


Abstract
The tumour suppressor p53 is a transcription factor that binds DNA in the vicinity of the genes it controls. The affinity of p53 for specific binding sites relative to other DNA sequences is an inherent driving force for specificity, all other things being equal. We measured the binding affinities of systematically mutated consensus p53 DNA-binding sequences using automated fluorescence anisotropy titrations. Based on measurements of the effects of every possible single base-pair substitution of a consensus sequence, we defined the DNA sequence with the highest affinity for full-length p53 and quantified the effects of deviation from it on the strength of protein–DNA interaction. The contributions of individual nucleotides were to a first approximation independent and additive. But, in some cases we observed significant deviations from additivity. Based on affinity data, we constructed a binding predictor that mirrored the existing p53 consensus sequence definition. We used it to search for high-affinity binding sites in the genome and to predict the effects of single-nucleotide polymorphisms in these sites. Although there was some correlation between the Kd and biological function, the spread of the Kds by itself was not sufficient to explain the activation of different pathways by changes in p53 concentration alone.
INTRODUCTION
The tumour suppressor p53 is responsible for inducing cell cycle arrest or apoptosis under DNA-damaging conditions in order to prevent dangerous accumulation of mutations (1–3). The regulation of transcription by p53 plays a critical role in cellular responses to carcinogenic stimuli. p53 recognizes a 20 bp DNA sequence consisting of two repeats of RRRCWWGYYY (4), separated by 0–13 bp. Transcription factors specifically recognize, on average, about half the nucleotides in the binding site. Therefore, for a binding site of 10 bp, one would expect about a thousand (45) variants, and for a binding site of 20 bp this number is about one million (410). Accordingly, one would expect to find a few thousand copies of 20 bp and millions of 10 bp binding sites in the human genome. It is remarkable that transcription regulation is as specific as it is, given the significant number of response elements for any one transcription factor in the genome. A driving force for recognition of specific binding sites is the affinity of the transcription factor for them in comparison with other DNA sequences present in the genome. Different sequences are recognized by transcription factors with different affinity. It is important to define the affinity landscape, the distribution of the affinity values among all potential binding sites, in order to understand the origins of specificity of a transcription factor. This would allow rationalization of the impact the variability of the binding sites or mutations/modifications of the transcription factor have on regulation of transcription. It is impractical to measure the affinity of every possible binding site. A way of identifying binding sites and accurately predicting the affinity of a transcription factor for a given sequence is needed in order to define the affinity landscape.
The DNA recognition preference of a protein is usually described by a ‘consensus’ sequence, which includes possible variants of this sequence. Such a definition does not quantify the detrimental effects of deviation from the consensus sequence, limiting its usefulness and predictive power. Position weight matrix representation (5) is also widely used to describe binding sites. The sites are defined based on the frequency of observing a particular base at a particular position within the binding site, derived from a set of known binding sites. The quality of position weight matrices crucially depends on the availability of large training datasets from experimentally observed binding sites. Such datasets can be derived from chromatin (ChIP) and DNA (DIP-Chip) immunoprecipitation assay, in vitro evolution (SELEX) or protein-binding microarray experiments (6–8). The logarithm of frequency of observing a particular sequence was shown to be proportional to the energy of interaction (9). Since the driving force for the transcription factor to recognize a specific DNA sequence is the energy of interaction, a number of approaches were developed to directly determine the contribution of individual nucleotide positions to the overall binding. These methods included measuring the affinity of a transcription factor for a set of DNA sequences using electrophoretic mobility shift assays (EMSA) (QuMFRA), captured luciferase activity assay or surface plasmon resonance (10–13). These powerful methods can identify the consensus sequence and probe the contribution of individual nucleotides in the DNA binding. Unfortunately, these methods do not provide an accurate estimate of the true protein–DNA affinity in solution because they rely on the surface immobilization of the DNA probes or detect the interactions in the gel media.
We developed an automation platform to multiplex titration experiments to measure affinities in solution with high accuracy. Using fluorescence anisotropy titrations, we measured the effects of every possible single base-pair substitution of a consensus sequence on the affinity of full-length p53 for DNA and quantified the contribution of individual nucleotides to the binding. The contributions of individual nucleotides were practically additive which allowed us to predict the affinity of any potential binding site for p53. We used this function to identify and predict the affinity of the tumour suppressor p53 for its potential binding sites in the genome, and to predict the effect of single nucleotide polymorphism (SNP) variations in these sites on p53 binding.
MATERIALS AND METHODS
We used the super-stable mutant of full-length p53 containing the following mutation in the core domain: M133L/V203A/N239Y/N268D (14,15) of p53 as pseudo-wt, which increased expression levels and sample stability. It was purified as described earlier (16). All other reagents were of highest grade available. Buffer conditions were 25 mM NaPi, 225 mM NaCl, 10% v/v glycerol, 5 mM DTT. Total ionic strength was 286 mM. BSA (0.2 mg/ml) was added to buffers to minimize non-specific binding of proteins at low concentrations when used with plastic ware. DNA oligonucleotides were ordered from Eurogentec, Belgium. Oligonucleotide concentration was quantified by absorbance and normalized to 1 mM using epMotion 5070 pipetting robot (Eppendorf AG, Germany) prior to annealing. They were annealed by heating to 95°C for 5 min and cooling at 1°C/min to room temperature in the PCR block (PTC-100, MJ Research, Inc., USA) and diluted to final concentration of 50 µM.
