

Abstract
Mast cells and macrophages infiltrate healing myocardial infarcts and may play an important role in regulating fibrous tissue deposition and extracellular matrix remodelling. This study examined the time-course of macrophage and mast cell accumulation in healing infarcts and studied the histological characteristics and protease expression profile of mast cells in a canine model of experimental infarction. Although macrophages were more numerous than mast cells in infarct granulation tissue, macrophage density decreased during maturation of the scar, whereas mast cell numbers remained persistently elevated. During the inflammatory phase of infarction, newly recruited leucocytes infiltrated the injured myocardium and appeared to be clustered in close proximity to degranulating cardiac mast cells. During the proliferative phase of healing, mast cells had decreased granular content and were localized close to infarct neovessels. In contrast, macrophages showed no selective localization. Mast cells in healing canine infarcts were alcian blue/safranin-positive cells that expressed both tryptase and chymase. In order to explain the pro-inflammatory and angiogenic actions of tryptase — the major secretory protein of mast cells — its effects on endothelial chemokine expression were examined. Chemokines are chemotactic cytokines that play an important role in leucocyte trafficking and angiogenesis and are highly induced in infarcts. Tryptase, a proteinase-activated receptor (PAR)-2 agonist, induced endothelial expression of the angiogenic chemokines CCL2/MCP-1 and CXCL8/IL-8, but not the angiostatic chemokine CXCL10/IP-10. Endothelial PAR-2 stimulation with the agonist peptide SLIGKV induced a similar chemokine expression profile. Mast cell tryptase may exert its angiogenic effects in part through selective stimulation of angiogenic chemokines.
Keywords: mast cell, infarction, myocardial ischaemia/reperfusion, macrophage, chemokine, tryptase, chymase, inflammation
Introduction
Healing of myocardial infarcts involves a dynamic interplay between a variety of cell types ultimately leading to formation of a scar [1]. Macrophages and mast cells infiltrate the infarcted myocardium and serve as a source of cytokines and growth factors, supporting the growth of neovessels and fibroblasts [2,3]. The proliferative phase of infarct healing is associated with formation of highly cellular granulation tissue and extracellular matrix deposition. Subsequently, inflammatory cells and fibroblasts undergo apoptosis and a mature scar is formed, composed of cross-linked collagen and only a small number of cells.
Mast cells synthesize cytokines, growth factors, and proteases and may regulate the healing process through the secretion of their granular contents [4,5]. Although the role of mast cells in tissue repair has been suggested by multiple studies [2,6], their exact contribution to infarct healing remains unclear. Mast cell-deficient mice have served as a valuable tool for elucidating the role of mast cells in biological processes [7,8]. Unfortunately, healing mouse infarcts, in contrast to canine infarcts [2] and human cardiomyopathic ventricles [9], contain a small mast cell population, suggesting that the mouse may not be a useful model for studying the role of mast cells in myocardial infarction [10]. We have previously demonstrated that mast cell accumulation in the proliferative phase of canine myocardial infarction is associated with upregulation of stem cell factor (SCF), a growth factor critical for mast cell differentiation and maturation [2]. The current study examines the time-course of mast cell and macrophage accumulation in canine infarcts. Furthermore, we studied the phenotypic characteristics of mast cells in healing infarcts. At all stages of healing, infarct mast cells contained both alcian blue- and safranin-positive granules and expressed the proteases tryptase and chymase. Tryptase, the most abundant protein in mast cell granules, has potent pro-inflammatory and angiogenic effects. These effects may be mediated through expression of chemokines, which play an important role in leucocyte recruitment and angiogenesis. Accordingly, we examined the effects of tryptase stimulation on chemokine synthesis by isolated canine endothelial cells.
Methods
Ischaemia/reperfusion protocols
All animal research protocols were approved by the Baylor College of Medicine Animal research committee. Healthy mongrel dogs (15–25 kg) of either sex were surgically instrumented as previously described [11,12]. Circumflex coronary artery occlusion was achieved by inflating a hydraulically activated coronary cuff occluder. After 1 h coronary occlusion, the cuff was deflated and the myocardium was reperfused. Reperfusion intervals ranged from 24 h to 28 days (24 h, n = 3; 7 days, n = 4; 14 days, n = 4; 28 days, n = 4). After the reperfusion periods, hearts were stopped and sectioned from apex to base into four transverse rings. Tissue samples were isolated from infarcted or normally perfused myocardium based on visual inspection. Myocardial segments were fixed in Carnoy's, or B*5 without formalin [13], and embedded in paraffin wax. The presence of a myocardial infarct was based on light-microscopic examination of haematoxylin–eosin-stained sections. Samples of control tissues were taken from the anterior septum and had normal morphology.
Generation of antibodies to human tryptase and canine chymase
β-Tryptase was purified from human lung extracts [14]. Aliquots of purified, denatured tryptase were combined with Freund's adjuvant and injected into rabbits. Resulting antisera were titred by ELISA against human lung tryptase as well as canine tryptase, which was purified from mastocytomas as described [15]. The IgG fraction was purified from antisera by ammonium sulphate precipitation and protein G affinity chromatography. Rabbit anti-human tryptase IgG binds selectively to human and canine tryptase as demonstrated by immunoblots of lung and mastocytoma extracts (not shown). Similarly, rabbit polyclonal antisera were raised against chymase purified from canine mastocytomas. These antisera bind selectively to chymase as previously demonstrated [16,17].
