

Abstract
Skeletal muscle transplantation strategies for muscle repair or gene therapy involve either the injection of proliferating myoblasts followed by fusion with host myofibers or implantation of ex vivo differentiated myofibers; however both implant procedures are associated with significant cell loss. Biodegradable porous, gas-foamed poly-lactide-co-glycolide (PLG) scaffolds have desirable characteristics for cell transfer and were used to study attachment, growth, differentiation and survival of human myogenic cells. Primary human myoblasts suspended in clinical grade extracellular matrixes (ECMs) and adhered to PLG scaffolds differentiated in vitro into high density tropomyosin positive myofibers. An immunodeficient NOD/SCID mouse implant model was used to study the transfer and in vivo survival of differentiated human myofibers on these scaffolds. Scaffold rigidity allowed the myofibers to be maintained under tension in vitro and following subcutaneous transplantation in vivo. Following implantation, myofiber density on the PLG scaffolds decreased linearly by 78% over a four week period. ECM composed of either Tisseel® fibrin or Zyderm® collagen type I did not significantly affect in vivo cell viability over the four week period. Varying PLG scaffold microsphere content (10 – 100%) also had little effect on cell survival in vivo. In contrast, when the residual NK cell population in the immunodeficient NOD/SCID mouse model was depleted with anti-asialo GM1 (ASGM1) antiserum, in vivo cell survival significantly increased from 22% to 34% after four weeks. With further improvements in cell survival, PLG scaffolds may prove useful for the implantation of primary human myofibers in future clinical applications.
Introduction
Skeletal muscle contains progenitor satellite cells, which are activated following injury and initiate muscle regeneration. Numerous attempts have been made to provide these cells exogenously to enhance the regeneration process and primary myoblasts can also be genetically engineered to secrete a therapeutic protein and implanted for ex vivo gene therapy [1]. Normally, transplanted proliferating myogenic cells undergo rapid and massive cell death within 48 hours following implantation into skeletal muscle, resulting in less than 5% of transplanted cells incorporating into the host myofibers [2–4]. Proliferating myogenic cells can also be tissue engineered ex vivo into contractile ‘organs’ containing postmitotic myofibers (reviewed in [5]) and may prove useful in muscle regenerative and gene therapy applications [6–9]. Bioartificial muscles with postmitotic muscle fibers as a protein secretion device have a better safety profile than injecting individual proliferating and migrating cells because of reversibility of implants in case of an adverse event [6–9]. Ex vivo differentiated myofibers exhibit improved cell survival kinetics when compared to implanted myoblasts, with a gradual loss of viability over 30–60 days after implantation [9, 10] partially due to poor myofiber tension maintenance [10]. Successful transplantation of skeletal muscle cells (either myoblasts or myofibers) for structural repair or ex vivo gene therapy applications will require optimization for long term cell survival. Recently, significant progress has been made in enhancing the survival and regenerative capacity of proliferating myogenic cells in vivo by transplanting with alginate scaffolds containing growth factors to stimulate the rapid migration of myoblasts away from the implant site [11, 12]. Three dimensional biodegradable polymer scaffolds have also been successfully utilized for growth, differentiation and implantation of numerous cell types, including rodent skeletal muscle cells [13, 14]. These scaffolds are highly porous, allowing rapid diffusion of nutrients to the transplanted cells and efficient offloading of therapeutic proteins from genetically engineered cells. In addition, they can be manufactured to release angiogenic/vasculogenic factors with varying release kinetics to stimulate in-growth of host vasculature [15] which enhances transplanted cell survival [16]. To be clinically useful as cell transplant vehicles, such scaffolds need to be biocompatible without toxic side effects and clinical grade components need to be used to attach cells to the scaffold.
In the present study we explored the use of biodegradable porous, gas-foamed poly-lactide-co-glycolide (PLG) scaffolds as an appropriate material [15, 17] to which primary human myoblasts could be attached using clinical grade extracellular matrix (ECM) carriers and differentiated into myofibers which could be maintained under tension. An immunodeficient NOD/SCID animal model was used to assess the in vivo characteristics of the constructs. We demonstrate that human myoblasts can be efficiently seeded and differentiated into postmitotic myofibers on gas foamed PLG scaffolds and maintained under tension when implanted subcutaneously. The PLG scaffolds improve human myofiber viability in vivo relative to non-scaffold implants and therefore may be useful for future clinical applications.
Materials and Methods
Preparation of PLG scaffolds
Salt-leached polymer scaffolds were fabricated from 85:15 poly(lactide-co-glycolide)(PLG)(Medisorb-Alkermes, Cambridge MA) and NaCl particles of 250–425 μm, expanded using high-pressure carbon dioxide, and leached to yield pore sizes of 360±85 μm as described previously [18, 19]. PLG microspheres were prepared by standard double emulsion [20]. Scaffolds 4 to 13 mm in diameter by 1.5 mm thick were fabricated from particulate PLG and 10% – 100% (wt/wt) microsphere PLG. Scaffolds were sterilized 30 min in 70% ethanol and rinsed in EBSS before cell seeding. Sterilization did not affect scaffold structure.
Muscle progenitor cell isolation, growth, and seeding
Primary human skeletal muscle cells isolated by needle biopsy [21] were expanded in myoblast growth medium [MGM; SkGM (Cambrex Bio Science, Walkersville, MD) plus 15% (v/v) fetal bovine serum]. Biopsies were performed on adult volunteers according to procedures approved by the Institutional Clinical Review Board of the Miriam Hospital. Cell preparations on average were 70% myogenic based on desmin-positive staining [22]. On 4.2, 7 and 13 mm diameter scaffolds, 1.5, 4 and 4–16 million myoblasts were seeded respectively. Cells were first mixed with either collagen ECM [0.1 to 0.4 mg/ml Type I bovine collagen (Zyderm®, Inamed Inc., Santa Barbara, CA)] or fibrin ECM [1 mg/ml fibrinogen+ 0.5U/ml thrombin (Tisseel®, Baxter Healthcare Corp., Glendale, CA)] in a volume of 15–20, 40 and 80–100 μl respectively and seeded on top of the scaffolds. When collagen was used, scaffolds were pre-filled with collagen ECM before cell seeding. Scaffolds were incubated at 37°C and 5% CO2 for 2 hours after seeding cells, flooded with MGM and returned to the incubator for 2 days. Myoblast fusion into postmitotic myofibers was induced by incubation in differentiation medium [high-glucose DMEM (Invitrogen, Carlsbad, CA) supplemented with insulin (10 μg/ml), bovine serum albumin (50 μg/ml), epidermal growth factor (10 ng/ml), and gentamicin (50 μg/ml)] for an additional 3–5 days. Human myoblasts were transduced with lentiviral vectors to express GFP, as described previously [9].