Fluorescence anisotropy measurements
DNA-binding experiments were done at 15°C in the Perkin Elmer LS50, Varian Eclipse and HoribaJobivYvon Fluoromax-3 fluorimeters using fluorescence anisotropy (16). The concentration of DNA was 1 nM for direct binding experiments and 20 nM for competition experiments. For competition experiments, 20 nM Alexa488 labelled reference DNA was mixed with p53 to a final concentration of 120 nM. A 50 μM stock of competitor DNA was added in small aliquots to compete the reference DNA from the complex. Data were analysed using laboratory software according to a competition model. Measurements were multiplexed by performing titrations on microtitre plates (Corning 3650) with Bravo 96-channel pipetting robot (Velocity11, USA) interfaced with Pherastar plate reader (BMG LABTECH GmbH, Germany) using 480/520 nm fluorescence polarization module using manufacturer-provided software. Titrations were done at room temperature 20°C. The sample volume during the titration was kept constant (200 µl) by aspirating the same volume of the sample prior to addition of an aliquot of the competitor DNA. In order to keep the concentration of reporter DNA and protein constant, competitor DNA was mixed with the same concentration of them as in the initial sample. By this process, only the concentration of the competitor DNA changes during the titration. To minimize the errors associated with handling small volumes (<1 µl), 2.5 µM stock of competitor DNA was used for the first part of the titration, switching to 25 µM for second part. Source microtitre plates were prepared using epMotion 5070 pipetting robot (Eppendorf AG, Germany). Data were processed and analysed using laboratory-developed software. Each titration was repeated at least three times.
Data analysis
The change of the affinity of interaction changes the amount of protein bound to this site as follows:
| 1 |
where [P] is the free concentration of the transcription factor and Kd is the affinity of interaction. While we do not know the exact value of [P] in the cell, the ChIP enrichment values are typically in the range of 1–10%, meaning that binding sites are only partially saturated in vivo. Therefore, we can assume that [P] < Kd. Under such conditions, the amount of protein bound would decrease as:
| 2 |
where n is the Hill constant of interaction which we have previously shown to be 2, for p53 (17). The importance of the co-operativity of interaction of the transcription factor with DNA cannot be stressed enough. It magnifies the effects of the change in the Kd on the relative amount of the protein–DNA complex formed. We converted the observed changes in the Kd to the sequence logo (18) (Figure 3).
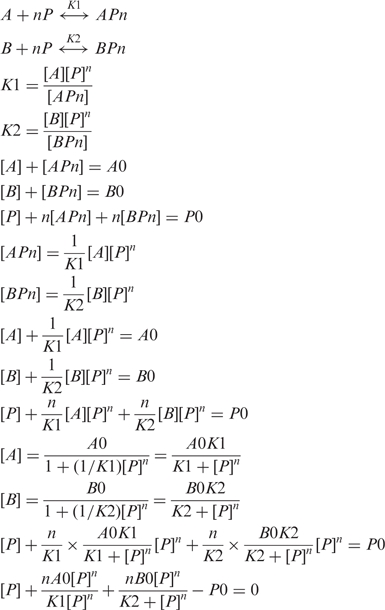 |
Figure 3.

Quantitative sequence logo of the p53 DNA-binding preferences. The height of the bars represents the number of times by which the amount of protein bound decreases, due to the base substitution. It shows the biggest possible effect caused by three alternative substitutions. The height of individual letters is proportional to the amount of transcription factor bound to individual sequence variants under identical conditions. The sequence of the second half-site is identical to the first one when read on the non-coding strand of DNA in the 5′–3′ direction. The sequence logo takes complementarity of DNA into account. The sequence with the highest affinity for p53 is GG(A/G)CATGCCCGGGCATG(T/C)CC.
Since the dissociation constants in the above derivation have dimensions of Mn, it is convenient to convert them into dimension of M so that the meaning of the Kd is concentration of protein at 50% saturation of the binding sites.
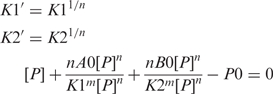 |
This equation could be solved numerically for [P]. Values of [A] and [APn] could be calculated from [P] and used to define a fitting function describing experimental data.
RESULTS
A ‘binding predictor’ for p53
The energy of interaction of a protein with DNA ΔG consists of the sum of the energies of interaction of the protein with individual base pairs ΔGi and is proportional to the logarithm of the Kd and a loss of entropy of association, TΔS.
By measuring the affinity of a protein for all four possible single-nucleotide permutations of the reference consensus sequence at every position, it is possible to assign a change ΔlogKd for every deviation from the consensus sequence. If the contributions of individual nucleotides are independent, the affinity of the protein for any given sequence of DNA can be calculated based on the affinity of the reference sequence and a sum of effects of substitution at every position.
Fluorescence anisotropy is the property of fluorescent molecules to retain the polarization of excitation light and reflects the tumbling rate of molecules in solution. It is ideal for studying protein–DNA interactions as the complex formed is larger and tumbles more slowly than the unbound oligonucleotide. The binding affinity for the fluorescein-labelled reference sequence was determined using direct fluorescence anisotropy titrations as previously described (17). In subsequent experiments, we used a competition assay (Figure 1) to improve the accuracy of determination of a difference in the affinity. The reference sequence labelled with fluorescein was mixed with p53 protein to form a complex, and unlabelled oligonucleotide was added to the cuvette in small aliquots, to compete the labelled oligonucleotide off. Analysis of the competition curve allowed accurate determination of the difference in the affinities of the two oligonucleotides (Supplementary Data).
Figure 1.

Fluorescence anisotropy R reflects the tumbling rate of molecules in solution. It is ideal for studying strong protein–DNA interactions as the complex formed is larger and tumbles more slowly than the unbound oligonucleotide. A displacement of the reference labelled oligonucleotide from the complex by the unlabelled oligonucleotide allows accurate measurement of the difference in the Kd between two sequences.
