

Abstract
Ranolazine is an antianginal agent that targets a number of ion channels in the heart, including cardiac voltage-gated Na+ channels. However, ranolazine block of muscle and neuronal Na+ channel isoforms has not been examined. We compared the state- and use-dependent ranolazine block of Na+ currents carried by muscle Nav1.4, cardiac Nav1.5, and neuronal Nav1.7 isoforms expressed in human embryonic kidney 293T cells. Resting and inactivated block of Na+ channels by ranolazine were generally weak, with a 50% inhibitory concentration (IC50) ≥ 60 μM. Use-dependent block of Na+ channel isoforms by ranolazine during repetitive pulses (+50 mV/10 ms at 5 Hz) was strong at 100 μM, up to 77% peak current reduction for Nav1.4, 67% for Nav1.5, and 83% for Nav1.7. In addition, we found conspicuous time-dependent block of inactivation-deficient Nav1.4, Nav1.5, and Nav1.7 Na+ currents by ranolazine with estimated IC50 values of 2.4, 6.2, and 1.7 μM, respectively. On- and off-rates of ranolazine were 8.2 μM−1 s−1 and 22 s−1, respectively, for Nav1.4 open channels and 7.1 μM−1 s−1 and 14 s−1, respectively, for Nav1.7 counterparts. A F1579K mutation at the local anesthetic receptor of inactivation-deficient Nav1.4 Na+ channels reduced the potency of ranolazine ~17-fold. We conclude that: 1) both muscle and neuronal Na+ channels are as sensitive to ranolazine block as their cardiac counterparts; 2) at its therapeutic plasma concentrations, ranolazine interacts predominantly with the open but not resting or inactivated Na+ channels; and 3) ranolazine block of open Na+ channels is via the conserved local anesthetic receptor albeit with a relatively slow on-rate.
Ranolazine is an antianginal and anti-ischemic drug that is used in patients with chronic angina (Pepine and Wolff, 1999). The therapeutic plasma concentration of ranolazine is in the range of 2 to 6 μM, but how ranolazine works against chronic angina remains unclear (Chaitman, 2006). Ranolazine displays a broad pharmacological profile on a number of targets and has complex electrophysiological effects in myocardium (Antzelevitch et al., 2004; Schram et al., 2004). Ranolazine inhibits cardiac ICa, human ether-a-go-go-related gene, and IsK currents with modest potencies. It is noteworthy that this drug seems to preferentially reduce the persistent late cardiac Nav1.5 Na+ currents with a 50% inhibitory concentration (IC50) of 15 μM (Fredj et al., 2006), whereas normal transient Na+ currents are less affected, with an IC50 of 135 μM. The binding site of ranolazine in human cardiac Na+ channels is probably the same as the local anesthetic (LA) receptor within the inner cavity of the Na+ channel, because a single mutation at this receptor (e.g., residue hNav1.5-F1760A) alters the ranolazine binding affinity drastically (Fredj et al., 2006).
Voltage-gated Na+ channels are essential for the generation and propagation of action potentials in excitable membranes. The Na+ channel protein contains four homologous domains (D1–D4), each with six transmembrane segments (S1–S6). Upon depolarization, Na+ channels go through rapid transitions from their resting to the open state, then to the inactivated state (Aldrich et al., 1983). It has been well recognized that genetic defects in Na+ channel inactivation that result in small sustained Na+ currents after the action potential firing could have devastating consequences, such as seizures, hyperkalemic periodic paralysis, paramyotonia, or LQT-3 syndrome (George, 2005; Cannon, 1996; Ashcroft, 2000). There are 9 isoforms (Nav1.1–1.9) found among mammalian excitable tissues, including central nervous system, peripheral nervous system, skeletal muscle, and heart (Goldin, 2001; Wood and Baker, 2001). Most therapeutic Na+ channel blockers are not isoform-selective and may have more than one clinical application. For example, lidocaine and phenytoin are used as an LA and an anticonvulsant, respectively, but they are also used as class 1b antiarrhythmic agents. In addition, overdoses of Na+ channel blockers can be fatal. For example, cardiac arrhythmias and sudden death have been reported after accidental injection of bupivacaine into blood vessels. Studying the effects of ranolazine in various Na+ channel isoforms may therefore provide information regarding potential benefits and/or adverse effects in other excitable tissues. To date, the action of ranolazine has not been examined in Na+ channel isoforms other than the cardiac channel.
