

Abstract
Under steady-state conditions, major histocompatibility complex (MHC) I molecules are associated with self-peptides that are collectively referred to as the MHC class I peptide (MIP) repertoire. Very little is known about the genesis and molecular composition of the MIP repertoire. We developed a novel high-throughput mass spectrometry approach that yields an accurate definition of the nature and relative abundance of unlabeled peptides presented by MHC I molecules. We identified 189 and 196 MHC I–associated peptides from normal and neoplastic mouse thymocytes, respectively. By integrating our peptidomic data with global profiling of the transcriptome, we reached two conclusions. The MIP repertoire of primary mouse thymocytes is biased toward peptides derived from highly abundant transcripts and is enriched in peptides derived from cyclins/cyclin-dependent kinases and helicases. Furthermore, we found that ∼25% of MHC I–associated peptides were differentially expressed on normal versus neoplastic thymocytes. Approximately half of those peptides are derived from molecules directly implicated in neoplastic transformation (e.g., components of the PI3K–AKT–mTOR pathway). In most cases, overexpression of MHC I peptides on cancer cells entailed posttranscriptional mechanisms. Our results show that high-throughput analysis and sequencing of MHC I–associated peptides yields unique insights into the genesis of the MIP repertoire in normal and neoplastic cells.
MHC class I molecules present short peptides at the cell surface, typically 8–11 mers, for scrutiny by CD8 T lymphocytes (1, 2). Generation of peptide–MHC I complexes is initiated by proteasomal degradation of source proteins in the cytosol (3). Peptides generated by the proteasome are translocated in the endoplasmic reticulum, where they are subjected to N-terminal trimming, and then incorporated in MHC I proteins and exported at the cell surface (4–7). Presentation of microbial peptides by MHC I is required to elicit CD8 T cell responses against intracellular pathogens (8). Under steady-state conditions, i.e., in the absence of infection, cell surface MHC I molecules are associated solely with self-peptides. These peptides, collectively referred to as the MHC class I peptide (MIP) repertoire (9), play vital roles. They shape the repertoire of developing thymocytes (10, 11), transmit survival signals to mature CD8 T cells (12), amplify responses against intracellular pathogens (13), allow immunosurveillance of neoplastic cells (14), and influence mating preferences in mice (15). The MIP repertoire is also involved in immunopathology because it can be targeted by autoreactive T cells that initiate autoimmune diseases and alloreactive T cells that cause graft rejection and graft-versus-host disease (16, 17).
Despite the tremendous importance of the MIP repertoire, we know very little about its genesis and molecular composition. Proteomic analysis of the MIP repertoire is a daunting task because estimates suggest that it encompasses thousands of peptides that are present in low copy numbers per cell (1, 18). Each MHC molecule recognizes peptides through a broadly defined consensus motif of amino acids serving as anchors to the appropriate binding pockets on the MHC molecules. Such motifs were first established by pool Edman sequencing of unfractionated peptide mixtures eluted from MHC molecules (19). Direct biochemical characterization of specific MHC I–associated peptides has typically involved immunoaffinity purification of MHC molecules after cell lysis, fractionation of the peptides by chromatography, and sequencing, initially by Edman's method and, more recently, by mass spectrometry (MS). Refinements in MS methods pioneered by Hunt et al. and Falk et al. represented major progress and led to the characterization of several MHC I–associated peptides (18, 20, 21), in spite of limitations such as low peptide yield, preferential loss of peptides with low affinity for MHC I, and contamination of the MHC molecules by cellular debris and detergents (22, 23).
Two high-throughput strategies were therefore implemented to provide comprehensive molecular definition of the MIP repertoire. The first is based on transfection of cell lines with expression vectors coding soluble secreted MHCs (lacking a functional transmembrane domain) and elution of peptides associated with secreted MHCs (24, 25). This interesting approach improves MHC I peptide recovery, but presents some limitations, as follows: (a) it cannot be used on freshly explanted cells; (b) cell transfection by itself may perturb the MIP repertoire (26); and (c) the MIP repertoire associated with soluble MHC corresponds to the repertoire of peptides that can bind the transfected MHC allele (what “can be presented”), but not necessarily to peptides that are normally presented at the cell surface (what “is presented”). The second approach hinges on chemical or metabolic labeling to provide quantitative profiles of MHC I–associated peptides (9, 27–29). Although chemical derivatization suffers from variable modification yields and unexpected side reaction products, metabolic labeling is only applicable to certain MHC I allelic products and to cell culture model systems (30, 31). Nonetheless, high-throughput peptide sequencing analyses have provided crucial insights into the structure of the MIP repertoire. First, the source proteins for the MIP repertoire are found in almost every compartment of the cell (25). Second, only a limited correlation is observed between the amounts of the MHC I–associated peptides presented by cells and the relative expression of source proteins from which these peptides are derived (28). A likely explanation for this discrepancy is that the MIP repertoire preferentially derives from defective ribosomal products (DRiPs) and short-lived proteins relative to slowly degraded proteins (2, 32, 33). Third, for peptides differentially expressed on normal versus neoplastic cells, no clear correlation was found between mRNA levels and corresponding MHC peptide levels (9).
The goal of our work was to understand the structure and genesis of the MIP repertoire. In accordance with a recent advocacy for “systems immunology” (34), we used novel bioinformatics tools based on peptide mapping and segmentation to obtain a global and accurate quantification of native unlabeled peptides present in the MIP repertoire (unpublished data) (35, 36). In particular, we addressed four specific questions. Does the MIP repertoire reflect the composition of the transcriptome? Are some gene families overrepresented in the MIP repertoire? What is the impact of neoplastic transformation on the MIP repertoire? Can a high-throughput, unlabeled-peptide–sequencing platform provide valuable insights for the identification of immunogenic tumor-associated epitopes?
RESULTS
Experimental approach for the identification and quantification of MHC I–associated peptides
Mild acid elution (MAE) of MHC I–associated peptides from living cells presents several advantages over immunoprecipitation of peptide–MHC I complexes. Because it involves fewer purification steps and no detergents, MAE yields ∼10 times more MHC I–associated peptides than the latter and introduces no bias linked to preferential loss of low-affinity peptides (37–39). However, MAE has never been used for high-throughput sequencing of the MIP repertoire because it is assumed that eluted peptides contain not only MHC I–associated peptides, but also “contaminant” peptides. We reasoned that this problem could be overcome by using MHC I–deficient cells as a negative control. Because β2-microglobulin (β2m) is essential for formation of stable peptide–MHC I complexes, β2m-deficient cells are MHC I deficient. We therefore compared peptides eluted from three cell populations, as follows: WT EL4 thymoma cell line, a β2m-deficient EL4 mutant cell line (C4.4-25−), and C4.4-25− cells transfected with a genomic clone of murine β2m (E50.16+) (40). Flow cytometry analysis showed that expression of H2Db, H2Kb, and Qa2 MHC I molecules were abrogated in β2m-deficient EL4 cells, but were restored in β2m transfectants (Fig. 1 A).
Figure 1.

Experimental design for identification and relative quantification of native unlabeled MHC I–associated peptides. (A) Cell surface MHC I expression on EL4 WT, β2m− mutant, and β2m+ transfectant cell lines. Cells were stained with antibodies against H2Db, H2Kb, and Qa2 (black) or the corresponding isotype control antibody (gray). (B) Peptides obtained by MAE were analyzed by on-line 2D-nanoLC-MS (three biological replicates for each cell population). Contour profiles of m/z versus retention time versus intensity were used to visualize differences between MS profiles (middle). A logarithmic intensity scale distinguishes between low (dark red) and highly (bright yellow) abundant species. Examples of peptides that were differentially expressed (blue line) or not (green line) between WT and β2m− EL4 cells are highlighted in boxes. (bottom) Heat map representation shows differential peptide expression between WT and β2m− EL4 cells where each horizontal line corresponds to a unique peptide cluster (n = 3,716). A logarithmic scale depicts peptides that are expressed at high (red) or low (green) level in each cell population. (C) Volcano plot representation showing reproducibly detected peptide ions across three replicate analyses. Peptide clusters (n = 1,236) highlighted in dashed box were considered as class I–restricted (P ≤ 0.05; fold change ≥ 5). Peptides that were sequenced by MS/MS are represented by colored dots: green for contaminants, and blue for MHC I–associated peptides.
Samples obtained by MAE of peptides from 8 × 107 cells were analyzed using an on-line 2D-nanoLC-MS system (see Materials and methods for more details). Each individual LC-MS run was visualized as a three-dimensional map where each isotopic profile (dot) corresponds to a given ion that is defined by its specific abundance, m/z, and retention time coordinates (Fig. 1 B). We first assessed the reproducibility of data obtained with our method using peptide eluates from WT EL4 cells. We found that 95% of peptide ions showed a variation of less than ±1.4- and ±2.2-fold in abundance across 2D-nanoLC-MS instrumental and biological replicates, respectively (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071985/DC1). Therefore, peptides were considered to be differentially expressed when the fold difference in abundance was ≥2.5 (P < 0.05). We next compared profiles from WT and β2m-deficient EL4 cells. Contour profiles obtained after computational analyses showed a higher level of peptide complexity for WT compared with β2m-deficient EL4 cells (Fig. 1 B). We assessed the proportion of MHC I–associated versus contaminant peptides using in-house peptide detection software (unpublished data). To identify and define individual peptide clusters, peptide lists generated by Mass Sense were compared through replicate injections from both WT- and β2m− mutant-derived samples using segmentation analyses with hierarchical clustering. Heat map representation was used to visualize differences in peptide cluster expression between WT- and β2m− mutant–derived samples (Fig. 1 B). Peptides that were recovered only from WT EL4 cells were considered to be part of the MIP repertoire. Because β2m-deficient cells express low amounts of incomplete MHC I molecules at the cell surface (41), we also considered that peptides significantly more abundant on WT relative to β2m-deficient cells were MHC I associated. In the latter case, we used a very stringent criterion; to be considered as MHC I associated, a peptide had to be at least five times more abundant on WT relative to β2m-deficient cells. Out of 3,716 unique peptide clusters that were reproducibly detected across three biological replicates, 1,487 peptide clusters were overexpressed or uniquely detected within the WT EL4 sample. Thus, we estimated that ∼40% of acid-eluted materials correspond to specific-MHC I peptides.
