

Summary
Peroxisome proliferator-activated receptor-γ (PPARγ) is a ligand activated transcription factor playing a critical role in metabolism. Thiazolidinediones, high affinity PPARγ ligands used clinically to treat type-II diabetes, have been reported to lower blood pressure and provide other cardiovascular benefits. Some mutations in PPARγ cause type-II diabetes and severe hypertension. We tested the hypothesis that PPARγ in vascular muscle plays a role in the regulation of vascular tone and blood pressure. Transgenic mice expressing dominant negative mutations in PPARγ under the control of a smooth muscle-specific promoter exhibit a loss of responsiveness to nitric oxide and striking alterations in contractility in the aorta, hypertrophy and inward remodeling in the cerebral microcirculation, and systolic hypertension. These results identify PPARγ as pivotal in vascular muscle as a regulator of vascular structure, vascular function and blood pressure, potentially explaining some of the cardioprotective effects of thiazolidinediones.
Introduction
The incidence of obesity and diabetes and their consequent cardiovascular sequelae has reached epidemic proportions, justifying the enormous effort to identify pathways and molecules involved in their pathogenesis. One molecule to emerge at the crossroads of metabolic and cardiovascular diseases is peroxisome proliferator activated receptor gamma (PPARγ). PPARγ is a member of the nuclear receptor superfamily of ligand-activated transcription factors that has a well recognized role in regulating metabolic processes. PPARγ is most highly expressed in adipose tissue (Chawla et al., 1994), regulates lipid metabolism in adipocytes (Lehrke and Lazar, 2005), and stimulates adipogenesis (Rosen et al., 1999). The importance of PPARγ in metabolism is underscored by studies reporting insulin resistance in models lacking PPARγ in adipose tissue, skeletal muscle, or liver (He et al., 2003; Hevener et al., 2003; Matsusue et al., 2003).
Perhaps the most important observation supporting the significance of PPARγ was the recognition that it was the molecular target of the thiazolidinedione (TZD) class of insulin sensitizer drugs used clinically to treat type-II non-insulin-dependent diabetes mellitus (Lehmann et al., 1995). In animal models, PPARγ activation by TZDs has been shown to have beneficial effects on cardiovascular endpoints. TZD treatment inhibited early atherosclerotic lesion formation in apolipoprotein-E and LDL receptor-deficient mice (Chen et al., 2001; Li et al., 2000), attenuated the rise in blood pressure (BP) in response to exogenously infused angiotensin-II (Diep et al., 2002), and reduced BP and improved vascular function in a normoglycemic model of chronic hypertension (Ryan et al., 2004). Although there is a substantial clinical literature reporting the BP lowering and cardioprotective effects of TZDs in type-II diabetes (Bennett et al., 2004; Dormandy et al., 2005), both have recently been challenged (Nissen and Wolski, 2007). A recent meta-analysis suggests the risk of cardiovascular events including myocardial infarction and death may be increased in patients taking rosiglitazone (Nissen and Wolski, 2007), although this view remains controversial (Singh et al., 2007). Accordingly, there is an urgent need to not only assess the beneficial and consequential effects of TZDs but to understand the fundamental mechanisms controlling metabolic and cardiovascular function of PPARγ itself.
The importance of PPARγ is corroborated by the severe consequences to individuals carrying certain PPARγ mutations. PPARγ mutations have been described in patients with familial partial lipodystrophy and other metabolic disturbances (Hegele et al., 2002; Savage et al., 2003); and heterozygous carriers of the V290M or the P467L dominant negative mutations in PPARγ exhibit severe early onset hypertension, insulin resistance and type-II diabetes (Barroso et al., 1999). The importance of PPARγ in the cardiovascular system is further demonstrated by the observation that heterozygous knock-in mice carrying one copy of PPARγ containing the dominant negative P465L mutation (equivalent to human P467L) exhibited abnormal fat distribution and elevated blood pressure (Tsai et al., 2004). Insulin resistance was observed in a similar mouse model expressing the P465L mutation on a genetic background of leptin-deficiency (Gray et al., 2006).
PPARγ is expressed in endothelium and smooth muscle in the blood vessel wall (Inoue et al., 1998; Marx et al., 1998). Consequently, we hypothesized that the beneficial cardiovascular effects of TZDs may be mediated by activation of vascular PPARγ and interference with its function by dominant negative mutations in these cell types may cause vascular dysfunction and hypertension. We tested the hypothesis that vascular muscle PPARγ is required to mediate vasodilator signals derived from the endothelium and to regulate vasoconstrictor mechanisms. We report that mice expressing the dominant negative P467L or V290M mutations in PPARγ in vascular muscle exhibit severely impaired aortic reactivity to endothelium-derived or exogenously applied nitric oxide mechanistically due to a defect in cGMP-mediated signaling. In addition, there are marked alterations in vasoconstrictor responses to several agonists, vascular hypertrophy and remodeling in the microcirculation, and systolic hypertension. These changes were due to the dominant negative activity imparted by mutant PPARγ since there were no defects in vascular reactivity to vasodilators or vasoconstrictors in mice expressing wildtype PPARγ in vascular muscle. This study provides evidence implicating a pivotal role for smooth muscle PPARγ in the regulation of vascular structure and function and provides a molecular explanation for some of the beneficial cardiovascular effects of TZD drugs.
Results
Analysis of Transgenic Mice Expressing PPARγ
Transgenic (Tg) mice were generated by expressing the protein coding sequence of wildtype (WT) human PPARγ1 or PPARγ1 carrying either the P467L or V290M muta0tions under the control of the smooth muscle myosin heavy chain promoter (Figure 1A) (Madsen et al., 1998). The mice are herein termed S-WT, S-P467L and S-V290M. Transgenic mice were born at the expected Mendelian ratio (365 vs 324 for S-P467L transgenic vs non-transgenic respectively, p=0.29 by χ2) and there was no difference in survival when compared to non-transgenic (NT) littermates up to 12 months of age.
Figure 1. Transgene Expression in S-PPARγ Transgenic Mice.