We set out to investigate whether ranolazine targets muscle Nav1.4 and neuronal Nav1.7 Na+ channel isoforms in a state-dependent manner and whether it elicits significant use-dependent block during repetitive pulses. We also studied the open Na+ channel block by ranolazine directly using inactivation-deficient Nav1.4 and Nav1.7 mutant Na+ channels (Wang et al., 2003b). We included blocking phenotypes of wild-type and, to a lesser extent, inactivation-deficient cardiac hNav1.5 Na+ channels for comparison. Earlier studies of the open Na+ channel block by ranolazine used the ΔKPQ and Y1795C hNav1.5 mutants (Fredj et al., 2006) as well as cardiac myocytes treated with ATX-II, a sea anemone toxin that increases persistent late Na+ currents (Song et al., 2004). However, most of these studies were hindered by the small size of the persistent late Na+ currents. Because inactivation-deficient rNav1.4 and hNav1.7 mutant channels used in this study had large persistent late Na+ currents, we were able to characterize for the first time the on- and off-rate kinetics regarding the ranolazine open-channel block.
Materials and Methods
Cultures of HEK293 Cells Stably Expressing Na+ Channels
Human embryonic kidney (HEK) 293 cell lines stably expressing rNav1.4 wild-type Na+ channels and inactivation-deficient Na+ channels (rNav1.4-L435W/L437C/A438W; L435L437A438→W435C437W438 or WCW mutant) were reestablished from frozen vials as described previously (Wang et al., 2004). The rat rNav1.4 clone was kindly provided by Dr. James Trimmer (University of California, Davis, CA). The rNav1.4-WCW construct and the phenotypes of this mutant were detailed previously (Wang et al., 2003b). Cultured HEK293 cells were maintained at 37°C in a 5% CO2 incubator in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Hy-Clone, Logan, UT), 1% penicillin and streptomycin solution (Sigma, St. Louis, MO), 3 mM taurine, and 25 mM HEPES (Invitrogen).
Na+ Channel cDNA, Mutant Constructs, and Transient Transfection of HEK293T Cells
We created an additional S6 mutant channel construct (rNav1.4-WCW/F1579K) in the rNav1.4-WCW cDNA background by site-directed mutagenesis as described previously (Wang et al., 2003b). This specific rNav1.4 residue (Phe1579) is located in the middle of the D4S6 segment and seems critical for LA binding (Ragsdale et al., 1994; Nau and Wang, 2004). The human hNav1.7 clone (Klugbauer et al., 1995) was kindly provided by Dr. Norbert Klugbauer (Albert-Ludwigs-Universität Freiburg, Freiburg, Germany). This Nav1.7 isoform is responsible for the excitability of peripheral nociceptive nerve fibers and has been implicated as an attractive target for novel analgesic drugs (Nassar et al., 2004; Wood et al., 2004). An inactivation-deficient hNav1.7-WCW mutant construct was created by triple substitutions at homologous positions (L396W/L398C/A399W). Human cardiac hNav1.5 Na+ channel was obtained from Dr. Roland Kallen (University of Pennsylvania, Philadelphia, PA), and its inactivation-deficient mutant construct, L409C/A410W (hNav1.5-CW), was created previously (Edrich et al., 2005). For transient transfection, HEK293T cells were grown to ~50% confluence and transfected by calcium phosphate precipitation. Transfection of mutant channels (5–10 μg) along with rat β1-pcDNA1 (10–20 μg) and reporter CD8-pih3m (1 μg) was adequate for current recording. The rat β1 subunit was included to increase the level of channel expression. Control experiments indicated that coexpression of the β1 subunit did not alter the binding affinities of ranolazine. Cells were replated 15 h after transfection in 35-mm dishes, maintained at 37°C in a 5% CO2 incubator, and used after 1 to 4 days. Transfection-positive cells were identified with immunobeads (CD8-Dynabeads; Dynal Biotech, Lake Success, NY).
Solutions and Chemicals
Cells were perfused with an extra-cellular solution containing 65 mM NaCl, 85 mM choline chloride, 2 mM CaCl2, and 10 mM HEPES (titrated with tetramethylammonium-OH to pH 7.4). The pipette (intracellular) solution consisted of 100 mM NaF, 30 mM NaCl, 10 mM EGTA, and 10 mM HEPES (titrated with cesium-OH to pH 7.2). Ranolazine-2HCl was purchased from Sigma (St. Louis, MO), dissolved in distilled water at 10 mM as stock solution, and stored at 4°C. Final drug concentrations up to 100 μM were made by serial dilution.
Electrophysiology and Data Acquisition
The whole-cell configuration of a patch-clamp technique (Hamill et al., 1981) was used to record Na+ currents in HEK293 cells at room temperature (22 ± 2°C). Electrode resistance typically ranged from 0.5 to 1.0 MΩ. Command voltages were elicited with pClamp9 software and delivered by Axopatch 200B (Molecular Devices, Sunnyvale, CA). Cells were held at −140 mV and dialyzed for 10 to 15 min before current recording. The capacitance and leak currents were cancelled with the patch-clamp device and by P/-4 subtraction. Liquid junction potential was not corrected. Peak currents at + 30 mV were 2 to 20 nA for the majority of cells. Access resistance was generally 1 to 2 MΩ under the whole-cell configuration; series resistance compensation of >85% typically resulted in voltage errors of ≤3 mV at +30 mV. Dose-response studies were mostly performed at ≤30 mV for the outward Na+ currents. Such recordings allowed us to avoid series resistance artifacts and to minimize changes in reversal potential resulting from inward Na+ ion loading (Cota and Armstrong, 1989). Curve-fitting was performed by OriginLab Corp. software (Northampton, MA). An unpaired Student’s t test was used to evaluate estimated parameters (mean ± S.E.M. or fitted value ± S.E. of the fit); P values of <0.05 were considered statistically significant.