We obtained MS/MS assignments for 881 of the 3,716 peptide clusters, all of which had a characteristic fragmentation associated to peptide backbone cleavages. Although these identifications correspond to 24% of the entire ion population detected, other MS/MS spectra were assigned to modified residues, de novo peptide sequences, or spectra of poor quality. Peptide coordinates were computationally related to their corresponding sequenced peptides. Out of 881 sequenced peptides, 383 had a consensus motif for H2Db, H2Kb, Qa1, or Qa2 MHC I molecules. Stringent validation criteria were next applied to select peptide ions showing mass accuracy of <30 parts per million (ppm), with MS/MS spectra displaying consistent sets of b- and/or y-type fragment ion series corresponding to the MHC I peptide sequence. This manual validation enabled the identification of 178 unique MS/MS corresponding to MHC I–associated peptides: 158 were detected uniquely in eluates from WT EL4 cells, whereas 20 were overexpressed at least 5-fold on WT relative to β2m-deficient EL4 cells (Fig. 1 C). To confirm that differences between WT and β2m-deficient EL4 cells were truly β2m dependent, we analyzed peptides recovered from E50.16+ cells (β2m-deficient EL4 cells transfected with a genomic clone of murine β2m). As expected, we observed that 95% of MHC I peptides were expressed at the cell surface of β2m+ transfectants. Absence of a few MHC I peptides on β2m transfectants is probably explained by the lower expression of H2Db and H2Kb on β2m transfectants compared with WT cells (Fig. 1 A). For the 178 sequenced MHC I–associated peptides, relative abundance in the three EL4 cell variants (WT, β2m− mutants, and β2m+ transfectants) is presented in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20071985/DC1). Data for a representative set of peptides is shown in Table I. Notably, although MHC I peptides were 8–13 mers, the length of contaminant peptides was more variable, ranging from 6 to 26 amino acids with a median value of 13 residues (unpublished data).
Table I.
Relative abundance of a representative set of peptides recovered in eluates from three EL4 cell variants: WT, β2m− mutants, and β2m+ transfectants
| Gene ID | Gene symbol | Sequence | WT | β2m− | β2m+ | WT/β2m− |
|---|---|---|---|---|---|---|
| 20848 | Stat3 | ATLVFHNL | 4,023,137 | 12,000 | 6,208,130 | 335 |
| 66165 | Bccip | KAPVNTAEL | 1,741,908 | 12,000 | 998,607 | 145 |
| 15016 | H2-Q5 | AMAPRTLLL | 5,436,819 | 12,000 | 12,941,105 | 453 |
| 67755 | Ddx47 | KTFLFSATM | 70,059 | 12,000 | 289,017 | 6 |
| 12649 | Chek1 | TGPSNVDKL | 6,545,497 | 112,308 | 14,733,333 | 58 |
| 267019 | Rps15a | VIVRFLTV | 514,406 | 12,000 | 12,000 | 43 |
| 71745 | Cul2 | VINSFVHV | 67,372 | 12,000 | 244,370 | 6 |
| 233489 | Picalm | NGVINAAFM | 265,420 | 12,000 | 438,191 | 22 |
| 26413 | Mapk1 | VGPRYTNL | 39,166,667 | 149,969 | 45,832,060 | 261 |
| 108655 | Foxp1 | QQLQQQHLL | 705,266 | 12,000 | 1,054,408 | 59 |
| 170755 | Sgk3 | YSIVNASVL | 255,048 | 38,491 | 151,335 | 7 |
| 326622 | Upf2 | SAVIFRTL | 244,247 | 12,000 | 1,102,686 | 20 |
| 12877 | Cpeb1 | SMLQNPLGNVL | 185,198 | 12,000 | 528,268 | 15 |
| 94176 | Dock2 | SMVQNRVFL | 895,081 | 12,000 | 671,603 | 75 |
| 16913 | Psmb8 | GGVVNMYHM | 3,332,728 | 12,000 | 2,116,510 | 277 |
Gene ID and Gene Symbol description refer to National Center for Biotechnology Information gene entries. Intensities for each EL4 variant cell line correspond to the average MS signal calculated from triplicate experiments. An intensity threshold value of 12,000 was fixed when no signal was detected. To be considered as MHC I associated, a peptide had to be at least five times more abundant on WT relative to β2m-deficient cells.
Definition of the MIP repertoire presented by discreteMHC I allelic products
Large databases of MHC I peptides and computational binding methods are now part of emerging biomedical resources (42–45). Information in these databases concerns mainly, though not exclusively, viral and bacterial-derived peptides presented by MHC I allelic products in humans and mice. Each MHC I peptide in Table S1 was manually classified according to restriction size and binding motif favored by Db, Kb, Qa1, and Qa2 class I molecules (19, 46, 47). From 178 MHC I peptides identified from variant EL4 cell lines, 92% were presented by classical MHC Ia allelic products H2Db and H2Kb, and 8% were presented by MHC Ib molecules Qa1 and Qa2 (Fig. 2 A and Table S2, available at http://www.jem.org/cgi/content/full/jem.20071985/DC1). When viewed in toto, peptide pools presented by H2Db, H2Kb, and Qa2 displayed the canonical binding motifs documented for these MHC I molecules (Fig. 2 B).
Figure 2.

Allelic distribution and binding scores of 178 MHC I–associated peptides eluted from EL4 cells. (A) Pie chart shows distribution of 178 MHC I–associated peptides eluted from EL4 cells. The smm, SYFPEITHI (H2Db and H2Kb), and Rankpep (Qa2) computational models were used to link individual peptides to MHC I allelic products. (B) Logo showing the profile motif for peptides presented by H2-Db, H2-Kb, and Qa2 molecules. Acidic (red), basic (blue), hydrophobic (black), and neutral (green) amino acids are illustrated. (C) Individual source proteins for peptides presented by H2Db and H2Kb (n = 164) were entered in the smm binding algorithm. We assessed the predicted MHC I binding affinity of all peptides contained in individual proteins. Pie charts show the proportion of peptides eluted from EL4 cells that ranked in the top 1% (blue), top 5% (green), top 10% (yellow), or below the 90th percentile of peptides (red).
A priori, two factors may determine whether specific peptides are presented by MHC I: peptide affinity for MHC molecules and the processing of source proteins along the MHC I presentation pathway. Protein attributes that are relevant to MHC I processing include rates of protein translation, DRiP formation, and degradation (32, 33, 48, 49). To evaluate the importance of peptide binding affinity, each of the 164 source proteins of peptides presented by H2Db (n = 96) or H2Kb (n = 68) was analyzed with the smm algorithm. We scored and ranked the predicted binding affinity of all peptides from individual proteins for H2Db or H2Kb (for example, a protein of 400 amino acids contains 392 nonapeptide sequences). We then asked how each peptide that we had sequenced by MS/MS would rank relative to other peptides from its source protein. Remarkably, 91% of H2Db-associated peptides eluted from EL4 cells ranked in the top 1% in terms of H2Db binding affinity (Fig. 2 C and Table S2). Similar results were obtained from the SYFPEITHI database (unpublished data). That some peptide sequences predicted to be top MHC I binders were not found in the MIP repertoire of EL4 cells is not surprising. This means either that proteases involved in MHC I processing do not generate these specific “degradation products” or that they are present in subthreshold amounts. For H2Kb, 85% of peptides ranked in the top 5% for predicted H2Kb binding affinity (Fig. 2 C and Table S2). These data support the concept that MHC I binding affinity plays a dominant role in shaping the content of the MIP repertoire.
Discrimination between MHC I–associated peptides and contaminant peptides using bioinformatic tools
In the aforementioned experiments, we used β2m-deficient cells as negative control to discriminate MHC I–associated peptides from contaminant peptides. The need for an MHC I–deficient negative control would nevertheless represent a significant hurdle for high-throughput sequencing of the MIP repertoire of primary cells. We therefore asked whether bioinformatic tools could be used to identify MHC I–associated peptides among a peptide mixture obtained by MAE. We first ranked the 164 H2Db- and H2Kb-associated peptides eluted from EL4 cells as a function of their MHC binding score estimated with smm and SYFPEITHI algorithms (Fig. 3, red bars). We found that ∼97% of MHC I–associated peptides were scored below an smm binding threshold of 1,000 nM (IC50) and above a SYFPEITHI binding threshold of 22 for H2Db and 13 for H2Kb (Fig. 3). The aforementioned thresholds therefore provided a 3% false-negative rate (3% of MHC I–associated peptides were wrongly classified as contaminants). To estimate the false-positive rate obtained with these thresholds, we analyzed the 215 contaminant peptides eluted from EL4 cells representing high-quality assignment (Mascot score >45, mass accuracy <30 ppm, MS/MS manually inspected) from a list of 498 candidates that were not significantly overexpressed on WT relative to β2m-deficient EL4 cells. Only 56 out of the 215 contaminant peptides had the canonical length for binding H2Db or H2Kb, and very few of them satisfied our smm and SYFPEITHI thresholds (Fig. 3, blue bars). Overall, the false-positive rate was <2% (<2% of contaminants were wrongly classified as MHC I–associated peptides). Similar results were obtained for Qa2 peptides using the Rankpep algorithm with a binding threshold of 120. We synthesized nine peptides across the predicted affinity spectrum and tested their ability to bind H2Kb or H2Db. Peptides classified as MHC I associated according to their smm score did bind to MHC I, whereas peptides classified as contaminants did not (Fig. S2). These experimental data further validate bioinformatic discrimination between MHC I–associated peptides and contaminant peptides. We therefore conclude that peptides obtained by MAE can be categorized as MHC I–associated versus contaminant peptides with 1–3% false-positive and -negative rates using publicly available algorithms.
Figure 3.

Discrimination between MHC I–associated peptides and contaminant peptides using bioinformatic tools. (A–E) For peptides eluted from EL4 cells, the y axis shows computed MHC binding scores determined with the smm (A and C), SYFPEITHI (B and D), and Rankpep (E) computational methods. The x axis cut at the selected binding thresholds. Each bar represents a sequenced peptide. Individual H2Db-, H2Kb-, and Qa2-associated peptides (red) and contaminant peptides (blue) were scored as illustrated.
Global portrayal of the MIP repertoire of primarymouse thymocytes
In the next series of experiments, peptides eluted from primary mouse thymocytes by MAE were analyzed by on-line 2D-nanoLC-MS/MS. Of the sequenced peptides, 189 were classified as MHC I associated based on the smm, SYFPEITHI, and Rankpep thresholds selected above: 84 H2Db-, 91 H2Kb-, 13 Qa2-, and 1 Qa1-associated peptides (Table S3, available at http://www.jem.org/cgi/content/full/jem.20071985/DC1). Based on analyses described in the previous paragraph, we estimate that the false-positive rate for the 189 thymocyte peptides is <2%. GO term enrichment analysis was performed on the 189 genes coding for those peptides by applying stringent criteria from 772 functional annotations (Fig. 4 A). In accordance with a study on HLA-B*1801–associated peptides (25), the MIP repertoire of primary thymocytes showed a modest but significant twofold enrichment in proteins located in the cytoplasm and the nucleus. More interestingly, we found an 11–19-fold enrichment in proteins related to cyclin and cyclin-dependent kinases (Ccng1, Cdkn1b, Chek1, Bccip, Cul2, Ccnd3, and CcnF), which regulate the cell cycle. We also found an 11–17-fold enrichment in helicases (Dhx15, Ddx5, Ddx6, Hells, and Ddx47), which are required for efficient and accurate replication, repair, and recombination of the genome.