Immunohistochemistry and histology
Sequential 3–5 μm sections were cut by microtomy. Immunostaining was performed using the ELITE rabbit, or mouse kit (Vector Laboratories, Burlingame, CA, USA). Segments used for immunohistochemistry were fixed with the B*5 fixative without formalin [13], which allows optimal preservation of fixation-sensitive antigens in paraffin wax-embedded tissues. Sections were pretreated with 3% H2O2, then incubated with 2% horse serum. Subsequently, the sections were incubated with the primary antibody for 2 h at room temperature. After rinsing with PBS, the slides were incubated for 30 min with the secondary antibody. The slides were then rinsed again with PBS and incubated for 30 min in ABC reagent. Peroxidase activity was detected using diaminobenzidine (Vector). The following primary antibodies were used: rabbit anti-tryptase antibody, rabbit anti-dog chymase antibody [16], mouse anti-CD31 antibody (Dako) [18], mouse anti-α-smooth muscle actin (α-SMA) antibody (Sigma) [19], anti-myeloid cell antibody Mac387 (Dako) [3,20] and anti-macrophage antibody PM-2K (Biogenesis, Kingston, NH, USA) [3]. The monoclonal antibody PM-2K identifies mature macrophages and has been previously used in canine tissues [3]. The Mac387 antibody labels myeloid cells (neutrophils and monocytes) but does not stain mature macrophages [20].
Mast cells were labelled using toluidine blue staining and alcian blue/safranin staining, using techniques previously described by our laboratory [21,22].
Quantitative histological analysis
Stained sections were photographed with a digital camera mounted on a Zeiss microscope. The density of mast cells and macrophages in infarcted and control areas was quantitated using Image Pro software. Semi-quantitative analysis of the granular content of mast cells was performed by assessment of the ‘granulation score’ for 20 mast cells from each infarcted and non-infarcted myocardial segment, scanned at 1000× magnification as follows: 3, fully granulated cell; 2, mild loss of granular content; 1, moderate granule loss; 0, markedly decreased granular content. The average granulation score was then quantitated for each segment. To confirm the reproducibility of the method, the ‘granulation score’ was compared in two toluidine blue-stained sections from each infarcted and non-infarcted segment. Comparison of the granulation score between the two sections gave a Pearson correlation coefficient of 0.95. Six infarcted and six non-infarcted segments obtained from four animals for each reperfusion interval were used for quantitation.
Endothelial cell isolation and stimulation
Canine jugular vein endothelial cells were obtained as previously described [23]. Endothelial cells were incubated with recombinant human skin tryptase (2.5–10 μg/ml, Promega), or the PAR-2 activating peptide SLIGKV (concentration: 10−3 m to 10−5 m, Bachem) for 2–16 h. At the end of the experiment, endothelial cells were used for RNA extraction.
RNA isolation
RNA isolation from stimulated endothelial cells was performed using the acid guanidinium phenol chloroform procedure. RNA (20 μg) was electrophoresed in 1% agarose gels containing formaldehyde, then transferred to a nylon membrane (Gene Screen Plus; New England Nuclear) by standard procedures.
Northern hybridization
Membranes were hybridized in QuikHyb (Stratagene, La Jolla, CA, USA) at 68 °C for 2 h with 1 × 106 dpm random hexamer 32P-labelled canine cDNA probes for CXCL8/interleukin (IL)-8 [24], CCL2/monocyte chemoattractant protein (MCP)-1 [25], and CXCL10/interferon-γ inducible protein (IP)-10 [26]. Quantitation was performed using densitometry as previously described [12]. Relative density was normalized to the intensity of the 28S rRNA.
Statistical analysis
Statistical analysis was performed using ANOVA followed by t-test corrected for multiple comparisons (Student–Newman–Keuls). Data were expressed as mean ± SEM. Statistical significance was set at 0.05.
Results
Time-course of mast cell and macrophage infiltration in canine infarcts
Mast cell density was significantly higher in canine infarcts after 7 days of reperfusion compared with non-infarcted areas from the same experiments (7-day infarct: 23.47 + 8.42 cells/mm2 vs. control: 7.05 + 1.33 cells/mm2; p < 0.05, n = 6) (Figure 1). The number of mast cells in healing infarcts remained elevated for at least 4 weeks of reperfusion (28-day infarct: 19.2 + 3.14 cells/mm2 vs. control: 4.22 + 1.12; p < 0.05, n = 6). In contrast, macrophage density in the infarct peaked after 14 days (14-day infarct: 647.5 + 79.7 cells/mm2 vs. control: 49.05 + 9.84 cells/mm2; p < 0.001, n = 6), then significantly decreased after 4 weeks of reperfusion (28-day infarct: 226.13 + 51.8 cells/mm2; p < 0.001 compared with 7- and 14-day infarcts) (Figure 2).
Figure 1.
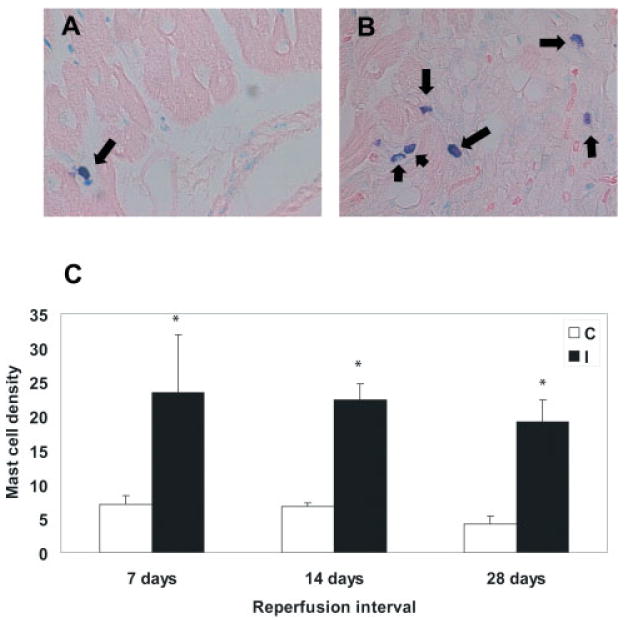
Time-course of mast cell accumulation in healing canine infarcts. (A) Toluidine blue staining identifies a resident mast cell population (arrow) in control canine myocardium. (B) After 7 days of reperfusion, a significant number of mast cells is noted in the infarcted heart (arrows). (C) Mast cell density in infarcted myocardial segments remained persistently elevated for at least 4 weeks after reperfusion of the ischaemic myocardium (*p < 0.05 compared with control segments). C, control; I, ischaemic
Figure 2.