Results
Resting and Inactivated Na+ Channel Block by Ranolazine
Voltage-gated Na+ channels have three distinct conformations: resting, open, and inactivated (Aldrich et al., 1983). To determine the blocking potency of ranolazine on the resting and inactivated rNav1.4 isoform, we used two different pulse protocols and applied the drug at various concentrations. Figure 2A shows that ranolazine even at 100 μM blocked less than 5% of peak rNav1.4 wild-type Na+ currents when the cell was held at −140 mV and stimulated by a brief test pulse at +30 mV once every 30 s. This weak blocking action of ranolazine was defined as the resting channel block. For the inactivated channel block, we used a conditioning pulse at −70 mV for 10s, which allowed ranolazine binding to reach its steady state (Wright et al., 1997); further increase in this 10-s duration would increase the slow inactivation in sodium channels. An interpulse of −140 mV for 95 ms was inserted before a brief +30 mV test pulse. During this interpulse, all drug-free Na+ channels recovered from their inactivated state. Figure 1B shows a greater block by 100 μM ranolazine, reaching ~60% peak current reduction. The reduction of peak currents at various concentrations was measured and used to construct the dose-response curves shown in Fig. 2C. The IC50 of ranolazine on inactivated rNav1.4 Na+ channels was estimated by the Hill equation with a value of 75.0 ± 3.5 μM (n = 6). Because a fraction of drug-bound inactivated Na+ channels could recover from block during the interpulse, we probably underestimated the inactivated block somewhat as shown before for lidocaine (Wright et al., 1997). Wild-type Nav1.5 and Nav1.7 Na+ channel isoforms were similarly examined using the identical protocols shown in Fig. 2, A and B, and their estimated IC50 values are listed in Table 1. The IC50 values of resting block by ranolazine in Na+ channel isoform could not be accurately estimated (Fig. 2C; dashed line), because they are larger than the concentration range used; whereas the inactivated block was ≥60 μM, or more than 10-fold above the therapeutic plasma concentration of ranolazine (2–6 μM) (Chaitman, 2006).
Fig. 2.
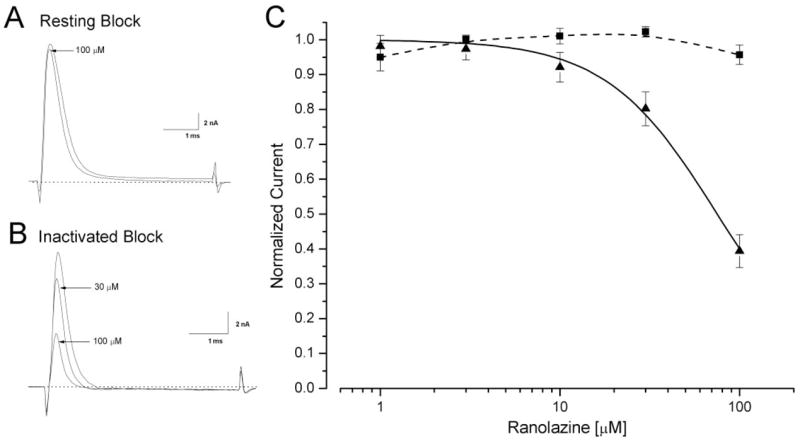
Block of resting and inactivated rNav1.4 Na+ channels at various ranolazine concentrations. A, representative current traces are superimposed before and after application of 100 μM ranolazine. Cells were held at −140 mV and received 5-ms test pulses of +30 mV at 30-s intervals. B, representative current traces are superimposed for ranolazine concentrations of 0, 30, and 100 μM. After a −70-mV conditioning pulse for 10 s, Na+ currents were evoked by the 5-ms test pulse at 30 mV, administered every 30 s. An interpulse of 95 ms at the holding potential was inserted to allow the recovery of the drug-free inactivated Na+ channels. C, a dose-response curve was constructed using data described in Fig. 2A (resting block: ■, n = 5) and 2B (inactivated block: ▲, n = 6). The peak current was measured, normalized to the control (0 μM), and plotted against the ranolazine concentration. The curve was fitted with the Hill equation (solid line). The IC50 value for resting block was >300 μM and could not be experimentally attained. The IC50 value for inactivated block was estimated at 75.0 ± 3.5 μM (Hill coefficient, 1.41 ± 0.10, n = 6).
Fig. 1.