Animal experiments
One to four scaffolds were implanted subcutaneously into the backs of adult immunodeficient NOD.CB17-Prkdcscid/J (NOD/SCID) mice (The Jackson Laboratory, Bar Harbor, ME). All animal procedures were approved by the Harvard Institutional Animal Care and Use Committee (IACUC) and conformed to the guiding principles of the American Physiological Society. In some experiments NK cells were depleted by intravenous injection of 50 μl anti-asialo GM1 (ASGM1) antiserum (Wako chemicals, Dallas, TX) 2 hours before implantation and 3, 7, 10, 14 and 21 days thereafter [23]. Control animals were injected with the same volume of normal rabbit serum (Invitrogen, Carlsbad, CA). To examine immunoglobulin leakiness in NOD/SCID mice, IgG levels were measured by IgG Elisa (Zeptometrix, Buffalo, NY) with C57/Bl6 mice included as a positive control.
Histology &Microscopy
Scaffold area covered with tropomyosin positive cells was quantified by counting at least 9 different fields per cell number condition. GFP expression was monitored by confocal microscopy (Leica TCS SP2 AOBS). The confocal focusing location was performed on random, non-overlapping regions of the sample. Depending on the size of the sample, this corresponded to n=3–10 regions, with 2–4 replicates. Confocal depth was set to 1 Airy unit. GFP area was determined with Metamorph v6.3. Where untransduced cells were used, cell viability was assessed with LIVE/DEAD stain (Invitrogen, Carlsbad, CA). For GFP transduced cells, viability was calculated by the average % GFP area at a specific timepoint divided by average % GFP area on the scaffolds pre-implant. For tropomyosin staining, scaffolds were fixed in 3.7% formaldehyde, permeabilized with methanol, incubated with mouse anti-tropomyosin antibody (Sigma, St. Louis, MO) 1:100, followed by a secondary biotinylated anti-mouse IgG and then a preformed avidin and biotinylated horse radish peroxidase complex (Vector Laboratories, Burlingame, CA). Development was by addition of DAB (3,3′-diaminobenzidene) to produce a brown precipitate.
Immunological assays
NK cell activity was measured using a standard chromium release assay as previously described [24]. Briefly, splenocytes were harvested at 2 or 4 weeks after scaffold implant. Target YAC-1 cells (American Type Culture Collection, Rockville, MD) were labeled with 51Cr and adjusted to 5 × 104 cells/ml. Various effector to target ratios were set up in triplicate in V-bottom plates. After incubation for 4 h of targets and effectors at 37 °C, 100 μl was removed from each well and the radioactivity present in each sample was determined using a gamma counter. The percentage-specific 51Cr release was calculated using the following formula: % specific release = [(X − S)/(T − S)] × 100, where X is the mean experimental release from triplicate wells, total release (T) was determined from wells receiving 51Cr-labeled YAC-1 target cells and 1N HCl, and spontaneous release (S) was determined from wells receiving 51Cr-labeled YAC-1 target cells in growth medium.
GFP protein quantification
Total protein was extracted from the scaffolds by sonication in 1 ml RIPA Buffer (RadioImmunoPrecipitation Assay). GFP content was measured in triplicate on 200 μl samples by fluorimetric analysis (excitation 485 nm-emission 528) on a Synergy HT microplate reader (Biotek Winooski, VT).
Statistics
Data are presented as mean ± SEM and statistical analyses were performed in Microsoft Excel for Mac using an unpaired t test. Bonferroni correction was applied when testing multiple groups.
Results
Differentiation of human muscle cells on PLG scaffolds in vitro
Cells were seeded at various densities (4–16 million) on 13 mm scaffolds, composed of particulate PLG and 10% (wt/wt) microsphere PLG. Human primary myoblasts were first mixed with an ECM solution and applied to the scaffolds; the ECM was required for retention of a majority of the cells on the PLG scaffold (data not shown). Two clinical grade products were utilized, either collagen type I (Zyderm®) or fibrin (Tisseel®) as the cell carrier. The pore size of 360±85 μm allowed for a quick penetration of the cell-ECM mix in the interconnected scaffold pores. When collagen I was used as the ECM, it took 10–30 minutes to solidify at 37°C, allowing the cell-ECM mix to penetrate throughout the scaffold and leak out. To prevent this in subsequent experiments, scaffolds were prefilled with cell-free collagen and allowed to solidify before adding the cell-ECM mix to the top of the scaffolds. When fibrin was used as the ECM, prefilling was not necessary due to a much faster clotting time (<90 seconds). In both cases, the cell-ECM mix formed a high density layer of myoblasts on top of the scaffolds. When scaffolds were incubated in differentiation medium for 4 days, a dense layer of postmitotic muscle fibers expressing sarcomeric tropomyosin was observed primarily on top of the scaffold (Fig. 1), indicating efficient differentiation into myofibers. Scaffold surface myofiber density was dependent on initial cell seeding number, with 4–8 million cells per 13 mm diameter scaffold giving optimal surface myofiber density. For scaffolds plated with 4 million cells, 54 ± 9 % of the scaffold area was covered with tropomyosin positive differentiated muscle fibers by day 8–10 postplating while 72 ± 9 % was tropomyosin positive when 16 million cells were used for the initial plating. Cell densities up to 16 million/scaffold did not influence their differentiation potential (Fig 1). As expected, myofibers were not aligned in any specific direction. Fiber density decreased from the surface of the scaffolds to the middle (data not shown). Plating the 4–8 million cells with either ECMs with these techniques resulted in the formation of an approximately 100 μm thick cell layer of well formed postmitotic muscle fibers on the surface of the scaffolds (Fig 1). With these protocols, myofibers were retained under tension in vitro on top of the gas foamed PLG scaffolds for at least several weeks. We attempted to seed syngeneic murine GFP transduced myoblasts on PLG scaffolds in a collagen:Matrigel®, collagen or fibrin ECM. In contrast to human myoblasts, the murine myofibers consistently rolled up into clumps and detached from the surface of the scaffold within 2–4 days of differentiation (data not shown). Scaffolds were also made containing 25%, 50% or 100% PLG microspheres and seeded with 16 million GFP expressing myoblasts. Differentiation was not affected by either varying the microsphere content of the scaffolds, or the composition of the carrier ECM. After 7–8 days in vitro, GFP myofiber retention was studied by confocal microscopy. Inclusion of a higher percentage microsphere PLG in the scaffolds improved the density of differentiated myofibers on the scaffold (Fig 2). For subsequent experiments, PLG scaffolds containing 50–100% microspheres were used with either fibrin or collagen ECM.