Figure 4.

Analyses of genes and transcripts coding MHC I–associated peptides eluted from primary thymocytes. (A) GO term enrichment analysis of 189 genes coding MHC I peptides eluted from thymocytes. Exact P values and global false-discovery rates were <0.05 for each listed GO term. Values in parentheses indicate the fold enrichment relative to the whole mouse genome. (B) We compared the relative abundance of two sets of thymic transcripts using previously reported microarray data (50): mRNAs coding MHC I peptides eluted from primary thymocytes (red), and 36,182 thymus-derived transcripts (gray). Original mRNA expression data on the x axis were plotted on a log2 scale. The y axes represent the number of transcripts for each sample set. (C) Frequency distributions for the two sets of thymic transcripts defined in B were plotted using a bin increment of 0.2. Three distinct mRNA expression groups are shown (low-, medium-, and high-abundance mRNA). Graph shows the proportion of mRNAs with low- (black), medium- (red), and high- (blue) abundance among the two sets of transcripts. *, P < 0.05; **, P < 0.0001. (D) Predicted MHC binding score (determined with smm) for peptides whose mRNA are expressed at low, medium, or high level. Spearman linear correlation coefficient (r) was calculated for H2Kb- (dashed line; white squares) and H2Db-associated (solid line; black squares) peptides.
MHC I–associated peptides derive preferentially from highly abundant mRNAs
We next sought to determine whether the MIP repertoire of thymocytes was molded at the mRNA level. To this end, we compared the relative abundance of two sets of thymic transcripts: (a) those encoding MHC I peptides eluted from primary thymocytes (Table S3), and (b) 36,182 transcripts encoded by the mouse genome (50). We found a dramatic enrichment in highly abundant mRNAs among transcripts coding for MHC I peptides (Fig. 4, B and C). Thus, although 9% of total mRNAs are expressed at high levels, 42% of those coding MHC I peptides did so (Fig. 4 C). Conversely, 62% of total mRNAs showed low abundance, but only 20% of those coding MHC I peptides were expressed at low levels. Nonetheless, some MHC I peptides are coded by low abundance mRNAs (Fig. 4, B and C). We hypothesized that low abundance mRNAs may contribute to the MIP repertoire because they code peptides with very high affinity for MHC I. Our data did not support that assertion. We found no correlation between the computed MHC I binding affinity of peptides and the abundance of their mRNAs (Fig. 4 D). MHC I peptides derived from low-abundance transcripts did not display superior computed MHC binding affinity. Although the ability to detect MHC I peptides is limited by the MS sensitivity (high attomole range) and dynamic range of detection (13 orders of magnitude on log2 scale of Fig. 4), our results demonstrate that MHC I peptides derived preferentially, but not exclusively, from highly abundant mRNAs.
Evidence that the MIP repertoire of thymocytes concealsa tissue-specific signature
Because MHC I peptides derived preferentially from highly abundant mRNAs, and abundance of discrete mRNAs varies among different tissues and organs, we hypothesized the MIP repertoire might harbor a tissue-specific signature. Using the SymAtlas database (http://symatlas.gnf.org/SymAtlas/) (50), we analyzed the relative expression in 58 mouse tissues of mRNAs encoding thymocyte MHC I–associated peptides (see Materials and methods for details on calculation). Remarkably, the mean expression of the 180 mRNAs coding thymocyte MHC I peptides was higher in the thymus than in all other tissues and organs (Fig. 5 A).
Figure 5.

Peptide source mRNAs expression patterns reveal an organ-specific signature in the MIP repertoire of thymocytes. (A) Comparison of normalized mean expression values (y axis) across 58 different tissues (x axis) including the thymus (red). Normalized mean expression values were calculated as follows: mean expression value from 180 peptide source genes/mean expression value of 36,182 transcripts for each particular tissue. Calculated values were ranked from left to right in a decreasing order. (B) Z scores were calculated for each of the 180 peptide source genes to identify transcripts preferentially expressed in the thymus. Graph shows frequency distributions of calculated z scores with a bin increment of 0.05. (C) Heat map shows the relative mRNA expression in 58 tissues of the 30 peptide source genes with highest thymic z scores. (D) High z score genes determine the thymus specificity of the gene set encoding MHC I–associated peptides. Genes preferentially overexpressed in the thymus (high thymic z scores; blue) were additively removed (x axis). Normalized mean expression values and thymus rank (y axis) were determined following removal of each individual gene. Removal of the 30 genes with a high thymic z score had a drastic impact on thymus rank. Removal of up to 70 randomly selected genes (100,000 permutations; green) had no significant impact on thymus rank.
Large-scale quantitative analyses of transcriptional expression profiles across different tissues have revealed broadly expressed and tissue-specific groupings (50, 51). To determine whether the MIP repertoire of thymocytes is imprinted by tissue-specific genes, we attributed a z score to individual source mRNAs (52). Genes with high z score (from ∼2.5 to 7) correspond to those that are preferentially overexpressed in the thymus. Among the 180 genes encoding thymocyte MHC I peptides, 30 showed a high z score (Fig. 5, B and C). The z score distribution of genes encoding thymocyte MHC I peptides is illustrated in Fig. 5 B, and a heat map representation of the 30 genes with high z scores is depicted in Fig. 5 C. Next, we estimated how additive removal of high z score genes would affect the thymus specificity of the gene set. Thymus specificity of the gene set was lost after removal of the 30 high z score genes (Fig. 5 D). In contrast, removal of up to 70 random genes did not affect the thymus specificity of the gene set. These results suggest that the MIP repertoire of thymocytes conceals a tissue-specific signature that is constituted by ∼30 genes, i.e., ∼17% of genes that are represented in the thymocytes' MIP repertoire.
Among the 30 genes with high thymic z score, 16 are expressed, albeit at lower levels than in other organs, whereas 14 genes are expressed almost exclusively in hematolymphoid organs: Rhoh, Centb1, Cxcr4, Depdc1a, Foxp1, Dnmt1, 9230105E10Rik, Cd3e, C330027C09Rik, Igtp, Mns1, Dock2, Actr2, and Vps13d (Fig. 5 C). In accordance with the concept that tissue-specific expression is indicative of gene function in mammals (51), we noted that hematolymphoid genes represented in the thymocytes' MIP repertoire play critical roles in T cell development. For example, Rhoh is important for positive thymocyte selection, Cxcr4 for migration of T cell progenitors, and CD3e for migration of T cell development (53–55). Further studies are needed to cogently assess the functional importance of genes that impart tissue specificity to the thymocytes' MIP repertoire. However, evidence suggests that many, if not all, of these genes are functionally important for thymocyte function. In conclusion, two major and related points can be made regarding the connection between the transcriptome and the MIP repertoire. First, the MIP repertoire is enriched in peptides derived from highly abundant transcripts. Second, our data suggest that thymocytes' MIP repertoire conceals a tissue-specific signature that derives from ∼17% of MHC I–associated peptides.
The MIP repertoire of normal versus neoplastic thymocytes
Ultimately, the genesis of MHC I peptides must be regulated by mRNA translation and protein degradation by the proteasome (2, 49), two processes that are profoundly perturbed in neoplastic cells (56, 57). To evaluate the impact of neoplastic transformation on the MIP repertoire, we compared the MIP repertoire of primary thymocytes from C57BL/6 female mice to that of neoplastic thymocytes. As a source of neoplastic thymocytes, we used in vivo grown EL4 cells. EL4 cells were originally derived from a C57BL/6 female mouse. Based on aforementioned experiments on the reproducibility of estimation of peptide abundance by MS analyses (Fig. S1), peptides were considered to be differentially expressed when the fold difference in abundance was ≥2.5 (P < 0.05). Table S3 and S4 (available at http://www.jem.org/cgi/content/full/jem.20071985/DC1) present the complete list of peptides eluted from primary thymocytes and in vivo grown EL4 cells and their computed MHC binding score. Overall, 25% of MHC I peptides were differentially expressed on normal versus neoplastic thymocytes (Table II and Fig. 6 A). Thus, 22 peptides were underexpressed and 21 were overexpressed on neoplastic relative to normal thymocytes (Table II). Differentially expressed peptides derived from genes implicated in several biological processes, such as cell cycle progression, apoptosis, signal transduction, cytoskeleton assembly, and differentiation, as well as regulation of transcription and translation (Table II). As an example, the X-linked lymphocyte-regulated 3 (Xlr3a/b) gene encoded a peptide found only on EL4 cells (Fig. 6 B and Table II). Xlr genes are important for T cell differentiation and are overexpressed in several lymphoid malignancies (58). In contrast, an MHC I peptide from cyclin-dependent kinase inhibitor 1B (Cdkn1b) was underexpressed on neoplastic cells (Fig. 6 C, Table II). Cdkn1b is known to act as a potent tumor suppressor gene in a variety of cancers (57). Remarkably, ∼50% of differentially expressed peptides derived from genes that are known to be involved in tumorigenesis (Table II and Table S5). For example, 10 differentially expressed peptides (Bach2, Cdkn1b, Cxcr4, Eif3s2, Eif3s10, Igtp, Pa2g4, Pi3kap1, Ptpn6, and Sgk) originated from genes related to the PI3K–AKT–mTOR pathway, which is the oncogenic signaling pathway most commonly targeted by genomic aberrations in cancer (59, 60).
Table II.