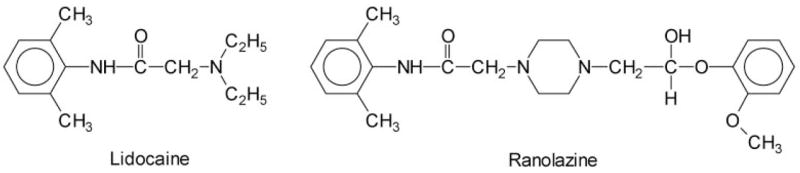
Chemical structures of ranolazine and lidocaine. Notice that ranolazine is larger and contains a lidocaine moiety.
TABLE 1. Estimates of IC50 values and Hill coefficients for resting, inactivated, and open states of Na+ channel isoforms.
Data were collected by pulse protocols described in Figs. 2, 6, and 9. Results were fitted with Hill equation; IC50 values are shown with Hill coefficients listed in parenthesis. The resting and inactivated Na+ channel block were determined in wild-type isoforms, whereas the peak and open sustained block were determined in inactivation-deficient rNav1.4-WCW, hNav1.5-CW, and hNav1.7-WCW mutant Na+ channels. Open-channel block of sustained rNav1.4-WCW/F1579K Na+ currents displayed a significant increase in its IC50 value compared with rNav1.4-WCW (P < 0.001). All data were derived from n = 5 except as indicated otherwise.
| Channel State | Wild-Type Channels
|
Mutant Channels
|
|||||
|---|---|---|---|---|---|---|---|
| rNav1.4 | HNav1.5 | hNav1.7 | rNav1.4-WCW | rNav1.4-WCW/F1579K | hNav1.5-CW | hNav1.7-WCW | |
| μM | μM | ||||||
| Resting | N.A. | N.A. | N.A. | 225.4 ± 16.3 (0.92 ± 0.05) | N.A. | 136.5 ± 196.2 (2.77 ± 12.15) | N.A. |
| Inactivated | 75.0 ± 3.5 (1.41 ± 0.10)a | 65.5 ± 9.4 (1.52 ± 0.32) | 59.9 ± 3.8 (1.16 ± 0.09) | ||||
| Open: Sustained | 2.4 ± 0.2 (1.15 ± 0.09) | 40.7 ± 1.3 (1.12 ± 0.04) | 6.2 ± 0.7 (1.28 ± 0.17) | 1.7 ± 0.1 (1.25 ± 0.08) | |||
N.A., not applicable for direct measurements, since their estimated IC50 values were >300 μM.
n = 6.
Use-Dependent Block of rNav1.4 Na+ Channels by Ranolazine at 100 μM
Because the open duration of Na+ channels is very brief, with a dwell time of ~1 ms (Aldrich et al., 1983), it is generally difficult to study the open channel block directly in wild-type Na+ channels. However, repetitive pulses often elicit use-dependent block of Na+ currents in the presence of LAs or class 1 antiarrhythmic agents. Figure 3A shows the use-dependent phenotype of rNav1.4 Na+ channels in the presence of 100 μM ranolazine. Peak currents were blocked during repetitive pulses of +50 mV for 10 ms applied at 5 Hz for a total of 60 pulses; steady-state block reached 77.2 ± 3.9% (n = 6). The development of this use-dependent block was rapid when 100 μM ranolazine was used, with a time constant of 3.43 pulses (Fig. 3B).
Fig. 3.
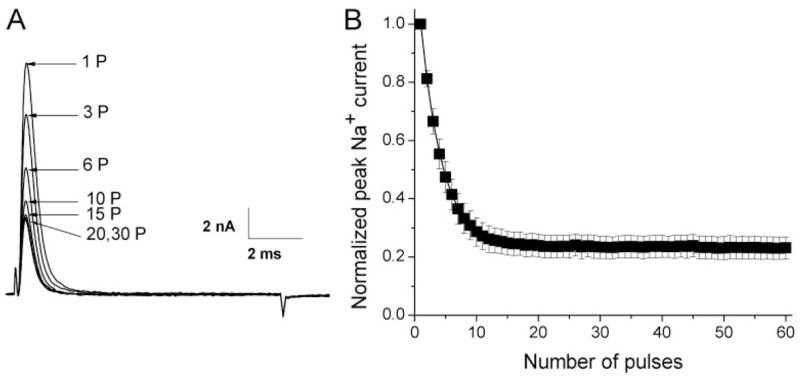
Use-dependent block of rNav1.4 wild-type Na+ currents by 100 μM ranolazine during repetitive pulses. A, repetitive pulses at + 50 mV for 10 ms were applied at 5 Hz for a total of 60 pulses. Representative traces of Na+ currents were recorded in the presence of 100 μM ranolazine; the number near the trace indicates the corresponding pulse number applied. B, peak currents were measured, normalized with respect to the peak amplitude at 1P, and plotted against the corresponding pulse. The curve (solid line) was best fitted by an exponential function with a time constant of τ = 3.43 ± 0.30 pulse (n = 6).
