

Abstract
Arrestins and G proteins-coupled receptor kinases (GRKs) regulate signaling and trafficking of G protein-coupled receptors. We investigated changes in the expression of arrestins and GRKs in the striatum of patients with Parkinson's disease without (PD) or with dementia (PDD) at post mortem using Western blotting and ribonuclease protection assay. Both PD and PDD groups had similar degree of dopamine depletion in all striatal regions. Arrestin proteins and mRNAs were increased in the PDD group throughout striatum. Protein and mRNA of GRK5, the major subtype in the human striatum, and GRK3 were also upregulated, whereas GRK2 and 6 were mostly unchanged. The PD group had lower concentration of arrestins and GRKs than the PDD group. There was no statistical link between the load of Alzheimer's pathology and the expression of these signaling proteins. Upregulation of arrestins and GRK in PDD may confer resistance to the therapeutic effects of levodopa often observed in these patients. In addition, increased arrestin and GRK concentrations may lead to dementia via perturbation of multiple signaling mechanisms.
Keywords: arrestin, G proteins-coupled receptor kinases, Parkinson's disease, Parkinson's disease with dementia, signaling mechanisms
1. Introduction
Parkinson's disease (PD) is an age-related neurodegenerative disorder caused by degeneration of dopaminergic neurons of the substantia nigra that provide dopamine (DA) to the brain, resulting in the depletion of DA in the striatum, the main recipient of dopaminergic input from the substantia nigra. Neurobiological mechanisms responsible for motor symptoms produced by the degeneration of dopaminergic neurons in PD are poorly understood. It is generally believed that dysregulation of DA receptor signaling in the basal ganglia plays a role in generating motor deficits [30,50,76,96], but the underlying molecular events are not known.
DA receptors belong to the G protein-coupled receptor (GPCR) superfamily. Upon persistent stimulation many GPCRs undergo desensitization via a two-step process: activation-dependent receptor phosphorylation by G protein-coupled receptor kinases (GRK) followed by the binding of arrestins that precludes further signaling via G proteins by shielding the cytoplasmic surface of receptors [59]. Arrestins promote receptor internalization via interaction with the internalization machinery of coated pits (reviewed in [34,36,61]). Internalized receptor can either be recycled back to the plasma membrane (resensitization) or degraded (receptor down-regulation). In addition to being key players in the GPCR desensitization and trafficking, arrestins are multifunctional signaling molecules translating GPCR activation into modulation of G protein-independent signaling pathways [8,9,62] and regulating the expression of specific genes [52]. Two arrestin subtypes, arrestin2 and arrestin3, are ubiquitous [6,13,31,32]. Five out of seven GRK subtypes, GRK2, 3, 4, 5, and 6, are widely expressed in the brain with largely overlapping cellular distribution [5,10,13,32,89]. In vitro data demonstrate that most GPCR subtypes can be phosphorylated by several GRKs [71,94] and bind both arrestins equally well [33,59,94]. Conversely, in vivo studies suggest that receptors may be preferentially phosphorylated by specific GRKs [26,44,45,57,95] and interact with specific arrestins [58,78]. Importantly, GPCRs phosphorylated by different GRKs may signal differently [54,92]. Thus, signaling via GPCRs may be differentially regulated by the changes in the cellular complement of arrestins and GRKs. Dysregulation of DA receptors following dopaminergic degeneration in PD may be linked to defects in their desensitization, trafficking, and signaling brought about by the alterations in the expression of specific arrestins and/or GRKs.
Approximately 30−40% of PD patients develop dementia as the disease progresses [24 and references therein]. Neuropathological basis of dementia in the idiopathic PD remains controversial. Many PD patients have substantial load of Alzheimer's histopathological features [15,17,48,49]. There is evidence that dementia in PD is associated more closely with cortical deposition of the hallmark component of Lewy bodies, α-synuclein, than with Alzheimer's pathology [2,43,68], although this conclusion is disputed by some authors [82]. A strong correlation between frequency of plaques and neurofibrillary tangles with neocortical Lewy bodies has been reported [4], suggesting common origin and/or cooperativity between the two types of pathology. Although dementia in idiopathic PD is very important for the disease's prognosis and management [23,49,88], underlying neurochemical mechanisms remain largely unexplored. Dementia associated with PD is resistant to L-DOPA treatment [88]. Importantly, dementia in PD strongly correlates with declining antiparkinsonian response to dopaminergic medication including direct DA agonists [4,16,51], which may reflect specific changes in DA receptor signaling that hamper their responsiveness. It is conceivable that specific alterations in mechanisms governing signaling via DA receptors and/or other GPCRs brought about by concomitant dementia pathology may be responsible. The GPCR regulation machinery is positioned at the intersection of important signaling pathways and is shared by many GPCRs. Therefore changes in availability and/or cellular complement of arrestins and/or GRKs induced by the activity of specific GPCRs may affect multiple GPCRs and signaling pathways, thereby propagating the pathology.
Here we report profound changes in the concentrations of arrestins and GRKs at post mortem in the basal ganglia of patients with PD and dementia as opposed to control and patients with PD only. Arrestin binding to GRK-phosphorylated GPCRs initiates second round of signaling via multiple mechanisms. Therefore we investigated changes in expression of other signaling molecules that may be affected by the modifications in arrestin/GRK expression.
2. Materials and Methods
2.1 Post mortem samples
Human brain tissue from 16 control and 21 PD patients was used for these studies. Postmortem brains were obtained via Sun Health Research Institute Brain Donor Program in Sun City, AZ. This is a community-based program located in a retirement community with high incidence of PD The staff of the Brain Donor Program keeps in contact with people signed up for the Program and is regularly informed by donors or family members about changes in the potential donor's health and family situation. Most live nearby in assisted living houses (with attending nurses) or retirement homes. Significant proportion of the donors is hospitalized in Boswell Hospital, where all autopsies for the Program are performed, or in the nearby Del E. Webb Hospital until their death. The staff of all medical and retirement establishments in the area is aware of the Program, and promptly notifies the Program's personnel when a death occurs. The Program keeps the autopsy team on stand-by at all times. Therefore, the autopsy is regularly performed within 2−3 hours post-mortem. Following the autopsy, the brains are cut coronally into 1-cm-thick slabs, rapidly frozen on dry ice, and stored at −80°C until used.
This study included all cases with appropriate diagnoses collected from the end of 1997 to the beginning of 2000, the approach that eliminates the selection bias. Clinical diagnosis of PD was made based on standard clinical criteria [28] and included a positive response to levodopa (L-DOPA) medication. Comprehensive medical records for all cases have been obtained and reviewed at the Institute. In most cases, the authors or staff of the Brain donor program contacted attending neurologists to collect additional information or verify clinical diagnosis. The clinical diagnosis of dementia was similarly determined and recorded. Of the 21 cases with the primary diagnosis of PD, 8 (38%) had no additional clinical diagnosis, whereas 13 were diagnosed with dementia (Table 1).
Table 1.
Record of patient characteristics
| Cases | Sex | Age, years | PMI, hours | Cause of death; additional diagnosis | Braak score | CERAD score | |
|---|---|---|---|---|---|---|---|
| Control | |||||||
| Co001 | M | 84 | 3 | Cardiac and/or respiratory failure | − | III | C |
| Co002 | F | 87 | 4 | Gastrointestinal hemorrage | − | III | 0 |
| Co003 | M | 90 | 2.5 | Cardiac and/or respiratory failure, acute renal failure | − | III | B |
| Co004 | M | 68 | 2 | Cardiac and/or respiratory failure; pneumonia | − | I | 0 |
| Co005 | M | 63 | 1.5 | Acute intracerebral hemorrage | − | II | 0 |
| Co006 | M | 83 | 3 | Cardiac and/or respiratory failure | − | III | 0 |
| Co007 | F | 82 | 2 | Lung cancer | − | II | A |
| Co008 | F | 73 | 1.5 | Ovarian cancer | − | I | 0 |
| Co009 | M | 78 | 2.25 | Post-operative colon resection for colon cancer | − | III | B |
| Co010 | F | 70 | 2 | Cardiac and/or respiratory failure; breast cancer | − | I | A |
| Co011 | M | 77 | 2.5 | Respiratory failure due to neck and head cancer | − | II | A |
| Co012 | M | 86 | 2.5 | Congestive heart failure, ischemic cardiomyopathy | − | II | 0 |
| Co013 | M | 91 | 3.5 | Heart failure; valvular heart disease | − | III | B |
| Co014 | F | 85 | 2.5 | Cardiac and/or respiratory failure; Cardiovascular accident | − | III | 0 |
| Co015 | M | 84 | 2.5 | Cardiac and/or respiratory failure | − | III | B |
| Co016 | M | 73 | 2 | Cardiac and/or respiratory failure; carcinoma of lung | − | III | 0 |
| N=16 | 11M+5F | 79.6±2.11 | 2.4±0.2 | ||||
| PD2 | Disease Duration, years | ||||||
| PD001 | M | 80 | 13 | End-stage Parkinson's disease | 8 | IV | A |
| PD002 | M | 81 | 2.5 | Small cell carcinoma of lung | 8 | I | 0 |
| PD003 | M | 77 | 4 | Cardiac and/or respiratory failure; severe myocardial infarction | 7.5 | III | A |
| PD004 | M | 64 | 3.75 | Complications of lung cancer | 5 | II | 0 |
| PD005 | F | 77 | 2.7 | End-stage Parkinson's disease | 21 | II | 0 |
| PD006 | F | 83 | 12 | Cardiovascular accident; Parkinson's disease | 21 | III | A |
| PD007 | M | 75 | 13.75 | Cardiac arrest | 34 | III | A |
| PD008 | F | 82 | 2.75 | Cardiopulmonary arrest | 16 | IV | B |
| N=8 | 5M+3F | 77.4±2.1 | 6.8±1.83* | 15.1±3.5 | |||
| PDD4 | |||||||
| PDD01 | M | 88 | 3.75 | Vascular dementia; multi-infarct dementia | 3.5 | I | B |
| PDD02 | M | 77 | 4 | End-stage Parkinson's disease; dehydration | 9 | IV | C |
| PDD03 | F | 86 | 3.5 | End-stage Parkinson's disease; failure to thrive | 10.5 | IV | B |
| PDD04 | F | 83 | 2.5 | End-stage Parkinson's disease | 8 | IV | C |
| PDD05 | F | 84 | 2 | End-stage Parkinson's disease | 13 | IV | B |
| PDD06 | M | 76 | 3.25 | End-stage Parkinson's disease; | 17 | IV | B |
| PDD07 | M | 89 | 3.25 | Pneumonia | 6 | III | A |
| PDD08 | F | 78 | 1.75 | Interstitial pneumonia | 23 | III | 0 |
| PDD09 | F | 84 | 2.5 | Failure to thrive secondary to Parkinson's disease | 7 | II | 0 |
| PDD10 | M | 73 | 4 | End stage Parkinson's disease | 17 | II | B |
| PDD11 | M | 85 | 2.75 | End-stage Parkinson's disease | 6 | III | A |
| PDD12 | M | 79 | 3 | End-stage Parkinson's disease | 11 | I | B |
| PDD13 | M | 64 | 4 | Failure to thrive secondary to Parkinson's disease | 15 | II | 0 |
| N=13 | 8M+5F | 80.5±1.9 | 3.1±0.2 | 11.3±1.5 |
Statistics for Age, postmortem interval (PMI), and disease duration are given as means±S.E.M.