Peptides differentially expressed on EL4 cells versus primary thymocytes
| Functional classification |
Gene symbol | Gene ID | Function | Sequence | P value | EL4/ Thy |
|---|---|---|---|---|---|---|
| Transcription | Pa2g4 a | 18813 | Growth regulation | AQFKFTVL | 0.02 | 4.6 |
| Top2a a | 21973 | DNA topoisomerase | NSMVLFDHV | 0.02 | 15.6 | |
| Dnmt1 a | 13433 | DNA methylation | LSLENGTHTL | 0.02 | 8.9 | |
| Pfdn5 a | 56612 | Protein binding/Folding | SMYVPGKL | 0.04 | 16.6 | |
| Per1 a | 18626 | Transcription regulator | YTLRNQDTF | 0.04 | 5.0 | |
| Foxp1 a | 108655 | Transcription factor | QQLQQQHLL | 0.01 | −4.3 | |
| Bach2 a | 12014 | Transcription factor | EQLEFIHDI | 0.02 | −20.2 | |
| Pbrm1 | 66923 | DNA binding | SQVYNDAHI | 0.02 | −3.5 | |
| Ddx5 a | 13207 | RNA helicase | NQAINPKLLQL | 0.02 | −5.7 | |
| Jhdm1d | 338523 | Histone demethylase | SSIQNGKYTL | 0.02 | −4.9 | |
| Cell differentiation | Ptpn6 a | 15170 | Key role in hematopoiesis | AQYKFIYV | 0.03 | 3.3 |
| Mark2 | 13728 | Maintenance of cell polarity | ASIQNGKDSL | 0.03 | 2.7 | |
| Pi3kap1 a | 83490 | Role in BCR-mediated Pi3K activation | YGLKNLTAL | 0.001 | −21.8 | |
| Xlr3a/b | 22446 | Role in lymphocyte development | VAAANREVL | 0.02 | 84.9 | |
| RhoH a | 74734 | Small GTPase/thymocyte maturation | YSVANHNSFL | 0.02 | −3.1 | |
| Cell cycle | Rcc2 | 108911 | Required in mitosis and cytokinesis | AAYRNLGQNL | 0.04 | 27.0 |
| Cdkn1b a | 12576 | Involved in G1 arrest | FGPVNHEEL | 0.003 | −3.1 | |
| Apoptosis | Sgk a | 20393 | Response to DNA damage stimulus | STLTYSRM | 0.02 | 9.7 |
| Pdcd10 a | 56426 | Role in apoptotic pathways | ILQTFKTVA | 0.001 | −4.3 | |
| Signal transduction | Cxcr4 a | 12767 | Receptor for the chemokine CXCL12/SDF1 |
VVFQFQHI | 0.003 | 2.6 |
| CD97 a | 26364 | Involved in adhesion and signaling process |
KLLSNINSVF | 0.03 | −4.6 | |
| Translation | Eif3s10 a | 13669 | Translation initiation factor | QSIEFSRL | 0.02 | 3.2 |
| Eif3s2 a | 54709 | Translation initiation factor | FGPINSVAF | 0.04 | 5.3 | |
| Transport | Tmed9 | 67511 | Intracellular protein transport | VIGNYRTQL | 0.02 | −6.9 |
| Copb1 | 70349 | ER to Golgi vesicle-mediated transport | IALRYVAL | 0.01 | 2.9 | |
| Cytoskeleton | Tmod1 | 21916 | Cytoskeleton organization | SSIVNKEGL | 0.004 | 10.5 |
| Mylc2b | 67938 | Cytoskeleton organization and biogenesis | SLGKNPTDAYL | 0.01 | 4.2 | |
| Mns1 | 17427 | Involved in cell division and motility | KIIEFANI | 0.01 | −12.4 | |
| Krt5 | 110308 | Strutural constituent of cytoskeleton | AAYMNKVEL | 0.04 | −106.7 | |
| Krt7 | 110310 | Strutural constituent of cytoskeleton | AAYTNKVEL | 0.003 | −121.5 | |
| Miscellaneous | Igtp a | 16145 | IFNγ-induced GTPase | IVAENTKTSL | 0.001 | −15.5 |
| Nup205 | 70699 | Outer membrane exporter porin | VNNEFEKL | 0.005 | −4.6 | |
| Dhx15 a | 13204 | RNA helicase | TLLNVYHAF | 0.02 | −11.5 | |
| Stk11ip | 71728 | Serine/Threonine kinase | SALRFLNL | 0.01 | −3.9 | |
| Pde2a | 207728 | Catalytic activity | IKNENQEVI | 0.02 | 16.0 | |
| Gtpbp4 a | 69237 | GTP binding | QILSDFPKL | 0.03 | 2.6 | |
| Narfl | 67563 | Prelamin recognition | VAYGFRNI | 0.01 | 8.7 | |
| Rmnd5a | 68477 | Hypothetic role for meiotic nuclear division |
WAVSNREML | 0.02 | −6.2 | |
| Unknown | Specc1 | 432572 | TSLAFESRL | 0.01 | 7.1 | |
| Ccdc41 | 77048 | AQVENVQRI | 0.01 | −2.8 | ||
| 2900073G15Rik | 67268 | SMGKNPTDEYL | 0.05 | 3.8 | ||
| D14Ertd436e | 218978 | SQHVNLDQL | 0.04 | −2.7 | ||
| 9230105E10Rik | 319236 | FISDVEHQL | 0.01 | −23.2 |
Gene ID and Gene Symbol description refer to National Center for Biotechnology Information gene entries. Fold change and P values were calculated from biological replicate experiments (n = 3). Functional classification is based upon bibliographic searches.
Genes that are involved in neoplastic transformation (see Table S6 [available at http://www.jem.org/cgi/content/full/jem.20071985/DC1] for references).
Figure 6.

Relative quantification of differentially expressed MHC I peptides and source mRNAs from thymocytes and EL4 cells. (A) Volcano plot representation illustrates MHC I peptides reproducibly detected across biological replicates (n = 3). Peptides over- and underexpressed on EL4 cells relative to thymocytes (P ≤ 0.05; fold change ≥ 2.5) were highlighted in blue and red, respectively. MS/MS spectra of circled peptides are shown in B and C. (B and C) Illustration of two differentially expressed MHC I peptides. Reconstructed ion chromatograms show differential abundance for m/z 471.772+ (VAAANREVL) and 521.262+ (FGPVNHEEL) in EL4 cells versus thymocytes. MS/MS spectra confirm MHC I peptide sequences and the identification of the cognate source proteins. (D) Scatter plot shows the correlation between relative expression of mRNA and that of MHC I peptide. Expression ratios for source mRNA (x axis) and MHC I peptide (y axis) between EL4 cells and thymocytes were plotted on a log2 scale for 47 pairs. A Spearman correlation coefficient was calculated from the linear regression. MHC I peptides overexpressed in EL4 cells or normal thymocytes are highlighted in blue and red, respectively; peptides that were not differentially expressed are shown in gray. Dashed box shows peptides whose overexpression on EL4 cells did not correlate with increased mRNA levels of their source protein.
Genesis of peptides overexpressed on tumor cells
An important question is whether differential expression of MHC I peptides on neoplastic relative to normal thymocytes correlates with changes in mRNA levels of source transcripts. To test this, we selected 19 peptides overexpressed on EL4 cells relative to primary thymocytes, 15 that were underexpressed, and 13 that were not differentially expressed. Then, we analyzed the level of expression of their source mRNAs in neoplastic versus primary thymocytes using quantitative real-time PCR. Scatterplot representation of the correlation between relative mRNA expression and relative MHC I peptide expression is depicted in Fig. 6 D. From the linear regression, a Spearman coefficient of 0.63 was calculated, showing a significant but moderate correlation between peptide and mRNA expression ratios. The strength of the correlation was conspicuously decreased by a set of 14 peptides that were more abundant on neoplastic cells, but whose mRNA levels were not overexpressed (Fig. 6 D, dotted box). Exclusion of these 14 genes increased the correlation coefficient to 0.78. Remarkably, for the 19 peptides overexpressed on EL4 cells, increased transcript levels were present in EL4 cells in only 5 cases (Fig. 6 D). Peptides overexpressed on neoplastic cells are particularly important because they can be used as targets in cancer immunotherapy (61, 62). Thus, the salient finding here is that 74% (14 of 19) of peptides overexpressed on EL4 cells would have been missed by studies of mRNA expression levels. An important implication is that, at least in our model, overexpression of MHC I–associated peptides on neoplastic cells generally entails posttranscriptional mechanisms.
Testing the immunogenicity of peptides overexpressedon neoplastic cells
Finally, we wished to determine whether peptides overexpressed on neoplastic thymocytes (in vivo grown EL4 cells) would be able to elicit specific CD8 T cell response. To test this, we used the following two peptides: (a) STLTYSRM, which is derived from serum/glucocorticoid-regulated kinase (Sgk) and presented by H2Kb, and (b) VAAANREVL derived from X-linked lymphocyte-regulated 3 (Xlr3a/b) and presented by H2Db. One important difference between the two peptides is that VAAANREVL was not found on primary thymocytes (fold difference relative to EL4 cells ≥ 85), whereas low levels of STLTYSRM were present on primary thymocytes (fold difference = 10; Table II and Table S6, available at http://www.jem.org/cgi/content/full/jem.20071985/DC1). TAP-deficient T2-Db and T2-Kb cells were first incubated with titrated amounts of synthetic peptides to evaluate their ability to bind and stabilize H2Kb and H2Db. In contrast to the Ld-restricted peptide RPQASGVYM, both STLTYSRM and VAAANREVL peptide loading resulted in an increase in cell surface expression of H2-Kb or H2-Db (Fig. S2), thereby confirming their ability to bind their respective MHC allele. We next immunized C57BL/6 mice with DCs coated or not coated with VAAANREVL and STLTYSRM synthetic peptides. Splenocytes from immunized mice were tested for in vitro cytotoxicity against primary thymocytes and EL4 target cells that were not loaded with exogenous peptides. Splenocytes from mice primed with unloaded DCs showed no cytotoxic activity. In contrast, splenocytes primed with coated DCs killed EL4 cells, but not primary thymocytes (Fig. 7). Thus, peptides overexpressed by 10- to ≥85-fold on neoplastic cells elicited specific cytotoxic activity against endogenously presented epitopes.
Figure 7.

Splenocytes primed against peptides overexpressed on EL4 cells selectively kill EL4 cells. Mice were immunized with DCs coated with STLTYSRM (A) or VAAANREVL (B) peptide. Splenocytes from primed mice were restimulated in vitro with the corresponding peptide for 6 d, and tested for in vitro cytotoxic activity against CFSE-labeled target cells (EL4 cells and primary mouse thymocytes) at different E/T ratios. Number of effectors represents the number of unfractionated splenocytes used in the cytotoxicity assay. Mice immunized with unloaded DCs were used as negative control. Data represent the mean ± the SD for four mice per group.
DISCUSSION
We have developed a novel method for high-throughput analysis of MHC I peptides and performed a comprehensive study of the MIP repertoire of normal and neoplastic thymocytes. Our studies yielded important insights into the genesis of the MIP repertoire and how it is modified by neoplastic transformation.
A novel method for high-throughput, MS-based analysisof MHC I peptides
In the last few years, high-throughput screening methods coupled with bioinformatics tools have helped to figure out the complexity of biological matrices. Thus, emerging technologies in mass spectrometry have led to the development of peptide detection algorithms that can be used, in combination with segmentation analyses, to compare unlabeled peptide populations (unpublished data) (36). This powerful approach allowed us to obtain an accurate quantification of native unlabeled MHC I peptides without any chemical or metabolic labeling modifications. Because preparation of samples does not require different purification steps, higher sensitivity can be achieved from limiting the amount of materials compared with chemical derivatization where peptide recovery can be affected by variable modification yields or side reaction products (31). Thus, by combining our MS strategy with MAE, we were able to generate a comprehensive portrayal of the MIP repertoire of EL4 cells from <108 cells. The ability to identify large numbers of MHC I peptides from limiting amounts of cells is a noteworthy advantage. Moreover, our quantification method can be used to analyze any type of cell population, whereas metabolic labeling strategies can only be applied to cell culture model systems. Nevertheless, low abundance peptides (<100 copies/cell) remain challenging to identify in view of the sensitivity of present high-throughput, MS-based methods. The portrayal of the MIP repertoire described in this study and others is far from complete, and low-abundance peptides that may be biologically relevant could remain elusive to current detection methods.