We then investigated the role of the pulse duration on use-dependent block by varying the duration from 0.5 to 20 ms. Figure 4A shows that repetitive pulses even with a duration as short as 1 ms elicits a nearly maximal use-dependent block of Na+ currents by 100 μM ranolazine compared with the duration of 10 ms shown in Fig. 3A. The level of the use-dependent block of Na+ currents was measured after repetitive pulses with different pulse duration, and plotted against the pulse duration (Fig. 4B). Significant use-dependent block was detected with a pulse duration of 0.5 ms. Maximal use-dependent block was achieved for a pulse duration as short as 2 ms and remained constant for duration of 4 to 20 ms (Fig. 4B). Without ranolazine, repetitive pulses produce little or no reduction of the peak currents. This result indicates that ranolazine blocked the open Na+ channel preferentially during repetitive pulses. The inactivated state of rNav1.4 Na+ channels did not seem to play a significant role in this use-dependent phenomenon; otherwise, longer pulse duration should enhance ranolazine binding and generate a larger use-dependent block.
Fig. 4.
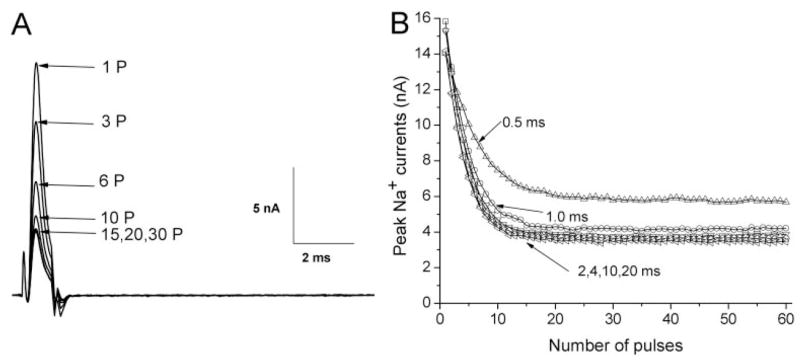
Duration dependence of the use-dependent block of rNav1.4 wild-type Na+ currents induced by 100 μM ranolazine. A, repetitive pulses were applied as described in Fig. 3A, except that the pulse duration was shortened to 1 ms. Representative traces of Na+ currents were recorded and labeled with the corresponding pulse number. B, repetitive pulses with durations of 0.5, 2, 4, 10, and 20 ms were applied, and Na+ currents were recorded as shown in Fig. 4A. Peak currents were measured and plotted against the pulse number. Notice that the block reaches the same level with pulse duration as short as 2 ms. Data in A and B were obtained from the same cell. Comparable results were recorded in 5 cells.
Use-Dependent Block by 100 μM Ranolazine in Various Na+ Channel Isoforms
Under identical ionic conditions, we asked whether the use-dependent phenotype induced by ranolazine could be present in other Na+ channel isoforms. Figure 5A shows that the use-dependent phenotype of the hNav1.7 neuronal Na+ channel isoform is nearly identical to that of the rNav1.4 muscle Na+ channel isoform. For comparison, the pulse duration was set at 10 ms. The steady-state block during repetitive pulses reached 83.3 ± 1.9% (n = 6) for the hNav1.7 isoform. For the hNav1.5 cardiac Na+ channel isoform, the steady-state block reached 66.8 ± 4.4% (n = 6) (Fig. 5B). These results demonstrated that ranolazine elicited a similar use-dependent phenotype in various Na+ channel isoforms, with a ranking order of hNav1.7 > rNav1.4 > hNav1.5. The reason for variations in the level of use-dependent block is unclear but could be related to gating differences in isoforms.
Fig. 5.
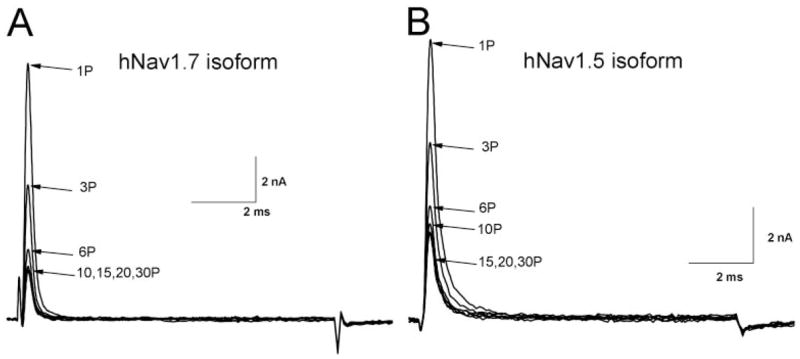
Use-dependent block of wild-type hNav1.7 and hNav1.5 Na+ channels by 100 μM ranolazine. Repetitive pulses of + 50 mV for 10-ms at 5 Hz were applied in the presence of 100 μM ranolazine. Representative current traces of wild-type hNav1.7 (A) and hNav1.5 (B) were recorded and superimposed, with the label indicating the corresponding pulse number. The degree of the steady-state block of each isoform by ranolazine after repetitive pulses is given under Results.