Fig 1. Differentiation of human skeletal myoblasts to myofibers on PLG scaffolds.
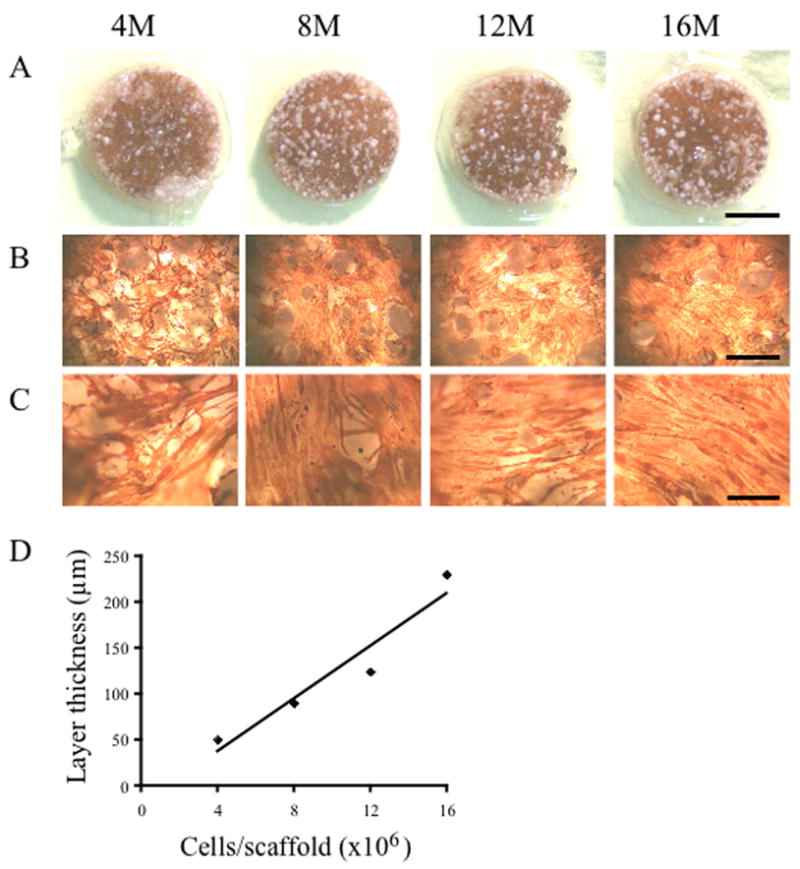
Various numbers (4, 8, 12 or 16 million cells, indicated on top of figure) of human myoblasts were seeded in 0.1 mg/ml collagen ECM and differentiated to myofibers on 13 mm PLG scaffolds containing 10% microspheres and prefilled with 0.1 mg/ml collagen ECM. After 5 days in differentiation medium, cells were stained for the expression of sarcomeric tropomyosin (brown color). Myofibers were short and not aligned in any specific direction. (A) Picture of entire scaffold. Scale bar represents 5 mm. (B) 2.5× Objective, scale bar represents 1 mm. (C) 10× Objective, scale bar represents 0.25 mm. (D) Cell layer thickness was measured on thin sections of paraffin embedded scaffolds and is linearly dependent on the initial number of seeded cells.
Fig 2. Growth of GFP human skeletal muscle cells on PLG scaffolds with varying microsphere content.
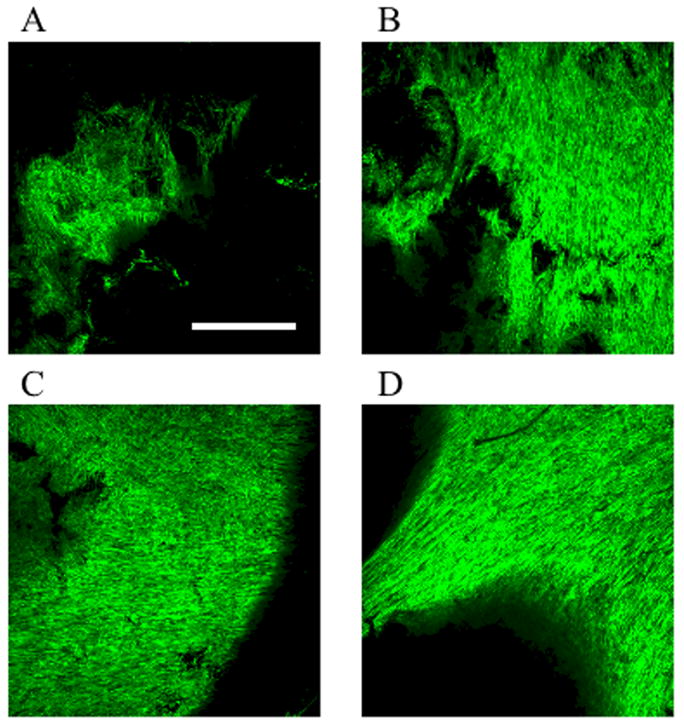
Sixteen million myoblasts in fibrin ECM were plated as described in Materials and Methods on the 13mm diameter scaffolds containing 10%, 25%, 50% or 100% (w/w) microspheres. After 7–8 days in vitro, cells were visualized by confocal microscopy. Images are composites of ten 1.7 μm sections of the top layer of the scaffolds. (A) 10%; (B) 25%; (C) 50%; (D) 100%. Scale bar represents 500 μm.