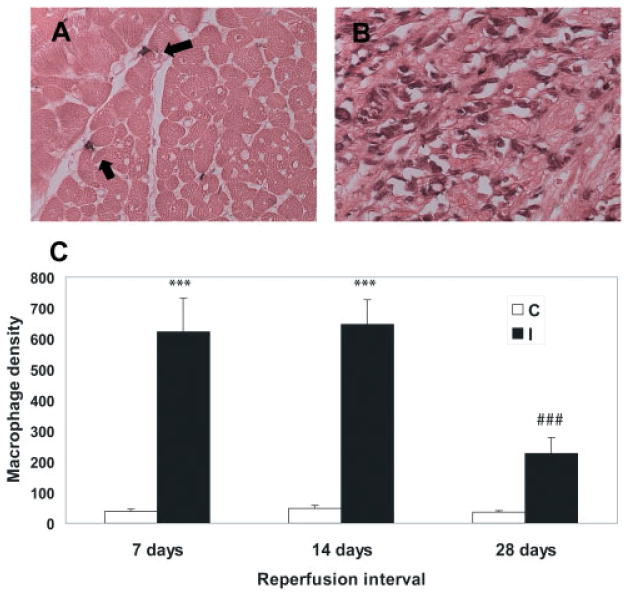
Time-course of macrophage accumulation in healing canine myocardial infarcts. (A) Immunohistochemistry with the monoclonal antibody PM-2K, a marker for mature macrophages, identifies macrophages in the non-infarcted heart (arrows). (B) Abundant macrophages are noted in infarcted myocardial segments after 14 days of reperfusion. (C) A marked increase in macrophage density was noted in infarcted segments after 7–14 days of reperfusion (***p < 0.001 vs. control segments). However, after 28 days of reperfusion, macrophage density significantly decreased (###p < 0.001 vs. 7- and 14-day infarcts. C, control; I, ischaemic
Morphological characteristics and protease expression of infarct mast cells
Mast cells in non-infarcted areas of the canine heart were elongated perivascular cells with a high granular content. Mast cells in infarcted and non-infarcted segments of the canine heart had both alcian blue-positive and safranin-positive granules (Figure 3A). Phenotypic characterization of infarct mast cells was performed using immunohistochemical staining with a rabbit anti-canine antibody to chymase [16] and a rabbit anti-human antibody to tryptase. Samples from the canine tongue and earlobe were used to demonstrate the cross-reactivity of the anti-tryptase antibody with canine species, and contained a large number of tryptase- and chymase-positive mast cells. Mast cells in control and infarcted areas contained both chymase and tryptase at all stages of infarct healing (Figure 4). Infarct mast cells exhibited significant granular loss (Figure 3C) after 7–28 days of reperfusion. The granular content in infarcted segments was significantly lower than in control myocardial segments after 7 (granulation score; infarct: 1.71 + 0.13 vs. control 2.83 + 0.07, n = 6; p < 0.01), 14 (infarct: 1.77 + 0.15 vs. control: 2.67 + 0.04; p < 0.01), and 28 days of reperfusion (infarct: 1.74 + 0.14 vs. control: 2.78 + 0.06, n = 6; p < 0.01). However, no significant differences in granular content were noted in infarcted segments over the course of infarct healing (7–28 days).
Figure 3.
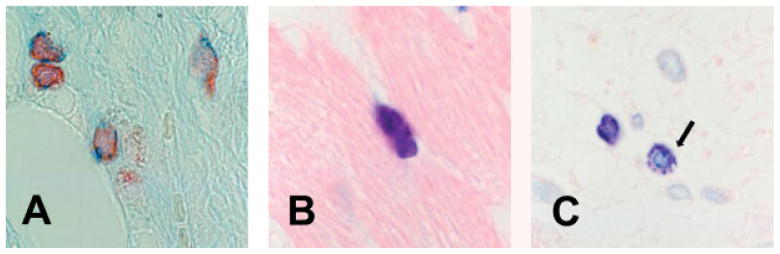
(A) Mast cells in the infarcted myocardium contain both alcian blue- and safranin-positive granules. (B) Mast cells in non-infarcted segments are packed with granules (toluidine blue staining). (C) In contrast, many mast cells in infarcted segments exhibit loss of granular contents (arrow) (14 days reperfusion, toluidine blue staining)
Figure 4.