We took advantage of EL4 variant cell lines (WT, β2m− mutant, and β2m+ transfectant) to unambiguously discriminate between MHC I–associated peptides and contaminants in peptide mixtures obtained by MAE. A large proportion of contaminants was made of long peptides derived from the C-terminal end of source proteins (unpublished data). We noted that some of these contaminants have previously been considered as MHC I–associated peptides in studies where peptides were obtained by MAE or immunoaffinity purification (23, 63). The need to have an MHC I–deficient negative control to distinguish MHC I peptides from contaminants would be a cumbersome limitation for analysis of primary cells. However, we showed that the use of computational models such as SYFPEITHI and smm obviated this need. Thus, thresholds used herein yielded false-positive and -negative rates of ∼2% in identification of MHC I peptides.
The MIP repertoire of primary cells
High-throughput analysis of peptides obtained by MAE provided us with a global portrayal of peptides presented by different MHC I allelic products (in this study: H2Db, H2Kb, Qa1, and Qa2). We found that, with rare exceptions, discrete MHC I molecules presented peptides derived from different sets of source proteins (Table S2 and S3). A corollary is that expression of multiple MHC I allelic products (a consequence of gene duplication and diversification [64]) favors representation of largely nonoverlapping sets of source proteins in the MIP repertoire. By integrating global profiling of the mouse protein-encoding transcriptome (50) with the MIP repertoire of thymocytes, we found that the thymocytes' MIP repertoire is enriched in peptides derived from highly abundant transcripts. Furthermore, our data suggest that the repertoire of MHC I–associated peptides conceals a tissue-specific signature that derives from ∼17% of genes represented in the MIP repertoire. Cogent evaluation of this exciting concept will require comprehensive analyses of MHC I–associated peptides eluted from various tissues and organs. Why would the MIP repertoire show a stronger correlation with the transcriptome (this study) than the proteome (28)? Probably because the proteome is enriched in slowly degraded proteins (with a mean t 1/2 of >1,000 min), whereas the MIP repertoire originates mainly from rapidly degraded proteins (with a mean t 1/2 of ∼10 min) that have recently been translated and degraded (28, 49). In other words, MHC I molecules sample what is being translated rather than what has been translated (49). Considering the pervasive roles of MIPs, particularly in CD8 T cell development and function, it will be extremely interesting to determine whether the MIP repertoire of specialized cell types does conceal a tissue-specific signature. Of particular interest are thymus cortical epithelial cells, which support thymocyte positive selection, and thymus medullary epithelial cells, which express promiscuous transcripts involved in tolerance induction. Besides, although the MIP repertoire is enriched in peptides derived from highly abundant mRNAs, some peptides presented by MHC class I molecules derive from low abundance mRNAs (Fig. 4). How can low-abundance transcripts successfully compete with more abundant transcripts for representation in the MIP repertoire? Attractive possibilities would be that successful transcripts generate more DRiPs than others or that they are translated by special “immunoribosomes” (33, 49).
The proteasome, which is the primary source of MHC I peptides, is much more ancient than the MHC. The MHC appeared in gnathostomes, whereas proteasomes are found in all eukaryotes. Fundamental functions of the proteasome are to regulate cell cycle, proliferation, and apoptosis (65). In line with this, we found that the MIP repertoire of primary thymocytes was enriched in peptides derived from cyclins, cyclin-dependent kinases, and helicases (Fig. 4 A). These highly conserved proteins regulate cell proliferation, cell cycle arrest, and apoptosis. Peptides derived from cyclin, cyclin-dependent kinase, and helicase gene families have also been found in other studies of MHC I peptides (9, 25, 63). We surmise that the presence of these peptide families in the MIP repertoire may represent an imprint of primordial functions of the proteasome. Nevertheless, an alternative hypothesis would be that overrepresentation of peptide-derived proteins regulating proliferation and apoptosis is a thymocyte-specific feature because thymocytes display particularly high rates of proliferation and apoptosis.
The MIP repertoire of neoplastic cells
Neoplastic transformation is associated with many genomic and proteomic changes. How these changes may impinge on the MIP repertoire and thereby be perceived by CD8 T cells, is a fundamental question in cancer immunology that can be addressed directly only by MS-based analysis of the MIP repertoire. Weinzierl et al. recently reported a seminal study in which they integrated mass spectrometry and microarray data on renal cell carcinomas and autologous normal kidney tissues (9). They found a poor correlation between changes in peptide expression and changes in the abundance of mRNA levels in normal versus cancer cells (r = 0.32). We found a better correlation between mRNA levels and expression of corresponding MHC I peptides in normal and neoplastic thymocytes (Fig. 6 D; r = 0.63). We surmise that the stronger correlation in our case may be caused by estimation of transcript levels with quantitative real-time PCR analyses rather than microarrays. Indeed, quantitative real-time PCR provides a more accurate estimation of quantitative differences than microarrays (66). However, our data support the main conclusion of Weinzierl et al., which is that a majority of peptides overexpressed on neoplastic cells (74% in our case) would have been missed by estimation of transcript levels. Together with data from Weinzierl et al., our results suggest that in general, overexpression of MHC I peptides on neoplastic cells is caused by posttranscriptional mechanisms and can be detected only by MS-based expression profiling approaches. Further studies on diverse cancer types are needed to test the generality of these observations. Relevant mechanisms may include dysregulation of microRNA expression, protein translation, and proteasomal degradation (67–69).
Our data suggest that the MIP repertoire gives a unique perspective into the mechanisms of carcinogenesis. Approximately 50% of peptides differentially expressed on normal versus neoplastic cells are coded by genes involved in neoplastic transformation (Table II and Table S5). It is quite remarkable that 10 of the differentially expressed peptides derived from genes related to the PI3K–AKT–mTOR pathway. The PI3K–AKT–mTOR pathway is the most prominent pathway regulating protein translation, cell growth, and cell proliferation, and is activated in most cancers (59). In addition, our data lead us to speculate that the MIP repertoire might give unique insights into acquired epigenetic abnormalities that are so important in cancer. Indeed, Dnmt1, which regulates DNA methylation and prevents genomic instability (70), is expressed at high levels in thymocytes (Fig. 5 C). We found that a Dnmt1-derived peptide was overexpressed ∼9-fold on neoplastic relative to primary thymocytes (Table II). This raises the enticing possibility that enhanced proteasomal degradation of Dnmt1 might be responsible for the genomic instability of EL4 cells. Finally, we were able to generate specific cytotoxic T cell responses against EL4 cells by priming WT mice with two peptides overexpressed on EL4 cells (Fig. 7). The finding that efficient cytotoxic responses could be elicited against endogenously expressed tumor peptides identified with our MS-based expression analyses validates the analytical potential of the present approach. Though further work is needed to evaluate the therapeutic value of vaccination with these peptides, our results strongly support the concept that high-throughput analysis of the MIP repertoire is a promising discovery platform for the identification of immunogenic tumor-associated peptides (27, 61, 62).
MATERIALS AND METHODS
Chemicals and materials.
Citric acid, aprotinin, iodoacetamide, and sodium phosphate dibasic (Na2HPO4) were purchased from Sigma-Aldrich; high-performance liquid chromatography–grade water, methanol (MeOH), and acetonitrile were obtained from Thermo Fisher Scientific; formic acid (FA) and ammonium acetate were purchased from EM Science; fused-silica capillaries were obtained from Polymicro Technologies; and Teflon and PEEK tubing were purchased from Supelco. The Jupiter Proteo C12 4 μm material used for packing homemade precolumn and column was obtained from Phenomenex. Strong cation exchange (SCX) material was purchased from PolyLC.
Cell lines and flow cytometry.
The EL4 (β2m+ WT), C4.4-25− (β2m− mutant), and E50.16+ (β2m+ transfectant of C4.4-25−) cell lines were provided by R. Glas (Karolinska Institutet, Karolinska University Hospital, Huddinge, Sweden). T2-Kb and T2-Db cell lines (gifts from S. Joyce, Vanderbilt University School of Medicine, Nashville, TN) were maintained as previously described (71). MHC class I molecules at the cell surface were stained with PE-conjugated anti–H-2Kb (clone AF6-88.5; BD Biosciences), FITC-conjugated anti–H-2Db (clone KH95; BD Biosciences), and biotin-conjugated anti–Qa-2 (clone 1–1-2; BD Biosciences) and analyzed on a BD LSR II flow cytometer using FACSDiva software (BD Biosciences).
Peptide extraction and mass spectrometry analysis.
Freshly isolated thymocytes were obtained from 4–6-wk-old C57BL/6 female mice purchased from The Jackson Laboratory. All mice were maintained under specific pathogen–free conditions according to the standards of the Canadian Council on Animal Care. Experimental protocols were approved by the Comité de Déontologie Animale of the Université de Montréal. Thymocytes were separated from stroma according to standard procedures (72). For isolation of EL4 cells grown in vivo, mice were injected intraperitoneally with 200,000 EL4 cells, and ascites fluid was harvested 17 d later. Purity of EL4 cells was assessed by flow cytometry (73). Three biological replicates were prepared and analyzed for normal thymocytes and EL4 cells. Cell surface MHC I peptides were isolated from viable cells as previously described, but using a slightly modified protocol (37). In brief, 4 ml of citrate-phosphate buffer at pH 3.3 (0.131 M citric acid/0.066 M Na2HPO4, NaCl 150 mM) containing aprotinin and iodoacetamide (1:100) was added to each flask, and cell pellets were resuspended by gentle pipetting for 1 min to denature MHC I complexes. Cell suspensions were then pelletted and the resulting supernatant was isolated. Peptides extracts were desalted using Oasis HLB cartridges (30 mg; Waters), and bound material was eluted with 1 ml H2O/80% MeOH/0.2% FA (vol/vol) and diluted to H2O/40% MeOH/0.2% FA (vol/vol). Peptides were then passed through ultrafiltration devices (Amicon Ultra; Millipore) to isolate peptides <5,000 Daltons and to remove β2m proteins. The resulting flowthrough was then lyophilized and stored at −30 or −80°C until analysis.
