the structure and function of adenovirus virus-associated RNA. J. Virol. 1992;66:2369–2377. doi: 10.1128/jvi.66.4.2369-2377.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pe’ery T, Mellits KH, Mathews MB. Mutational analysis of the central domain of Adenovirus Virus-Associated RNA mandates a revision of the proposed secondary structure. J. Virol. 1993;67:3534–3543. doi: 10.1128/jvi.67.6.3534-3543.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahman A, Malhotra P, Dhar R, Kewalramani T, Thimmapaya B. Effect of single base substitutions in the central domain of Virus Associated RNA-I on its function. J. Virol. 1995;69:4299–4307. doi: 10.1128/jvi.69.7.4299-4307.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laing LG, Draper DE. Thermodynamics of RNA folding in a conserved ribosomal RNA domain. J. Mol. Biol. 1994;237:560–576. doi: 10.1006/jmbi.1994.1255. [DOI] [PubMed] [Google Scholar]
- 26.Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends. NAR Methods. 2003;31:e82. doi: 10.1093/nar/gng082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pe’ery T, Mathews MB. Synthesis and purification of single-stranded RNA for use in experiments with PKR and in cell-free translation systems. Methods Comp. Methods Enzymol. 1997;11:371–381. doi: 10.1006/meth.1996.0435. [DOI] [PubMed] [Google Scholar]
- 28.Conn GL. Expression of active RNA-activated protein kinase (PKR) in bacteria. Biotechniques. 2003;35:682–683. doi: 10.2144/03354bm03. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.