Muscle fiber survival on the scaffolds in vivo
Nontransduced or GFP-transduced myoblasts were differentiated into myofibers on 13 mm scaffolds containing 100% PLG microspheres and implanted subcutaneously in the dorsal region of immunodeficient NOD/SCID mice for up to one month. For non-GFP cells, living cells were stained green with membrane permeable calcein AM and dead cells were stained red by membrane impermeant ethidium homodimer-1. Cell survival at various depths was determined by confocal microscopy. At the surface of the scaffold, many GFP positive cells could be observed (Fig 3A). Longitudinal myofiber morphology was retained on the scaffolds even after 30 days, indicating that the fibers could be kept under tension on the scaffolds (Fig 3A). At 50–70 μm depth, many dead cells were observed (Fig 3B) and at 150 μm depth exclusively dead cells were seen (Fig 3C).
Fig 3. Myoblast cell survival in vivo at various depths in the scaffold.
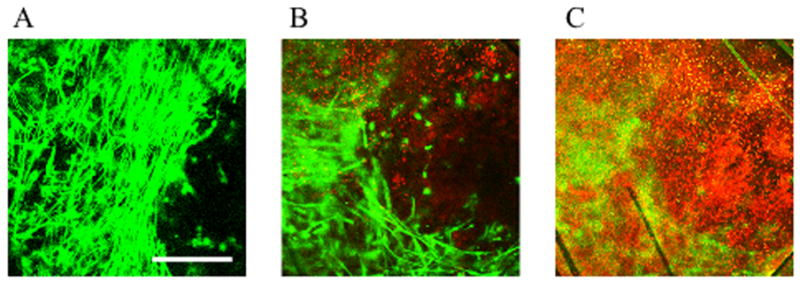
Myoblasts were seeded on the PLG scaffolds in a fibrin ECM, differentiated to myofibers and implanted subcutaneously in the back of NOD/SCID mice for 1 month. Living cells were stained with membrane permeable calcein AM (green) and dead cells were stained red by membrane impermeant ethidium homodimer-1. Cell survival at various depths was imaged by confocal microscopy. (A) Surface of scaffold, (B) 50–70 μm depth, (C) 150 μm depth. Scale bar represents 250 μm.
Primary human skeletal myoblasts stably transduced with GFP were used to track in vivo myofiber survival by confocal microscopy. Myoblasts were embedded in a fibrin or zyderm ECM and seeded on 4.2 mm scaffolds, composed of either 50 % or 100 % microspheres. Scaffolds were implanted subcutaneously in the dorsal region of NOD/SCID mice and explanted for analysis at 2 and 4 weeks in vivo. Scaffolds were imaged by confocal microscopy (3–10 fields/scaffold, 4 scaffolds/group) pre-implant and at 2 or 4 weeks in vivo and GFP area was quantified with Metamorph software. Results are summarized in Fig. 4. To calculate cell survival at a specific timepoint, GFP area at that time point was normalized to GFP area pre-implant. At 2 weeks in vivo, myofibers embedded in a zyderm ECM survived better than myofibers in the fibrin ECM (viability 77±8% vs 52±8%), however this difference was not statistically significant (t-test with Bonferroni correction). There was no significant difference in cell survival between the 50% and 100% microspheres scaffold compositions. At 4 weeks, cell survival for the different conditions was similar for both collagen and fibrin ECM. Average survival in scaffolds at 4 weeks with fibrin ECM was 24±3%, and 21±4% in the collagen ECM. Overall survival of the muscle cells was 66±6% at 2 weeks and 22±2% at 4 weeks.
Fig 4. Cell survival as a function of ECM and scaffold composition.
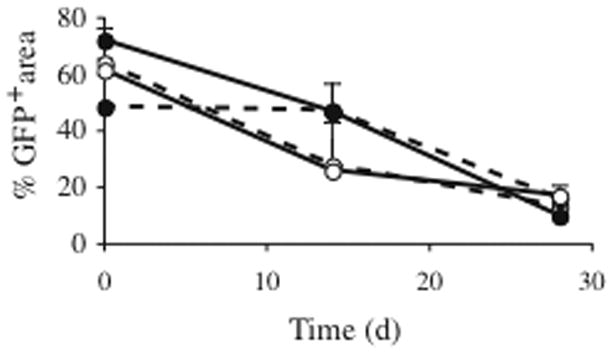
Scaffolds (4.2 mm diameter) composed of either 100 % microspheres (solid lines) or 50 % microspheres (dotted lines) and seeded with 1.5 million human GFP human myoblasts were implanted subcutaneously in the back of NOD/SCID mice. Cells were embedded in a fibrin ECM (open circles) or collagen ECM (filled circles). Datapoints represent GFP area as quantified by confocal analysis pre-implant and after 2 or 4 weeks in vivo. At 2 weeks in vivo, myofibers embedded in a collagen ECM on average covered more area than myofibers in the fibrin ECM; however this difference was not statistically significant. At 4 weeks, GFP area for all conditions was 10–17% (22±2% survival when area was normalized to preimplant GFP area), with no statistical differences for the tested conditions.
Degradation of PLG
Beginning 2 weeks after scaffold implantation in vivo, a transparent material was observed covering or attached to parts of the PLG scaffolds. This material was observed independently of microsphere content or use of fibrin or collagen ECM and increased with time in vivo. Since the material was also observed on scaffolds not containing cells (Fig 5A), it cannot be attributed to the implanted myofibers. The material appeared to have a crystalline structure by light microscopy (Fig 5B–C). Interestingly, the size of these microscopic structures was similar to the size of the NaCl crystals which were used to create the pores of the scaffolds (250–425 μm). The clear film did not dissolve in chloroform, a solvent for PLG, and was also resistant to collagenase treatment. Its impact on muscle cell survival is unknown.
Fig 5. Degradation products.
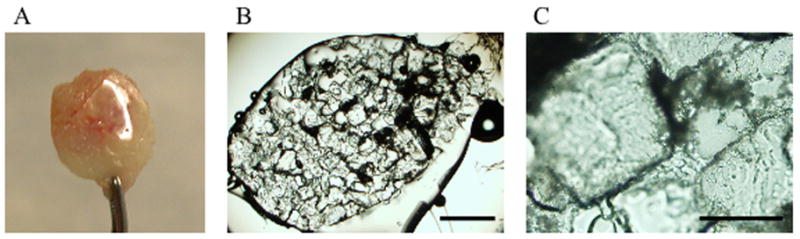
(A) PLG scaffold prefilled with 0.4 mg/ml collagen ECM, not containing cells was implanted subcutaneously in a NOD/SCID mouse for 47 days. A transparent product covers part of the scaffold. (B) Brightfield microscopic examination revealed a crystalline structure. Scalebar represents 1 mm. (C) Detail of the crystalline structure in the transparent film. Scalebar represent 250 μm.