Characterization of mast cells in canine infarcts using immunohistochemistry for chymase and tryptase. Serial section staining identifies infarct mast cells as metachromatic cells (B: toluidine blue staining) expressing tryptase (A) and chymase (C) (1 h ischaemia/7 days reperfusion)
Mast cells are localized in areas of leucocyte infiltration during the pro-inflammatory phase and are associated with neovessels in the healing wound
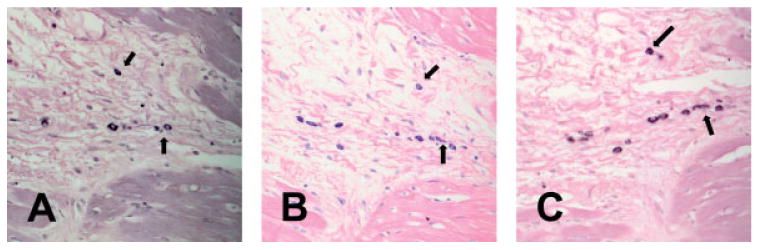
We have previously used Mac-387 immunohistochemistry to label newly recruited leucocytes (monocytes and neutrophils) and distinguish them from mature macrophages in the canine and human heart [3,20]. This antibody detects an epitope on the calcium-binding protein MRP14, which is highly expressed in monocytes and neutrophils, but downregulated in mature macrophages [27]. After 24 h of reperfusion, infiltrating leucocytes were clustered in close proximity to degranulating mast cells (Figure 5). In contrast, mature macrophages were uniformly distributed in the infarcted area and did not demonstrate a close spatial association with newly recruited leucocytes (Figure 5C). After 7 days of reperfusion the injured cardiomyocytes were replaced with highly vascular granulation tissue (Figure 6). At this stage α-SMA-positive myofibroblasts were predominantly localized in the infarct border zone, whereas the centre of the wound exhibited a high capillary density. Mast cells were selectively localized in close proximity to vascular structures, in particular those with a developed muscular wall (Figure 6). In contrast, macrophages were similarly distributed in all areas of the infarct and had no selectively perivascular location.
Figure 5.
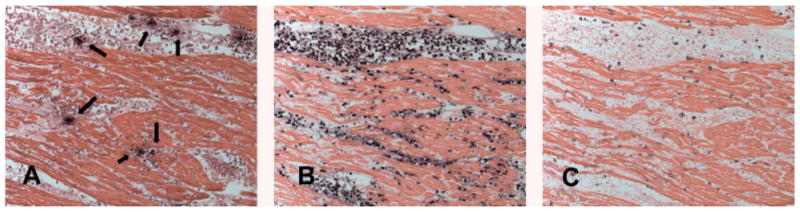
Serial section staining demonstrates that infiltrating leucocytes are clustered around mast cells in the inflammatory phase of myocardial infarction (1 h ischaemia/24 h reperfusion). (A) Chymase staining identifies mast cells in the infarct (arrows). (B) Immunohistochemistry with the monoclonal antibody Mac387 labels newly recruited myeloid cells (neutrophils and monocytes) in the infarct. This antibody does not stain mature macrophages [3]. Note that infiltrating leucocytes are clustered around mast cells. (C) Immunohistochemistry with the monoclonal antibody PM-2K stains mature macrophages in the infarct
Figure 6.
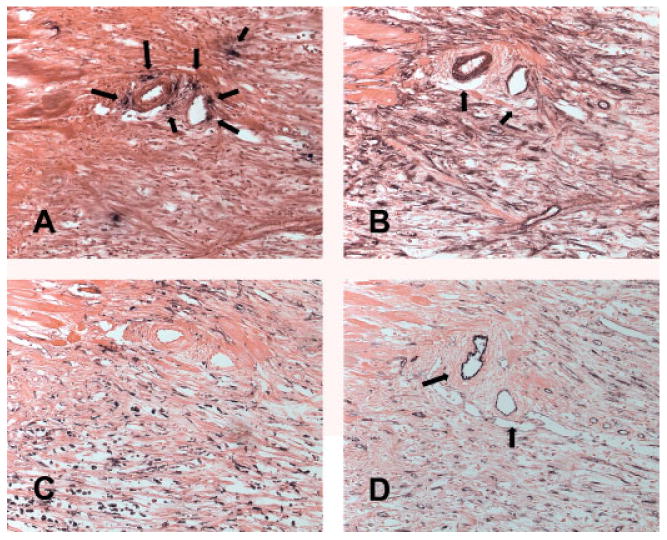
Mast cell localization in the proliferative phase of myocardial infarction (1 h ischaemia/7 days reperfusion). Chymase staining (A) identifies mast cells in the infarct. Note that mast cells are located in close proximity to vessels, identified by α-SMA staining of their muscular coat (arrows, B) and by CD31 staining (D). (C) PM-2K immunohistochemistry labels infarct macrophages. No selective localization of macrophages in the perivascular area is noted
Mast cell tryptase and PAR-2 activation increase endothelial expression of the angiogenic chemokines IL-8 and MCP-1, but not of the angiostatic chemokine IP-10
Unstimulated canine endothelial cells demonstrated negligible mRNA expression of the chemokines IL-8, MCP-1, and IP-10. Incubation with human recombinant tryptase (10 μg/ml) induced a rapid but transient upregulation of the angiogenic chemokines MCP-1 and IL-8 in canine endothelial cells (Figure 7). MCP-1 and IL-8 mRNA levels peaked after 4 h and returned to baseline after 16 h of incubation. In contrast, expression of the angiostatic chemokine IP-10 was not found in tryptase-stimulated endothelial cells. Because many of the pro-inflammatory actions of tryptase are presumed to be due to PAR-2 activation, we examined the effects of the PAR-2 activating peptide SLIGKV on endothelial chemokine synthesis. Although low peptide concentration (10−5 m and 10−4 m) had no effect on endothelial chemokine expression, high concentration (10−3 m) induced upregulation of IL-8 and MCP-1, but did not stimulate IP-10 synthesis in canine endothelial cells. In contrast to the transient nature of tryptase-induced chemokine synthesis, PAR-2 activation caused a persistent upregulation of IL-8 and MCP-1 mRNA levels (Figure 8).