After initial experiments, we have chosen a sequence CGCGGACATGTCCGGACATGTCCCGC as a reference sequence (Table 1). It consists of the two identical copies of the GGACATGTCC half-site, which is representative of a consensus sequence RRRCWWGYYY, flanked by the CGC and CGC triplet to improve the annealing properties of the oligonucleotide. We measured the changes in the affinity (Figure 2) caused by all possible substitutions within the reference oligonucleotide, including unlabelled reference sequence. Nucleotide substitutions at some positions (e.g. 4) had larger effects than at other positions (e.g. 1 or 2). Different nucleotide substitutions at the same position also had different effects on the affinity of interaction (e.g. a base change A>C at position 3 compared to A>T). The substitution of the T>C at position 8 resulted in increased affinity, although the effect was negligible. This defined the sequence with the highest affinity for p53 as GG(A/G)CATGCCCGGGCATG(T/C)CC. The complete definition of the binding predictor matrix is presented in Table 2, and its graphical representation in Figure 3 (see Appendix 1 for details).
Table 1.
Oligonucleotide sequences used to construct binding predictor
| Sequence No. | Sequence | Substitution |
|---|---|---|
| 600 | CGC GGACATGTCC GGACATGTCC CGC | Ref sequence |
| 601 | CGC AGACATGTCC GGACATGTCC CGC | 1A |
| 602 | CGC TGACATGTCC GGACATGTCC CGC | 1T |
| 603 | CGC CGACATGTCC GGACATGTCC CGC | 1C |
| 604 | CGC GAACATGTCC GGACATGTCC CGC | 2A |
| 605 | CGC GTACATGTCC GGACATGTCC CGC | 2T |
| 606 | CGC GCACATGTCC GGACATGTCC CGC | 2C |
| 607 | CGC GGTCATGTCC GGACATGTCC CGC | 3T |
| 608 | CGC GGGCATGTCC GGACATGTCC CGC | 3G |
| 609 | CGC GGCCATGTCC GGACATGTCC CGC | 3C |
| 610 | CGC GGAAATGTCC GGACATGTCC CGC | 4A |
| 611 | CGC GGATATGTCC GGACATGTCC CGC | 4T |
| 612 | CGC GGAGATGTCC GGACATGTCC CGC | 4G |
| 613 | CGC GGACTTGTCC GGACATGTCC CGC | 5T |
| 614 | CGC GGACGTGTCC GGACATGTCC CGC | 5G |
| 615 | CGC GGACCTGTCC GGACATGTCC CGC | 5C |
| 616 | CGC GGACAAGTCC GGACATGTCC CGC | 6A |
| 617 | CGC GGACAGGTCC GGACATGTCC CGC | 6G |
| 618 | CGC GGACACGTCC GGACATGTCC CGC | 6C |
| 619 | CGC GGACATATCC GGACATGTCC CGC | 7A |
| 620 | CGC GGACATTTCC GGACATGTCC CGC | 7T |
| 621 | CGC GGACATCTCC GGACATGTCC CGC | 7C |
| 622 | CGC GGACATGACC GGACATGTCC CGC | 8A |
| 623 | CGC GGACATGGCC GGACATGTCC CGC | 8G |
| 624 | CGC GGACATGCCC GGACATGTCC CGC | 8C |
| 625 | CGC GGACATGTAC GGACATGTCC CGC | 9A |
| 626 | CGC GGACATGTTC GGACATGTCC CGC | 9T |
| 627 | CGC GGACATGTGC GGACATGTCC CGC | 9G |
| 628 | CGC GGACATGTCA GGACATGTCC CGC | 10A |
| 629 | CGC GGACATGTCT GGACATGTCC CGC | 10T |
| 630 | CGC GGACATGTCG GGACATGTCC CGC | 10G |
The sequence for the coding strand is shown. Oligonucleotides were annealed with corresponding complementary oligonucleotides to form double-stranded oligonucleotides.
Figure 2.

The effects of individual substitutions in the reference sequence on the affinity of p53 for its response element. Positive bars correspond to weaker interactions. Only first 10 bp of 20 bp site are shown because of the mirror symmetry of the response element. See Table 1 for description of individual sequences. Reference sequence bound p53 with the logKd of −7.51 (30 nM).
Table 2.
Binding predictor matrix definition
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Highest affinity sequence | G | G | A/G | C | A | T | G | C | C | C | G | G | G | C | A | T | G | T/A | C | C |
| A | 0.05 | 0.03 | 0.00 | 0.59 | 0.00 | 0.16 | 0.55 | 0.25 | 0.15 | 0.12 | 0.03 | 0.10 | 0.05 | 0.62 | 0.00 | 0.21 | 0.31 | 0.15 | 0.10 | 0.07 |
| T | 0.07 | 0.10 | 0.15 | 0.31 | 0.21 | 0.00 | 0.62 | 0.05 | 0.10 | 0.03 | 0.12 | 0.15 | 0.25 | 0.55 | 0.16 | 0.00 | 0.59 | 0.00 | 0.03 | 0.05 |
| G | 0.00 | 0.00 | 0.00 | 0.55 | 0.28 | 0.35 | 0.00 | 0.47 | 0.29 | 0.13 | 0.00 | 0.00 | 0.00 | 0.61 | 0.32 | 0.47 | 0.00 | 0.40 | 0.18 | 0.11 |
| C | 0.11 | 0.18 | 0.40 | 0.00 | 0.47 | 0.32 | 0.61 | 0.00 | 0.00 | 0.00 | 0.13 | 0.29 | 0.47 | 0.00 | 0.35 | 0.28 | 0.55 | 0.00 | 0.00 | 0.00 |
Each cell contains a difference ΔlogKd between the oligonucleotide containing a single substitution indicated in a row header and the tightest binding sequence. The highest affinity sequence shown in the top row had a logKd of −7.61.