Effect of NK cell depletion
NOD/SCID mice are deficient in T and B cell function, complement deficient and have low levels of natural killer (NK) cells [25]. We examined whether these residual NK cells contributed to the decreased cell survival in vivo. All mice were initially tested for leakiness by immunoglobulin G ELISA and found to have IgG levels < 0.5 μg/ml plasma, whereas C57/Bl6 controls had IgG levels >5000 μg/ml (data not shown). Seven mm diameter PLG scaffolds containing 100 % microspheres were seeded with 4 million GFP myoblasts in a fibrin ECM and after differentiation implanted subcutaneously in NOD/SCID animals. Half of the animals received intravenous (iv) infusions of anti-ASGM1 antibody, effectively depleting the residual NK cells as determined by a cytotoxicity test (data not shown), whereas the control group received isotype serum injections. GFP area on the scaffolds was quantified by confocal microscopy pre-implant and after 2 or 4 weeks in vivo (Fig. 6A). No significant difference in myofiber survival was apparent at week 2 (NK depleted: 57±5%; control: 55±3%), but at 4 weeks, myofiber survival in the NK depleted group was significantly higher than control: 34±2% in the NK depleted group versus 22±2% in the control group (p<0.05).
Fig 6. Effect of NK cell depletion on cell viability.
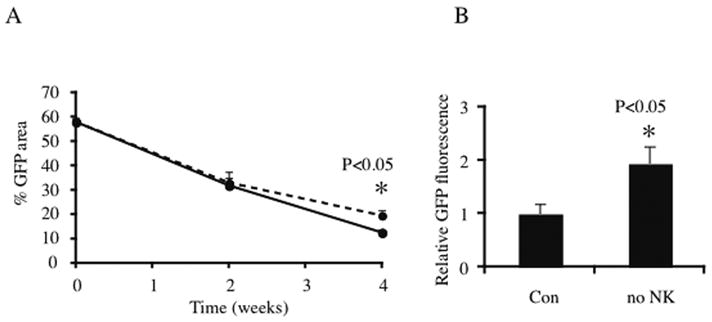
100 % microsphere, 7mm diameter PLG scaffolds were seeded with 4 million GFP myoblasts in a fibrin ECM and implanted subcutaneously in NOD/SCID animals. (A) GFP area on the scaffolds was quantified by confocal microscopy pre-implant and after 2 or 4 weeks in vivo. In the NK cell depleted group (dotted line), animals received intravenous injections of anti-ASGM1 antibody, while control group (solid line) received an isotype serum control. No significant difference was apparent at week 2, but at week 4, myofiber survival in the NK depleted group was significantly (p<0.05) higher than control. (B) Scaffolds in the NK depleted animals contained 2-fold as much GFP protein compared to the control group (p<0.05) after 4 weeks in vivo.
To confirm these results, total protein was extracted from the scaffolds at week 4 and GFP fluorescence from the extracts was assessed by fluorimetric analysis. Data were normalized to the fluorescence from scaffolds in the control group. Relative GFP content of the scaffolds in the NK depleted animals was 2 fold higher than controls (p<0.05, Fig 6B).
Discussion
In this study, we investigated the potential of biodegradable PLG gas-foamed scaffolds as structures for skeletal myoblast seeding, differentiation and in vivo myofiber survival. Since PLG with high lactic acid content (85:15) exhibits superior mechanical properties [26], this copolymer was chosen to ensure the scaffold would remain intact for the duration of the in vivo studies. The matrix of the scaffolds was formed by foaming PLG with NaCl in CO2 to obtain a highly porous (94.6±0.9 %) structure with largely open, interconnected pores [18, 26, 27]. Pore size of the PLG scaffolds was 360±85 μm [18], which allows for an even distribution of seeded cells, infiltration by host cells and vascularization [28]. These scaffolds were described to be suitable for cell adherence, proliferation and formation of new three-dimensional tissues [16, 18].
For gene therapy applications, skeletal muscle myofibers have a very strong protein production capacity, are non-dividing and non-migratory and thus have a favorable safety perspective compared to proliferating migratory myogenic cells since the attached cells would be retrievable if an adverse event occurs [6, 10, 22]. For clinical use, the components used for implantation need to be FDA approved. PLG has been used in the clinic for more than 3 decades in biodegradable sutures and has more recently also been used for fracture fixation devices and drug delivery systems. It has good biocompatibility and implantation profiles in bone or soft tissues, causing none or only a mild inflammatory response which dimishes with time [29]. To retain cells in or on scaffolds an extracellular matrix carrier such as Matrigel™ is often being used. Tisseel® and Zyderm® are 2 FDA approved ECM products used commonly as a tissue glue and bulking agent, respectively, and were used in this study.
Human myoblasts could be efficiently seeded on PLG scaffolds, and both the fibrin or collagen ECM could be used to retain the cells on the scaffold. Myoblasts could subsequently be differentiated into postmitotic myofibers. When fibrin was employed as an ECM, myoblasts did not penetrate deeply into the scaffold pores due to the rapid solidification of the fibrin ECM (less than 90 seconds). In contrast, the gelling time of collagen ECM was significantly longer (10–30 minutes) and resulted in a penetration of cells deeper into the pores of the scaffolds, and seeping out of the cell-ECM mixture. Subsequently, scaffolds were prefilled with collagen ECM to limit cell-ECM penetration into the scaffolds and cell-ECM loss during the seeding process. Using these procedures, myofibers formed upon incubation in a differentiation medium, and were randomly oriented in a well defined layer on top of the scaffold, with good myofiber retention in vitro for at least four weeks. The gas foamed PLG scaffold thus appears to be an adequate method for transferring differentiated myofibers under tension for in vivo studies.