Figure 7.
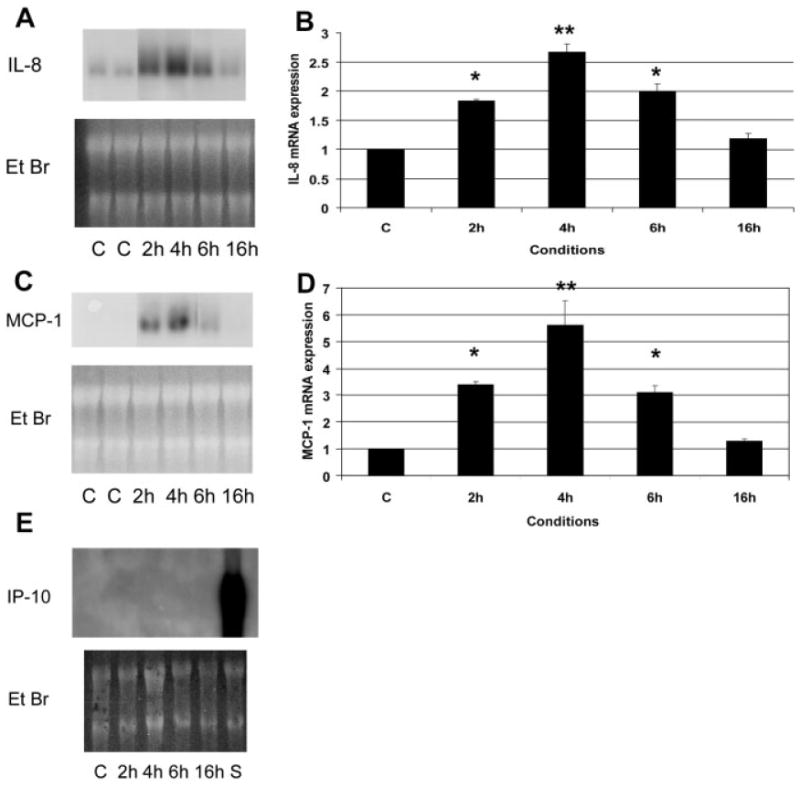
Tryptase induces chemokine synthesis by isolated canine jugular vein endothelial cells. (A, B) Tryptase stimulation (10 μg/ml) induces transient IL-8 mRNA synthesis peaking after 4 h. (C, D) Tryptase also upregulates MCP-1 mRNA expression in canine venous endothelial cells (E) In contrast, tryptase does not induce synthesis of the angiostatic chemokine IP-10. A sample from the spleen of an endotoxin-stimulated animal (S) is used as a positive control. C, control; EtBr, ethidium bromide (*p < 0.05 vs C, **p < 0.01 vs C)
Figure 8.
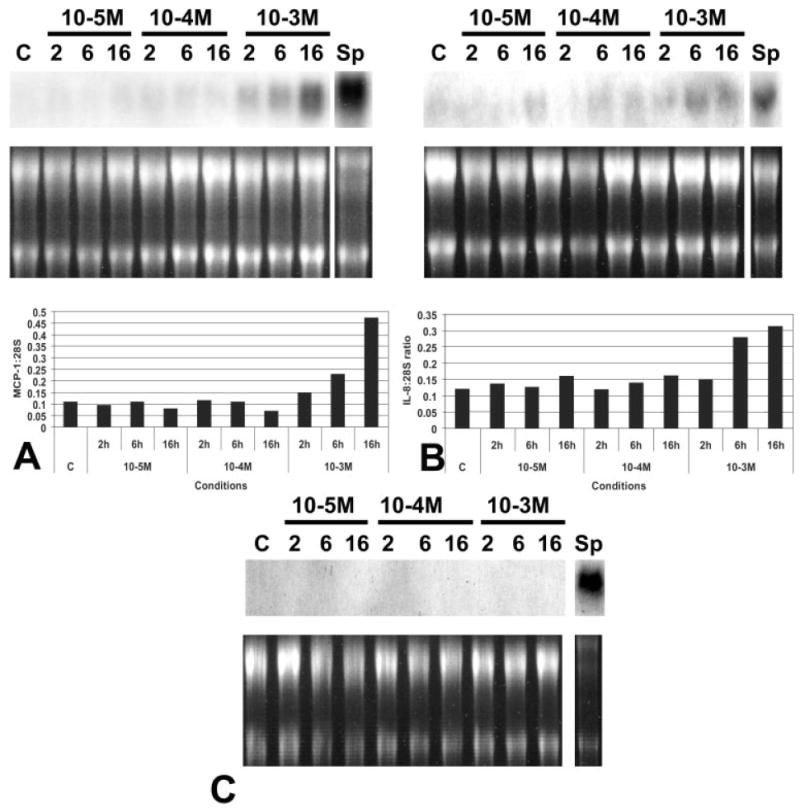
Stimulation with the PAR-2 activating peptide SLIGKV induces expression of angiogenic chemokines in canine venous endothelial cells. (A) Incubation with high concentration (10−3 m) SLIGKV induces endothelial MCP-1 mRNA synthesis after 6 h of reperfusion. MCP-1 induction is prolonged. A sample from the spleen of an endotoxin-stimulated animal (Sp) is shown as a positive control. (B) SLIGKV also induces IL-8 expression, but has no effect on endothelial IP-10 mRNA synthesis (C). Findings are representative of two experiments with similar results
Discussion
Myocardial infarction is associated with an inflammatory response ultimately leading to healing and formation of a scar [1]. Macrophages, lymphocytes, and mast cells infiltrate the infarcted myocardium and may regulate the healing process by releasing proteases, cytokines, and growth factors [10]. Canine myocardial infarction is associated with degranulation of resident cardiac mast cells and release of mast cell-derived TNF-α and histamine, initiating the cytokine cascade responsible for inflammatory leucocyte recruitment in the infarcted myocardium [11]. In addition, during the proliferative phase of infarct healing, mast cells accumulate in the infarcted area and may contribute to fibroblast growth and angiogenesis. Although several studies have suggested a role for mast cells in infarct healing, their specific contribution to the cellular events associated with post-infarction inflammation and scar formation remains unclear for two main reasons. First, many mast cell-derived mediators can also be secreted by other cell types, such as macrophages, fibroblasts, and lymphocytes, which are present in large numbers in the infarcted ventricle. Second, while increased mast cell density is noted in canine myocardial infarcts and in hearts from patients with ischaemic cardiomyopathy, murine myocardial infarcts contain only few mast cells [10], suggesting that mast cell-deficient mice may not be suitable to investigate the functional role of mast cells in infarct healing.