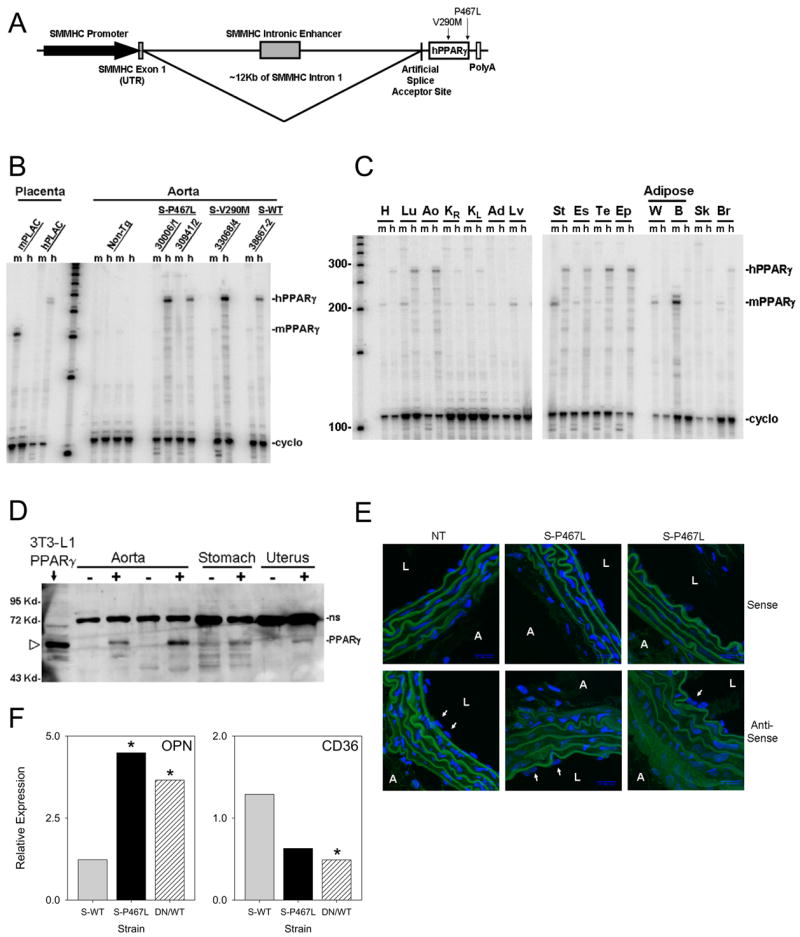
A. Schematic of the S-PPARγ transgenes containing 16 kb of 5′ region of the rat smooth muscle myosin heavy chain (SMMHC) gene including 4 kb of the promoter, exon 1, and 12 kb of intron 1 containing a smooth muscle-specific enhancer, and an artificial splice acceptor site. B. RNase protection assay (RPA) verifying transgene expression in aorta. Probes specific to mouse (m) or human (h) PPARγ differentiate endogenous from transgene PPARγ expression. Specificity was verified using mouse or human placental (PLAC) RNA. C. Representative tissue RPA from S-P467L mice using probes from B. Tissues include heart (H), lung (Lu), aorta (Ao), right (KR) and left (KL) kidney, adrenal gland (Ad), liver (Lv), stomach (St), esophagus (Es), testes (Te), epididymis (Ep), white (W) and brown (B) adipose tissue, skeletal muscle (Sk), and brain (Br). D. Western blot of tissues from NT (−) and S-P467L (+) mice. NS indicates a non-specific band detected by the 2° antisera even in the absence of the 1° antisera. The position of the PPARγ band is indicated by the open arrowhead identified both by size and its overexpression in 3T3-L1 cells transfected with CMV-P467L expression vector. E. Fluorescent in situ hybridization on slices of thoracic aorta from S-P467L and NT littermates were hybridized with a sense or anti-sense RNA probe directed at the unique 3′ end of the transgene. Hybridization was visualized using FITC-labeled anti-DIG antibody, and cell nuclei were visualized using DAPI. L- lumen; A- adventitia; arrows indicate endothelial cell nuclei. F. OPN and CD36 gene expression in aorta of NT, S-WT and S-P467L measured by real-time RT-PCR. *, P<0.05.
RNase protection assays (RPA) utilizing probes specific to either mouse or human PPARγ revealed that the level of transgene expression in the aorta was elevated compared to endogenous PPARγ mRNA (Figure 1B). Transgene over-expression was also evident in the stomach and uterus, both rich in smooth muscle (Figure S1). Expression of the transgene in S-P467L line 30006/1 was observed in nearly all tissues tested consistent with the presence of blood vessels (Figure 1C). Importantly however, whereas hPPARγ was primarily over-expressed in tissues high in smooth muscle content, transgene expression was virtually undetectable in tissues high in endogenous PPARγ such as the white and brown adipose tissue and skeletal muscle (Figure 1C). Consistent with the absence of transgene expression in fat and skeletal muscle there was no difference in body weight and adipose tissue weights of S-P467L transgenic mice compared with their NT littermates, nor was there a difference in levels of fasting blood glucose, fasting plasma insulin, or serum leptin (Table S1). Therefore, expression of dominant negative PPARγ in vascular smooth muscle did not cause overt systemic metabolic dysfunction. PPARγ protein was evident in the aorta, stomach and uterus from S-P467L line 30006/1 mice (Figure 1D).
In situ hybridization was performed to confirm cell-specific expression of the S-P467L transgene in the aorta (Figure 1E). Positive staining was observed in the cytoplasm of smooth muscle cells but not in the endothelium from S-P467L mice. There was no staining with the sense probe, and staining was eliminated in sections incubated without probe or without the secondary antisera (Figure S2).
Dominant negative activity of PPARγ was verified by examining expression of two of its target genes, osteopontin (OPN) and the fatty acid transporter CD36. OPN is a secreted extracellular matrix protein synthesized by macrophages, endothelial and smooth muscle cells (Giachelli et al., 1995). Its expression is decreased upon PPARγ activation in THP-1 cells and in whole aorta of mice treated with rosiglitazone (Oyama et al., 2002). Consistent with dominant negative activity, we found significantly elevated OPN expression in aorta of S-P467L mice, but not in S-WT mice (Figure 1F). CD36 expression is increased in response to PPARγ activation (Feng et al., 2000). In contrast to OPN, we found CD36 to be decreased in S-P467L but unaltered in S-WT mice (Figure 1F).
Interference with PPARγ Causes Impaired Vasodilation
Endothelial dysfunction is an initial step in the pathogenesis of many cardiovascular diseases including hypertension. To determine the effect of smooth muscle-specific expression of dominant negative PPARγ on endothelial function, we studied aorta using vascular rings pre-contracted with prostaglandin-F2a (PGF2a) and measured relaxation to increasing doses of acetylcholine (Ach), an endothelium-dependent agonist. The response of S-P467L aorta to Ach was markedly impaired compared to that of NT littermates (Figure 2A). Ach stimulates the endothelium to produce nitric oxide (NO) from endothelial NO synthase (eNOS) which then diffuses to the vascular muscle to cause relaxation. Since endothelial dysfunction can result from decreased NO bioavailability, we tested the responsiveness of S-P467L aorta to exogenous nitric oxide using the NO donor sodium nitroprusside (SNP). Like Ach, the vasodilator response of S-P467L aorta to SNP was significantly impaired (Figure 2B). To determine whether the observed impairment in vascular relaxation was due to generalized smooth muscle dysfunction, we used the endothelium-independent and NO-independent vasodilator papaverine (PAP). We observed a slight, but statistically significant rightward shift in the dose-response curve in aorta from S-P467L to submaximal concentrations of PAP (Figure 2C). However, the aorta from S-P467L and NT controls were able to completely relax at maximal concentrations of PAP. Although the dose response to PAP suggests some generalized deficit in smooth muscle function, this impairment was modest and cannot explain the magnitude of the impaired response to Ach or SNP.