Since the second half-site, positions 11–20, mirrors the first half-site, only the first one will be discussed. The flanking base pairs at positions 1, 2 and 10 had little overall effect on DNA binding as compared to other positions. Position 1 does not form specific interactions with DNA, and has little effects on overall binding. p53 makes contacts with the nucleotide in position 2 via K120, but the exact nature of this contact depends on the DNA sequence (19). Non-specific recognition of oligonucleotides is also true at the position 3, with preferred A/T/G at the position 3 and C/T/A at position 8. Overall effects on binding at positions 3 and 8 are larger than at positions 1/10, suggesting that these interactions, formed by C277, play an important role in the DNA binding of p53. There is a marked preference for a C at position 4 and G at position 7. The G on the non-coding stand opposite to the conserved C is recognized by R280. Large changes in Kd induced by nucleotide substitutions at this position suggest that these interactions are invariant. Even though the nucleotide at position 5 does not make contacts with p53 in the crystal structure, there is a marked preference for A at this position, and T at position 6, which may be caused by DNA geometry constraints. This preference is consistent with p53 transactivation data (20). Interestingly, the exact contributions of the nucleotides of the outer quarter site (positions 1–5) differed from those of the inner quarter site (positions 6–10) (Table 2). This difference may be explained by domain–domain interactions present in the two inner p53 core domains and absent in the two outer ones (19).
Our data could be translated into consensus sequence of a NDRCATGYYY or NNDCWWGYHN half-site, depending on the threshold value selected. It is less stringent than the existing definition of the p53 consensus sequence RRRCWWGYYY (4). The most obvious differences come from the selectivity of the flanking position 1, 2 and 10, and a preference for AT at positions 5/6. However, most known p53-binding sites deviate from the consensus sequence (20). Our affinity data correspond well to the rules describing the transactivation activity of p53 response elements: changes of conserved C or G dramatically affect activity; AT in the middle provides the strongest activity; the effects of deviation in the flanking RRR and YYY regions are stronger the closer they are to the central CWWG motif. The consensus sequence definition (4) is derived from only 20 sequences, which were found to bind p53. While it certainly captures the essence of the DNA-binding preferences of p53, relatively small sample size and under-representation of weaker binding sequences may explain the differences observed.
Taking this into account, the existing definition of the p53 consensus sequence (4,21), as well as empirical rules describing active p53 response elements based on transactivation ability of p53 (20), are mirrored remarkably well by the quantitative sequence logo. The main improvement is quantification of the effect of sequence variation on protein-recruiting ability.
Predicting the affinity of protein–DNA interactions
Assigning a change in the affinity ΔlogKd to every deviation from the reference sequence offers a convenient way of estimating the affinity of a transcription factor for any given DNA sequence:
| 3 |
where x is the DNA sequence, logKd(ref) is the affinity of the reference sequence, and ΔlogKd(i,N) is the change in the Kd induced by a nucleotide N substitution in the ith position. The error of prediction accumulates over all base pairs of the binding site. With an average error of logKd determination of ∼0.05, one would expect an error of prediction of ∼0.25.
We tested the accuracy of the logKd prediction by comparing predicted affinities with experimentally measured affinities for naturally occurring response elements (Figure 4 and Table 3). The native response elements tested differed from the reference sequence by 3–13 individual nucleotide substitutions. Some of the sequences also had an extra base pair inserts between two half-sites (Table 3). The predicted values were calculated using laboratory-developed software according to the binding predictor positional matrix in Table 2 and Equation (3). Equation (3) implies independent contributions of the individual nucleotide positions to the overall binding affinity. This assumption has been shown to hold reasonably well (22), even though there are examples where nucleotides were not independent (23,24).
Figure 4.

Correspondence of the measured and predicted affinities of naturally occurring response elements. Black line represents ‘perfect’ prediction.
Table 3.
Oligonucleotide sequences used to test predicted logKds
| Name of the promoter | Sequence(29) | logKd (expected) | logKd (predicted) |
|---|---|---|---|
| Cyclin G | CGCAGACCTGCCCGGGCAAGCCTCGC | −7.02 | −6.83 |
| 14-3-3s | CGCAGGCATGTGCCACCATGCCCCGC | −6.85 | −6.52 |
| CDK1NA (p21) 3′ site | CGCGAAGAAGACTGGGCATGTCTCGC | −6.9 | −6.54 |
| p53R2 | CGCTGACATGCCCAGGCATGTCTCGC | −7.35 | −7.46 |
| PCNA | CGCGAACAAGTCCGGGCATATGTCGC | −6.82 | −6.83 |
| Maspin | CGCGAACATGTTGGAGGCCTTTTGCGC | −6.89 | −6.19 |
| PUMA BS2 | CGCCTGCAAGTCCTGACTTGTCCCGC | −6.88 | −6.86 |
| Noxa | CGCAGGCTTGCCCCGGCAAGTTGCGC | −6.93 | −6.87 |
| p53AIP1 | CGCTCTCTTGCCCGGGCTTGTCGCGC | −6.92 | −6.73 |
| Bax A | CGCTCACAAGTTAGAGACAAGCCTCGC | −6.67 | −6.59 |
| IGF-BP3 | CGCAAACAAGCCACCAACATGCTTCGC | −6.77 | −6.89 |
| p53DINP1 | CGCGAACTTGGGGGAACATGTTTCGC | −6.78 | −6.25 |
| PUMA | CGCCTCCTTGCCTTGGGCTAGGCCCGC | −6.8 | −5.99 |
| rad51 | AAACTCGCGCAGGATCAAGCTCT | −5.96 | −6.12 |
| PA26 | GGACAAGTCTCAACAAGTTC | −6.47 | −6.85 |
| Bax B | AGACAAGCCTGGGCGTGGGC | −6.24 | −6.47 |
| MMP2 | AGACAAGCCTGAACTTGTCT | −6.39 | −7.01 |
| CDK1NA (p21) 5′ site | CAACATGTTGGGACATGTTC | −7.14 | −7.11 |
| MDM2_RE1 | GGTCAAGTTGGGACACGTCC | −6.47 | −6.69 |
| MDM2_RE2 | GAGCTAAGTCCTGACATGTCT | −6.41 | −6.42 |
There was a clear correlation between the experimentally measured and the predicted affinity values. Interestingly, the observed deviations were greater than experimental error and corresponding errors in predicted affinity values.