Time-Dependent Block in rNav1.4 Inactivation-Deficient Na+ Currents by Ranolazine
To assess the block of open Na+ channels by ranolazine directly, we chose inactivation-deficient rNav1.4-WCW mutant Na+ channels. Without the drug, most inactivation-deficient Na+ currents were maintained during a 50-ms depolarization at +30 mV (Fig. 6A). To construct the dose-response curve, we measured the Na+ current sustained at the end of the 50-ms pulse under various ranolazine concentrations, normalized with respect to the current amplitude without drug, and plotted against the corresponding drug concentration (Fig. 6B). The estimated IC50 value for this open-channel block was 2.4 ± 0.2 μM (□; n = 5) with a Hill coefficient of 1.15 ± 0.09 (n = 5). Reduction in peak currents by ranolazine was also measured for the resting block and plotted against the concentration (■); this procedure yielded an estimated IC50 value of 225.4 ± 16.3 μM with a Hill coefficient of 0.92 ± 0.05. It is noteworthy that we probably overestimated the inhibition for the resting block (Fig. 6B, dashed line) of inactivation-deficient Na+ channels, because the ranolazine open-channel block clearly occurred before the current trace reached its peak (Fig. 6A).
Fig. 6.
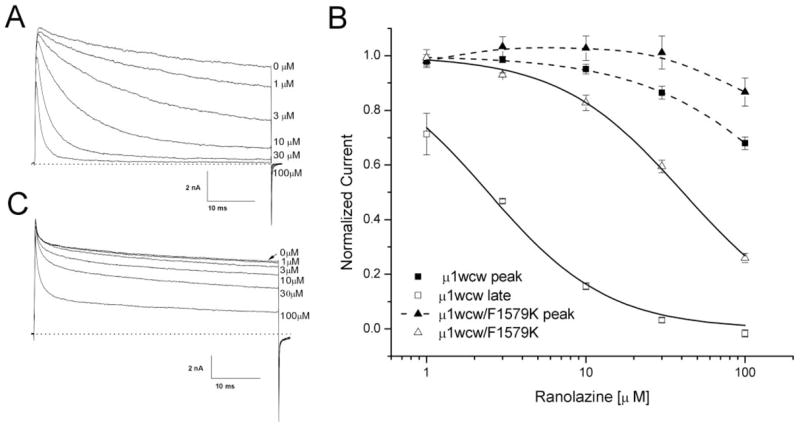
Block of inactivation-deficient rNav1.4-WCW and rNav1.4-WCW/F1579K Na+ channels. A, representative current traces of inactivation-deficient rNav1.4-WCW Na+ channel block were superimposed at various ranolazine concentrations. Cells were held at −140 mV and received 50-ms test pulses of +30 mV at 30-s intervals. B, a dose-response curve was constructed from the data presented in Fig. 6A. Both peak (■) and late (□) currents were measured, normalized to the control (0 μM), and plotted against ranolazine concentration. The curve was fitted with the Hill equation (solid lines). The IC50 value for the peak current block was estimated 225.4 ± 16.3 μM (Hill coefficient, 0.92 ± 0.05) (n = 5) and the IC50 value for rNav1.4-WCW of the late current was 2.4 ± 0.2 μM (1.15 ± 0.09) (n = 5). C, representative current traces of rNav1.4-F1579K inactivation-deficient Na+ channel block were superimposed at various ranolazine concentrations. Currents were evoked using the pulse-protocol represented in A. Peak and late Na+ currents were measured, normalized, and plotted against concentration as shown in B (triangles). The estimated IC50 value for rNav1.4-F1579K inactivation-deficient channel block of the late current was 40.8 ± 1.3 μM (Hill coefficient, 1.12 ± 0.04) (n = 5).
Fredj et al. (2006) found that ranolazine block of sustained cardiac Na+ currents was significantly reduced by mutation of hNav1.5-F1760A, which was reported to form the receptor for LAs. To investigate whether the open-channel block by ranolazine in the rNav1.4 Na+ channel takes place through the LA receptor, we created an inactivation-deficient rNav1.4-WCW/F1579K mutant. Figure 6C shows the reduced ranolazine block of rNav1.4-WCW/F1579K currents at various drug concentrations. The steady-state block of the peak and the sustained mutant currents was measured and plotted against the concentration (Fig. 6B, △, ▲, and Table 1). The IC50 of rNav1.4-WCW/F1579K was 16.7-fold larger than that of rNav1.4-WCW (40.7 ± 1.3 μM, n = 5 versus 2.4 ± 0.2 μM, n = 5). This result indicates that the ranolazine block in inactivation-deficient rNav1.4-WCW mutant Na+ channels occurs through the LA receptor.