PD — Parkinson's disease.
- p<0.05 as compared to Control and PDD groups according to one-way ANOVA with Group as main factor followed by Bonferroni/Dunn test.
PDD — Parkinson's disease with dementia.
The neuropathologic diagnosis procedure was described in detail elsewhere [51]. Briefly, fixed and frozen sections were cut at 40μm thickness and stained with hematoxylin and eosin, Luxol Fast Blue, or with α-synuclein antibodies to evaluate the presence of Lewy body-related neuritis. Additional sections were stained with Campbell-Switzer, Gallyas, and Tioflavine-S methods to estimate the abundance of senile plaques and neurofibrillary tangles [18]. Only cases with definite PD diagnosed according to published criteria [28] were included in the study. All cases (control and PD) were assigned a CERAD score for plaques [73] and a Braak score for tangles [18]. Cases with plaques and tangles were assigned a diagnostic probability for AD according to the recent NIA-Reagan Institute criteria [1]. Cases diagnosed with dementia with Lewy bodies were excluded from this study. Differential diagnosis of PD with dementia and dementia with Lewy bodies remains complicated, because at later stages clinical and neuropathological features of the two disorders are similar [93,102]. Dementia with Lewy bodies diagnosed based on published consensus criteria [70]. In particular, any case with PD preceding dementia for less than a year (the one year rule) was diagnosed as dementia with Lewy bodies and only the cases with PD preceding dementia for at least one year were classified as PD with dementia. Control cases were cognitively intact and have no clinical diagnosis of PD, AD, Lewy body dementia, or any other diagnostic category. Neuropathologically, control cases had low burden of Alzheimer's pathology: all have Braak scores ≤3 and CERAD score B or less (only one case had C) (Table 1). These data are in agreement with published results regarding Alzheimer's pathology in cognitively intact elderly [56]. Of 21 PD cases, 8 (38%) had no additional clinical diagnosis, whereas 13 (62%) were additionally diagnosed with dementia. All PD cases, including those with dementia, met criteria for definite PD [70] (Table 1). Five out of 13 cases in the group with dementia and one out of 8 cases of nondemented PD patients were also assigned intermediate probability of AD according to NIA-Reagan Institute criteria (Braak score III-IV; CERAD score B) [1], whereas remaining cases showed low load of Alzheimer's pathology (Table 1).
Pharmacological treatment received by PD patients varied substantially from individual to individual, but there was no consistent difference in the treatment pattern and/or doses between the PD and PD with dementia groups. All PD cases with or without dementia had been treated with levodopa/carbidopa (Sinemet® 25/100) and many - with DA agonists, most often with pramipexole (Mirapex®). Doses of Sinemet® (25/100 levodopa/carbidopa) varied from 0.5 to 6 tablets a day. Other, nondopaminergic, antiparkinsonian drugs had also been used such as tolcapone and amantadine. Psychotropic drugs used included benzodiazepines (temazepam, clonazepam, lorazepam), antidepressants, and antipsychotics (mostly clozapine). The patients, including control cases, had also been treated with non-psychotropic drugs for various ailments. There is no evidence that patients with PD and dementia had been treated with cholinesterase inhibitors.
2.2. Western Blotting
The brain areas of interest, caudate nucleus (CN) and putamen (Pu), and ventral striatum (VSTR; comprises nucleus accumbens and ventral-most part of the putamen) were outlined precisely on 100 μm-thick coronal sections, and the tissue was scraped into 200 μl of Lysis solution (Ambion, Austin, TX). The samples were weighed with submilligram precision to determine the weight of the added tissue, and then appropriate volume of Lysis solution was added to make 1:7 w/v homogenate. The Lysis solution effectively lyses the tissue while inhibiting enzymatic activity. The advantage of the procedure is that the same lysates can be used to measure protein and mRNA by RNAse protection assay (RPA). For Western blots, samples (7.5 mg tissue wet weight/ml) were prepared by methanol precipitation as described [13]. Electrophoresis and transfer onto Immobilon-P (Millipore, Bedford, MA, USA) membrane, and detection were performed essentially as described [13]. Arrestin2 and GRK2 were detected using the equivalent of 8.9 μg tissue per lane, arrestin3, GRK3, and GRK5 - 35.7, and GRK6 - 120 μg tissue per lane. Arrestins were detected with arrestin2- [75] (1:6,000) or arrestin3-specific [79] (1:700) affinity-purified rabbit polyclonal antibodies. We used rabbit polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) to quantify GRK2 (catalog # sc-562; 1:500), GRK3 (catalog # sc-563; 1:300), and GRK5 (catalog # sc-565; 1:500). For GRK6, mouse monoclonal antibody A16/17 (1:1,000) (Upstate Biotechnology, Waltham, MA) was used. To detect total ERK1, 2 (p44/42), Akt, and GSK3α, the antibodies from Cell Signaling Technology (Beverly, MA) were used at 1:1,000 dilution. Tyrosine hydroxylase was measured with rabbit antibody (1:6,000) and dopamine transporter – with rat monoclonal antibody (1:1,000; both from Chemicon International, Temecula, CA). Blots were incubated overnight at 4°C with appropriate primary antibodies followed by horseradish peroxidase-conjugated goat anti-rabbit or rabbit anti-mouse secondary antibodies (Jackson Immunoresearch Laboratories, West Grove, PA) at 1:25,000 dilution (1 h at RT) and SuperSignal enhanced chemiluminescence reagent WestPico (Pierce, Rockford, IL, USA). Upon development, the blots were exposed to X-ray film for appropriate periods of time. For quantification of arrestins, dilutions of standards containing 1:1 mix of Escherichia coli-expressed purified bovine arrestin2 and arrestin3 in sample buffer were loaded onto each gel along with samples, as described previously [13,31,32]. For quantification of GRKs, we used bovine GRK2 and GRK3 [53], human GRK5 [60] and GRK6 [65], purified as described in the given references. Appropriate dilutions of GRK proteins were loaded onto each gel alongside the samples to generate calibration curves [13,32]. For quantification of ERK, we used total MAPK protein standards supplied by the manufacturer. For other proteins, purified standards were not available. Therefore, we used serial dilutions of tissue samples to make sure that all samples are in the linear range and expressed the values in arbitrary units. Calibration curves generated with arrestin and GRK standards on each blot allowed for quantification of the proteins in absolute units (fmol/mg of total protein), which is necessary for comparison of expression levels of different proteins. Each sample was analyzed on 3−5 independent blots.
2.3. RNAse protection assay (RPA)
The brain tissue samples collected in Lysis solution were stored at −80°C until used. The probes for the RPA were labeled with [32P]UTP in a standard in vitro transcription reaction. Arrestin2 and arrestin3 were measured in the same hybridization reaction and all 4 GRKs – in another reaction. The probes for arrestin2 and arrestin3 were 212 and 172 bases long, respectively, and corresponded to bases 1025−1236 and 479−650 (GeneBank accession numbers NM_004041 and NM_004313, respectively). Both probes were designed to yield the same protected fragment with all known splice variants of respective arrestins [83,98]. The probe for GRK2 was 214 bp (1381−1594 nucleotides, accession #NM_001619), for GRK3 − 241 bp (315−555 nucleotides; accession # NM_005160), for GRK5 − 158 bp (602−760; accession #L15388), and for GRK6 − 186 bp (419−604; accession #NM_002082). RPA was performed essentially as described previously [31,32] using Direct Protect Kit (Ambion, Austin, TX, USA). The advantage of the direct protect procedure is that it requires no RNA isolation. Hybridization occurs directly in Lysis buffer used for tissue homogenization. The technique is very sensitive and reliable, since inevitable and often uneven loss of RNA during isolation is avoided. Due to its simplicity, the procedure is particularly suitable for processing a large number of samples. The probes were purified on 6% polyacrylamide gel, and the bands corresponding to full-length transcripts were excised and eluted by incubating the gel slices with Probe Elution Buffer overnight at 37°C. Purified probes (200,000 cpm/tube for each mRNA species) were hybridized with 45 μl of lysate (containing 5.625 mg tissue) overnight at 37°C. Upon completion of hybridization, the samples were subjected to digestion with RNAse A/T1, treated with proteinase K, precipitated with isopropanol, dried, and dissolved in gel loading buffer. Protected fragments were separated by electrophoresis on 0.75 mm (Bio-Rad, Hercules, CA, USA) 6% polyacrylamide/8 M urea gels. Gels were dried and exposed to Kodak MS film for 3 days at −80°C. A calibration curve was constructed using unlabeled sense mRNA (5−100 pg) for arrestins and GRKs synthesized in vitro with full size cDNAs as templates. A series of calibration samples was included in each experiment and run alongside experimental samples on each gel. This procedure allowed for determination of absolute expression levels of each message and direct comparison of concentrations of different messages. Each sample was measure in 2−3 independent RPA reactions.