Figure 2. Dominant Negative PPARγ Causes Impaired Vasodilation.

Isometric tension studies on thoracic aorta segments of S-P467L line 30006/1 mice (filled circle) and NT littermates (grey circle) (6–8 months of age) in response to increasing doses of Ach (A), SNP (B), and PAP (C) after the aorta was precontracted with PGF2α (5–10μM). Similar studies were performed in S-V290M line 33068/4 (filled diamonds), S-WT line 38667/2 (filled square) and NT (grey triangle) mice in response to Ach (D), SNP (E) and PAP (F). *, P<0.05 at the indicated concentration. Error bars represent SEM.
Importantly, the same impaired response to Ach, SNP and PAP was replicated in the aorta derived from independent transgenic mice expressing the V290M dominant negative variant of PPARγ, but not mice expressing wildtype PPARγ (Figure 2D–F). This was verified by comparing Ach-mediated relaxation in aorta of S-WT and a line of S-P467L mice (30941/2) expressing similar levels of PPARγ mRNA (Figure 1B) and protein in aorta (Figure S3). This strongly suggests that the vascular dysfunction observed in S-P467L and S-V290M mice is due to dominant negative activity imparted by the P467L and V290M mutations in PPARγ.
In vascular muscle, NO stimulates soluble guanylate cyclase (sGC) to produce cGMP. To determine the mechanism causing the impaired response to NO and to specifically test whether the impairment is at the level of sGC, we examined the response of S-P467L aorta to the cGMP analog, 8-Br-cGMP. We observed a significant reduction in the relaxation of S-P467L aorta to 8-Br-cGMP (Figure 3A), suggesting the PPARγ-mediated defect in smooth muscle is downstream of sGC. cGMP stimulates a series of intracellular signals through activation of the cGMP-dependent protein kinase (PKG). There was no change in the levels of PKG1 expression in the aorta comparing S-P467L to NT mice (Figure 3B). However, there was 11.9-fold (P<0.001) induction in the expression of PKG2 mRNA, an isoform of PKG normally found in renal and adrenal cells, but not smooth muscle (Figure 3B). Interestingly, neither PKG1 (0.95±0.04) nor PKG2 (1.0±0.03) mRNAs were altered in aorta from normal C57BL/6 mice in response to the PPARγ agonist Rosiglitazone suggesting they are not direct targets of PPARγ (unpublished data, GEO Database Accession GSE8949). This suggests the signaling defect may lie downstream of PKG, a finding consistent with the loss of Ach and cGMP-mediated relaxation in PKG1-deficient mice (Pfeifer et al., 1998). The upregulation of the atypical isoform may be an attempt, albeit unproductive, to compensate for the defect in cGMP-dependent signaling in response to PPARγ interference.
Figure 3. Mechanism of Impaired Vasodilation in S-P467L Mice.
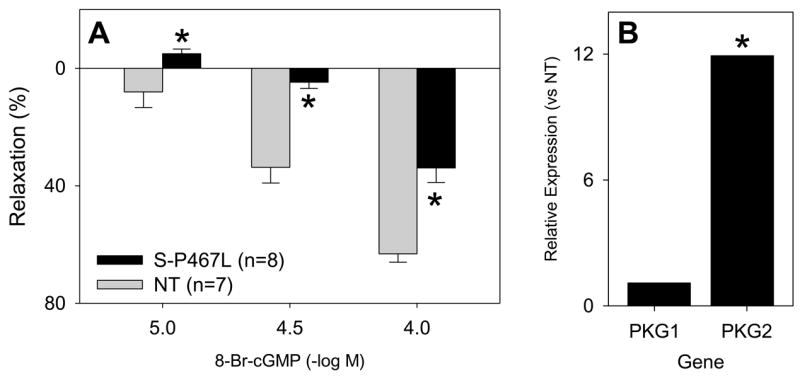
A. Isometric tension studies on thoracic aorta segments of S-P467L line 30006/1 mice (filled bar) and their NT littermates (grey bar) in response to increasing doses of the cGMP analog 8-Br-cGMP. *, P<0.05 at the indicated concentration. B. Gene expression of PKG1 and PKG2 in aorta comparing S-P467L to NT. *, P<0.001. Error bars represent SEM.
Interference with PPARγ Causes Altered Vasoconstriction
The peptide endothelin-1 (ET-1) is an extremely potent vasoconstrictor released by the endothelium implicated in many models of hypertension (Schiffrin, 2001). PPARγ activation by TZDs has been shown to inhibit induction of ET-1 expression by thrombin in endothelial cells (Delerive et al., 1999). Consequently, we hypothesized that the vasoconstrictor response to ET-1 in aorta may be augmented in S-P467L mice. ET-1 produces little contraction in aorta from normal mice (Figure 4A). Remarkably however, we observed a significant hyperresponsiveness (over 7-fold) to the constrictor effect of ET-1 in aorta from S-P467L (Figure 4A). Importantly, a similar effect was observed in aorta from mice expressing the S-V290M construct (Figure 4B) and to a lesser extent in a second independent S-P467L line exhibiting a lower level of transgene expression (Figure S3). However, there was no enhancement in ET-1-mediated vasoconstriction in mice expressing wildtype PPARγ (Figure 4B, Figure S3) suggesting the phenotype we observed in S-P467L and S-V290M is due to dominant negative effects of PPARγ.
Figure 4. Dominant Negative PPARγ Causes Increased Contraction to ET-1.

A. Aortic ring studies of S-P467L line 30006/1 mice (filled circle) and their NT littermates (grey circle) were performed in response to increasing doses of endothelin-1. B. The same studies as A were performed on aortic segments from NT controls (grey triangle), S-V290M (closed diamond), and S-WT (filled square). C–E. The response of S-P467L aorta to ET-1 in the presence of ETA receptor antagonist (1 μmol/L, C), ETB receptor antagonist (1 μmol/L, D), or the rho kinase inhibitor Y27632 (3 μmol/L, E) was determined. *, P<0.05 between transgenic and NT. **, P<0.05 antagonist vs no antagonist. F. Relative gene expression of ETA receptor, ETB receptor and preproET-1 mRNA in aorta comparing S-P467L to NT. G. Plasma ET-1 levels in NT (grey bar) and S-P467L (filled bar) mice. *, P=0.02. Error bars represent SEM.