Non-specific binding of p53 to DNA may skew the correlation between measured and predicted values. It is 2–3 orders of magnitude weaker than specific binding (25). In our experiments, the weakest binding sequence had an affinity 1.5 orders of magnitude weaker than the reference sequence. The affinities of the majority of sequences were within one order of magnitude of the reference sequence. In addition, we did not observe clustering of the sequences with weak experimentally measured affinity, suggesting that non-specific binding was not the reason for the observed deviations.
Such deviations would also be observed if the contributions of the individual nucleotides were not completely independent. The identity of the neighbouring nucleotides may influence the contribution of a particular nucleotide. Such behaviour would result in deviation from the additive model. The accuracy of predictions was better for high-affinity sites (logKd < −6.9) than for lower affinity sites (logKd from −6.9 to −6.0), reflecting the fact that they contain fewer deviations from the reference sequence. The average deviation was 0.35 for all sequences tested (70% were within this range). In extreme cases, there were differences of up to 0.8 logKd units.
The presence of a spacer between the two half-sites adds to the complexity of the p53 response elements. Based on recent data (20,21), most functional p53 sites have an insert length of 0 or 1. In our calculations, we have assumed that the presence of an insert does not affect the affinity. The affinities of the IGF-BP3 and Bax A response elements with a one base-pair insert were correctly predicted, supporting this assumption. We did not test sites with longer spacers between half-sites.
The observed deviations from the additive model suggest that the contributions of individual nucleotides are not 100% independent. The first-order additive model is accurate for high-affinty sites (logKd < −6.9) and is a useful approximation for lower affinity sites (logKd > −6.9), providing accurate estimates in 70% of cases. The usefulness of such prediction for the lower affinity sites depends on the context in which they are used. The binding predictor software, together with the definition of the p53-binding matrix, is available for download from our website (www.mrc-cpe.cam.ac.uk).
Search for high-affinity response elements in the genomic DNA sequence
Ability to predict the affinity of the transcription factor for any DNA sequence offers a convenient way of searching for high-affinity sites in the genome. We calculated the affinity for a binding site located at every nucleotide position in the genome. A small portion of a genomic sequence surrounding known p53-binding site in the promoter of a p21 gene is shown in Figure 5. Two features are evident from this figure. The known p53 response element is easy to identify as the tightest binding site. p53 could also bind at any position within this stretch of DNA, with a significantly lower affinity. This is not surprising as p53, and transcription factors in general, do form both specific and non-specific (backbone) contacts with DNA, and have affinity for non-specific DNA. The exact number of identified high-affinity binding sites in the human genome increases exponentially with increase in the cutoff value for the logKd selected (Figure 6). We located over 10 000 high-affinity sites with a predicted logKd < −6.9, and over 200 000 weaker sites (logKd < −6.5). The error in the logKd prediction for the low-affinity sequences will not significantly affect the total number of sites identified because the number of sites with their logKd value either under-estimated or over-estimated will be comparable.
Figure 5.

A plot of p53 affinity for DNA as a function of bp position. A 50 bp region around the 5′ p53-binding site, 2209 bp upsream of the CDK1NA (p21) gene is shown (Chr6: 36752204:36752223 in NCBI 36.2 genome release). The circle marks the known p53-binding site.
Figure 6.

Number of putative p53-binding sites of the length of 20 or 21 bp identified in the human genome (release 36.2) grows exponentially with increasing cutoff value.
A well-documented site in the promoter region of CDKN1A (p21), known p53 target, was in the top 6000 sites ranked according to predicted affinity. Interestingly, a promoter region of p73, a paralogue of p53, also contained a high-affinity site. Some other known p53 targets, such as MDM2 and BAX, on the other hand, contained binding sites with weaker affinities. An analysis of 100 documented p53-binding sites (summarized in 26), presented in Table S1, suggested that there are large variations in the logKd values for sites regulating genes with similar functions. The binding sites in the promoter regions of genes controlling DNA repair (−7.1 ± 0.3) and the death receptor (−7.0 ± 0.2) pathways contained higher affinity sites as compared to apoptotic pathway (−6.6 ± 0.3). However, the distribution of logKd values predicted for various pathways overlapped (Table S1). Moreover, the human genome contains a large number of putative sites with similar affinity. One order of magnitude change from the highest affinity sequence results in over 200 000 predicted binding sites in the genome, and a p53-binding site in the promoter region of almost every gene. A list of all the p53-binding sites identified in the genome, and a list of the corresponding genes which have at least one binding site within 100 kb distance is available from our website.