2.4. Data Analysis
For Western blots and RPA gels, the gray values of the bands were measured on X-ray film using Versadoc 4 (Bio-Rad, Hercules, CA). The optical densities of the bands corresponding to arrestins and GRKs proteins or their messages were converted into ng of the respective protein or pg of mRNA per gram of tissue (wet weight) using the calibration curves. To facilitate comparison of the expression levels of different arrestin and GRK subtypes, control protein values were then converted into pmol/g tissue using the following molecular weight values (in kDa): arrestin2 − 46, arrestin3 − 45, GRK2 − 79.5, GRK3 − 79, GRK5 − 67.7, GRK6 − 66. Control mRNA values were converted into fmol/g tissue using the following molecular weight values (in kDa) calculated based on the length of respective mRNAs: arrestin2 − 429, arrestin3 − 461, GRK2 − 718, GRK3 − 682, GRK5 − 844, GRK6 − 940. Calibration curves were fitted to linear equations using Prism 4.0 (GraphPad Software, San Diego, CA). For the statistical analysis, StatView software (SAS Institute, Cary, NC) was used. The data were analyzed by two-way repeated measure ANOVA with Group (Control, PD, PD with dementia) and Brain Region (CN, Pu, VSTR) as main effects: Group as a between group factor and Brain Region as a repeated measure factor. If main effects were significant, the data were then analyzed separately for each brain region by ANOVA with Group as main factor followed by Bonferroni/Dunn post-hoc test with correction for multiple comparisons. The value of α<0.05 was considered significant. Age and PMI were treated as co-variates, and Sex as blocking factor. For the factor analysis, we used principal component method for factor extraction and oblique solution to determine factor loadings.
3. Results
3.1. Cohort characterization
We have analyzed the expression levels of arrestins and GRKs at post mortem in the basal ganglia of control cases as compared to patients with Parkinson's disease. The cases of PD with dementia were analyzed separately from cases with PD only (designated as PDD group). We have grouped the cases based on clinical diagnosis rather than on any neuropathological feature, for two reasons: first, we were interested in the neurochemical mechanisms of dementia in PD; second, neuropathological substrate of dementia in PD remains controversial (see for example [63]).
The PDD group had significantly longer PMI than control or PD group (Table 1) but there were no significant differences in age among groups. There was no significant difference in duration of illness between PD and PDD groups, although there was a clear tendency toward shorter duration of illness in the PDD group (p=0.07). Using a largely overlapping cohort of cases we have previously determined that there was no difference between PD and PDD groups in the degree of loss of dopaminergic nigral neurons [51]. Here we confirmed using Western blot technique that TH was severely reduced in the basal ganglia region in both groups with PD (Fig. 1A). There was even more striking loss of DAT in all basal ganglia regions in both PD groups (Fig. 1B). There were no significant differences in the degree of loss of DAT expression between the two PD groups. The loss of both TH and DAT immunoreactivity was the most pronounced in Pu and the least – in CN.
Figure 1.

Comparison of concentrations of tyrosine hydroxylase (TH) and dopamine transporter (DAT) the caudate nucleus (CN), putamen (Pu) and ventral striatum (ventralmost part of the putamen and nucleus accumbens; VSTR) of control subjects and patients with Parkinson's disease without (PD) or with dementia (PDD). Bar graphs represent the mean±S.E. of the amount (arbitrary units) of TH (A) and DAT (B) in the striatal measured by quantitative Western blotting as described in Materials and Methods. The data were statistically analyzed separately for individual brain regions by one-way ANOVA with Group (control, PD, PDD) as main factor followed by Bonferroni/Dunn post hoc test. * - p<0.05, ** - p<0.01, * - p<0.001 to control.
3.2. Arrestin and GRK expression in control human brain
We have demonstrated previously that the antibodies to arrestin2 and arrestin3 selectively label the respective arrestin subtypes in the rat [31] or monkey [13] brain. The antibodies also proved to be selective for human arrestins. The arrestin2 antibody labels only the band corresponding to arrestin2 (Fig. 2A). Only the long arrestin2 splice variant [98] was detected in the human brain. The arrestin3-specific antibody also has much higher affinity to human arrestin3 than to arrestin2, but because the concentration of endogenous arrestin2 substantially exceeds that of arrestin3, a faint band corresponding to arrestin2 is detectable in the tissue samples. Both long and short splice variant of arrestin3 [31,98] were detected and analyzed together (Fig. 2B). There are five non-visual GRKs, GRK2, 3, 4, 5, and 6. GRK4 has a very limited distribution and is found mostly in testis [97]; therefore, it was not analyzed in this work. First, we tested the antibodies for selectivity and sensitivity to human GRK subtypes using recombinant purified GRK proteins as standards. The antibody directed against GRK2 was selective for GRK2 and labeled one band on the blot (Fig. 2C). GRK2 and 3 are structurally very similar, and the GRK3 antibody detects both GRK2 and 3 in the rat, albeit with different sensitivity. The antibody also detects bovine GRK2 as well as GRK3 used here as standards, with the sensitivity lower for GRK2 than for GRK3. However, unlike the situation in the rat, in human samples only one band corresponding to GRK3 is detected, similar to what we previously observed in the monkey [13] (Fig. 2D). The antibody to GRK5 was selective for the GRK5 subtype, labeling one major band on the blot corresponding to purified GRK5 used as standard (Fig. 2E). The antibody used to detect GRK6 is a monoclonal antibody that labels GRK4, 5, and 6. GRK4 is virtually undetectable in the striatum, and GRK5 is well separated by size from GRK6 as previously seen in the monkey brain [13] (Fig. 2F)
Figure 2.

Representative Western blots showing antibody specificity and expression of arrestins and GRKs in the caudate nucleus (CN) of a control subject (Co), patient with Parkinson's disease (PD), and patient with Parkinson's disease and dementia (PDD). The following antibodies were used as described in Materials and Methods: A - to arrestin 2; in B – to arrestin3; C – to GRK2; D – to GRK3; E – to GRK5; F – to GRK6. Note the difference in the concentration range of standards used for arrestin2 (A) and arrestin3 (B). In D, the antibody (sc-563) used to measure GRK3 cross-reacts with purified GRK2, but its sensitivity towards GRK2 is much lower than to GRK3, and no bands corresponding to GRK2 are seen in tissue samples. In C, the anti-GRK2 antibody (sc-562) used to detect GRK2 has much higher sensitivity to GRK2 than sc-563 antibody (D) and it does not cross-react with GRK3. In F, arrowheads points to the band corresponding to GRK6. The thicker lower band is unrelated to GRKs. Note much lower sensitivity of this antibody as compared to other antibodies used in the study.
Figure 3 demonstrates detection of arrestin and GRK mRNAs by RPA. GRK2 mRNA and mRNA of both arrestin subtypes in a single hybridization reaction with specific RNA probes that we designed to separate well by size to allow for simultaneous detection of multiple mRNA species (Fig. 3A). GRKs 3, 5, and 6 were measured in a separate reaction in a similar manner (Fig. 3 B).
Figure 3.
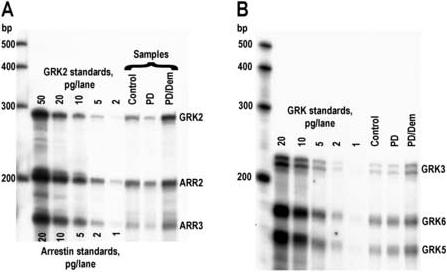
Representative image of ribonuclease protection assay (RPA) performed with samples derived from control and diseased (PD – Parkinson's disease; PDD – Parkinson's disease with dementia) caudate nucleus. RPA was performed as described in Materials and Methods. Samples containing the equivalent of 5.6 mg tissue (wet weight) were loaded per lane. Several left lanes on each gel were loaded with indicated amounts of cold full-length RNAs synthesized in in vitro transcription reaction to serve as standards. A – RPA for arrestin2, arrestin3, and GRK2; B – RPA for GRK3, 5, and 6. Note the difference in the scale between GRK2 and arrestins and different sensitivity of different probes. As seen in B, the probe to GRK3 consistently gives two protected bands both in standards and tissue samples about ten base pairs apart. Both bands correspond to GRK3 and were analyzed together.
Two non-visual arrestins are ubiquitously expressed throughout the brain [31]. We have previously shown that arrestin2 is the major arrestin subtype in the rat and monkey brain, with more than 10-fold excess over arrestin3 protein [13,31,32]. The same relationship was also observed for the control human brain, with the concentration of the arrestin2 protein 10−18-times higher than that of arrestin3 (Fig. 4A). However, the concentration of arrestin2 mRNA was only slightly higher than that of arrestin3, suggesting differential post-transcriptional regulation and/or differences in protein stability (Fig. 4B). As shown in Fig.4A, GRK5 and 6 are the major GRK subtypes in the human striatum, with almost mirror distribution among the striatal subdivisions. Interestingly, concentrations of GRK mRNAs did not match protein concentrations. The mRNA/protein ratios for GRK subtypes 2 and 3 were much higher than for GRK5 and GRK6 (Fig. 4A,B). GRK2 displayed the highest mRNA/protein ratio of all subtypes, probably, due to a very short half-life (60 min) and fast turnover of the GRK2 protein [85].
Figure 4.

Comparison of protein and mRNA concentrations of arrestin and GRK subtypes in the human brain. Bar graphs represent the mean±S.E. of the amount of arrestins and GRKs proteins (A) or mRNAs (B) in the striatal subterritories of the control human brain.
3.3. Arrestins are upregulated in Parkinson's disease with dementia
We found that the concentrations of both mRNA and protein for arrestin-2 and arrestin-3 were significantly altered in patients with Parkinson's disease. For ARR2 protein, two-way repeated measure ANOVA with Group as between factor and Brain Region as within group factor yielded a significant effect of Groups (F(2,64)=10.0, p<0.001), but no significant effect of Brain Region or Group × Region interaction, suggesting the regional pattern of expression remained similar between groups. More detailed analysis demonstrated that the arrestin2 protein was significantly elevated in the PDD group in CN (by 92.3%) and VSTR (by 82.8%) as compared to both control and PD groups, whereas in Pu there was no significant difference (Fig. 5A). Conversely, the levels of arrestin2 protein in the PD group were not different from that in control. The concentration of arrestin2 mRNA was significantly elevated in the PDD group as compared to control in Pu (47.5%; F(2,34)=4.22, p<0.05) and VSTR (by 58.7%: F(2,34)=8.55, p<0.002); but in CN the only difference was between PD and PDD groups (F(2,34)=4.45, p<0.02) (Fig. 5B).