To examine the mechanism for this hypercontractility we first examined which receptor subtype, ETA or ETB mediated the ET-1 response. The ETA receptor is mainly expressed in vascular muscle and is thought to mediate contraction, while the ETB receptor is expressed on both endothelial cells, where it mediates relaxation through the production of NO, and on vascular muscle where it mediates contraction. We tested the response of these vessels in the presence of BQ-123, an ETA receptor antagonist, and BQ-788, an ETB receptor antagonist. We found that the response is primarily mediated by the ETA receptor (Figure 4C). However, blocking the ETB receptor resulted in slight, but significant inhibition of the contractile response at mid-range doses (Figure 4D). The response to both receptor blockers together was not different from ETA blocker alone (data not shown). ET-1 peptide levels in plasma of S-P467L mice were unchanged (Figure 4G), whereas in aorta, preproendothelin-1 mRNA levels were reduced 40% (Figure 4F). At the receptor level, there was no increase in the level of ETA or ETB receptor mRNA in the aorta of S-P467L mice (Figure 4F). This data suggests that the increase in ET-1-mediated contraction is most likely due to a post-receptor mechanism. Consequently, we evaluated if the ET-1 response is dependent upon Rho Kinase activation. The Rho kinase-specific inhibitor Y27632 had the same inhibitory effect as ETA receptor blockade suggesting the response to be dependent upon Rho Kinase activity (Figure 4E).
To further examine the mechanism for the hypercontractile response to ET-1, we evaluated if it was due to a general reprogramming of the vessel wall or was selective for ET-1. Angiotensin-II causes minimal contraction in thoracic and abdominal aorta and there was no increase in contraction in aorta from S-P467L mice (data not shown). There was no difference in the constrictor response to the receptor-independent vasoconstrictor, potassium chloride (KCl) (Figure 5A) or PGF2α, the agent used to precontract the vessels prior to Ach and SNP (Figure 5B). Similar to ET-1, there was a modest hypercontraction to serotonin (5-HT) (Figure 5C) that was also observed in S-V290M but not S-WT mice (Figure 5D). It is noteworthy that there was a modest decrease in contraction of the aorta to the a-adrenergic agonist phenylephrine (PE) in S-P467L mice (Figure 5E). These data strongly suggest that interference with PPARγ causes an agonist-selective alteration in vasoconstriction and not a general reprogramming of the vessel wall to be hypercontractile. Consequently, PPARγ regulates vascular tone by modulating a number of both vasodilator and vasoconstrictor pathways.
Figure 5. Dominant Negative PPARγ Causes Altered Contraction to Agonists.

The contractile response of thoracic aorta to KCl (A), PGF2α (B), 5-HT (C, D), and phenylephrine (E) was assessed. S-P467L aorta (filled circle), NT littermates (grey circle). In D, S-V290M (filled diamond), S-WT (filled square) and NT (grey triangle) was examined. *, P<0.05 transgenic vs. NT. Error bars represent SEM.
Interference with PPARγ Causes Systolic Hypertension
One of the main features of patients that carry the V290M or P467L mutations in PPARγ is early onset hypertension (Barroso et al., 1999). Therefore, we measured BP of S-P467L and NT littermate mice using radiotelemetry. The normal diurnal variation in systolic, mean, and diastolic BP and heart rate (HR) was maintained in S-P467L and NT mice (Figure 6A–D). Systolic BP values throughout each 24 hour period (over 9 consecutive days) were higher in S-P467L than NT mice (Figure 6E) whereas the increase in mean BP was only significant during the day (Figure 6F). There was no difference in diastolic BP (Figure 6G) or the pulse pressure (Figure 6H). These data suggest the presentation of isolated systolic hypertension, a very common form of hypertension observed in the elderly associated with altered vascular compliance (Wallace et al., 2007). Interestingly, HR was also significantly increased in S-P467L mice (Figure 6D and I). To determine if this increase in HR was pathological we performed echocardiography on S-P467L and NT mice. There was no difference in end diastolic or systolic volumes, stroke volume, cardiac output or ejection fraction (Table S2) and the heart weight to body weight ratio was unchanged (Figure 6J). Considering that P467L PPARγ was not over-expressed compared to endogenous PPARγ in the heart the increase in HR is more likely a result of the vascular dysfunction or alterations in vascular compliance than a result of myocyte dysfunction.
Figure 6. Dominant Negative PPARγ Causes Hypertension.

Hourly systolic (A), mean (B), diastolic (C) blood pressure, and heart rate (D) tracing of S-P467L mice (filled circle, N=7) and their NT littermates (grey circle, N=9) over 9 consecutive 24 hour periods was examined. The hourly blood pressure for each 60 minute period across all 9 days of measurement were combined to provide the average hourly BP and HR recording. Daytime (6am to 6pm) and nighttime (6pm to 6am) averages for SBP (E), MBP (F), DBP (G), pulse pressure (PP, H) and HR (I) over 9 days. J. Heart weight to body weight ratio. NT, grey bar; S-P467L, black bar. *, P<0.05 vs. NT. Error bars represent SEM.
Interference with PPARγ Causes Vascular Hypertrophy and Remodeling
We next examined the structure of cerebral arterioles, resistance vessels that exhibit hypertrophy and inward remodeling in response to hypertension. Before deactivation with EDTA, the diameter of cerebral arterioles was not different in S-P467L (36±3 μm) and NT (33±4 μm) mice. During maximal dilation with EDTA, internal diameter was significantly less in cerebral arterioles in S-P467L mice than in NT mice at all levels of arteriolar pressure (Figure 7E). While internal and external diameter was less, cross-sectional area of the vessel wall was significantly greater in S-P467L mice (Figure 7A–C). Wall thickness was concomitantly increased in S-P467L mice (Figure 7D). The stress-strain curve in cerebral arterioles in S-P467L mice was shifted to the right of the curve in NT mice (Figure 7F). Thus, cerebral arterioles in S-P467L mice underwent hypertrophy with an increase in distensibility and remodeling with a reduction in external diameter.
Figure 7. Structural and Mechanical Changes in Cerebral Arterioles in S-P467L Mice.

Internal (A) and external (B) diameter, cross-sectional area (CSA) of the vessel wall (C) and wall thickness (D) of cerebral arterioles after maximal dilation. E. Pressure-internal diameter relationships in arterioles during maximal dilation. F. Stress-strain relationships in arterioles during maximal dilation. NT (grey bar and circle); S-P467L (closed bar and circle). D, cerebral arteriolar diameter; D0, original arteriolar diameter. *, P<0.05 vs. NT. Error bars represent SEM.