To extract the equivalent amount of MHC I peptide from EL4 cells and normal thymocytes, we assessed the amount of MHC I molecules on both cell populations using previously described methods (74). Equivalent amount of MHC I material from neoplastic (8 × 107) and normal (2.3 × 109) thymocytes were separated by on-line 2D separation (SCX/C12 Jupiter Proteo 4μm) using an Eksigent nanoLC-2D system. Samples were diluted in H2O/2% acetonitrile/0.2% FA before LC-MS analyses. The homemade SCX column (0.3 mm i.d. × 45 mm) was connected directly to the switching valve. During sample loading, the SCX column was positioned off-line of C12 precolumn to remove interfering species. Salt fractions (10 μl each) were loaded on SCX column at 5 μl/min for 6 min to sequentially elute peptides onto the C12 precolumn using pulsed fractions of 0, 75, 300, and 1,000 mM of ammonium acetate (pH 3.0). A 69-min gradient from 3–60% acetonitrile (0.2% FA) was used to elute peptides from a homemade reversed-phase column (150 μm i.d. × 100 mm) with a flow rate set at 600 nanoliter/min. On-line 2D-nanoLC-MS system was used to provide enhanced selectivity, as well as a higher capacity to detect low-abundance MHC I peptides. To achieve high mass accuracy and sensitivity from a limited amount of starting material, abundance profiles and tandem MS experiments were performed simultaneously using a LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ion source (1.5–1.7 kV). MS scans were acquired in the FT mode (Orbitrap) with a resolution set at 60,000 (m/z 400). Each full MS spectrum was followed by three MS/MS spectra (four scan events), where the three most abundant multiply charged ions were selected for MS/MS sequencing. Tandem MS experiments were performed using collision-induced dissociation in the linear ion trap (IT mode). Target ions already selected for MS/MS fragmentation were dynamically excluded for 80 s, and a minimal intensity value of 10,000 was fixed for precursor ion selection.
Peptide detection and clustering.
Raw data files generated from the Orbitrap were processed using in-house peptide detection software (Mass Sense) to identify all ions according to their corresponding m/z values, charge state, retention time, and intensity. From this process, lists of detected peptides ions were generated to define individual LC-MS analysis. A user-defined intensity threshold above the background noise (typically between 5,000 and 15,000) was fixed to limit false-positive identification. Segmentation analyses were performed across sample sets using hierarchical clustering to generate lists of nonredundant peptide clusters (35). User-specified tolerances were fixed to, typically, ±0.04 m/z, ±1.5 min, and ±1 fraction for 2D-LC-MS Orbitrap experiments. Identification files from Mascot were converted in Excel format (Microsoft), and sequenced peptides were aligned with their corresponding peptide clusters using in-house clustering software.
MS/MS sequencing and protein identification.
The data were searched against International Protein Index mouse database using the Mascot (Matrix Science) search engine. The mass tolerance on precursor and fragment ions was set to ±0.1 and ±0.4 Daltons, respectively. Searches were performed without enzyme specificity. All relevant hits were inspected manually for mass accuracy and to validate sequence assignment according to the observation of consistent b- and y-types fragment ion series. From MHC I peptide structures identified, 85% were sequenced at least twice across replicate injections, and all had mass accuracy within 30 ppm of the theoretical values (without the use of a lock mass). To estimate the false-positive rate across non–MHC I–associated peptide identification, β2m-deficient EL4 samples were searched against a concatenated forward/reverse International Protein Index mouse database to establish a cutoff score threshold (>45) for a false-positive rate of <2%. All identified MHC I peptides were blasted against nonredundant NCBInr (restricted to mouse entries) to identify their corresponding source proteins (Gene ID).
Quantitative real-time PCR.
Freshly isolated thymocytes and EL4 cells were homogenized in TRIzol RNA preparation reagent (Invitrogen), and total RNA was isolated as instructed by the manufacturer. 1 μg of total RNA was converted to cDNA using random hexamer priming (Thermoscript RT-PCR System; Invitrogen). Gene expression level was determined using primer and probe sets from Universal ProbeLibrary (Table S7, available at http://www.jem.org/cgi/content/full/jem.20071985/DC1). PCR reactions for 384-well plate formats were performed using 2 μl of cDNA samples (50 ng), 5 μl of the TaqMan PCR Master Mix (Applied Biosystems), 2 μM of each primer, and 1 μM of the Universal TaqMan probe in a total volume of 10 μl. The ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) was used to detect the amplification level and was programmed to an initial step of 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. All reactions were run in triplicate, and the mean values were used for quantification. The mouse GAPDH or 18S ribosomal RNAs were used as endogenous controls.
Microarray dataset cross comparison.
Normalized mRNA expression data (50) (http://symatlas.gnf.org/SymAtlas/) used in this study were visualized with TIGR MultiExperiment Viewer (http://www.tigr.org/software/microarray.shtml). Normalized mean expression values were calculated as follows for each tissue: average expression value of 180 peptide source genes/average expression value of 36,182 transcripts.
Expression Analysis Systematic Explorer analysis and statistical methods.
The Expression Analysis Systematic Explorer (EASE) software (75) was downloaded from the Database for Annotation, Visualization, and Integrated Discovery (http://david.abcc.ncifcrf.gov/ease/ease.jsp) (76). For gene enrichment analysis, statistical and corrected P values were calculated from the Fisher exact test and the global false-discovery rate, respectively.
Z score transformation.
Raw intensity data for each tissue were log2 transformed and used for the calculation of z scores. Z scores were calculated by subtracting the overall average tissue intensity (for a single gene) from the raw intensity data within a given tissue, and dividing that result by the SD of the overall tissue intensities: z score = (intensity GTh − mean intensity GT1…T n)/SD GT1…T n, where GTh is any gene on the microarray from the thymus tissue and T1…Tn represents the aggregate measure of all tissues.
Peptide-binding and in vitro cytolytic assay.
Peptides were synthesized by the Centre Hospitalier de l'Université Laval Research Center (Quebec) and purified by high performance liquid chromatography (purity > 90%). Rescue of class I expression in T2-Db and T2-Kb cells by allele-specific peptides was determined as previously described (77). Bone marrow–derived DCs were generated as previously described (78). On day 9 of culture, peptides were added at a concentration of 2 × 10−6 M and incubated with DCs for 3 h at 37°C. For mouse immunization, 106 peptide-pulsed DCs were injected i.v. in C57BL/6 females at day 0 and 7. At day 14, splenocytes were harvested from the spleens of immunized mice and depleted of red blood cells using 0.83% NH4CL. Cells were plated at 5 × 106 cells/well in 24-well plates and restimulated with 2 × 10−6 M peptide at 37°C. After 6 d, cytotoxicity was evaluated by a CFSE-based assay (79). The percentage of specific lysis was calculated as follows: (number of remaining CFSE+ cells after incubation of target cells alone − number of remaining CFSE+ cells after incubation with effector cells/number of CFSE+ cells after incubation of target cells alone) × 100.
Online supplemental material.
Fig. S1 illustrates that 95% of peptide ions showed a variation of less than ±1.4- and ± 2.2-fold in abundance across 2D-nanoLC-MS instrumental and biological replicates, respectively. Fig. S2 depicts experimental data that validate bioinformatic discrimination between MHC I–associated peptides and contaminant peptides. Table S1 shows the relative abundance of peptides eluted from WT, β2m−, and β2m+ EL4 cell lines. Table S2 shows the computed MHC binding affinity of MHC I–associated peptides eluted from EL4 cells. Table S3 shows the computed MHC binding affinity of MHC I–associated peptides eluted from primary mouse thymocytes. Table S4 shows the computed MHC binding affinity of MHC I–associated peptides eluted from in vivo grown EL4 cells. Table S5 is a list (with references) of cancer-related genes coding for MHC I peptides differentially expressed in neoplastic (EL4) versus normal thymocytes. Table S6 shows the relative abundance of MHC I–associated peptides eluted from primary thymocytes versus in vivo grown EL4 cells. Table S7 is a list of primers used for quantitative RT-PCR analyses.
Supplemental Material
Acknowledgments
We are grateful to the staff of the following core facilities at the Institute for Research in Immunology and Cancer for their outstanding support: Animal facility, Bioinformatics, Flow Cytometry, and Proteomics. We thank Dr. R. Glas for kindly providing us with WT EL4, C4.4-25−, and E50.16+ cell lines.
This work was supported by funds from the Canadian Cancer Society and the Terry Fox Foundation through the National Cancer Institute of Canada. M.-H. Fortier and E. Caron are supported by training grants from the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research, respectively. C. Perreault and P. Thibault hold Canada Research Chairs in Immunobiology and Proteomics and Bioanalytical Spectrometry, respectively.
The authors have no conflicting financial interests.
Abbreviations used: 2D-nanoLC-MS, two-dimensional liquid chromatography nanoelectrospray MS; β2m, β2-microglobulin; DRiP, defective ribosomal product; FA, formic acid; MAE, mild acid elution; MIP, MHC class I peptide; MS, mass spectrometry; MS/MS, tandem MS; ppm, parts per million; SCX, strong cation exchange.
M.-H. Fortier and E. Caron, and C. Perreault and P. Thibault contributed equally to this work.