Figure 5.

Expression of arrestin2 protein (A) and mRNA (B) and arrestin3 protein (C) and mRNA (D) in the striatum of control subjects and subjects with Parkinson's disease (PD) or with Parkinson's disease and dementia (PDD). Bar graphs show the results of quantitative analysis of Western blots as means±S.E. The data were statistically analyzed separately for individual brain regions by one-way ANOVA with Group (control, PD, PDD) as main factor followed by Bonferroni/Dunn post hoc test. * - p<0.05, ** - p<0.01 to control; # - p<0.05; & - p<0.01 to PD group.
The expression of arrestin3 protein and mRNA was also altered. Two-way repeated measure ANOVA revealed significant effects of Group (F(2,64)=7.58, p=0.02) and Brain Region (F(2,66)=5.2, p<0.01) for the concentration of arrestin3 protein but no interaction effect. Regional analysis demonstrated that arrestin3 was significantly upregulated in the PDD group as compared to both control and PD groups in CN (F(2,34)=4.9, p<0.02; by 60% to control) and VSTR (F(2,34)=4.7, p<0.02, by 77% to control) but not in Pu (Fig. 5C). Similarly to arrestin2, arrestin3 mRNA was increased in the PDD group in all striatal subdivisions (in CN only the difference between PD and DP/Dem was significant) (Fig. 5D). No effects of Age, PMI, or Sex on the arrestin protein or mRNA expression were detected.
3.4. Expression of GRK3 and GRK5 is increased in Parkinson's disease with dementia
We have compared expression levels of four GRK subtypes in control and both PD groups first by two-way ANOVA with Brain Region as a repeated measure factor and Group as between group factor followed by individual analysis in each striatal subdivision. We detected a strong tendency for differences in the levels of GRK2 protein (p=0.067) among groups owning to a somewhat elevated level in the PDD group and slightly reduced in the PD group, but there were no significant differences by brain region (Fig. 6A). Similarly, there were no changes in the concentration of the GRK2 mRNA in any of the region examined (Fig. 6B). In contrast, the expression of GRK3 protein was significantly influenced by Group (F(2,64)=7.27, p<0.01). The effect was due to a higher concentration of GRK3 protein in the PDD group contrasting with a reduction in GRK3 concentration in the PD group. Among regions, VSTR displayed significant difference between PDD and PD group (by 161.5%), but neither of the groups were different from control (Fig. 6 C). The concentration of GRK3 mRNA was impacted in all regions, with effect of Group highly significant (F(2,64)=5.8, p<0.01) by two-way ANOVA. In contrast with GRK3 protein, the concentration of GRK3 mRNA was significantly increased in the CN of the PDD group as compared to both control (by 64%) and PD groups (by 129%), and in Pu – as compared to control (by 76%). For VSTR, there were no significant differences among groups (Fig. 6D).
Figure 6.

Expression of GRK2 protein (A) and mRNA (B) and GRK3 protein (C) and mRNA (D) in the experimental groups. The data were statistically analyzed separately for individual brain regions by one-way ANOVA with Group (control, PD, PDD) as main factor followed by Bonferroni/Dunn post hoc test. * - p<0.05 control; # - p<0.05; & -p<0.01 to PD group.
The expression of GRK5 was significantly affected by Group on both protein and mRNA levels as evidenced by two-way ANOVA analysis with Brain Region as repeated measure factor (F(2,64)=3.72, p<0.05, for protein and F(2,64)=8.69, p<0.002, for mRNA). In CN, there was substantial increase in both protein and mRNA in PDD group as compared to control and PD groups (Fig. 7A, B). The mRNA concentration was also higher in Pu of the PDD group as compared to control, but the protein concentration was not significantly altered in this region. There were no significant differences in the expression levels of GRK6 protein among groups (Fig. 7C). Interestingly, GRK6 mRNA was significantly affected by Group in the two-way ANOVA analysis (F(2,64)=5.27, p<0.02). The detailed analysis by region showed that GRK6 mRNA was upregulated in the PDD group in CN as compared to the PD group (by 61%) (Fig.7D). Similar tendency was detected in VSTR, but there was no significance (p=0.096). No effects of Age, PMI, or Sex on the GRK proteins or mRNAs expression were detected.
Figure 7.

Expression of GRK5 protein (A) and mRNA (B) and GRK6 protein (C) and mRNA (D) in the experimental groups. The data were statistically analyzed separately for individual brain regions by one-way ANOVA with Group (control, PD, PDD) as main factor followed by Bonferroni/Dunn post hoc test. * - p<0.05 control; # - p<0.05; & -p<0.01 to PD group. For GRK5 mRNA in VSTR, there was a significant F value by one-way ANOVA (F(2,34)=3.3, p<0.05) but Bonferroni/Dunn post hoc test yielded no significant differences.
3.5. Changes in expression of other signaling proteins
Because of the profound difference in the expression of arrestins and GRKs between the two PD groups, we have analyzed the expression of four other proteins involved in arrestin/GRK-mediated signaling to test if a similar pattern would emerge. The expression level of DARPP-32, a protein abundant in striatal medium spiny neurons that serves as a regulator of dopamine receptor signaling, was not significantly different among groups in any of the brain region examined (Fig. 8A). The DARPP-32 expression varied greatly among individuals in all groups, perhaps impacted by drug treatment, which cannot be controlled for. The PD group tended to have lower concentration of DARPP-32 protein than the other two groups, but due to large variability the power was insufficient to reliably detect these differences. We cannot rule out a reduction in DARPP-32 expression in the PD group, but there was certainly no upregulation of DARPP-32 expression in the PDD group as compared to control.
Figure 8.

Expression of DARPP-32 (A), ERK (B), Akt (C), and GSK3α (D) in the striatal subdivisions of control subjects and patients with Parkinson's disease (PD) or Parkinson's disease with dementia (PDD). The data were statistically analyzed separately for individual brain regions by one-way ANOVA with Group (control, PD, PDD) as main factor followed by Bonferroni/Dunn post hoc test. * - p<0.05, ** - p<0.01 to control; & -p<0.01 to PD group.
In contrast to DARPP-32, there were significant differences for ERK1/2, Akt and GSK3α. Two-way repeated measure ANOVA analysis detected Group (F(2,64)=4.8, p<0.02) differences for protein levels of ERK1/2 (both isoforms analyzed together) but not for Brain region. However the PDD group showed a highly significant elevation in the CN as compared to the control (by 93%) as well as to the PD (by 133%) group (Fig. 8B). We have also analyzed the protein levels for Akt and its common substrate GSK3α. The concentration of Akt protein was significantly different among groups as determined by two-way repeated measure ANOVA (F(2,64)=9.4, p<0.001). The difference was due to elevated Akt in the PDD group in all striatal regions (by 54% in CN, 53% in Pu, 82% in VSTR) (Fig. 8C). The concentration of GSK3α protein was also significantly different between groups when compared across regions (F(2,64)=4.9, p<0.02). Among individual regions, the difference was significant only in VSTR (F(2,34)=4.9, p<0.02), where the PDD group showed elevated expression as compared to control (by 87%) (Fig. 8D).
3.6. Relationship between expression of signaling molecules and Alzheimer's pathology
We have performed factor analysis on all variables in the study in order to establish whether there was a statistically valid relationship between the expression levels of signaling proteins measured in this study and the load of Alzheimer's pathology. The analysis yielded two factors. The variables related to the expression of signaling molecules are represented by one factor (Factor 1; Table 2) with large loadings of that factor (over 0.75) for all variables. Variables related to the load of Alzheimer's pathology (number of senile plaques and neurofibrillary tangles) were represented by a second distinct factor (Factor 2). Among all signaling proteins studied, only the expression of DARPP-32 had substantial loading (0.4) on the second factor. The analysis provided evidence that the concentrations of signaling proteins varies across subjects in a manner distinct from that of the number of plaques and tangles.
Table 2.
Results of factor analysis performed on the concentrations of signaling proteins and the load of Alzheimer's pathology across all samples. Rotated factors (Factor 1 and Factor 2) and corresponding factor loadings (oblique solution) for each measure extracted by principal component analysis are shown.
| Factor 1 | Factor 2 | |
|---|---|---|
| ARR2 | 0.844a | 0.204 |
| ARR3 | 0.910 | 0.105 |
| GRK2 | 0.848 | 0.073 |
| GRK3 | 0.812 | −0.052 |
| GRK5 | 0.888 | −0.004 |
| ERK | 0.904 | 0.013 |
| Akt | 0.872 | 0.052 |
| GSK3α | 0.813 | −0.048 |
| DARPP-32 | 0.783 | −0.423b |
| Senile plaques | 0.002 | 0.782 |
| Neurofibrillary tangles | −0.055 | 0.795 |
— Bold underlined indicate high loadings of a factor.
— Note that DARPP-32 is the only signaling protein with a relatively high loading of Factor2 (underlined).
4. Discussion
4.1. Upregulation of arrestin and GRK expression in Parkinson's disease with dementia
Our data indicate that a subgroup of patients with PD that had been clinically diagnosed with dementia has higher levels of the two non-visual arrestins and two of GRK subtypes, GRK3, and GRK5, in the striatum when compared with control and PD patients without dementia. Within the striatum, upregulation of both mRNA and protein levels for the arrestins was the most pronounced in CN and VSTR. Increase in the arrestins' and GRKs' protein expression was accompanied by elevation in their mRNA concentrations in multiple striatal regions, often even in those that did not show protein upregulation (Table 3). The pattern of arrestin and GRK expression in the striatum of patients with PD but without clinical diagnosis of dementia was surprisingly different. There was absolutely no evidence of elevated expression of any of the proteins in any striatal region examined. We also detected no significant differences between the PD and control groups, although some decrease in GRK3 protein in VSTR and arrestins' mRNA in CN can be inferred from the fact that the values in the PDD group were significantly higher than that in the PD group but not in control. A conspicuous feature of the PDD group was the uniformity of values as compared to both control and PD groups, in which a wide range of values for all arrestin/GRK proteins, particularly for GRK2, were detected. Changes in the expression of other signaling proteins we examined (except DARPP-32) followed a pattern similar to that of arrestins and GRKs: we detected a significant upregulation of ERK1/2 in CN, Akt in CN and VSTR, and GSK3α in VSTR in the PD group with dementia as compared to control and PD groups (Table 3). Interestingly, no changes in the expression of any of the signaling proteins were noted in Pu, although the degree of dopaminergic loss was the highest in that striatal region (Table 3).