Discussion
According to the American Diabetes Association, approximately 65% of diabetic patients will suffer severe cardiovascular consequences such as myocardial infarction and stroke and as many as 73% are hypertensive. Thus, gaining an understanding of the basic mechanisms linking metabolic and cardiovascular function is critical. PPARγ has become an extremely attractive candidate as it stands at the crossroads of metabolic and cardiovascular regulation. Although PPARγ is expressed in many cardiovascular tissues, its expression in the endothelium and vascular muscle has been of particular interest because of the antihypertensive, antiatherogenic and antiinflammatory actions of TZDs (reviewed in (Blaschke et al., 2006)). TZDs increase NO synthase and decrease ET-1 expression in cultured endothelial cells (Delerive et al., 1999; Goya et al., 2006). They also decrease proliferation, migration, and production of fibrotic factors, block calcium channel activity, and increase insulin receptor signaling in cultured vascular muscle (Nakamura et al., 1998; Pandey et al., 2007; Gao et al., 2007). With the increasing clinical use of TZDs as anti-diabetes medications, and the plethora of TZD-treatment studies in animals, a protective role for PPARγ in preventing or diminishing adverse cardiovascular sequelae associated with type-II diabetes has emerged. It is particularly noteworthy that TZDs can lower BP despite promoting sodium and water retention by renal tubules (a serious side effect of TZD drugs) suggesting that their antihypertensive effects are profound and independent of renal sodium handling.
There has been only one study which has directly interrogated the specific importance of vascular PPARγ in vivo in a manner which eliminates the PPARγ-independent actions of TZDs (Feinstein et al., 2005). This was accomplished using mice with a cre-loxP-mediated endothelial-specific PPARγ deficiency (Nicol et al., 2005). These mice were normotensive at baseline but developed hypertension after being fed a high fat diet. Consequently, this finding and the report that individuals carrying dominant negative mutations in PPARγ exhibit severe early onset hypertension (Barroso et al., 1999), led us to hypothesize that the relative importance of PPARγ in regulating BP and vascular tone may be greater in the vascular muscle than the endothelium. We sought to address this concept directly by interfering with PPARγ activity by overexpressing the same dominant negative mutations in PPARγ which cause hypertension in humans. Smooth muscle selectivity of the mutant PPARγ expression was achieved by employing the smooth muscle myosin heavy chain promoter.
Overexpression of P467L PPARγ in vascular muscle resulted in a robust phenotype. Aorta of these animals exhibited a severely impaired response to NO and its downstream mediators. These findings suggest that the effect exerted by PPARγ interference occurs downstream of soluble guanylate cyclase, perhaps at the level or below PKG (the cGMP-dependent protein kinase). That PKG1 and PKG2 do not appear to be direct targets of PPARγ and that transcription of the atypical isoform of PKG (PKG2) was markedly increased supports the hypothesis that the deficit may be downstream. Concomitantly, aorta from these mice exhibited a marked hypercontractile response to ET-1 that was mediated by the ETA receptor and dependent upon the rho kinase pathway. Again, the defect causing hypercontraction is likely to be post-receptor as there was no difference in ETA, ETB, preproET-1 mRNA nor plasma ET-1 levels. Hyperresponsiveness to ET-1 is often seen in vessels from animal models of hypertension (Miki et al., 1998). Like ET-1, there was a modest increase in the contractile response to 5-HT, the contraction to which is augmented in hypertension (Watts, 2002). That both ET-1- and 5-HT-mediated contraction is increased in vessels from eNOS−/− mice is consistent with these results (Zhou et al., 2006; Budzyn et al., 2004).
Unlike ET-1 and 5-HT, the contractile responses to KCl, a receptor-independent vasoconstrictor, and PGF2α a receptor dependent vasoconstrictor were unaltered, whereas the response to PE was modestly reduced in aorta from S-P467L mice. PGF2α was used to pre-contract aorta prior to treatment with Ach, SNP and PAP; and therefore, the alteration in vasodilator responses cannot be accounted for by changes in PGF2α-mediated contraction. While these data suggest that the hypercontractile phenotype to ET-1 and 5-HT is selective to specific agonists and not a generalized change in the vasoconstrictor state of the vessel, it does not explain the paradoxical decrease in contraction to the a-adrenergic agonist. Indeed, like ET-1 and 5-HT, PE-induced contractions are augmented in eNOS−/− mice (Budzyn et al., 2004). Conversely, PE-induced contractions are reduced in vessels from rats chronically infused with ET-1, and the sensitivity to PE is reduced in ET-1 infused rats treated with the NOS inhibitor L-NNA (Thorin et al., 1998). This may be analogous to the S-P467L mice where ET-1 responsiveness is increased and NO-responsiveness is severely impaired.
One of the most important findings from this study was that there was a similar vascular dysfunction in mice overexpressing another dominant negative mutation in PPARγ (V290M), but not in mice expressing wildtype PPARγ in vascular muscle. These data suggest that the V290M or P467L mutations act through a similar interfering mechanism and that the vascular dysfunction is a result of PPARγ interference rather than mere PPARγ overexpression. Moreover, our data suggest that the extent of vascular impairment seems to be affected by the ‘dose’ of dominant negative PPARγ. Dominant negative mutations in PPARγ have been identified in the DNA binding and ligand binding domains of the protein. The mutations affecting the DNA binding domain are thought to retain their interaction with retinoid X receptor (RXR) and other co-activators but are transcriptionally defective because they cannot bind to the PPARγ response element (PPRE) (Agostini et al., 2006). They may act dominantly by squelching the availability of RXR or other co-activators thus preventing their association with normal PPARγ. The ligand binding domain mutants, including P467L, although they retain DNA binding, are transcriptionally defective because the mutations destabilize a region of the protein required for co-activator recruitment in response to ligand binding (Agostini et al., 2004). Current data suggests they act dominantly because of increased interaction with co-repressors and reduced cycling of the transcription complex on the DNA presumably preventing the interaction of the PPRE with normal PPARγ/RXR (Li and Leff, 2007). If the reduced cycling rate of RXR causes it to be in limiting amounts then both the DNA-binding domain and ligand-binding domain mutants in PPARγ may have far reaching effects on other members of the ligand-activated transcription factor family that interact with RXR. Consequently, we cannot rule out that the vascular dysfunction in our model is the result of such effects; effects which may also occur in human subjects carrying these mutations. In addition to the classical trans-activation pathway, PPARγ exerts effects on inflammatory genes such as inducible NO synthase by a more complex ligand-dependent but PPRE-independent trans-repression mechanism (Pascual et al., 2005). Our observation that dominant negative PPARγ causes a robust increase in OPN expression suggests that these mutations may also affect this trans-repression activity.