Following implantation of the PLG scaffolds subcutaneously in the NOD-SCID mice for several weeks, we observed a clear plastic-like product around the implants, which was seen when either collagen or fibrin ECM was used. To our knowledge, such material has not been reported before. Generation of this substance, which consists of small adjacent crystals, is not attributable to myofibers, since it also appeared when scaffolds were implanted without cells. PLG undergoes hydrolysis to produce the original monomers, lactic acid and glycolic acid, which are by-products of various metabolic pathways in the body. Polymer impurity is a possible explanation for the occurence of this PLG degradation product. A much lower incidence of late-stage foreign body reactions has been observed with purified melt processed PLA (polylactide) than with bulk PLA, which may have contained a relatively high level of residual lactide [30]. Partial recrystallization of lactide monomers may have led to formation of this material on the surface of the implants. Poly-l-lactic acid polymer has a crystallinity which is in agreement with our observations. It has also been suggested that the occurrence of inflammatory reactions may depend on the anatomical region, the capacity of the tissue to clear the degradation products, and the volume of polymer implanted [31]. The contribution of this material on myofiber loss from the scaffold with time in vivo as seen in the present study is unknown.
In a previous study, human myofibers were embedded only in a collagen ECM and implanted subcutaneously in NOD/SCID mice without tension [9]. Cell viability decreased 91±4 % over 4 weeks, and could not be improved by delivery of VEGF165 to stimulate vascularization of the implant site [9]. It was hypothesized that loss of cell viability may be partly explained by the lack of tension on the myofibers. Since human myofibers could be kept under tension on PLG scaffolds in vivo we were able to study the effect of tensioning on cell survival. Overall cell survival of the tensioned myofibers was 66±6% at 2 weeks and 22±2% at 4 weeks, which is 2 fold higher than untensioned myofibers as reported previously [9]. Using either fibrin or collagen type I as the ECM, or varying the percentage of PLG microspheres did not affect this cell viability.
NOD/SCID mice have become a standard model for in vivo evaluation of human hematopoietic stem cell function [32]. They are not only deficient in T and B cells, but also exhibit macrophage dysfunction and an absence of circulating complement [25]. NOD/SCID mice are often referred to as immunodeficient, yet there is still a low level of natural killer cells present and active, which may be involved in rejection of xenogenic cell transplants [33–36]. Depletion of NK activity by antibody treatment [36] or use of β2-microglobulin-deficient NOD/SCID mice [35] was demonstrated to improve engraftment of human cells. Antibody depletion of NK cells led to 50% higher muscle cell survival at 4 weeks after implantation. Even with NK depletion, nearly 70% of muscle cells died by 4 weeks in vivo. Thus, other components of the innate immune system may still affect the survival of the xenogeneic human cells. When human mesenchymal stem cells were implanted on poly(ε-caprolactone) in a NOD/SCID model, host macrophages along with neutrophils and mast cells slowly invaded the implants and the majority of the implanted cells were eliminated by 2 weeks [33]. We thus cannot exclude that cell loss is partly due to other innate immune functions, such as macrophage activity. Macrophages have been detected in the spleens of NOD/SCID mice, however they are incapable of secreting IL-1 in response to stimulation with lipopolysaccharides and therefore are thought to be functionally immature [25, 37]. Use of other immunodeficient animal models may enhance xenogeneic cell survival. Gamma c−/RAG2− [38] and NOD/SCID/gamma c− mice [39] are completely T, B and NK cell negative and have been found to improve survival of implanted human myoblasts compared to SCID mice [40]. Nevertheless it was observed that human hematopoietic stem cells still needed exogenous cytokine administration in these animals for an enhanced engraftment [41].
An alternative hypothesis for poor cell survival in the xenogeneic setting may be a lack of one or more local growth/survival factors. Murine (growth factor) proteins may not be efficiently recognized by human cell receptors and therefore may contribute to impaired growth/survival of the xenogeneic cells. The use of syngeneic (or autologous for use in human patients) cells is warranted for optimal cell survival and avoiding immune responses. Indeed, implanting syngeneic myofibers in the NOD/SCID animal model resulted in much better cell survival, even in the absence of tension [9]. Delivery of recombinant growth factors at defined times after cell implantation may be beneficial to further increase cell viability and protein secretion capacity. Anabolic growth factors such as insulin-like growth factor 1 (IGF-1) [9, 42] or growth hormone (GH) [6] can stimulate myofiber hypertrophy or attenuate atrophy. The use of angiogenic molecules such as VEGF may be beneficial to rapidly induce a functional blood vessel supply to the transplanted myofibers [8, 9]. Delivery of multiple growth factors can be precisely controlled by incorporation of PLG microspheres containing growth factors in the scaffolds [15, 26, 43]. HGF and FGF2, which have been used to stimulate myoblast migration in other studies [12] may not be optimal for the post-mitotic muscle fibers. In a therapeutic approach where genetically modified myofibers attached to a scaffold are used for long-term protein secretion, cell migration would be an undesirable feature since retrievability of genetically engineered tissue in case of an adverse response is one of the key advantages of using non-migratory myofibers.
Survival of myofibers on the PLG scaffolds compares favorably to use of other biomaterials for in vivo muscle cell survival. Alginate hydrogels containing cell adhesion peptides had a murine myoblast survival of 16% over a 24 hour time period [11]. Optimal scaffold design with sustained delivery of HGF and FGF2 increased viability to 40% at 96 hours. Cell viability on PLG scaffolds as described in this paper compares favorably with the alginate scaffolds, even without growth factor integration.
Conclusions
Biodegradable polymer scaffolds based on PLG provide a reliable method for the successful transfer of ex vivo differentiated postmitotic human muscle fibers in vivo. Using these scaffolds, myofibers remain under tension, doubling their long-term viability. Therefore, these PLG scaffolds may be important for future use in human muscle repair or gene therapy applications.