Table 3.
Summary of changes in the expression levels of arrestins, GRKs, and other signaling proteins in the striatal subdivisions in Parkinson's disease and Parkinson's disease with dementia at postmortem1.
|
Caudate Nucleus |
Putamen |
Ventral Striatum |
||||
|---|---|---|---|---|---|---|
| |
Protein |
mRNA |
Protein |
mRNA |
Protein |
mRNA |
|
ARR2 |
PDD 2 2Co,PD |
PDD PD PD |
 3 3 |
PDD Co Co |
PDD Co,PD |
PDD Co Co |
|
ARR3 |
PDD Co,PD |
PDD PD PD |
 |
PDD Co Co |
PDD Co,PD |
PDD Co Co |
|
GRK2 |
 |
 |
 |
 |
 |
 |
|
GRK3 |
 |
PDD Co,PD |
 |
PDD Co Co |
PDD PD PD |
 |
|
GRK5 |
PDD Co,PD |
PDD Co,PD |
 |
PDD Co Co |
 |
 |
|
GRK6 |
 |
PDD PD PD |
 |
 |
 |
 |
|
ERK |
PDD Co,PD |
|
 |
|
 |
|
|
Akt |
PDD Co Co |
|
 |
|
PDD Co Co |
|
|
GSK3α |
 |
|
 |
|
PDD Co Co |
|
| DARPP-32 | ||||||
Empty cells mean values were not measured.
Indicates that the value in the PDD (Parkinson's disease with dementia) group was significantly higher than in groups shown after the arrow (here as compared to both control (Co) and Parkinson's disease (PD) groups) according to Bonferroni post hoc test following ANOVA with Group as main factor.
The symbol indicates that there were no significant differences.
What could be the reason for such substantial differences in expression of signaling proteins between these two groups of PD patients? There were no significant differences between the groups in age or sex composition. The postmortem interval was significantly shorter in PDD group (the same as in control). However, the expression of arrestins and GRKs (as well as of other signaling proteins) was not influenced by any of these factors. There was a tendency for the PDD group to have shorter disease duration (time between diagnosis and death), which suggests a faster, more severe disease progression. This is in agreement with published data demonstrating negative impact of dementia in PD on survival [49,88]. The question of whether the presence of dementia reflects greater damage to the dopaminergic system has been addressed in our previous study that used a cohort of patients largely overlapping with the one used here [51]. We found that the loss of DA terminals as measured by the concentrations of DA transporter binding sites, TH immunoreactivity in the basal ganglia, and the number of TH positive neurons in the dorsal, ventral and medial DA cell groups were similar in both subgroups of PD patients. In this study, we confirmed a similar loss of DA transporter and TH in the striatum of both PD groups. According to published reports [21,40], there is no difference in vivo in the rate or extent of dopaminergic degeneration between patients with PD and PD with dementia. Our post mortem results generally support that conclusion. Ito and colleagues [47] reported that patients with PD and dementia had lower 18-fluoro-DOPA uptake in the ventral striatum and right caudate nucleus as compared to patients with PD only. Interestingly, we also have detected a more pronounced loss of TH in the ventral striatum in PDD group, although the difference was very small. Additionally, in the present study the caudate nucleus and ventral striatum were the regions where most of the detected changes in the arrestin/GRK expression occurred (Table 3), in spite of lesser dopaminergic loss in these regions as compared to the putamen. Therefore, specific mesolimbic and caudate dopaminergic dysfunction may be linked to dementia in PD and may also determine the changes in arrestin/GRK concentrations seen in that group.
Some cases of PDD had Alzheimer's disease-like pathology (plaques and tangles) to varying degrees, which could be the reason for their dementia as well as for changes in arrestin/GRK expression we detected. However, other cases had no such pathology, and their values were within the same range. We performed factor analysis on all variables and found that expression levels of signaling molecules studied are represented by one factor, whereas loads of senile plaques and neurofibrillary tangles are represented by a different factor (Table 2). Therefore, the load of Alzheimer's pathology is not predictive of the expression levels of arrestins and GRKs. Since the plaque/tangle density and the arrestin/GRK expression are statistically uncorrelated, they seem unlikely to arise from a common cause or to have any causal connections. Consequently, such contrasting patterns of changes in the arrestin/GRK expression in the PD and PD with dementia groups is unlikely to be due to the presence of Alzheimer's pathology.
Therefore, we conclude that deficits in signaling mechanisms specific for PD patients with dementia may be responsible for the pattern of arrestin/GRK expression in these patients. The concentrations of arrestin/GRKs in cultured cells and in vivo are altered by drugs that directly or indirectly cause persistent stimulation or blockade of GPCRs [7,22,25,29,42,45,72,80,99], which implies the involvement of signaling mechanism in the regulation of the arrestin/GRK expression. The concentration GRK2, a short-lived protein, is regulated by synthesis as well as degradation, both of which are responsive to various signaling pathways (reviewed in [85,86]). The activity of the GRK2 promoter is enhanced by stimulation of Gq-coupled GPCRs [91], whereas Gi-coupled receptors promote degradation of GRK2 in arrestin-dependent manner [84]. Judging by their higher protein/mRNA ratios, other GRKs seem to be more stable than GRK2 and are likely to be controlled mostly by posttranscriptional mechanisms, but specific information is lacking. Dementia in Alzheimer' disease and PD is associated with similar cholinergic deficits, which are substantially more severe in PD patients with dementia than in cognitively intact PD patients [24 and references therein,40]. Cholinesterase inhibitors used to treat Alzheimer's disease also seem to be effective against PD-associated dementia [67], the fact that argues for the central role of cholinergic dysfunctions in PD with dementia. It is possible that the pattern of arrestin/GRK expression in PD with dementia contrasting with that in PD is brought about by cholinergic and other signaling deficits combined with dopaminergic depletion.
Another important consideration is antemortem drug treatment the patients had received. The patients' records indicated that all had been treated with levodopa/carbidopa and many – with DA agonists. Individual doses of levodopa varied greatly within each of the two PD groups. Patients from both groups had also been treated with other drugs, including psychotropic drugs such as antidepressants, benzodiazepines, and antipsychotics. Importantly, there seems to be no substantial difference in the pattern of drug treatment between the two PD groups. Antemortem drug treatment is important, because, as discussed above, drug acting at GPCRs or signal transduction may alter the expression of arrestins and GRKs. Specific information, however, is sparse, because most drug classes have never been tested for their effects on the arrestin/GRK expression. Depression is a common non-motor feature of PD, and PD patients are often treated with antidepressants [64,69]. Limiter research performed so far indicate that antidepressants may downregulate GRK2 in the prefrontal cortex of depressed patients at postmortem [29], and the effects may differ between tricyclic and SSRI (selective serotonin reuptake inhibitor) antidepressants [72]. Our data demonstrated that L-DOPA reduced the expression of arrestin2 and GRK2 and 6 upregulated in the basal ganglia of MPTP-treated monkeys [13]. At present, it is unknown whether benzodiazepines or antipsychotics can alter the arrestin/GRK expression. Animal studies are urgently needed to feel this gap.
4.2. Functional significance of changes in arrestin and GRK expression
When dopaminergic neurons die in PD, a number of compensatory events occur in postsynaptic neurons to maintain dopaminergic signaling at a normal level (reviewed in [11,12]). If the compensation for DA depletion was aimed at restoring a normal level of G protein-mediated signaling via DA receptors, a down-regulation of arrestins and GRKs in the PD brain should be expected. It is well established that reduction of the arrestin and/or GRK concentration invariably reduces GPCR desensitization and internalization and enhances G protein-mediated signaling [3,14,27,41,58,75,103]. Therefore, decreased concentrations of arrestin/GRKs in the DA-depleted basal ganglia would impede desensitization of DA receptors and help sustain the G protein-mediated signaling. In this study, we did observe a tendency towards a down-regulation of GRK proteins in the striatum of PD patients. In contrast, in PD patients with dementia, which displayed loss of dopaminergic innervation in the striatum at least as extensive as in the PD group, the opposite happened: the expression of arrestins and GRKs in the striatum was enhanced.
What effect might elevated arresin/GRK levels have on receptor signaling? A wealth of data shows that an increase in arrestin/GRK concentration facilitates desensitization and internalization of various GPCRs and dampens G protein-mediated signaling [45,55,71,74]. Overexpression of arrestins and GRKs induces excessive degradation of GPCRs in vitro [81] and in vivo [46], leading to sustained receptor down-regulation. Earlier we found that D3 DA dopamine receptors were consistently downregulated in the PD group with dementia across the striatal and pallidal subterritories, whereas in non-demented PD cases there was a slight elevation in D3 receptor binding in the rostral basal ganglia [51]. This result matches the pattern in arrestin/GRK expression observed in this study. Down-regulation of D3 receptors in the PDD group was posttranscriptional as evidenced by lack of changes in the D3 mRNA expression. The elevated arrestin/GRK expression might lead to the D3 receptors down-regulation via enhanced internalization. The DA D3 receptor is resistant to GRK-mediated phosphorylation, and its trafficking is very sensitive to the availability of arrestins and GRKs [55]. However, D2 DA receptor binding was increased in both PD groups to a similar extent [51], which appears inconsistent with the contrasting changes in arrestin/GRK expression seen in these groups. PD patients with dementia often respond poorly to L-DOPA, as shown by us [51] and others [16,19]. It is important to note that persistent elevation of arrestin and GRK concentrations, whatever the cause, can lead to reduced signaling via GPCRs, including DA receptors, and, therefore may be a contributing factor to poor therapeutic response to L-DOPA regardless of changes in DA receptor numbers.