Another important finding in this study is the lack of effect of S-P467L PPARγ expression on metabolic parameters, particularly body weight, adipose tissue distribution, and glucose homeostasis. These data suggest that PPARγ in smooth muscle does not affect adipocyte metabolism or whole body glucose homeostasis. Because expression of dominant negative PPARγ in adipose tissue, skeletal muscle or liver would have been expected to produce metabolic disturbances, particularly lipodystrophy and insulin resistance, the absence of these findings in the face of severe vascular dysfunction further supports the specificity of our model. As PPARγ has recently been reported to modulate insulin receptor signaling in smooth muscle (Pandey et al., 2007), future studies will have to address whether the defects observed in the vasculature are limited to the signaling machinery regulating vasorelaxation and contraction or whether there are other cellular metabolic defects as well.
Since PPARγ is a transcription factor, the vascular protection afforded by its ligand-mediated activation most likely results from a change in the gene expression profile in vascular muscle. In type-II diabetes, administration of TZDs causes increased PPARγ activity which alters the expression of genes involved in fatty acid metabolism and glucose utilization thus resulting in increased insulin sensitivity. Prevailing evidence is that in the vascular wall, activation of PPARγ shifts the balance from a pro-inflammatory, pro-atherogenic and pro-vasoconstrictive state toward an anti-inflammatory, anti-atherogenic and pro-vasodilatory state. Consequently during a metabolic disturbance, endogenous PPARγ ligands increase, activating vascular PPARγ in an attempt to counterbalance potential damaging effects. Interfering with PPARγ function in the vascular muscle, as in the mice described herein, prevents this protective effect from occurring thus leading to impaired vascular function and hypertension. In conclusion, the data presented herein provide strong evidence supporting a critical role for vascular muscle PPARγ in the regulation of vascular tone.
Experimental Procedures
Generation of Transgenic Mice
Generation of the transgene construct is described in Supplemental Experimental Procedures. The transgene was excised, purified and microinjected into pronuclei of B6SJL mice (C57BL/6J × SJL/J). Transgenic mice were maintained by backcross breeding to C57BL/6J. Genotyping was performed by PCR of tail DNA using primers 5′-TATCTTCTAACTGGGTGGTGGTG-3′ and 5′-GAGGAGAGTTACTTGGTCGTTCA-3′. All mice were fed standard mouse chow (7013, Teklad Premier Laboratory Diets) and water ad libitum. Care of the mice used in the experiments met the standards set forth by the NIH in their guidelines for the care and use of experimental animals. All procedures were approved by the University Animal Care and Use Committee at the University of Iowa.
RNase Protection Assay
Mouse total RNA was isolated from tissues using Tri-Reagent (Molecular Research Center Inc., Cincinnati, OH) as described by the manufacturer. The RPAIII Kit (Ambion, Austin, TX) was used to assay transgene and endogenous PPARγ expression using cyclophilin (105 nucleotides) as a loading control. Probe construction is detailed in Supplemental Experimental Procedures.
Western Blots
30 μg protein were run a 10% SDS-polyacrylamide gel. Following transfer, the membrane was immunoblotted using serum (1:2000) from rabbit GN-16,963 immunized with the hPPARγ peptide TPHYEDIPFTRTDPVVADYKC (Sigma). HRP-conjugated Donkey a-Rabbit IgG (Santa Cruz) was used (1:50,000) as a secondary antibody prior to protein detection using ECL Plus kit (Amersham Biosciences).
Real Time RT-PCR
Total aortic RNA was purified using RNeasy spin columns (RNeasy Mini Kit, Qiagen), eluted from the column in 30μl and the concentration was established using a NanoDrop ND-1000 (NanoDrop Technologies). cDNA was generated by reverse transcription PCR using Superscript III (Invitrogen) from 400ng of total RNA using oligo dT priming (18dT). The reverse transcription was performed at 50°C for 2 hours, the enzyme was heat inactivated at 70°C for 15min and real-time PCR (qPCR) was performed using the TaqMan system (Applied Biosystems). The equivalent of 10ng of reverse transcribed RNA was PCR amplified using 2X TaqMan Universal PCR Master Mix (Applied Biosystems). Applied Biosystems Assays numbers and procedures are provided in the Supplemental Experimental Procedures.
In situ Hybridization
10 μm cryosections of thoracic aorta were placed on SuperFrost/Plus slides (Fisher Scientific), stored at −80°C overnight and allowed to dry for 10 min at room temperature (RT) before a 10 min 4% paraformaldehyde fixation. The prehybridization mix consisted of 50% deionized formamide, 5XSSC, 40 μg/ml of herring sperm DNA (prewarmed at +80°C for 5 min) and 10 X Denhardt’s solution. The prehybridization was performed in the microwave (Biowave, Ted Pella) at 45°C, 650W with 5 min microwave irradiation followed by 2 min incubation without irradiation and another 5 min pulse. The hybridization was carried out using the same mix described above with 0.25 ng/μl of digoxigenin (DIG)-labeled probe (see Supplemental Experimental Procedures) for a total amount of 5ng of probe per section. The post-hybridization washes were done in the microwave at 250W (three washes with a 1 min pulse at room temperature) in 5XSSC. The stringency washes were 2XSSC at 250W for 3 min with irradiation, 2 min without and 3 min with irradiation at +50°C and 0.5XSS. The slides were allowed to cool to RT before transfer in 1XPBS pH7.4. Sections were blocked with 2.5% BSA and 0.4% cold water fish skin gelatin in 1XPBS pH7.4 for 1 hour at RT before overnight incubation at 4°C with sheep anti-DIG (Fab fragments)-fluorescein conjugated antibody at 1/200 (1μg/ml) (Roche Diagnostics GmbH). The slides were washed 6 times, 3 min each, in PBS at RT and coverslipped under Vecta shield with DAPI (Vector Laboratories) before imaging using a Bio-Rad Radiance 2100MP Confocal Microscope.
Drugs and Reagents
Ach, SNP, PAP, KCl, 5-HT, PE, BQ-123, and BQ-788 were obtained from Sigma (St. Louis, MO), 8-Br-cGMP was from Fisher Scientific, PGF2α was from Pfizer pharmaceutical, Y-27632 was from Calbiochem (San Diego, CA). All reagents were dissolved in physiological saline. ET-1 was purchased from Peninsula Laboratories Inc., San Carlos, CA and dissolved in water. Plasma ET-1 was determined as described in Supplemental Experimental Procedures.