References
- 1.Rammensee, H.G., K. Falk, and O. Rotzschke. 1993. Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 11:213–244. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell, J.W., E. Reits, and J. Neefjes. 2003. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 3:952–961. [DOI] [PubMed] [Google Scholar]
- 3.Rock, K.L., I.A. York, T. Saric, and A.L. Goldberg. 2002. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 80:1–70. [DOI] [PubMed] [Google Scholar]
- 4.Heemels, M.T., and H. Ploegh. 1995. Generation, translocation, and presentation of MHC class I-restricted peptides. Annu. Rev. Biochem. 64:463–491. [DOI] [PubMed] [Google Scholar]
- 5.Pamer, E., and P. Cresswell. 1998. Mechanisms of MHC class I–restricted antigen processing. Annu. Rev. Immunol. 16:323–358. [DOI] [PubMed] [Google Scholar]
- 6.Hammer, G.E., T. Kanaseki, and N. Shastri. 2007. 4. The final touches make perfect the peptide-MHC class I repertoire. Immunity. 26:397–406. [DOI] [PubMed] [Google Scholar]
- 7.Zhang, Y., and D.B. Williams. 2006. Assembly of MHC class I molecules within the endoplasmic reticulum. Immunol. Res. 35:151–162. [DOI] [PubMed] [Google Scholar]
- 8.Wong, P., and E.G. Pamer. 2003. CD8 T cell responses to infectious pathogens. Annu. Rev. Immunol. 21:29–70. [DOI] [PubMed] [Google Scholar]
- 9.Weinzierl, A.O., C. Lemmel, O. Schoor, M. Muller, T. Kruger, D. Wernet, J. Hennenlotter, A. Stenzl, K. Klingel, H.G. Rammensee, and S. Stevanovic. 2007. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol. Cell. Proteomics. 6:102–113. [DOI] [PubMed] [Google Scholar]
- 10.Starr, T.K., S.C. Jameson, and K.A. Hogquist. 2003. Positive and negative T cell selection. Annu. Rev. Immunol. 21:139–176. [DOI] [PubMed] [Google Scholar]
- 11.Huseby, E.S., J. White, F. Crawford, T. Vass, D. Becker, C. Pinilla, P. Marrack, and J.W. Kappler. 2005. How the T cell repertoire becomes peptide and MHC specific. Cell. 122:247–260. [DOI] [PubMed] [Google Scholar]
- 12.Marrack, P., and J. Kappler. 2004. Control of T cell viability. Annu. Rev. Immunol. 22:765–787. [DOI] [PubMed] [Google Scholar]
- 13.Anikeeva, N., T. Lebedeva, A.R. Clapp, E.R. Goldman, M.L. Dustin, H. Mattoussi, and Y. Sykulev. 2006. Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response. Proc. Natl. Acad. Sci. USA. 103:16846–16851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn, G.P., L.J. Old, and R.D. Schreiber. 2004. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 21:137–148. [DOI] [PubMed] [Google Scholar]
- 15.Slev, P.R., A.C. Nelson, and W.K. Potts. 2006. Sensory neurons with MHC-like peptide binding properties: disease consequences. Curr. Opin. Immunol. 18:608–616. [DOI] [PubMed] [Google Scholar]
- 16.Perreault, C., F. Decary, S. Brochu, M. Gyger, R. Belanger, and D. Roy. 1990. Minor histocompatibility antigens. Blood. 76:1269–1280. [PubMed] [Google Scholar]
- 17.Liblau, R.S., F.S. Wong, L.T. Mars, and P. Santamaria. 2002. Autoreactive CD8 T cells in organ-specific autoimmunity: emerging targets for therapeutic intervention. Immunity. 17:1–6. [DOI] [PubMed] [Google Scholar]
- 18.Hunt, D.F., R.A. Henderson, J. Shabanowitz, K. Sakaguchi, H. Michel, N. Sevilir, A.L. Cox, E. Appella, and V.H. Engelhard. 1992. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science. 255:1261–1263. [DOI] [PubMed] [Google Scholar]
- 19.Falk, K., O. Rotzschke, S. Stevanovic, G. Jung, and H.G. Rammensee. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 351:290–296. [DOI] [PubMed] [Google Scholar]
- 20.Huczko, E.L., W.M. Bodnar, D. Benjamin, K. Sakaguchi, N.Z. Zhu, J. Shabanowitz, R.A. Henderson, E. Appella, D.F. Hunt, and V.H. Engelhard. 1993. Characteristics of endogenous peptides eluted from the class I MHC molecule HLA-B7 determined by mass spectrometry and computer modeling. J. Immunol. 151:2572–2587. [PubMed] [Google Scholar]
- 21.McBride, K., C. Baron, S. Picard, S. Martin, D. Boismenu, A. Bell, J. Bergeron, and C. Perreault. 2002. The model B6 dom1 minor histocompatibility antigen is encoded by a mouse homolog of the yeast STT3 gene. Immunogenetics. 54:562–569. [DOI] [PubMed] [Google Scholar]
- 22.Admon, A., E. Barnea, and T. Ziv. 2003. Tumor antigens and proteomics from the point of view of the major histocompatibility complex peptides. Mol. Cell. Proteomics. 2:388–398. [DOI] [PubMed] [Google Scholar]
- 23.Gebreselassie, D., H. Spiegel, and S. Vukmanovic. 2006. Sampling of major histocompatibility complex class I-associated peptidome suggests relatively looser global association of HLA-B*5101 with peptides. Hum. Immunol. 67:894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnea, E., I. Beer, R. Patoka, T. Ziv, O. Kessler, E. Tzehoval, L. Eisenbach, N. Zavazava, and A. Admon. 2002. Analysis of endogenous peptides bound by soluble MHC class I molecules: a novel approach for identifying tumor-specific antigens. Eur. J. Immunol. 32:213–222. [DOI] [PubMed] [Google Scholar]
- 25.Hickman, H.D., A.D. Luis, R. Buchli, S.R. Few, M. Sathiamurthy, R.S. VanGundy, C.F. Giberson, and W.H. Hildebrand. 2004. Toward a definition of self: proteomic evaluation of the class I peptide repertoire. J. Immunol. 172:2944–2952. [DOI] [PubMed] [Google Scholar]
- 26.Hickman, H.D., A.D. Luis, W. Bardet, R. Buchli, C.L. Battson, M.H. Shearer, K.W. Jackson, R.C. Kennedy, and W.H. Hildebrand. 2003. Cutting edge: class I presentation of host peptides following HIV infection. J. Immunol. 171:22–26. [DOI] [PubMed] [Google Scholar]
- 27.Lemmel, C., S. Weik, U. Eberle, J. Dengjel, T. Kratt, H.D. Becker, H.G. Rammensee, and S. Stevanovic. 2004. Differential quantitative analysis of MHC ligands by mass spectrometry using stable isotope labeling. Nat. Biotechnol. 22:450–454. [DOI] [PubMed] [Google Scholar]
- 28.Milner, E., E. Barnea, I. Beer, and A. Admon. 2006. The turnover kinetics of major histocompatibility complex peptides of human cancer cells. Mol. Cell. Proteomics. 5:357–365. [DOI] [PubMed] [Google Scholar]
- 29.Meiring, H.D., E.C. Soethout, M.C. Poelen, D. Mooibroek, R. Hoogerbrugge, H. Timmermans, C.J. Boog, A.J. Heck, A.P. de Jong, and C.A. van Els. 2006. Stable isotope tagging of epitopes: a highly selective strategy for the identification of major histocompatibility complex class I-associated peptides induced upon viral infection. Mol. Cell. Proteomics. 5:902–913. [DOI] [PubMed] [Google Scholar]
- 30.Mann, M. 2006. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 7:952–958. [DOI] [PubMed] [Google Scholar]
- 31.Ong, S.E., L.J. Foster, and M. Mann. 2003. Mass spectrometric-based approaches in quantitative proteomics. Methods. 29:124–130. [DOI] [PubMed] [Google Scholar]
- 32.Caron, E., R. Charbonneau, G. Huppe, S. Brochu, and C. Perreault. 2005. The structure and location of SIMP/STT3B account for its prominent imprint on the MHC I immunopeptidome. Int. Immunol. 17:1583–1596. [DOI] [PubMed] [Google Scholar]
- 33.Yewdell, J.W., and C.V. Nicchitta. 2006. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol. 27:368–373. [DOI] [PubMed] [Google Scholar]
- 34.Benoist, C., R.N. Germain, and D. Mathis. 2006. A plaidoyer for ‘systems immunology’. Immunol. Rev. 210:229–234. [DOI] [PubMed] [Google Scholar]
- 35.Fortier, M.H., E. Bonneil, P. Goodley, and P. Thibault. 2005. Integrated microfluidic device for mass spectrometry-based proteomics and its application to biomarker discovery programs. Anal. Chem. 77:1631–1640. [DOI] [PubMed] [Google Scholar]
- 36.Kearney, P., and P. Thibault. 2003. Bioinformatics meets proteomics–bridging the gap between mass spectrometry data analysis and cell biology. J. Bioinform. Comput. Biol. 1:183–200. [DOI] [PubMed] [Google Scholar]
- 37.Storkus, W.J., H.J. Zeh III, R.D. Salter, and M.T. Lotze. 1993. Identification of T-cell epitopes: rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immunother. Emphasis Tumor Immunol. 14:94–103. [PubMed] [Google Scholar]
- 38.Perreault, C., J. Jutras, D.C. Roy, J.G. Filep, and S. Brochu. 1996. Identification of an immunodominant mouse minor histocompatibility antigen (MiHA). T cell response to a single dominant MiHA causes graft-versus-host disease. J. Clin. Invest. 98:622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purcell, A.W. 2004. Isolation and characterization of naturally processed MHC-bound peptides from the surface of antigen-presenting cells. Methods Mol. Biol. 251:291–306. [DOI] [PubMed] [Google Scholar]
- 40.Glas, R., K. Sturmhofel, G.J. Hammerling, K. Karre, and H.G. Ljunggren. 1992. Restoration of a tumorigenic phenotype by beta 2-microglobulin transfection to EL-4 mutant cells. J. Exp. Med. 175:843–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams, D.B., B.H. Barber, R.A. Flavell, and H. Allen. 1989. Role of beta 2-microglobulin in the intracellular transport and surface expression of murine class I histocompatibility molecules. J. Immunol. 142:2796–2806. [PubMed] [Google Scholar]