Predicting the effect of SNPs
SNP is a natural nucleotide variation between individuals. If such variation is in the binding site of a transcription factor, it may affect the transcription control. SNP represent a special case for the binding predictor. Since it is a single-nucleotide substitution, the accuracy of prediction is very good because the error does not accumulate over the whole binding site. We analysed the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/) and identified 19 355 SNPs which are located in putative p53-binding sites, where at least one of the polymorphic variants fulfils our criteria for a high-affinity binding site (logKd < −6.5) and was predicted to affect the p53 binding by more than a factor of 3 [Equation (2)]. Out of these, 5142 were predicted to affect the binding of p53 by more than a factor of 10. In a recently reported example, a C to T SNP variation in the flt-1 promoter of the VEGF system makes it responsive to p53 and incorporates it into p53 pathway (27). We predicted a change in the logKd from −6.46 to −6.78, which corresponds to a 4-fold increase in the amount of p53 bound to this site. The predicted increase is in excellent agreement with the experimental data reported (27). A database of SNPs with p53 recognition sites and predicted effects is on our website.
DISCUSSION
Matrices are widely used to describe the relationship between a nucleotide sequence and its functional activity (5,28). The elements of the matrix corresponding to the bases in the sequence are added to calculate a score, and any sequence could be assigned 1. The quality of this relationship definition and the predictive value of the matrix are determined by the accuracy of the method used to assign individual values. We experimentally measured the effect of single-nucleotide substitutions on the p53–DNA affinity. Fluorescence anisotropy titrations provided accurate affinity values of interaction in solution. We used these values to construct the ‘binding predictor’ positional matrix. The elements of the matrix are the differences in the logKd value caused by a particular nucleotide substitution. The logKd value is proportional to the binding energy of interaction. The calculated score is the logarithm of the dissociation constant.
In order to have a manageable number of measurements, we minimized the sequence space coverage. We restricted it to the single-point mutants of the representative consensus sequence, assuming the case of independent and additive contributions. It is possible that the change in the neighbouring nucleotides could affect the nucleotide binding preferences of the position in question. We compared, therefore, the calculated values with experimentally determined values for a set of known response elements containing multiple nucleotide substitution (Figure 4, Table 3).
We experimentally tested sequences with logKd up to −6.0. The deviation of predicted values from experimentally measured ones was greater than that of the expected. This suggests that the individual nucleotides do not make completely independent contributions to the overall binding. For example, the PUMA response element was predicted to have much weaker affinity than the experimentally measured one by logKd of 0.8. The average deviation of predicted ones from measured values was 0.35 for all sequences tested, which means that the affinity of 70% of sequences was predicted within a factor of 2. The accuracy of predictions was better for high-affinity sites (logKd < −6.9) than for lower affinity sites (logKd from −6.9 to −6.0), reflecting the fact that they contain fewer deviations from the reference sequence. The quality of prediction may be improved by using more fine-grained models, e.g. measuring the effect of every possible dinucleotide substitution, at the expense of the exponentially increasing number of measurements necessary. Overall, the first-order additive model is accurate for high-affinity sites (logKd < −6.9) and provides a useful approximation for lower affinity sites (logKd > −6.9).
The dissociation constants for native response elements measured in this study differ from those we reported earlier (29). There were changes in absolute values, as well as changes in the relative values of dissociation constants (Figure S1). Most notably, the range in affinity values reported is smaller in the present study. However, we used different buffer conditions, a different format of binding experiments, and different protein constructs. The changes in absolute values can be explained by the changes in the buffer conditions. Protein–DNA interactions are electrostatically driven and are very sensitive to changes in the ionic strength. In our present study, we used buffer conditions with higher ionic strength (I = 286 mM versus I = 225 mM), which resulted in lower absolute affinity values. We used a competition format for binding experiments instead of direct binding. This resulted in greater accuracy of measurements. The dynamic range of the competition experiments (1.5–2 orders of magnitude relative to the reference sequence) was sufficient to reliably measure the values reported. In the present study, we used full-length p53 as opposed to the construct containing the DNA binding and tetramerization domain we had used earlier (29). The presence of the flanking N-terminal and C-terminal domains could result in the altered DNA-binding preferences of the protein, and in particular, in increased binding to the weaker response elements. The current measurements reflect the DNA-binding properties of full-length p53.
Ability to predict the affinity of p53 to any DNA sequence permits the search for high-affinity sites in the genome by calculating the affinity for every position, the very property that governs the transcription factor binding to response elements. The number of predicted p53-binding sites increases exponentially with an increase in the affinity cutoff value (Figure 6). We analysed the positioning of the identified putative binding sites relative to the transcription start sites. The sites were evenly distributed in the genome, with no marked tendency to cluster around transcription start sites. Interestingly, most established binding sites are located close to the transcription start sites (30). This apparent difference could be explained by involvement of other cellular factors in the selection of functional binding sites from all possible putative binding sites.
As with any other ranking functions, it is important to decide what affinity value represents a genuine binding site, and what values are irrelevant or not important. For example, only the highest affinity sites representing the desired total number (e.g. top 500) may be included, and the corresponding affinity cutoff value could be determined (Figure 6). Alternatively, the affinity values can be compared with the affinity of the previously documented binding sites. The set of binding sites identified experimentally using chromatin immunoprecipitation assay (ChIP) can also be compared with the set of putative binding sites selected based on their affinity.