The current study examines the time-course of mast cell and macrophage infiltration in healing canine infarcts. We demonstrate that, although macrophages are much more numerous than mast cells in the infarcted heart during the proliferative phase of healing, mast cell density remains elevated in mature infarcts, while the number of macrophages significantly decreases. Infarct mast cells have alcian blue- and safranin-positive granules, express both chymase and tryptase, and undergo progressive degranulation during infarct healing. Mast cell tryptase is the most abundant protein in mast cell granules and has potent pro-inflammatory and angiogenic properties. For this reason, we investigated its in vitro effects on chemokine expression by isolated canine endothelial cells. We found that tryptase induced transient endothelial upregulation of the angiogenic chemokines IL-8 and MCP-1, but not of the angiostatic chemokine IP-10. These findings may offer new insight into the mechanistic basis of tryptase-induced angiogenesis.
Recruitment of neutrophils and mononuclear cells is a prominent and early event in reperfused myocardial infarcts and is followed by an increase in the number of mast cells. During the maturation phase of healing, however, the number of macrophages decreases (Figure 2) as many of these cells undergo apoptosis, whereas mast cell density remains high (Figure 1). In contrast to other myeloid cells, mast cells have a long life-span and express members of the bcl-2 family that may promote their survival [28]. The persistent presence of mast cells in infarcts 1 month after coronary occlusion, when macrophage density is significantly decreased, suggests that mast cells may play an important role in scar maturation.
A wide body of evidence suggests an important role for mast cells in wound healing [6,29]. This is based, in most cases, on investigations demonstrating increased numbers of mast cells in healing wounds and on their capability of synthesizing, storing, and releasing pro-inflammatory, fibrogenic, and angiogenic mediators. Mast cells are potentially important sources of cytokines (such as TNF-α), growth factors (such as TGF-β, VEGF, and bFGF), the serine proteases chymase and tryptase, and matrix metalloproteinases [16]. Although mast cells are capable of modulating fibrosis and angiogenesis [30–33], both critical events in tissue repair, direct evidence supporting a crucial role for mast cells in healing wounds is lacking. A recent study suggested that mast cell-deficient kitw/kitw–v mice have normal cutaneous healing; however, mast cell deficiency decreased neutrophil infiltration during the inflammatory phase [34]. We have recently demonstrated that mice, in contrast to large mammals [2], do not have increased numbers of mast cells in healing myocardial infarcts [10]; for this reason mast cell-deficient mice may not be a suitable model for examining the role of mast cells in cardiac repair.
Infarct mast cells were identified as alcian blue/safranin-positive cells, expressing both chymase and tryptase at all stages of healing (Figures 3 and 4). Chymase and tryptase may be critical mediators in scar formation. Chymase may regulate fibrous tissue deposition through activation of angiotensin II [35], and TGF-β [36]. In addition, canine chymase activates gelatinase B [37] and may modulate extracellular matrix remodelling. Tryptase has recently emerged as an important and versatile mediator. In view of the strategic perivascular location of mast cells, release of their granular contents during inflammatory processes is likely to expose neighbouring endothelial cells to high concentrations of tryptase. Compton and co-workers demonstrated that tryptase induced endothelial IL-8 and IL-1β expression without upregulating expression of the adhesion molecules ICAM-1, VCAM-1, and E-selectin [38]. Other studies indicated that tryptase also has potent angiogenic effects, stimulating capillary growth [30]. In order to investigate the pro-inflammatory and angiogenic actions of tryptase we examined its effects on endothelial cell chemokine expression. Chemokines are critical regulators of leucocyte trafficking [39], but also modulate the angiogenic response [40]. The CXC chemokine CXCL8/IL-8 and the CC chemokine CCL2/MCP-1 have potent angiogenic properties [41,42]. In contrast, CXC chemokines lacking the ELR motif, such as CXCL10/IP-10, inhibit bFGF or IL-8-mediated angiogenesis [43,44]. Our experiments indicated that tryptase selectively induces the angiogenic chemokines MCP-1 and IL-8, but not the angiostatic chemokine IP-10 (Figure 7). In contrast, less selective pro-inflammatory mediators such as TNF-α and M-CSF markedly induce expression of all three chemokines in isolated canine venous endothelial cells [23,26]. The selective upregulation of angiogenic chemokines may be in part responsible for the angiogenic and pro-inflammatory effects of tryptase.