- 42.Rammensee, H., J. Bachmann, N.P. Emmerich, O.A. Bachor, and S. Stevanovic. 1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 50:213–219. [DOI] [PubMed] [Google Scholar]
- 43.Moutaftsi, M., B. Peters, V. Pasquetto, D.C. Tscharke, J. Sidney, H.H. Bui, H. Grey, and A. Sette. 2006. A consensus epitope prediction approach identifies the breadth of murine T CD8+-cell responses to vaccinia virus. Nat. Biotechnol. 24:817–819. [DOI] [PubMed] [Google Scholar]
- 44.Peters, B., H.H. Bui, S. Frankild, M. Nielson, C. Lundegaard, E. Kostem, D. Basch, K. Lamberth, M. Harndahl, W. Fleri, et al. 2006. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput. Biol. 2:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters, B., and A. Sette. 2007. Integrating epitope data into the emerging web of biomedical knowledge resources. Nat. Rev. Immunol. 7:485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aldrich, C.J., A. DeCloux, A.S. Woods, R.J. Cotter, M.J. Soloski, and J. Forman. 1994. Identification of a Tap-dependent leader peptide recognized by alloreactive T cells specific for a class Ib antigen. Cell. 79:649–658. [DOI] [PubMed] [Google Scholar]
- 47.Rotzschke, O., K. Falk, S. Stevanovic, B. Grahovac, M.J. Soloski, G. Jung, and H.G. Rammensee. 1993. Qa-2 molecules are peptide receptors of higher stringency than ordinary class I molecules. Nature. 361:642–644. [DOI] [PubMed] [Google Scholar]
- 48.Qian, S.B., M.F. Princiotta, J.R. Bennink, and J.W. Yewdell. 2006. Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J. Biol. Chem. 281:392–400. [DOI] [PubMed] [Google Scholar]
- 49.Qian, S.B., E. Reits, J. Neefjes, J.M. Deslich, J.R. Bennink, and J.W. Yewdell. 2006. Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J. Immunol. 177:227–233. [DOI] [PubMed] [Google Scholar]
- 50.Su, A.I., T. Wiltshire, S. Batalov, H. Lapp, K.A. Ching, D. Block, J. Zhang, R. Soden, M. Hayakawa, G. Kreiman, et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 101:6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang, W., Q.D. Morris, R. Chang, O. Shai, M.A. Bakowski, N. Mitsakakis, N. Mohammad, M.D. Robinson, R. Zirngibl, E. Somogyi, et al. 2004. The functional landscape of mouse gene expression. J. Biol. 3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Virtaneva, K., F.A. Wright, S.M. Tanner, B. Yuan, W.J. Lemon, M.A. Caligiuri, C.D. Bloomfield, A. de La Chapelle, and R. Krahe. 2001. Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics. Proc. Natl. Acad. Sci. USA. 98:1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dorn, T., U. Kuhn, G. Bungartz, S. Stiller, M. Bauer, J. Ellwart, T. Peters, K. Scharffetter-Kochanek, M. Semmrich, M. Laschinger, et al. 2007. RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood. 109:2346–2355. [DOI] [PubMed] [Google Scholar]
- 54.Plotkin, J., S.E. Prockop, A. Lepique, and H.T. Petrie. 2003. Critical role for CXCR4 signaling in progenitor localization and T cell differentiation in the postnatal thymus. J. Immunol. 171:4521–4527. [DOI] [PubMed] [Google Scholar]
- 55.Malissen, M., A. Gillet, L. Ardouin, G. Bouvier, J. Trucy, P. Ferrier, E. Vivier, and B. Malissen. 1995. Altered T cell development in mice with a targeted mutation of the CD3-epsilon gene. EMBO J. 14:4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richter, J.D., and N. Sonenberg. 2005. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 433:477–480. [DOI] [PubMed] [Google Scholar]
- 57.Nakayama, K.I., and K. Nakayama. 2006. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 6:369–381. [DOI] [PubMed] [Google Scholar]
- 58.Siegel, J.N., C.A. Turner, D.M. Klinman, M. Wilkinson, A.D. Steinberg, C.L. MacLeod, W.E. Paul, M.M. Davis, and D.I. Cohen. 1987. Sequence analysis and expression of an X-linked, lymphocyte-regulated gene family (XLR). J. Exp. Med. 166:1702–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hennessy, B.T., D.L. Smith, P.T. Ram, Y. Lu, and G.B. Mills. 2005. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4:988–1004. [DOI] [PubMed] [Google Scholar]
- 60.Mamane, Y., E. Petroulakis, O. LeBacquer, and N. Sonenberg. October 2006. 16. mTOR, translation initiation and cancer. Oncogene. 25:6416–6422. [DOI] [PubMed] [Google Scholar]
- 61.Singh-Jasuja, H., N.P. Emmerich, and H.G. Rammensee. 2004. The Tubingen approach: identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol. Immunother. 53:187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Purcell, A.W., J. McCluskey, and J. Rossjohn. 2007. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6:404–414. [DOI] [PubMed] [Google Scholar]
- 63.Suri, A., J.J. Walters, M.G. Levisetti, M.L. Gross, and E.R. Unanue. 2006. Identification of naturally processed peptides bound to the class I MHC molecule H-2Kd of normal and TAP-deficient cells. Eur. J. Immunol. 36:544–557. [DOI] [PubMed] [Google Scholar]
- 64.Gleimer, M., and P. Parham. 2003. Stress management: MHC class I and class I-like molecules as reporters of cellular stress. Immunity. 19:469–477. [DOI] [PubMed] [Google Scholar]
- 65.Glickman, M.H., and A. Ciechanover. 2002. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82:373–428. [DOI] [PubMed] [Google Scholar]
- 66.Matos, M., R. Park, D. Mathis, and C. Benoist. 2004. Progression to islet destruction in a cyclophosphamide-induced transgenic model: a microarray overview. Diabetes. 53:2310–2321. [DOI] [PubMed] [Google Scholar]
- 67.Ruggero, D., and P.P. Pandolfi. 2003. Does the ribosome translate cancer? Nat. Rev. Cancer. 3:179–192. [DOI] [PubMed] [Google Scholar]
- 68.Nalepa, G., M. Rolfe, and J.W. Harper. 2006. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 5:596–613. [DOI] [PubMed] [Google Scholar]
- 69.Lu, J., G. Getz, E.A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B.L. Ebert, R.H. Mak, A.A. Ferrando, et al. 2005. MicroRNA expression profiles classify human cancers. Nature. 435:834–838. [DOI] [PubMed] [Google Scholar]
- 70.Eden, A., F. Gaudet, A. Waghmare, and R. Jaenisch. 2003. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 300:455. [DOI] [PubMed] [Google Scholar]
- 71.Yoshimura, Y., R. Yadav, G.J. Christianson, W.U. Ajayi, D.C. Roopenian, and S. Joyce. 2004. Duration of alloantigen presentation and avidity of T cell antigen recognition correlate with immunodominance of CTL response to minor histocompatibility antigens. J. Immunol. 172:6666–6674. [DOI] [PubMed] [Google Scholar]
- 72.Louis, I., G. Dulude, S. Corneau, S. Brochu, C. Boileau, C. Meunier, C. Côté, N. Labrecque, and C. Perreault. 2003. Changes in the lymph node microenvironment induced by Oncostatin M. Blood. 102:1397–1404. [DOI] [PubMed] [Google Scholar]
- 73.Meunier, M.C., G. Roy-Proulx, N. Labrecque, and C. Perreault. 2003. Tissue distribution of target antigen has a decisive influence on the outcome of adoptive cancer immunotherapy. Blood. 101:766–770. [DOI] [PubMed] [Google Scholar]
- 74.Pion, S., P. Fontaine, C. Baron, M. Gyger, and C. Perreault. 1995. Immunodominant minor histocompatibility antigens expressed by mouse leukemic cells can serve as effective targets for T cell immunotherapy. J. Clin. Invest. 95:1561–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hosack, D.A., G. Dennis Jr., B.T. Sherman, H.C. Lane, and R.A. Lempicki. 2003. Identifying biological themes within lists of genes with EASE. Genome Biol. 4:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dennis, G. Jr., B.T. Sherman, D.A. Hosack, J. Yang, W. Gao, H.C. Lane, and R.A. Lempicki. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4:P3. [PubMed] [Google Scholar]
- 77.Pion, S., P. Fontaine, M. Desaulniers, J. Jutras, J.G. Filep, and C. Perreault. 1997. On the mechanisms of immunodominance in cytotoxic T lymphocyte responses to minor histocompatibility antigens. Eur. J. Immunol. 27:421–430. [DOI] [PubMed] [Google Scholar]
- 78.Galea-Lauri, J., J.W. Wells, D. Darling, P. Harrison, and F. Farzaneh. 2004. Strategies for antigen choice and priming of dendritic cells influence the polarization and efficacy of antitumor T-cell responses in dendritic cell-based cancer vaccination. Cancer Immunol. Immunother. 53:963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jedema, I., N.M. van der Werff, R.M. Barge, R. Willemze, and J.H. Falkenburg. 2004. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood. 103:2677–2682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
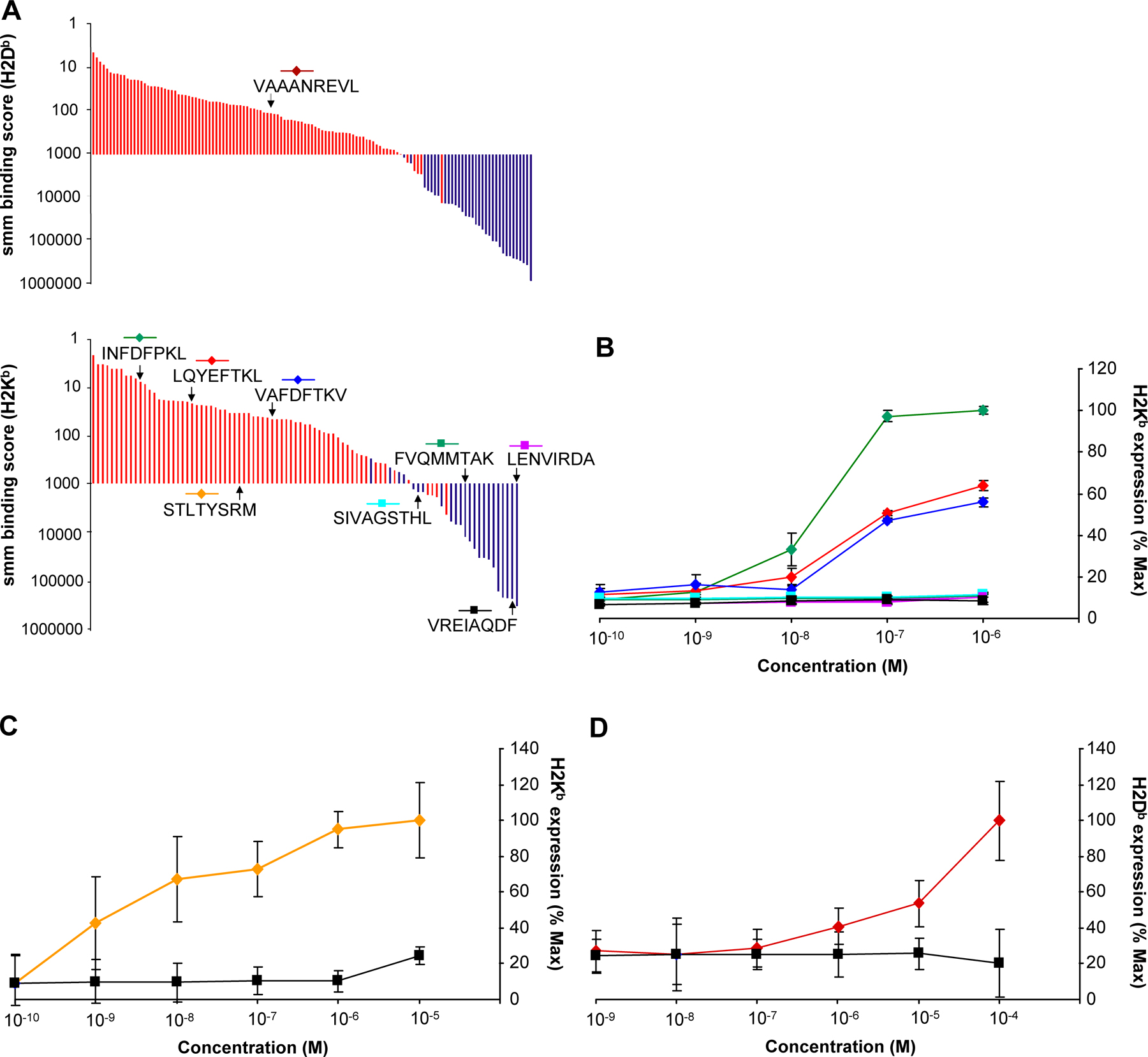{kind=link}
{kind=link}