Acknowledgments
The authors would like to thank Claudia Fishbach and Will Yuen for help with the scaffold preparation and Gilbert DiLeone for assistance with the NK assay. This research was funded through a Fellowship from the Belgian American Educational Foundation (LT), a NIH Ruth L. Kirschstein Senior Fellowship (HV), The Miriam Hospital, EUFP6 INTHER LSHB-CT-2005018961 and Fund for Scientific Research, Flanders (FWO). The authors confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Appendix : Abbreviations
ASGM1: anti-asialo GM1
DAB: 3,3′-diaminobenzidene
ECM: extracellular matrix
ELISA: enzyme linked immunosorbent assay
FGF2: fibroblast growth factor 2
GFP: green fluorescent protein
GH: growth hormone
HGF: hepatocyte growth factor
IGF-1: insulin-like growth factor 1
IgG: immunoglobulin
MGM: myoblast growth medium
NK: natural killer
NOD/SCID: non-obese diabetic/severe combined immunodeficiency
PLA: polylactide
PLG: poly-lactide-co-glycolide
RIPA: radioimmunoprecipitation assay
SEM: standard error of the mean
VEGF: vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blau HM, Springer ML. Muscle-Mediated Gene-Therapy. New England Journal of Medicine. 1995 DEC 7;333(23):1554–1556. doi: 10.1056/NEJM199512073332308. [DOI] [PubMed] [Google Scholar]
- 2.Skuk D, Caron NJ, Goulet M, Roy B, Tremblay JP. Resetting the problem of cell death following muscle-derived cell transplantation: detection, dynamics and mechanisms. J Neuropathol Exp Neurol. 2003 Sep;62(9):951–967. doi: 10.1093/jnen/62.9.951. [DOI] [PubMed] [Google Scholar]
- 3.Beauchamp JR, Morgan JE, Pagel CN, Partridge TA. Dynamics of myoblast transplantation reveal a discrete minority of precursors with stem cell-like properties as the myogenic source. J Cell Biol. 1999 Mar 22;144(6):1113–1122. doi: 10.1083/jcb.144.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qu Z, Balkir L, van Deutekom JC, Robbins PD, Pruchnic R, Huard J. Development of approaches to improve cell survival in myoblast transfer therapy. J Cell Biol. 1998 Sep 7;142(5):1257–1267. doi: 10.1083/jcb.142.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosnik P, Dennis JE, Vandenburgh H. Tissue engineering skeletal muscle. In: Guilah F, editor. Functional Tissue Engineering. New York: Springer; 2003. pp. 377–392. [Google Scholar]
- 6.Vandenburgh H, Del Tatto M, Shansky J, Goldstein L, Russell K, Genes N, et al. Attenuation of skeletal muscle wasting with recombinant human growth hormone secreted from a tissue-engineered bioartificial muscle. Hum Gene Ther. 1998 Nov 20;9(17):2555–2564. doi: 10.1089/hum.1998.9.17-2555. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Shansky J, Del Tatto M, Ferland P, McGuire S, Marszalkowski J, et al. Therapeutic potential of implanted tissue-engineered bioartificial muscles delivering recombinant proteins to the sheep heart. Ann N Y Acad Sci. 2002 Jun;961:78–82. doi: 10.1111/j.1749-6632.2002.tb03055.x. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y, Shansky J, Del Tatto M, Ferland P, Wang X, Vandenburgh H. Recombinant vascular endothelial growth factor secreted from tissue-engineered bioartificial muscles promotes localized angiogenesis. Circulation. 2001 Jul 31;104(5):594–599. doi: 10.1161/hc3101.092215. [DOI] [PubMed] [Google Scholar]
- 9.Thorrez L, Vandenburgh H, Callewaert N, Mertens N, Shansky J, Wang L, et al. Angiogenesis enhances factor ix delivery and persistence from retrievable human bioengineered muscle implants. Mol Ther. 2006 Sep;14(3):442–451. doi: 10.1016/j.ymthe.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 10.Vandenburgh H, Del Tatto M, Shansky J, Lemaire J, Chang A, Payumo F, et al. Tissue-engineered skeletal muscle organoids for reversible gene therapy. Hum Gene Ther. 1996 Nov 10;7(17):2195–2200. doi: 10.1089/hum.1996.7.17-2195. [DOI] [PubMed] [Google Scholar]
- 11.Hill E, Boontheekul T, Mooney DJ. Designing scaffolds to enhance transplanted myoblast survival and migration. Tissue Eng. 2006 May;12(5):1295–1304. doi: 10.1089/ten.2006.12.1295. [DOI] [PubMed] [Google Scholar]
- 12.Hill E, Boontheekul T, Mooney DJ. Regulating activation of transplanted cells controls tissue regeneration. Proc Natl Acad Sci U S A. 2006 Feb 21;103(8):2494–2499. doi: 10.1073/pnas.0506004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riboldi SA, Sampaolesi M, Neuenschwander P, Cossu G, Mantero S. Electrospun degradable polyesterurethane membranes: potential scaffolds for skeletal muscle tissue engineering. Biomaterials. 2005 Aug;26(22):4606–4615. doi: 10.1016/j.biomaterials.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 14.Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, et al. Engineering vascularized skeletal muscle tissue. Nat Biotechnol. 2005 Jul;23(7):879–884. doi: 10.1038/nbt1109. [DOI] [PubMed] [Google Scholar]
- 15.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Polymeric system for dual growth factor delivery. Nat Biotechnol. 2001 Nov;19(11):1029–1034. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 16.Smith MK, Peters MC, Richardson TP, Garbern JC, Mooney DJ. Locally enhanced angiogenesis promotes transplanted cell survival. Tissue Eng. 2004 Jan–Feb;10(1–2):63–71. doi: 10.1089/107632704322791709. [DOI] [PubMed] [Google Scholar]
- 17.Lee KY, Peters MC, Anderson KW, Mooney DJ. Controlled growth factor release from synthetic extracellular matrices. Nature. 2000 Dec 21–28;408(6815):998–1000. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 18.Harris LD, Kim BS, Mooney DJ. Open pore biodegradable matrices formed with gas foaming. J Biomed Mater Res. 1998 Dec 5;42(3):396–402. doi: 10.1002/(sici)1097-4636(19981205)42:3<396::aid-jbm7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 19.Mooney DJ, Baldwin DF, Suh NP, Vacanti JP, Langer R. Novel approach to fabricate porous sponges of poly(D, L-lactic-co-glycolic acid) without the use of organic solvents. Biomaterials. 1996 Jul;17(14):1417–1422. doi: 10.1016/0142-9612(96)87284-x. [DOI] [PubMed] [Google Scholar]