Although upregulation of arrestins and GRKs inhibits G protein-mediated signaling, other signaling pathways facilitated by arrestin binding to GPCRs may be enhanced. Arrestins, in addition to their “negative” role in G protein-mediated signaling, have “positive” signaling functions. Arrestins serve as a link between GPCRs and the mitogen-activated protein kinase (MAPK) signaling pathways (reviewed in [37,87]). Arrestins work as scaffolds recruiting a number of components of several MAPK cascades to activated GPCRs, thereby mediating activation of the MAPK pathways. In particular, both non-visual arrestins have been shown to activate ERK [66,87,100]. Over-expression of arrestins potentiates arrestin-mediated ERK activation [100,101]. Conversely, reduction of the arrestin concentration by siRNAs inhibits ERK activation [3]. Therefore, the increase in arrestin concentration in the PD with dementia group might facilitate stimulation of the MAPK pathways by GPCRs. We did not directly examine ERK phosphorylation levels in our samples. Proteins are normally rapidly dephosphorylated, and after prolonged post mortem intervals the data would be difficult to interpret. However, an increase in the concentration of total ERK protein in the PD group with dementia may be viewed as evidence of the activation of this pathway, possibly, linked to the upregulation of arrestins.
Recently arrestin3 has been implicated in regulation of Akt pathway [8,9]. Persistent activation of D2 DA receptors inhibits Akt. This reduces phosphorylation of its usual substrate glycogen synthase kinase3 (GSK3α,β thereby increasing its activity [8]. Arrestin3 mediates this action of DA by forming a signaling complex comprising Akt and protein phosphatase2 that dephosphorylates Akt [9]. Arrestin3 knockout in mice inhibits DA-mediated behavior and abolishes DA-mediated regulation of Akt [9]. It is still unknown whether only arrestin3 or both non-visual arrestin subtypes regulate DA-dependent Akt signaling. Conceivably, the upregulation of Akt and GSK3α expression seen in the striatum of PD patients with dementia is linked to the elevated concentrations of arrestins. Another interesting aspect is that arrestin2 has recently been shown to act as a messenger between activated GPCRs and the nucleus and directly activates specific promoters such as p27 and c-fos by facilitating local histone H4 acetylation [52]. Arrestin2 up-regulation results in enhanced activation of such promoters, and arrestin2 knockdown by siRNA suppresses transcription of these genes [52]. Therefore, it is possible that elevated arrestin expression in the brain of patients with Parkinson's disease and dementia may modulate activation and/or expression of other signaling molecules either via cytoplasmic signaling mechanisms or via interaction with promoters of specific genes.
Arrestin activity is often contingent upon receptor phosphorylation (reviewed in [35,36]). Therefore, changes in the concentration of GRKs modulate arrestin-mediated signaling. Different subclasses of GRKs have differential role in arrestin-dependent cellular functions [54,92]. GRK2 and 3 largely mediate “classic” GRK functions, such as receptor phosphorylation, arrestin recruitment, and receptor endocytosis. However, arrestin-mediated ERK activation requires GRK5 or 6. ERK activation is enhanced by overexpression of GRK5 or 6 and inhibited by their knockdown. Manipulations of GRK2/3 concentrations have the opposite effects. So far, this functional antagonism has been demonstrated for two GPCRs, angiotensin II receptor [54] and V2 vasopressin receptor [92], and only in cultured cells. It is unclear to what extent these findings are applicable to the in vivo situation in general and to neuronal signaling mechanisms in particular. However, it seems likely that changes in GRK expression observed in PD with dementia cases contribute to specific alterations in arrestin-mediated signaling and, possibly, affect other downstream signaling pathways.
GRKs were traditionally considered selective for GPCRs. However, GRKs phosphorylate other substrates and therefore may have functions not directly related to GPCR regulation. GRK2 and GRK5 bind microtubules and phosphorylate tubulin, possibly promoting microtubule assembly [20]. Visual arrestin has been shown to bind microtubules [38], and this ability plays a role in arrestin-mediated visual adaptation [77]. Non-visual arrestins also bind microtubules, demonstrating even higher affinity [39]. It is conceivable that coordinated action of arrestins and GRKs at microtubules is important for function of neuronal cytoskeleton, and that it may be altered by enhanced expression of arrestins and GRKs in PD patients with dementia. GRK2 and GRK5 also phosphorylate synucleins, with GRK5 preferring α-synuclein as a substrate [90]. GRK5-mediated phosphorylation of α-synuclein inhibits its interaction with phospholipids and phospholipase D2 acting as negative control of α-synuclein function in vesicle trafficking [90]. It is possible that upregulation of GRK5 in the brains of PD patients with dementia modifies the α-synuclein activity and/or metabolism, possibly promoting Lewy body formation. Interestingly, we have observed higher levels of cortical and striatal insoluble α-synuclein in the PDD group as compared to PD and control (Joyce, Borwege and Osredkar, unpublished observation). It is important to note that α-synuclein phosphorylation by GRK5 is controlled by GPCR activity [90] and, therefore, is likely to be affected by neurotransmitter deficits and drug treatment regiment.
Previously, we have examined the alterations in arrestin/GRK expression in the MPTP-treated Macaque monkeys [13]. Non-human primates treated with neurotoxin MPTP to induce dopaminergic degeneration are considered the golden standard of animal models of PD. We have found that MPTP lesion significantly upregulated arrestin2 and two GRKs, GRK2 and 6, in most basal ganglia regions. Chronic L-DOPA treatment brings the expression of all proteins back to normal regardless whether dyskinesia develops in response to L-DOPA. Increased expression of arrestins and GRKs in MPTP-treated monkeys was accompanied by enhanced ERK activation and elevated total ERK concentration. In fact, MPTP-treated drug-naive monkeys resemble the PD with dementia group of human patients in terms of changes in arrestin/GRK concentrations, whereas the PD group is similar to L-DOPA-treated MPTP monkeys. Patients in both groups of this cohort had been treated with dopaminergic drugs during their illness. The only difference is that the PD group as a rule retained good therapeutic response to L-DOPA whereas most PDD patients became resistant to the medication. It is tempting to speculate that PD patients with dementia are resistant to L-DOPA on the therapeutic level because they are resistant to the drug on the molecular level. L-DOPA may down-regulate arrestin/GRK expression in the brain of some patients keeping them responsive to medication. At the same time those with persistently elevated arrestin/GRK expression become resistant to the therapeutic drug effects. Dementia seen in these patients may be linked to abnormally enhanced arrestin/GRK expression.
Many limitations inherent to postmortem studies often make it difficult to understand functional significance of postmortem findings. Any human disease exists as a plethora of individual forms that limited postmortem cohorts fail to represent fully. Postmortem specimens are often derived from elderly patients at late stages of the disease and invariably suffering from other disorders. Therefore, it is hardly possible to follow the progression of the disease or identify its unique mechanisms. Patients are treated antemortem with a wide variety of drugs that are likely to interfere with postmortem measurements. Inevitable postmortem delay may induce drastic changes in molecular structure. Specimens in the present study have remarkably short postmortem delay but it is still much longer than in animal experiments. Most importantly, postmortem studies cannot prove causal relationship between the disease symptoms and molecular features, because targeted experimental manipulations are not possible. Therefore, experimental analysis of the functional role of arrestins and GRKs in PD is best accomplished in animal models of PD that are amenable to experimental and genetic manipulations. However, there is no way to model in animals the combination of PD with dementia, which is not uncommon in human patients. The unique heuristic value of postmortem studies lies in the ability to study a real human disease at structural and molecular resolution not achievable by any other method applicable to the human brain.
Substantial changes in the expression levels of arrestins and GRKs in the striatum of patients with PDD described here suggests the involvement of these regulatory proteins in PD and/or dementia pathology and identifies them as novel targets for therapeutic intervention. It is possible that enhanced expression of the components of the GPCR regulation machinery seen in the PD with dementia cohort may be induced by dopaminergic denervation similarly to what happens in the MPTP monkey. In patients in which dopaminergic drugs fail to suppress arrestin/GRK expression, elevated concentration of these proteins may confer resistance to therapeutic effects of dopaminergic drugs and propagate pathology by interfering with the functions of the cytoskeleton and/or that of α-synuclein. Deciphering signaling mechanisms leading to the enhanced expression of arrestins and GRKs as well as downstream signaling events unleashed by high concentrations of arrestins and GRKs is critical for understanding of the mechanisms of the PD-associated dementia and for designing effective treatment strategies.
5. Acknowledgements
We are grateful to the Sun Health Research Institute Brain Donation Program of Sun City, Arizona, (Dr. Thomas Beach, Director) for the provision of human brain material. The Brain Donation Program is supported by the National Institute on Aging (P30 AG19610), Arizona Alzheimer's Disease Core Center, the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center) and the Arizona Biomedical Research Commission (contract 0011, Arizona Parkinson's Disease Center). We are grateful to Dr. R.J. Lefkowitz (Duke University) for the generous gift of purified GRK3. We are indebted to Drs. B. Rapoport (University of California San Francisco, CA) for the gift of the cDNA clone of human arrestin3 and A. De Blasi (University of Rome La Sapienza, Italy) for cDNA clones of human arrestin2, GRK2, and GRK3. This work was supported by NIH grants MH62654 and NS045117 (to EVG), E11500 and GM63097 (to VVG), GM44944 and GM47417 (to JLB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5.1. Disclosure Statement. There is no actual or potential conflict of interest.