All the known actions of tryptase on cells appear to be mediated via its catalytic activity [45]. Tryptase activates the PAR-2 receptor through proteolytic cleavage of the amino terminus of the receptor revealing a tethered ligand, capable of activating itself and initiating signal transduction [46,47]. We found that, much like tryptase stimulation, PAR-2 activation with an agonist peptide induced expression of MCP-1 and IL-8, but not IP-10 (Figure 8). The similar chemokine expression profile is consistent with the hypothesis that a PAR-2-related mechanism is responsible for tryptase-induced chemokine upregulation. PAR-2 agonists induce inflammation and angiogenesis in vivo and in vitro [48–50]. Whether these effects are mediated through chemokine induction remains unknown.
We suggest that mast cell-derived mediators may be important in regulating post-infarction inflammation and cardiac repair. Mast cells secrete cytokines and growth factors, capable of modulating inflammatory leucocyte recruitment, fibrosis and angiogenesis. They also release unique products such as the serine proteases chymase and tryptase. Tryptase exerts pro-inflammatory and angiogenic effects on endothelial cells, seemingly mediated through PAR-2 activation; these actions may be in part related to endothelial induction of angiogenic chemokines. Large animal studies using specific tryptase inhibitors may be useful to clarify the biological significance of the pleiotropic effects of tryptase on healing infarcts.
Acknowledgments
This work was supported by National Institutes of Health grants HL-42550 (MLE, NGF, LHM), HL-24136 (GHC) a grant-in-aid from the American Heart Association, Texas affiliate (NGF), and the DeBakey Heart Center. The authors wish to thank Lisa Thurmon, Alida Evans, and Stephanie Butcher for their outstanding technical assistance and Concepcion Mata and Sharon Malinowski for their expert secretarial assistance in preparing the manuscript.
References
- 1.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG, Perrard JL, Mendoza LH, et al. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 3.Frangogiannis NG, Mendoza LH, Ren G, et al. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol. 2003;285:H483–492. doi: 10.1152/ajpheart.01016.2002. [DOI] [PubMed] [Google Scholar]
- 4.Mekori YA, Metcalfe DD. Mast cells in innate immunity. Immunol Rev. 2000;173:131–140. doi: 10.1034/j.1600-065x.2000.917305.x. [DOI] [PubMed] [Google Scholar]
- 5.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 6.Trautmann A, Toksoy A, Engelhardt E, Brocker EB, Gillitzer R. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol. 2000;190:100–106. doi: 10.1002/(SICI)1096-9896(200001)190:1<100::AID-PATH496>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Wershil BK, Galli SJ. The analysis of mast cell function in vivo using mast cell-deficient mice. Adv Exp Med Biol. 1994;347:39–54. doi: 10.1007/978-1-4615-2427-4_5. [DOI] [PubMed] [Google Scholar]
- 8.Lazarus B, Messina A, Barker JE, Hurley JV, Romeo R, Morrison WA, Knight KR. The role of mast cells in ischaemia-reperfusion injury in murine skeletal muscle. J Pathol. 2000;191:443–448. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH666>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 9.Patella V, Marino I, Arbustini E, et al. Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation. 1998;97:971–978. doi: 10.1161/01.cir.97.10.971. [DOI] [PubMed] [Google Scholar]
- 10.Dewald O, Ren G, Duerr GD, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frangogiannis NG, Lindsey ML, Michael LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 12.Frangogiannis NG, Mendoza LH, Lindsey ML, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–2808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 13.Beckstead JH. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. J Histochem Cytochem. 1994;42:1127–1134. doi: 10.1177/42.8.8027531. [DOI] [PubMed] [Google Scholar]
- 14.Hartmann T, Ruoss SJ, Raymond WW, Seuwen K, Caughey GH. Human tryptase as a potent, cell-specific mitogen: role of signaling pathways in synergistic responses. Am J Physiol. 1992;262:L528–534. doi: 10.1152/ajplung.1992.262.5.L528. [DOI] [PubMed] [Google Scholar]
- 15.Caughey GH, Viro NF, Ramachandran J, Lazarus SC, Borson DB, Nadel JA. Dog mastocytoma tryptase: affinity purification, characterization, and amino-terminal sequence. Arch Biochem Biophys. 1987;258:555–563. doi: 10.1016/0003-9861(87)90377-8. [DOI] [PubMed] [Google Scholar]
- 16.Fang KC, Wolters PJ, Steinhoff M, Bidgol A, Blount JL, Caughey GH. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-beta. J Immunol. 1999;162:5528–5535. [PubMed] [Google Scholar]
- 17.Wolters PJ, Laig-Webster M, Caughey GH. Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways. Am J Respir Cell Mol Biol. 2000;22:183–190. doi: 10.1165/ajrcmb.22.2.3767. [DOI] [PubMed] [Google Scholar]
- 18.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. 2002;50:71–79. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 19.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 20.Frangogiannis NG, Shimoni S, Chang SM, et al. Evidence for an active inflammatory process in the hibernating human myocardium. Am J Pathol. 2002;160:1425–1433. doi: 10.1016/S0002-9440(10)62568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frangogiannis NG, Burns AR, Michael LH, Entman ML. Histochemical and morphological characteristics of canine cardiac mast cells. Histochem J. 1999;31:221–229. doi: 10.1023/a:1003541332070. [DOI] [PubMed] [Google Scholar]
- 22.Gersch C, Dewald O, Zoerlein M, Michael LH, Entman ML, Frangogiannis NG. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol. 2002;118:41–49. doi: 10.1007/s00418-002-0425-z. [DOI] [PubMed] [Google Scholar]