- 20.Cohen S, Yoshioka T, Lucarelli M, Hwang LH, Langer R. Controlled delivery systems for proteins based on poly(lactic/glycolic acid) microspheres. Pharm Res. 1991 Jun;8(6):713–720. doi: 10.1023/a:1015841715384. [DOI] [PubMed] [Google Scholar]
- 21.Hennessey JV, Chromiak JA, Della Ventura S, Guertin J, MacLean DB. Increase in percutaneous muscle biopsy yield with a suction-enhancement technique. J Appl Physiol. 1997 Jun;82(6):1739–1742. doi: 10.1152/jappl.1997.82.6.1739. [DOI] [PubMed] [Google Scholar]
- 22.Powell C, Shansky J, Del Tatto M, Forman DE, Hennessey J, Sullivan K, et al. Tissue-engineered human bioartificial muscles expressing a foreign recombinant protein for gene therapy. Hum Gene Ther. 1999 Mar 1;10(4):565–577. doi: 10.1089/10430349950018643. [DOI] [PubMed] [Google Scholar]
- 23.Yamanokuchi S, Ikai I, Nishitai R, Matsushita T, Sugimoto S, Shiotani T, et al. Asialo GM1 positive CD8+ T cells induce skin allograft rejection in the absence of the secondary lymphoid organs. J Surg Res. 2005 Nov;129(1):57–63. doi: 10.1016/j.jss.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Fast LD. Identification of a single non-H-2 gene regulating graft-versus-host disease response. J Immunol. 1990 Jun 1;144(11):4177–4182. [PubMed] [Google Scholar]
- 25.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154(1):180–191. [PubMed] [Google Scholar]
- 26.Sheridan MH, Shea LD, Peters MC, Mooney DJ. Bioabsorbable polymer scaffolds for tissue engineering capable of sustained growth factor delivery. J Control Release. 2000 Feb 14;64(1–3):91–102. doi: 10.1016/s0168-3659(99)00138-8. [DOI] [PubMed] [Google Scholar]
- 27.Mooney DJ, Sano K, Kaufmann PM, Majahod K, Schloo B, Vacanti JP, et al. Long-term engraftment of hepatocytes transplanted on biodegradable polymer sponges. J Biomed Mater Res. 1997 Dec 5;37(3):413–420. doi: 10.1002/(sici)1097-4636(19971205)37:3<413::aid-jbm12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 28.Peters MC, Polverini PJ, Mooney DJ. Engineering vascular networks in porous polymer matrices. J Biomed Mater Res. 2002 Jun 15;60(4):668–678. doi: 10.1002/jbm.10134. [DOI] [PubMed] [Google Scholar]
- 29.Avgoustakis K. In: Polylactic-Co-Glycolic Acid (PLGA) Bowlin GL, editor. Encyclopedia of Biomaterials and Biomedical Engineering; Dekker: 2005. [Google Scholar]
- 30.Perrin DE, English JP. Polyglycolide and polylactide. In: Domb AJ, Kost J, Wiseman DM, editors. Handbook of Biodegradable Polymers. Amsterdam: Harwood Academic Publishers; 1997. pp. 3–27. [Google Scholar]
- 31.Athanasiou KA, Niederauer GG, Agrawal CM. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials. 1996 Jan;17(2):93–102. doi: 10.1016/0142-9612(96)85754-1. [DOI] [PubMed] [Google Scholar]
- 32.Greiner DL, Hesselton RA, Shultz LD. SCID mouse models of human stem cell engraftment. Stem Cells. 1998;16(3):166–177. doi: 10.1002/stem.160166. [DOI] [PubMed] [Google Scholar]
- 33.Xia Z, Ye H, Choong C, Ferguson DJ, Platt N, Cui Z, et al. Macrophagic response to human mesenchymal stem cell and poly(epsilon-caprolactone) implantation in nonobese diabetic/severe combined immunodeficient mice. J Biomed Mater Res A. 2004 Dec 1;71(3):538–548. doi: 10.1002/jbm.a.30185. [DOI] [PubMed] [Google Scholar]
- 34.Lin Y, Chen Y, Zeng Y, Wang T, Zeng S. Lymphocyte phenotyping and NK cell activity analysis in pregnant NOD/SCID mice. J Reprod Immunol. 2005 Dec;68(1–2):39–51. doi: 10.1016/j.jri.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Christianson SW, Greiner DL, Hesselton RA, Leif JH, Wagar EJ, Schweitzer IB, et al. Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J Immunol. 1997 Apr 15;158(8):3578–3586. [PubMed] [Google Scholar]
- 36.Yoshino H, Ueda T, Kawahata M, Kobayashi K, Ebihara Y, Manabe A, et al. Natural killer cell depletion by anti-asialo GM1 antiserum treatment enhances human hematopoietic stem cell engraftment in NOD/Shi-scid mice. Bone Marrow Transplant. 2000 Dec;26(11):1211–1216. doi: 10.1038/sj.bmt.1702702. [DOI] [PubMed] [Google Scholar]
- 37.Serreze DV, Gaedeke JW, Leiter EH. Hematopoietic stem-cell defects underlying abnormal macrophage development and maturation in NOD/Lt mice: defective regulation of cytokine receptors and protein kinase C. Proc Natl Acad Sci U S A. 1993 Oct 15;90(20):9625–9629. doi: 10.1073/pnas.90.20.9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldman JP, Blundell MP, Lopes L, Kinnon C, Di Santo JP, Thrasher AJ. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br J Haematol. 1998 Nov;103(2):335–342. doi: 10.1046/j.1365-2141.1998.00980.x. [DOI] [PubMed] [Google Scholar]
- 39.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002 Nov 1;100(9):3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 40.Cooper RN, Irintchev A, Di Santo JP, Zweyer M, Morgan JE, Partridge TA, et al. A new immunodeficient mouse model for human myoblast transplantation. Hum Gene Ther. 2001 May 1;12(7):823–831. doi: 10.1089/104303401750148784. [DOI] [PubMed] [Google Scholar]
- 41.Mazurier F, Fontanellas A, Salesse S, Taine L, Landriau S, Moreau-Gaudry F, et al. A novel immunodeficient mouse model--RAG2 x common cytokine receptor gamma chain double mutants--requiring exogenous cytokine administration for human hematopoietic stem cell engraftment. J Interferon Cytokine Res. 1999 May;19(5):533–541. doi: 10.1089/107999099313983. [DOI] [PubMed] [Google Scholar]
- 42.Shansky J, Creswick B, Lee P, Wang X, Vandenburgh H. Paracrine release of insulin-like growth factor 1 from a bioengineered tissue stimulates skeletal muscle growth in vitro. Tissue Eng. 2006 Jul;12(7):1833–1841. doi: 10.1089/ten.2006.12.1833. [DOI] [PubMed] [Google Scholar]
- 43.Ennett AB, Kaigler D, Mooney DJ. Temporally regulated delivery of VEGF in vitro and in vivo. J Biomed Mater Res A. 2006 Oct;79(1):176–184. doi: 10.1002/jbm.a.30771. [DOI] [PubMed] [Google Scholar]