Kinetic Parameters of Ranolazine Open-Channel Block
With ranolazine, we clearly observed a conspicuous decaying of inactivation-deficient Na+ currents. This time-dependent block of Na+ currents was also concentration-dependent; the higher the concentration, the faster the time-dependent block. After normalization to correct the intrinsic current decay, the rate of the remaining current decay at each drug concentration was well fitTED by a single exponential function. The 1/τ values were plotted against the ranolazine concentration and fitted by a linear regression (Fig. 7). The y-intercept yielded a value of 22 ± 2 s−1 (n = 5), which corresponded to the dissociation time constant, k−1, for ranolazine. The slope was 8.2 ± 0. 2 μM−1 s−1 (n = 5), which corresponded to the association time constant, k1. As a result, the equilibrium dissociation constant (KD) was estimated to be 2.68 μM (KD = k−1/k1). This calculated KD value is very close to the IC50value (2.4 ± 0.2 μM) derived from the dose-response curves (Fig. 6B, Table 1).
Fig. 7.
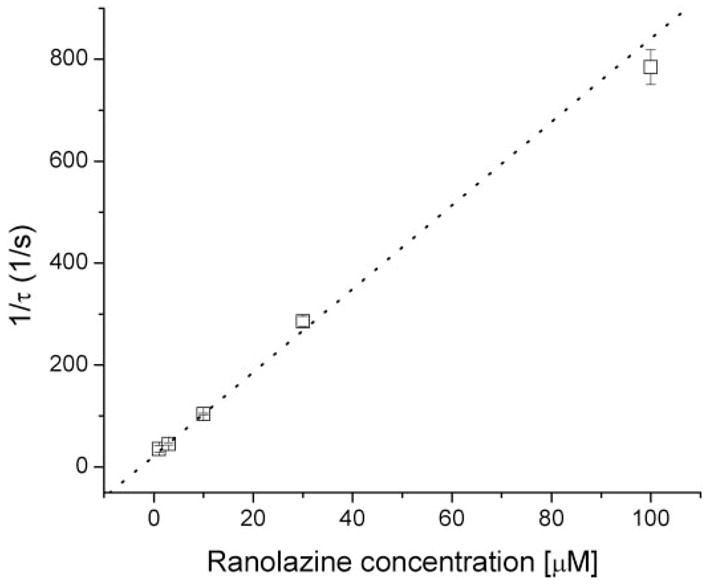
Time-dependent block of inactivation-deficient rNav1.4-WCW Na+ currents at various ranolazine concentrations. The decaying phase of Na+ currents in the presence of ranolazine shown in Fig. 6A were each normalized with respect to the control current trace without drug and fitted by a single exponential function. Normalization was applied to remove the slow-decaying component found in the control. The inverse of the time constant, τ, was plotted against ranolazine concentration (y-intercept: 22.1 ± 2.3, n = 5; slope: 8.2 ± 0.2; linear correlation coefficient r = 0.997).
Recovery Time Course from the Time-Dependent Block of RNav1.4 Inactivation-Deficient Na+ Currents by Ranolazine
After the time-dependent block by 100 μM ranolazine developed fully during a +50-mV/50-ms conditioning pulse, the recovery time course from this open-channel block at the holding potential was measured with various time intervals (Fig. 8A, inset). Figure 8A shows the current traces corresponding to various time intervals. The recovery time course can be fitted with a single exponential function (Fig. 8B, solid line) with a time constant of 558.2 ± 40.7 ms (n = 5).
Fig. 8.
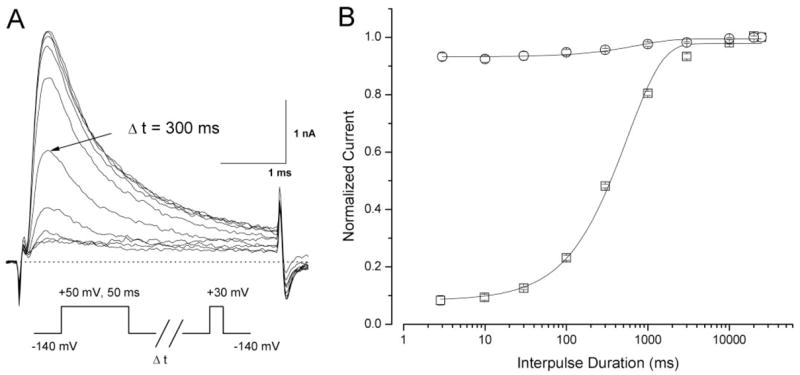
Recovery time course from ranolazine open-channel block at 100 μM. A, a conditioning pulse of +50 mV for 50 ms was applied to induce the time-dependent block of rNav1.4-WCW Na+ currents at 100 μM ranolazine. Representative traces of rNav1.4-WCW Na+ currents were evoked by a +30-mV test pulse after an interpulse at −140 mV with an increasing duration and superimposed. The pulse protocol is shown in the inset. B, normalized peak current after drug treatment (100 μM ranolazine; squares) was plotted against the interpulse duration and fitted by an exponential function with a τ value of 558.2 ± 40.7 ms (n = 5). The control data before drug treatment (circles; n = 5) was shown for comparison; most currents (>90%) reappeared within 3 ms.