References
- 1.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 2.Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson's disease: a prospective, community-based study. Ann Neurol. 2005;58:773–776. doi: 10.1002/ana.20635. [DOI] [PubMed] [Google Scholar]
- 3.Ahn S, Nelson CD, Garrison TR, Miller WE. Lefkowitz RJ. Desensitization, internalization, and signaling functions of beta-arrestins demonstrated by RNA interference. Proc Natl Acad Sci U S A. 2003;100:1740–1744. doi: 10.1073/pnas.262789099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol. 2002;59:102–112. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- 5.Arriza JL, Dawson TM, Simerly RB, Martin LJ, Caron MG, Snyder SH, Lefkowitz RJ. The G-protein-coupled receptor kinases βARK1 and βARK2 are widely distributed at synapses in rat brain. J Neurosci. 1992;12:4045–4055. doi: 10.1523/JNEUROSCI.12-10-04045.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 7.Bawa T, Altememi GF, Eikenburg DC, Standifer KM. Desensitization of alpha 2A-adrenoceptor signaling by modest levels of adrenaline is facilitated by beta 2-adrenoceptor-dependent GRK3 up-regulation. Br J Pharmacol. 2003;138:921–931. doi: 10.1038/sj.bjp.0705127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Nat Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 10.Benovic JL, Gomez J. Molecular cloning and expression of GRK6. A new member of the G protein-coupled receptor kinase family. J Biol Chem. 1993;268:19521–19527. [PubMed] [Google Scholar]
- 11.Bezard E, Gross CE. Compensatory mechanisms in experimental and human parkinsonism : towards a dynamic approach. Prog Neurobiol. 1998;55:93–116. doi: 10.1016/s0301-0082(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 12.Bezard E, Gross CE, Brotchie JM. Presymptomatic compensation in Parkinson's disease is not dopamine-mediated. Trends Neurosci. 2003;26:215–221. doi: 10.1016/S0166-2236(03)00038-9. [DOI] [PubMed] [Google Scholar]
- 13.Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis. 2005;18:323–335. doi: 10.1016/j.nbd.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 15.Boller F, Mizutani T, Roessmann U, Gambetti P. Parkinson disease, dementia, and Alzheimer disease: clinicopathological correlations. Ann Neurol. 1980;7:329–335. doi: 10.1002/ana.410070408. [DOI] [PubMed] [Google Scholar]
- 16.Bonelli SB, Ransmayr G, Steffelbauer M, Lukas T, Lampl C, Deibl M. L-dopa responsiveness in dementia with Lewy bodies, Parkinson disease with and without dementia. Neurology. 2004;63:376–378. doi: 10.1212/01.wnl.0000130194.84594.96. [DOI] [PubMed] [Google Scholar]
- 17.Braak H, Braak E, Yilmazer D, De Vos RAI, Jansen ENH, Bohl J. New aspects of pathology in Parkinson's disease with concomitant incipient Alzheimer's disease. J Neural Transm. 1996;(Suppl 48):1–6. doi: 10.1007/978-3-7091-7494-4_1. [DOI] [PubMed] [Google Scholar]
- 18.Braak H, Braak E. Staging of Alzheimer-related cortical destruction. Int Psychogeriatr. 1997;9(Suppl 1):257–261. [PubMed] [Google Scholar]
- 19.Burn DJ, Rowan EN, Minett T, Sanders J, Myint P, Richardson J, Thomas A, Newby J, Reid J, O'Brien JT, McKeith JG. Extrapyramidal features in Parkinson's disease with and without dementia and dementia with Lewy bodies: A cross-sectional comparative stud. Mov Disord. 2003;18:884–889. doi: 10.1002/mds.10455. [DOI] [PubMed] [Google Scholar]
- 20.Carman CV, Som T, Kim CM, Benovic JL. Binding and phosphorylation of tubulin by G protein-coupled receptor kinases. J Biol Chem. 1998;273:20308–20316. doi: 10.1074/jbc.273.32.20308. [DOI] [PubMed] [Google Scholar]
- 21.Colloby SJ, Williams ED, Burn DJ, Lloyd JJ, McKeith IG, O'Brien JT. Progression of dopaminergic degeneration in dementia with Lewy bodies and Parkinson's disease with and without dementia assessed using 123I-FP-CIT SPECT. Eur J Nucl Med Mol Imaging. 2005;32:1176–1185. doi: 10.1007/s00259-005-1830-z. [DOI] [PubMed] [Google Scholar]
- 22.Diaz A, Pazos A, Florez J, Ayesta FJ, Santana V, Hurle MA. Regulation of mu-opioid receptors, G-protein-coupled receptor kinases and beta-arrestin 2 in the rat brain after chronic opioid receptor antagonism. Neuroscience. 2002;112:345–353. doi: 10.1016/s0306-4522(02)00073-8. [DOI] [PubMed] [Google Scholar]
- 23.e Lau LM, Schipper CM, Hofman A, Koudstaal PJ, Breteler MM. Prognosis of Parkinson disease: risk of dementia and mortality: the Rotterdam Study. Arch Neurol. 2005;62:1265–1269. doi: 10.1001/archneur.62.8.1265. [DOI] [PubMed] [Google Scholar]
- 24.Emre M. Dementia associated with Parkinson's disease. Lancet. 2003;2:229–237. doi: 10.1016/s1474-4422(03)00351-x. [DOI] [PubMed] [Google Scholar]
- 25.Fan X, Zhang J, Zhang X, Yue W, Ma L. Acute and chronic morphine treatments and morphine withdrawal differentially regulate GRK2 and GRK5 gene expression in rat brain. Neuropharmacology. 2002;43:809–816. doi: 10.1016/s0028-3908(02)00147-8. [DOI] [PubMed] [Google Scholar]
- 26.Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron. 1999;24:1029–1036. doi: 10.1016/s0896-6273(00)81048-x. [DOI] [PubMed] [Google Scholar]
- 27.Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, Torres GE, Kim KM, Lefkowitz RJ, Caron MG, Premont RT. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron. 2003;38:291–303. doi: 10.1016/s0896-6273(03)00192-2. [DOI] [PubMed] [Google Scholar]
- 28.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 29.Grange-Midroit M, Garcia-Sevilla JA, Ferrer-Alcon M, La Harpe R, Huguelet P, Guimon J. Regulation of GRK 2 and 6, beta-arrestin-2 and associated proteins in the prefrontal cortex of drug-free and antidepressant drug-treated subjects with major depression. Brain Res Mol Brain Res. 2003;111:31–41. doi: 10.1016/s0169-328x(02)00667-8. [DOI] [PubMed] [Google Scholar]
- 30.Guigoni C, Aubert I, Li Q, Gurevich VV, Benovic JL, Ferry S, Mach U, Stark H, Leriche L, Hakansson K, Bioulac BH, Gross CE, Sokoloff P, Fisone G, Gurevich EV, Bloch B, Bezard E. Pathogenesis of levodopa-induced dyskinesia: focus on D1 and D3 dopamine receptors. Parkinsonism Relat Disord. 2005;11(Suppl 1):S25–29. doi: 10.1016/j.parkreldis.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- 32.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- 33.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 34.Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 35.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. TIPS. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Gurevich VV, Gurevich EV. Structural basis of arrestin regulation of G protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall RA, Lefkowitz RJ. Regulation of G protein-coupled receptor signaling by scaffold proteins. Circ Res. 2002;91:672–680. doi: 10.1161/01.res.0000037000.74258.03. [DOI] [PubMed] [Google Scholar]
- 38.Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanson SM, Francis DJ, Vishnivetskiy SA, Raman D, Van Eps N, Hubbell WL, Klug CS, Gurevich VV. Arrestin binding to microtubules involves a distinct conformational change. FASEB J. 2006;20:A110–A110. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilker R, Thomas AV, Klein JC, Weisenbach S, Kalbe E, Burghaus L, Jacobs AH, Herholz K, Heiss WD. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65:1716–1722. doi: 10.1212/01.wnl.0000191154.78131.f6. [DOI] [PubMed] [Google Scholar]
- 41.Horie K, Insel PA. Retrovirally mediated transfer of a G protein-coupled receptor kinase (GRK) dominant-negative mutant enhances endogenous calcitonin receptor signaling in chinese hamster ovary cells. GRK inhibition enhances expression of receptors and receptor mRNA. J Biol Chem. 2000;275:29433–29440. doi: 10.1074/jbc.M003413200. [DOI] [PubMed] [Google Scholar]
- 42.Hurle MA. Changes in the expression of G protein-coupled receptor kinases and beta-arrestin 2 in rat brain during opioid tolerance and supersensitivity. J Neurochem. 2001;77:486–492. doi: 10.1046/j.1471-4159.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 43.Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM-Y, Clark CM, Glosser G, Stern MB, S.M. G, Arnold SE. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54:1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 44.Iaccarino G, Rockman HA, Shotwell KF, Tomhave ED, Koch WJ. Myocardial overexpression of GRK3 in transgenic mice: evidence for in vivo selectivity of GRKs. Am J Physiol. 1998;275:1298–1306. doi: 10.1152/ajpheart.1998.275.4.H1298. [DOI] [PubMed] [Google Scholar]
- 45.Iaccarino G, Tomhave ED, Lefkowitz RJ, Koch WJ. Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by β-adrenergic receptor stimulation and blockade. Circulation. 1998;98:1783–1789. doi: 10.1161/01.cir.98.17.1783. [DOI] [PubMed] [Google Scholar]
- 46.Iaccarino G, Lefkowitz RJ, Koch WJ. Myocardial G protein-coupled receptor kinases: implications for heart failure therapy. Proc Assoc Am Physicians. 1999;111:399–405. doi: 10.1111/paa.1999.111.5.399. [DOI] [PubMed] [Google Scholar]
- 47.Ito K, Nagano-Saito A, Kato T, Arahata Y, Nakamura A, Kawasumi Y, Hatano K, Abe Y, Yamada T, Kachi T, Brooks DJ. Striatal and extrastriatal dysfunction in Parkinson's disease with dementia: a 6-[18F]fluoro-L-dopa PET study. Brain. 2002;125:1358–1365. doi: 10.1093/brain/awf134. [DOI] [PubMed] [Google Scholar]
- 48.Jellinger KA. Morphological substrates of dementia in parkinsonism. A critical update. J Neural Transm. 1997;51:57–82. doi: 10.1007/978-3-7091-6846-2_6. [DOI] [PubMed] [Google Scholar]
- 49.Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson's disease. J Neural Transm. 2002;109:329–339. doi: 10.1007/s007020200027. [DOI] [PubMed] [Google Scholar]
- 50.Joyce JN, Ryoo H, Gurevich EV, Adler C, Beach T. Ventral striatal D(3) receptors and Parkinson's Disease. Parkinsonism Relat Disord. 2001;7:225–230. doi: 10.1016/s1353-8020(00)00060-2. [DOI] [PubMed] [Google Scholar]