- 23.Frangogiannis NG, Mendoza LH, Smith CW, Michael LH, Entman ML. Induction of the synthesis of the C-X-C chemokine interferon-gamma-inducible protein-10 in experimental canine endotoxemia. Cell Tissue Res. 2000;302:365–376. doi: 10.1007/s004410000274. [DOI] [PubMed] [Google Scholar]
- 24.Kukielka GL, Smith CW, LaRosa GJ, et al. Interleukin-8 gene induction in the myocardium after ischemia and reperfusion in vivo. J Clin Invest. 1995;95:89–103. doi: 10.1172/JCI117680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar AG, Ballantyne CM, Michael LH, et al. Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation. 1997;95:693–700. doi: 10.1161/01.cir.95.3.693. [DOI] [PubMed] [Google Scholar]
- 26.Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001;15:1428–1430. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 27.Hashimoto S, Suzuki T, Dong HY, Yamazaki N, Matsushima K. Serial analysis of gene expression in human monocytes and macrophages. Blood. 1999;94:837–844. [PubMed] [Google Scholar]
- 28.Baghestanian M, Jordan JH, Kiener HP, et al. Activation of human mast cells through stem cell factor receptor (KIT) is associated with expression of bcl-2. Int Arch Allergy Immunol. 2002;129:228–236. doi: 10.1159/000066773. [DOI] [PubMed] [Google Scholar]
- 29.Artuc M, Hermes B, Steckelings UM, Grutzkau A, Henz BM. Mast cells and their mediators in cutaneous wound healing: active participants or innocent bystanders? Exp Dermatol. 1999;8:1–16. doi: 10.1111/j.1600-0625.1999.tb00342.x. [DOI] [PubMed] [Google Scholar]
- 30.Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, Gruber BL. Human mast cells stimulate vascular tube formation: tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–2700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trautmann A, Krohne G, Brocker EB, Klein CE. Human mast cells augment fibroblast proliferation by heterotypic cell-cell adhesion and action of IL-4. J Immunol. 1998;160:5053–5057. [PubMed] [Google Scholar]
- 32.Berton A, Levi-Schaffer F, Emonard H, Garbuzenko E, Gillery P, Maquart FX. Activation of fibroblasts in collagen lattices by mast cell extract: a model of fibrosis. Clin Exp Allergy. 2000;30:485–492. doi: 10.1046/j.1365-2222.2000.00737.x. [DOI] [PubMed] [Google Scholar]
- 33.Coussens LM, Raymond WW, Bergers G, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egozi EI, Ferreira AM, Burns AL, Gamelli RL, Dipietro LA. Mast cells modulate the inflammatory but not the proliferative response in healing wounds. Wound Repair Regen. 2003;11:46–54. doi: 10.1046/j.1524-475x.2003.11108.x. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto T, Wada A, Tsutamoto T, Ohnishi M, Isono T, Kinoshita M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003;107:2555–2558. doi: 10.1161/01.CIR.0000074041.81728.79. [DOI] [PubMed] [Google Scholar]
- 36.Lindstedt KA, Wang Y, Shiota N, et al. Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. Faseb J. 2001;15:1377–1388. doi: 10.1096/fj.00-0273com. [DOI] [PubMed] [Google Scholar]
- 37.Fang KC, Raymond WW, Blount JL, Caughey GH. Dog mast cell alpha-chymase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92 bonds of the catalytic domain. J Biol Chem. 1997;272:25 628–25 635. doi: 10.1074/jbc.272.41.25628. [DOI] [PubMed] [Google Scholar]
- 38.Compton SJ, Cairns JA, Holgate ST, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1 beta and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol. 1998;161:1939–1946. [PubMed] [Google Scholar]
- 39.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 40.Strieter RM, Polverini PJ, Arenberg DA, et al. Role of C-X-C chemokines as regulators of angiogenesis in lung cancer. J Leukoc Biol. 1995;57:752–762. doi: 10.1002/jlb.57.5.752. [DOI] [PubMed] [Google Scholar]
- 41.Koch AE, Polverini PJ, Kunkel SL, et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 42.Salcedo R, Ponce ML, Young HA, et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96:34–40. [PubMed] [Google Scholar]
- 43.Angiolillo AL, Sgadari C, Taub DD, et al. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182:155–162. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strieter RM, Polverini PJ, Kunkel SL, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27 348–27 357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 45.Frungieri MB, Weidinger S, Meineke V, Kohn FM, Mayerhofer A. Proliferative action of mast-cell tryptase is mediated by PAR2, COX2, prostaglandins, and PPARgamma: possible relevance to human fibrotic disorders. Proc Natl Acad Sci USA. 2002;99:15 072–15 077. doi: 10.1073/pnas.232422999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molino M, Barnathan ES, Numerof R, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 47.Cairns JA. Mast cell tryptase and its role in tissue remodelling. Clin Exp Allergy. 1998;28:1460–1463. doi: 10.1046/j.1365-2222.1998.00467.x. [DOI] [PubMed] [Google Scholar]
- 48.Steinhoff M, Vergnolle N, Young SH, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 49.Shpacovitch VM, Brzoska T, Buddenkotte J, et al. Agonists of proteinase-activated receptor 2 induce cytokine release and activation of nuclear transcription factor kappaB in human dermal microvascular endothelial cells. J Invest Dermatol. 2002;118:380–385. doi: 10.1046/j.0022-202x.2001.01658.x. [DOI] [PubMed] [Google Scholar]
- 50.Milia AF, Salis MB, Stacca T, et al. Protease-activated receptor-2 stimulates angiogenesis and accelerates hemodynamic recovery in a mouse model of hindlimb ischemia. Circ Res. 2002;91:346–352. doi: 10.1161/01.res.0000031958.92781.9e. [DOI] [PubMed] [Google Scholar]