Aortic Ring Preparation
Male and female mice 6–8 months of age were given a lethal dose of pentobarbital (50 mg/mouse IP), the thoracic aorta was quickly removed and placed in Krebs’ buffer with the following composition (mmol/L): NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, glucose 11. Connective tissue and adventitial fat were removed and the vessel was cut into 4 equal pieces 4–5 mm in length. Vascular rings were suspended in an organ bath containing 20 mL Krebs’ solution maintained at 37°C. The rings were connected to a force transducer via steel hooks to measure isometric tension. Resting tension was increased stepwise to reach 0.5g and the rings were allowed to equilibrate for 45 minutes. After an initial sub-maximal pre-contraction (40–50% of max) with PGF2α (5–10 μM) and relaxation with Ach (100μM) and SNP (100μM) to establish the functionality of the vessel, dose response curves were preformed. Vessels were contracted sub-maximally with PGF2α and after a stable contraction plateau was reached, dose-response curves were obtained for Ach (0.01–30μM), SNP (0.01–30μM), PAP (1nM–100μM), and 8-Br-cGMP (1–100μM). Contractile response in the aorta to ET-1 (0.1nM–0.3μM), 5-HT (0.01–30μM), PE (1nM–30μM), and KCl (4mM–160mM) were examined. For ET-1 receptor blockade, two aortic rings were incubated with 1μM BQ-123, BQ-788, or 3μM Y-27632 while the other two segments were incubated with vehicle for 30min followed by ET-1. Data was collected using PowerLab 8SP and analyzed using Chart5 software (AD Instruments).
Blood Pressure Analysis
Blood pressure was measured through radiotelemetry as previously described (Lavoie et al., 2006). Mice 4–6 months of age were anesthetized with ketamine:xylazine (87.5mg:12.5mg/kg), the catheter (Data Sciences International) was placed into the left common carotid artery, and the transmitter subcutaneously along the left flank. Mice recovered for 10 days, after which heart rate and arterial pressure were continuously recorded (sampling every 5-minute for 10-second intervals) for 9 days. Data were collected and stored using Dataquest ART.
Measurements of vascular structure
Measurements of pressure and diameter in arterioles on the cerebrum were obtained as described previously (Baumbach et al., 2002) and detailed in the Supplemental Experimental Procedures.
Statistical Analysis
All data are expressed as mean±SEM. Data was excluded only when values were outside the mean ± 2SD range. Comparisons were made with 2-way repeated measures ANOVA using a Tukey post hoc test using SigmaStat software. Student’s t-test was used where appropriate. P<0.05 was considered significant.
Gene Expression Profiling
Microarray data (array platform-GPL1261) is available in MIAME format at the Gene Expression Omnibus Database (GEO) using accession number GSE8949.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL48058, HL61446, and HL55006 to CDS; HL38901, HL62984 and NS24621 to FMF, T32 GM008629 to CMH as part of the Genetics Training Program), American Heart Association (0415460Z to AMB). S-PPARγ mice were generated and maintained at the University of Iowa Transgenic Animal Facility, supported by the Carver College of Medicine. We would like to thank Drs. Kamal Rahmouni, Michael J. Ryan and Justin Grobe for advice, Chantal Allamargot in the Central Microscopy Facility for performing in situ hybridization, Robert Bianco for initial construct generation, Debbie Davis for mouse surgery, Robert Weiss (funded by NIH grant RR017369) for performing echocardiography and Gary Owens for the gift of the promoter. We gratefully acknowledge the generous research support of the Roy J. Carver Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agostini M, Gurnell M, Savage DB, Wood EM, Smith AG, Rajanayagam O, Garnes KT, Levinson SH, Xu HE, Schwabe JW, Willson TM, O’Rahilly S, Chatterjee VK. Tyrosine agonists reverse the molecular defects associated with dominant-negative mutations in human peroxisome proliferator-activated receptor gamma. Endocrinology. 2004;145:1527–1538. doi: 10.1210/en.2003-1271. [DOI] [PubMed] [Google Scholar]
- Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, Rajanayagam O, Semple R, Luan J, Bath L, Zalin A, Labib M, Kumar S, Simpson H, Blom D, Marais D, Schwabe J, Barroso I, Trembath R, Wareham N, Nagy L, Gurnell M, O’Rahilly S, Chatterjee K. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab. 2006;4:303–311. doi: 10.1016/j.cmet.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O’Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–3. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- Baumbach GL, Sigmund CD, Bottiglieri T, Lentz SR. Structure of cerebral arterioles in cystathionine beta-synthase-deficient mice. Circ Res. 2002;91:931–937. doi: 10.1161/01.res.0000041408.64867.1d. [DOI] [PubMed] [Google Scholar]
- Bennett SM, Agrawal A, Elasha H, Heise M, Jones NP, Walker M, Wilding JP. Rosiglitazone improves insulin sensitivity, glucose tolerance and ambulatory blood pressure in subjects with impaired glucose tolerance. Diabet Med. 2004;21:415–422. doi: 10.1111/j.1464-5491.2004.01155.x. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Spanheimer R, Khan M, Law RE. Vascular effects of TZDs: new implications. Vascul Pharmacol. 2006;45:3–18. doi: 10.1016/j.vph.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Budzyn K, Marley PD, Sobey CG. Chronic mevastatin modulates receptor-dependent vascular contraction in eNOS-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2004;287:R342–R348. doi: 10.1152/ajpregu.00156.2004. [DOI] [PubMed] [Google Scholar]
- Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA. Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology. 1994;135:798–800. doi: 10.1210/endo.135.2.8033830. [DOI] [PubMed] [Google Scholar]
- Chen Z, Ishibashi S, Perrey S, Osuga J, Gotoda T, Kitamine T, Tamura Y, Okazaki H, Yahagi N, Iizuka Y, Shionoiri F, Ohashi K, Harada K, Shimano H, Nagai R, Yamada N. Troglitazone inhibits atherosclerosis in apolipoprotein E-knockout mice: pleiotropic effects on CD36 expression and HDL. Arterioscler Thromb Vasc Biol. 2001;21:372–377. doi: 10.1161/01.atv.21.3.372. [DOI] [PubMed] [Google Scholar]
- Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin- induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, Neves MF, Schiffrin EL. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation. 2002;105:2296–2302. doi: 10.1161/01.cir.0000016049.86468.23. [DOI] [PubMed] [Google Scholar]
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Dello RC. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–188. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM, Jr, Hajjar DP, Nicholson AC. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res. 2000;41:688–696. [PubMed] [Google Scholar]