- 51.Joyce JN, Ryoo HL, Beach TB, Caviness JN, Stacy M, Gurevich EV, Reiser M, Adler CH. Loss of response to levodopa in Parkinson's disease and co-occurrence with dementia: role of D3 and not D2 receptors. Brain Res. 2002;955:138–152. doi: 10.1016/s0006-8993(02)03396-6. [DOI] [PubMed] [Google Scholar]
- 52.Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, Zhang M, Bao G, Wang F, Zhang X, Yang R, Fan F, Chen X, Pei G, Ma L. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–847. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 53.Kim CM, Dion SB, Onorato JJ, Benovic JL. Expression and characterization of two beta-adrenergic receptor kinase isoforms using the baculovirus expression system. Receptor. 1993;3:39–55. [PubMed] [Google Scholar]
- 54.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Nat Acad Sci USA. 2005;102:142–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem. 2001;276:37409–37414. doi: 10.1074/jbc.M106728200. [DOI] [PubMed] [Google Scholar]
- 56.Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, Smith GE, Dickson DW, Johnson KA, Petersen LE, McDonald WC, Braak H, Petersen RC. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62:1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 57.Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 58.Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. Beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci U S A. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319. doi: 10.1146/annurev.pharmtox.38.1.289. [DOI] [PubMed] [Google Scholar]
- 60.Kunapuli P, Onorato JJ, Hosey MM, Benovic JL. Expression, purification, and characterization of the G protein-coupled receptor kinase GRK5. J Biol Chem. 1994;269:1099–1105. [PubMed] [Google Scholar]
- 61.Lefkowitz RJ, Whalen EJ. Beta-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162–168. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 62.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 63.Levy G. Dementia in Parkinson's disease challenges the “gold standard”. Ann Neurol. 2005;58:663–665. doi: 10.1002/ana.20698. [DOI] [PubMed] [Google Scholar]
- 64.Lieberman A. Depression in Parkinson's disease -- a review. Acta Neurol Scand. 2006;113:1–8. doi: 10.1111/j.1600-0404.2006.00536.x. [DOI] [PubMed] [Google Scholar]
- 65.Loudon RP, Benovic JL. Expression, purification, and characterization of the G protein-coupled receptor kinase GRK6. J Biol Chem. 1994;269:22691–22697. [PubMed] [Google Scholar]
- 66.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Nat Acad Sci USA. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maidment I, Fox C, Boustani M. Cholinesterase inhibitors for Parkinson's disease dementia. Cochrane Database Syst Rev. 2006;1:CD004747. doi: 10.1002/14651858.CD004747.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathologica. 2000;100:285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- 69.McDonald WM, Holtzheimer PEr, Byrd EH. The diagnosis and treatment of depression in Parkinson's disease. Curr Treat Opinions Neurol. 2006;8:245–255. doi: 10.1007/s11940-006-0015-9. [DOI] [PubMed] [Google Scholar]
- 70.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 71.Ménard L, Ferguson SS, Barak LS, Bertrand L, Premont RT, Colapietro AM, Lefkowitz RJ, Caron MG. Members of the G protein-coupled receptor kinase family that phosphorylate the β2-adrenergic receptor facilitate sequestration. Biochemistry. 1996;35:4155–4160. doi: 10.1021/bi952961+. [DOI] [PubMed] [Google Scholar]
- 72.Miralles A, Asensio VJ, Garcia-Sevilla JA. Acute treatment with the cyclic antidepressant desipramine, but not fluoxetine, increases membrane-associated G protein-coupled receptor kinases 2/3 in rat brain. Neuropharmacology. 2002;43:1249–1257. doi: 10.1016/s0028-3908(02)00306-4. [DOI] [PubMed] [Google Scholar]
- 73.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 74.Mundell SJ, Luty JS, Willets J, Benovic JL, Kelly E. Enhanced expression of G protein-coupled receptor kinase 2 selectively increases the sensitivity of A2A adenosine receptors to agonist-induced desensitization. Br J Pharmacol. 1998;125:347–356. doi: 10.1038/sj.bjp.0702081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mundell SJ, Loudon RP, Benovic JL. Characterization of G protein-coupled receptor regulation in antisense mRNA-expressing cells with reduced arrestin levels. Biochemistry. 1999;38:8723–8732. doi: 10.1021/bi990361v. [DOI] [PubMed] [Google Scholar]
- 76.Muriel MP, Bernard V, Levey AI, Laribi O, Abrous DN, Agid Y, Bloch B, Hirsch EC. Levodopa induces a cytoplasmic localization of D1 dopamine receptors in striatal neurons in Parkinson's disease. Ann Neurol. 1999;46:103–111. doi: 10.1002/1531-8249(199907)46:1<103::aid-ana15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 77.Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, barrestin1, and barrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 79.Orsini MJ, Benovic JL. Characterization of dominant negative arrestins that inhibit beta-2-adrenergic receptor internalization by distinct mechanisms. J Biol Chem. 1998;273:34616–34622. doi: 10.1074/jbc.273.51.34616. [DOI] [PubMed] [Google Scholar]
- 80.Ozaita A, Escriba PV, Ventayol P, Murga C, Mayor FJ, Garcia-Sevilla JA. Regulation of G protein-coupled receptor kinase 2 in brains of opiate-treated rats and human opiate addicts. J Neurochem. 1998:1249–1257. doi: 10.1046/j.1471-4159.1998.70031249.x. [DOI] [PubMed] [Google Scholar]
- 81.Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin × receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- 82.Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57:82–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- 83.Parruti G, Peracchia F, Sallese M, Ambrosini G, Masini M, Rotilio D, De Blasi A. Molecular analysis of human beta-arrestin-1: cloning, tissue distribution, and regulation of expression. Identification of two isoforms generated by alternative splicing. J Biol Chem. 1993;268:9753–9761. [PubMed] [Google Scholar]
- 84.Penela P, Elorza A, Sarnago S, Mayor FJ. Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J. 2001;20:5129–5138. doi: 10.1093/emboj/20.18.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Penela P, Ribas C, Mayor FJ. Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal. 2003;15:973–981. doi: 10.1016/s0898-6568(03)00099-8. [DOI] [PubMed] [Google Scholar]
- 86.Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor FJ. Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69:46–56. doi: 10.1016/j.cardiores.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 87.Pierce K, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 2001;20:1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- 88.Poewe W. Treatment of dementia with Lewy bodies and Parkinson's disease dementia. Mov Disord. 2005;20:S77–S82. doi: 10.1002/mds.20544. [DOI] [PubMed] [Google Scholar]
- 89.Premont RT, Koch WJ, Inglese J, Lefkowitz RJ. Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem. 1994;269:6832–6841. [PubMed] [Google Scholar]
- 90.Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- 91.Ramos-Ruiz R, Penela P, Penn RB, Mayor FJ. Analysis of the human G protein-coupled receptor kinase 2 (GRK2) gene promoter: regulation by signal transduction systems in aortic smooth muscle cells. Circulation. 2000;101:2083–2089. doi: 10.1161/01.cir.101.17.2083. [DOI] [PubMed] [Google Scholar]
- 92.Ren XR, Reiter E, Ahn S, Kim J, Chen W, Lefkowitz RJ. Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc Nat Acad Sci USA. 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richard IH, Papka M, Rubio A, Kurlan R. Parkinson's disease and dementia with Lewy bodies: one disease or two? Mov Disord. 2002;17:1161–1165. doi: 10.1002/mds.10274. [DOI] [PubMed] [Google Scholar]
- 94.Richardson RM, Hosey MM. Agonist-induced phosphorylation and desensitization of human m2 muscarinic cholinergic receptors in Sf9 insect cells. J Biol Chem. 1993;267:22249–22255. [PubMed] [Google Scholar]
- 95.Rockman HA, Choi DJ, Rahman NU, Akhter SA, Lefkowitz RJ, Koch WJ. Receptor-specific in vivo desensitization by the G protein-coupled receptor kinase-5 in transgenic mice. Proc Nat Acad Sci USA. 1996;93:9954–9959. doi: 10.1073/pnas.93.18.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ryoo HL, Pierrotti D, Joyce JN. Dopamine D3 receptor is decreased and D2 receptor is elevated in the striatum of Parkinson's disease. Mov Disord. 1998;13:788–797. doi: 10.1002/mds.870130506. [DOI] [PubMed] [Google Scholar]
- 97.Sallese M, Mariggio S, Collodel G, Moretti E, Piomboni P, Baccetti B, De Blasi A. G protein-coupled receptor kinase GRK4. Molecular analysis of the four isoforms and ultrastructural localization in spermatozoa and germinal cells. J Biol Chem. 1997;272:10188–10195. doi: 10.1074/jbc.272.15.10188. [DOI] [PubMed] [Google Scholar]
- 98.Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, Benovic JL. Polypeptide variants of beta-arrestin and arrestin3. J Biol Chem. 1993;268:15640–15648. [PubMed] [Google Scholar]
- 99.Terwilliger RZ, Ortiz J, Guitart X, Nestler EJ. Chronic morphine administration increases beta-adrenergic receptor kinase (beta ARK) levels in the rat locus coeruleus. J Neurochem. 1994;63:1983–1986. doi: 10.1046/j.1471-4159.1994.63051983.x. [DOI] [PubMed] [Google Scholar]
- 100.Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- 101.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 102.Tsuboi Y, Dickson DW. Dementia with Lewy bodies and Parkinson's disease with dementia: are they different? Parkinsonism Relat Disord. 2005;11(Suppl 1):S47–51. doi: 10.1016/j.parkreldis.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 103.Willets JM, Parent JL, Benovic JL, Kelly E. Selective reduction in A2 adenosine receptor desensitization following antisense-induced suppression of G protein-coupled receptor kinase 2 expression. J Neurochem. 1999;73:1781–1789. doi: 10.1046/j.1471-4159.1999.0731781.x. [DOI] [PubMed] [Google Scholar]