The examination of p53-binding sites using ChIP of 1% of the human genome, as part of the ENCODE project, identified 37 p53-binding sites, which can be extrapolated to ∼3700 sites genome wide (31). The analysis of p53 binding to chromosomes 21 and 22 (circa 3% of the genome) (32) identified 48 binding sites, which can be extrapolated to 1500 binding sites genome wide. Genome-wide analysis identified 542 p53-binding sites (21). The same number of binding sites could be selected by imposing a logKd cutoff value of −7.1, −7.2 and −7.3, on our data (Figure 6). The difference in the number of binding sites identified may be explained by a difference in the stringency of selection criteria employed. However, the 236 sites with insert length of 0 between half-sites identified in the genome-wide ChIP experiment (21) have logKd values in the range −7.5 to −6.93. We identified over 8500 putative binding sites with similar affinity in the genome. Furthermore, there was only 7% overlap between our highest affinity 230 putative binding sites and the 236 ChIP-identified sites.
These cutoff values compare well with affinities of the p53 response elements controlling known p53 target genes p21 (−7.11) and p53R2 (−7.46). However, such a stringent selection criterion misses a number of known p53 target sites. Mdm2, the negative regulator of p53 and a known p53 target gene, has multiple binding sites for p53 with a logKd in the range around −6.69. Multiple weak binding sites can behave as one strong site if binding to any of them results in increased transcription. Unfortunately, it is not known if the binding sites in mdm2 promoter are synergetic. The known p53-binding site in the promoter region of the BAX gene has an even weaker affinity with a logKd of −6.59. Given the large number of sites with comparable affinity in the genome (circa 200 000 with an insert length of 0 or 1), specific transcription activation of mdm2 and BAX by p53 cannot be explained based on the affinity of p53 to DNA alone.
Many of known experimentally verified p53-binding sites had the predicted value of the Kd 1–1.5 orders of magnitude weaker than the sequence with highest affinity (Table S1). The Kd value may play a role in the activation of the target gene: the genes with tighter binding sites should respond stronger to the changes in p53 concentration caused by carcinogenic stress. Indeed, there is some correlation between the biological pathway of the gene and the strength of the site (Table S1). The binding sites controlling genes involved in DNA repair and the death receptor pathway were predicted to be the strongest, and sites involved in apoptosis and negative regulation were the weakest. The sites controlling genes cell cycle and mitosis had intermediate logKd values. Large variations in the logKd values resulted in the overlap of the distribution of affinities of sites controlling different pathways. It is very unlikely that changes in p53 concentration alone could explain the specificity of activation of different pathways in response to carcinogenic stress, e.g. cell cycle arrest versus apoptosis. In addition, there are hundreds of thousands putative sites of similar affinity in the human genome.
It is clear that p53 recognizes sites in DNA whose sequence resembles the canonical p53-binding site, and for which it has a marked preference compared with the surrounding sequence. The fact that only a small fraction of all possible sites with affinities similar to those identified in the ChIP experiment is occupied in the cell (and a small fraction needs to be occupied for transcription regulation to have any specificity), suggests that there are other factors controlling the selection of binding sites. Changes in the chromatin state, for example, may result in only a small percentage of potential p53-binding sites being accessible at a given time. The alternative possibility is that p53 functions together with another transcription factor(s) which has a binding site in close proximity (33). Even weak protein–protein interactions would dramatically increase the apparent affinity of both p53 and its partner for DNA and provide the basis for selection. Such co-operative binding could provide a mechanism for selection of one site over other potential binding sites.
With over 10 million individual SNPs documented in the human genome, it is not surprising that some of them are in the binding sites of transcription factors. The SNP may affect the binding of a transcription factor, disturb the functioning of a particular pathway, and pre-dispose an individual to a development of a disease. The binding predictor is ideally suited for predicting the effect of SNP in the binding site of p53. The relative effect of the SNP will remain the same even if the absolute affinity of a transcription factor for DNA is affected by other cellular factors. Assuming that 1% of the predicted binding sites are physiologically relevant, we estimated that around 200 SNPs would affect the p53 binding (>3-fold difference), and around 50 would affect it dramatically (>10-fold). It is a significant proportion of the estimated 500–3000 p53-binding sites existing in the cell.
The systematic study of the protein–DNA interactions by single-point mutations in consensus DNA sequence accessed by accurate measurements of affinity in solution using fluorescence anisotropy is a powerful way to describe the DNA binding preferences of a transcription factor. Using this method, we defined the DNA sequence with the highest affinity for p53 and quantified the effect of deviations from this sequence on the strength of the interaction. This method allows prediction of p53-binding affinity for any potential response element in the genome, all other things being equal. The binding predictor can identify potential p53-binding sites in the genome and predict the effects of SNPs on these interactions, and is applicable to the study of the DNA-binding specificity of any transcription factor.
ACKNOWLEDGEMENTS
We thank Caroline Blair for protein purification, Andreas Joerger and Caroline Blair for their valuable comments on the manuscript, Dasha Veprintseva for help with initial experiments, and Jennifer Jordan and Michael Resnick for providing the sequences of some of the response elements included in this study. This research was supported by Cancer Research UK, the Medical Research Council and by EC FP6 funding. This publication reflects the authors’ views and not necessarily those of the EC. The Community is not liable for any use that may be made of the information. Funding to pay the Open Access publication charges for this article was provided by the Medical Research Council.
Conflict of interest statement. None declared.
APPENDIX 1
Sequence logo construction
The relative height of individual nucleotide bar at each position reflects the amount of protein bound to the sequence relative to the highest affinity sequence as defined by Equation (2)
| A.1 |
Where i is the position of the nucleotide, N is its identity (A,T,G,C), n is the Hill coefficient and ΔlogKd(i,N) is the change in the logKd taken from the binding matrix (Table 2).