- Gao DF, Niu XL, Hao GH, Peng N, Wei J, Ning N, Wang NP. Rosiglitazone inhibits angiotensin II-induced CTGF expression in vascular smooth muscle cells - role of PPAR-gamma in vascular fibrosis. Biochem Pharmacol. 2007;73:185–197. doi: 10.1016/j.bcp.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Giachelli CM, Liaw L, Murry CE, Schwartz SM, Almeida M. Osteopontin expression in cardiovascular diseases. Ann N Y Acad Sci. 1995;760:109–126. doi: 10.1111/j.1749-6632.1995.tb44624.x. [DOI] [PubMed] [Google Scholar]
- Goya K, Sumitani S, Otsuki M, Xu X, Yamamoto H, Kurebayashi S, Saito H, Kouhara H, Kasayama S. The thiazolidinedione drug troglitazone up-regulates nitric oxide synthase expression in vascular endothelial cells. J Diabetes Complications. 2006;20:336–342. doi: 10.1016/j.jdiacomp.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Gray SL, Nora ED, Grosse J, Manieri M, Stoeger T, Medina-Gomez G, Burling K, Wattler S, Russ A, Yeo GS, Chatterjee VK, O’Rahilly S, Voshol PJ, Cinti S, Vidal-Puig A. Leptin deficiency unmasks the deleterious effects of impaired peroxisome proliferator-activated receptor gamma function (P465L PPARgamma) in mice. Diabetes. 2006;55:2669–2677. doi: 10.2337/db06-0389. [DOI] [PubMed] [Google Scholar]
- He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T. PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes. 2002;51:3586–3590. doi: 10.2337/diabetes.51.12.3586. [DOI] [PubMed] [Google Scholar]
- Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, Wilkes J, Evans RM, Olefsky J. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003;9:1491–1497. doi: 10.1038/nm956. [DOI] [PubMed] [Google Scholar]
- Inoue I, Shino K, Noji S, Awata T, Katayama S. Expression of peroxisome proliferator-activated receptor alpha (PPAR alpha) in primary cultures of human vascular endothelial cells. Biochem Biophys Res Commun. 1998;246:370–374. doi: 10.1006/bbrc.1998.8622. [DOI] [PubMed] [Google Scholar]
- Lavoie JL, Liu X, Bianco RA, Beltz TG, Johnson AK, Sigmund CD. Evidence supporting a functional role for intracellular renin in the brain. Hypertension. 2006;47:461–466. doi: 10.1161/01.HYP.0000203308.52919.dc. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator–activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Leff T. Altered promoter recycling rates contribute to dominant-negative activity of human peroxisome proliferator-activated receptor-gamma mutations associated with diabetes. Mol Endocrinol. 2007;21:857–864. doi: 10.1210/me.2006-0401. [DOI] [PubMed] [Google Scholar]
- Madsen CS, Regan CP, Hungerford JE, White SL, Manabe I, Owens GK. Smooth muscle-specific expression of the smooth muscle myosin heavy chain gene in transgenic mice requires 5′-flanking and first intronic DNA sequence. Circ Res. 1998;82:908–917. doi: 10.1161/01.res.82.8.908. [DOI] [PubMed] [Google Scholar]
- Marx N, Schonbeck U, Lazar MA, Libby P, Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res. 1998;83:1097–1103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B, Jr, Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki S, Takeda K, Kiyama M, Hatta T, Morimoto S, Kawa T, Itoh H, Nakata T, Sasaki S, Nakagawa M. Augmented response of endothelin-A and endothelin-B receptor stimulation in coronary arteries of hypertensive hearts. J Cardiovasc Pharmacol. 1998;31(Suppl 1):S94–S98. doi: 10.1097/00005344-199800001-00029. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Ohya Y, Onaka U, Fujii K, Abe I, Fujishima M. Inhibitory action of insulin-sensitizing agents on calcium channels in smooth muscle cells from resistance arteries of guinea-pig. Br J Pharmacol. 1998;123:675–682. doi: 10.1038/sj.bjp.0701669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol CJ, Adachi M, Akiyama TE, Gonzalez FJ. PPARgamma in endothelial cells influences high fat diet-induced hypertension. Am J Hypertens. 2005;18:549–556. doi: 10.1016/j.amjhyper.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Oyama Y, Akuzawa N, Nagai R, Kurabayashi M. PPARgamma ligand inhibits osteopontin gene expression through interference with binding of nuclear factors to A/T-rich sequence in THP-1 cells. Circ Res. 2002;90:348–355. doi: 10.1161/hh0302.105098. [DOI] [PubMed] [Google Scholar]
- Pandey NR, Benkirane K, Amiri F, Schiffrin EL. Effects of PPAR-gamma knockdown and hyperglycemia on insulin signaling in vascular smooth muscle cells from hypertensive rats. J Cardiovasc Pharmacol. 2007;49:346–354. doi: 10.1097/FJC.0b013e31804654d7. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C, Wang GX, Korth M, Aszodi A, Andersson KE, Krombach F, Mayerhofer A, Ruth P, Fassler R, Hofmann F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. PPAR(gamma) agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension. 2004;43:661–666. doi: 10.1161/01.HYP.0000116303.71408.c2. [DOI] [PubMed] [Google Scholar]
- Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M, Gurnell M, Williams RL, Umpleby AM, Thomas EL, Bell JD, Dixon AK, Dunne F, Boiani R, Cinti S, Vidal-Puig A, Karpe F, Chatterjee VK, O’Rahilly S. Human Metabolic Syndrome Resulting From Dominant-Negative Mutations in the Nuclear Receptor Peroxisome Proliferator-Activated Receptor-{gamma} Diabetes. 2003;52:910–917. doi: 10.2337/diabetes.52.4.910. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL. Role of endothelin-1 in hypertension and vascular disease. Am J Hypertens. 2001;14:83S–89S. doi: 10.1016/s0895-7061(01)02074-x. [DOI] [PubMed] [Google Scholar]
- Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA. 2007;298:1189–1195. doi: 10.1001/jama.298.10.1189. [DOI] [PubMed] [Google Scholar]
- Thorin E, Cernacek P, Dupuis J. Endothelin-1 regulates tone of isolated small arteries in the rat: effect of hyperendothelinemia. Hypertension. 1998;31:1035–1041. doi: 10.1161/01.hyp.31.4.1035. [DOI] [PubMed] [Google Scholar]
- Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR, Kim JK, Maeda N. Hypertension and abnormal fat distribution but not insulin resistance in mice with P465L PPARgamma. J Clin Invest. 2004;114:240–249. doi: 10.1172/JCI20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SM, Yasmin McEniery CM, Maki-Petaja KM, Booth AD, Cockcroft JR, Wilkinson IB. Isolated systolic hypertension is characterized by increased aortic stiffness and endothelial dysfunction. Hypertension. 2007;50:228–233. doi: 10.1161/HYPERTENSIONAHA.107.089391. [DOI] [PubMed] [Google Scholar]
- Watts SW. Serotonin-induced contraction in mesenteric resistance arteries: signaling and changes in deoxycorticosterone acetate-salt hypertension. Hypertension. 2002;39:825–829. doi: 10.1161/hy0302.104668. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Mitra S, Varadharaj S, Parinandi N, Zweier JL, Flavahan NA. Increased expression of cyclooxygenase-2 mediates enhanced contraction to endothelin ETA receptor stimulation in endothelial nitric oxide synthase knockout mice. Circ Res. 2006;98:1439–1445. doi: 10.1161/01.RES.0000224120.52792.10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.