The overall height of the bar at each position is determined by the nucleotide substitution which causes the largest change in the logKd and results in the smallest amount of protein bound.
| A.2 |
Combining Equations (A.1) and (A.2), the heights of the individual nucleotide bars are calculated as follows:
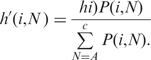 |
REFERENCES
- 1.Hainaut P, Wiman KG. 25 Years of P53 Research. New York: Springer; 2005. [Google Scholar]
- 2.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Lane DP. p53 in health and disease. Nat. Rev. Mol. Cell. Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 4.El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat. Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 5.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Smith LT, Plass C, Huang TH. ChIP-chip comes of age for genome-wide functional analysis. Cancer Res. 2006;66:6899–6902. doi: 10.1158/0008-5472.CAN-06-0276. [DOI] [PubMed] [Google Scholar]
- 7.Bulyk ML. DNA microarray technologies for measuring protein–DNA interactions. Curr. Opin. Biotechnol. 2006;17:422–430. doi: 10.1016/j.copbio.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Noll DM, Lieb JD, Clarke ND. DIP-chip: rapid and accurate determination of DNA-binding specificity. Genome Res. 2005;15:421–427. doi: 10.1101/gr.3256505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 1987;193:723–750. doi: 10.1016/0022-2836(87)90354-8. [DOI] [PubMed] [Google Scholar]
- 10.Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 11.Linnell J, Mott R, Field S, Kwiatkowski DP, Ragoussis J, Udalova IA. Quantitative high-throughput analysis of transcription factor binding specificities. Nucleic Acids Res. 2004;32:e44. doi: 10.1093/nar/gnh042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Clarke ND. Rationalization of gene regulation by a eukaryotic transcription factor: calculation of regulatory region occupancy from predicted binding affinities. J. Mol. Biol. 2002;323:1–8. doi: 10.1016/s0022-2836(02)00894-x. [DOI] [PubMed] [Google Scholar]
- 13.Udalova IA, Mott R, Field D, Kwiatkowski D. Quantitative prediction of NF-kappa B DNA–protein interactions. Proc. Natl Acad. Sci. USA. 2002;99:8167–8172. doi: 10.1073/pnas.102674699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikolova PV, Henckel J, Lane DP, Fersht AR. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl Acad. Sci. USA. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joerger AC, Allen MD, Fersht AR. Crystal structure of a superstable mutant of human p53 core domain: insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 2004;279:1291–1296. doi: 10.1074/jbc.M309732200. [DOI] [PubMed] [Google Scholar]
- 16.Veprintsev DB, Freund SM, Andreeva A, Rutledge SE, Tidow H, Canadillas JM, Blair CM, Fersht AR. Core domain interactions in full-length p53 in solution. Proc. Natl Acad. Sci. USA. 2006;103:2115–2119. doi: 10.1073/pnas.0511130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinberg RL, Veprintsev DB, Fersht AR. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004;341:1145–1159. doi: 10.1016/j.jmb.2004.06.071. [DOI] [PubMed] [Google Scholar]
- 18.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Tomso DJ, Inga A, Menendez D, Pittman GS, Campbell MR, Storici F, Bell DA, Resnick MA. Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proc. Natl Acad. Sci. USA. 2005;102:6431–6436. doi: 10.1073/pnas.0501721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 22.Benos PV, Bulyk ML, Stormo GD. Additivity in protein–DNA interactions: how good an approximation is it? Nucleic Acids Res. 2002;30:4442–4451. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Man TK, Stormo GD. Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res. 2001;29:2471–2478. doi: 10.1093/nar/29.12.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Stormo GD. Quantitative analysis of EGR proteins binding to DNA: assessing additivity in both the binding site and the protein. BMC Bioinformatics. 2005;6:176. doi: 10.1186/1471-2105-6-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ang HC, Joerger AC, Mayer S, Fersht AR. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006;281:21934–21941. doi: 10.1074/jbc.M604209200. [DOI] [PubMed] [Google Scholar]
- 26.Ma B, Pan Y, Zheng J, Levine AJ, Nussinov R. Sequence analysis of p53 response-elements suggests multiple binding modes of the p53 tetramer to DNA targets. Nucleic Acids Res. 2007;35:2986–3001. doi: 10.1093/nar/gkm192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menendez D, Krysiak O, Inga A, Krysiak B, Resnick MA, Schonfelder G. A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc. Natl Acad. Sci. USA. 2006;103:1406–1411. doi: 10.1073/pnas.0508103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stormo GD, Schneider TD, Gold L. Quantitative analysis of the relationship between nucleotide sequence and functional activity. Nucleic Acids Res. 1986;14:6661–6679. doi: 10.1093/nar/14.16.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005;348:589–596. doi: 10.1016/j.jmb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 30.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaneshiro K, Tsutsumi S, Tsuji S, Shirahige K, Aburatani H. An integrated map of p53-binding sites and histone modification in the human ENCODE regions. Genomics. 2007;89:178–188. doi: 10.1016/j.ygeno.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Qian H, Wang T, Naumovski L, Lopez CD, Brachmann RK. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene. 2002;21:7901–7911. doi: 10.1038/sj.onc.1205974. [DOI] [PubMed] [Google Scholar]
- 35.Lim YP, Lim TT, Chan YL, Song AC, Yeo BH, Vojtesek B, Coomber D, Rajagopal G, Lane D. The p53 knowledgebase: an integrated information resource for p53 research. Oncogene. 2007;26:1517–1521. doi: 10.1038/sj.onc.1209952. [DOI] [PubMed] [Google Scholar]