Time-Dependent Block in hNav1.7 and hNav1.5 Inactivation-Deficient Na+ Currents by Ranolazine
To determine whether other inactivation-deficient Na+ isoforms display similar ranolazine-blocking phenotypes, we studied homologous inactivation-deficient mutant Na+ channels first using the hNav1.7 clone. Figure 9A shows that ranolazine induces time-dependent block of inactivation-deficient hNav1.7-WCW Na+ currents in a manner comparable with that shown in Fig. 6A for inactivation-deficient rNav1.4-WCW mutant channels. At higher ranolazine concentrations, again, the rates of the time-dependent block are proportionally faster. The 1/τ values versus ranolazine concentrations also follow a linear relationship (Fig. 9B). The k−1 and k1 values are calculated to be 14.1 ± 1.1 s−1 and 7.1 μM−1 s−1, respectively, with a calculated KD value of 1.99 μM. This KD value is comparable with the IC50 value determined from the dose-response curve (1.7 ± 0.1 μM, n = 5; Table 1). Our data thus demonstrate that ranolazine also targets preferentially the open state of the hNav1.7 Na+ channel isoform. For the inactivation-deficient cardiac hNav1.5-CW Na+ channels, the IC50 value was estimated as 6.2 ± 0.7 μM (n = 5; Table 1). However, ranolazine seemed to increase the hNav1.5-CW inactivation-deficient Na+ currents at 1 μM in approximately 50% of cells. Such unusual increase of inactivation-deficient cardiac Na+ currents made the IC50 estimate less reliable. The increase in peak cardiac Na+ currents by ranolazine was puzzling but resembled earlier hNav1.5-CW experiments using cells treated with low concentrations of propaferone (Edrich et al., 2005).
Fig. 9.

Time-dependent block of inactivation-deficient hNav1.7-WCW Na+ currents at various ranolazine concentrations. A, representative traces of inactivation-deficient hNav1.7-WCW Na+ currents were superimposed before and after application of ranolazine at various concentrations. Cells were held at −140 mV and received 5-ms test pulses of +30 mV at 30-s intervals. B, the decaying phase of Na+ currents at various ranolazine concentrations were each normalized with the control current without drug and fitted with a single exponential function as described in Fig. 7. Normalization was necessary because of the presence of a slow-decaying component in the control trace. The inverse of the time constant, τ, was plotted against ranolazine concentration (y-intercept, 14.1 ± 1.1 (n = 5); slope, 7.1 ± 0.5; r = 0.986).
Discussion
We investigated the common actions of ranolazine on muscle Nav1.4 and neuronal Nav1.7 Na+ channel isoforms heterologously expressed in HEK293T cells. We found that ranolazine block is highly state- and use-dependent in these Na+ channel isoforms. In particular, ranolazine seemed to target the open state of Na+ channels selectively with IC50 values of 1.7 to 2.4 μM, which are within its therapeutic plasma concentrations (2–6 μM) (Chaitman, 2006) but 10-fold lower than IC50 values for the inactivated states (≥60 μM). In contrast, resting block of Na+ channel isoforms by ranolazine is minimal (Table 1; ≫100 μM). The significance of common state- and use-dependent block by ranolazine in Nav1.4 and Nav1.7 Na+ channel isoforms is discussed below.
Ranolazine Block of Na+ Channels Occurred via the Local Anesthetic Receptor
Ranolazine is a derivative of lidocaine (Fig. 1), with its N-di-ethyl group cross-linked by an amine group to form a piperazine ring. The molecular weight of ranolazine (427.5) is 1.82-fold higher than that of lidocaine (234.3). Lidocaine is commonly used as an LA, and its receptor has been well studied in various Na+ channel isoforms (Catterall and Mackie, 2001; Nau and Wang, 2004). The LA receptor is formed by residues from multiple S6 segments and seems highly conserved among Na+ channel isoforms. Site-directed mutagenesis of residue rNav1.4-F1579K at D4S6 suggests that ranolazine interacts with the phenylalanine residue at the D4S6 segment (Fig. 6, B and C), which forms a part of the LA receptor. Alternatively, the open state of this F1579K mutant could become less accessible for ranolazine, because lysine substitution of Phe1579 reduced the single channel conductance (McNulty et al., 2007). This interpretation is less likely because substitution of alanine at the same site also reduces ranolazine binding drastically (Fredj et al., 2006). It is perhaps not surprising that ranolazine with a lidocaine moiety will target the conserved LA receptor in Na+ channel isoforms. However, because ranolazine is much larger than lidocaine, additional interactions between ranolazine and the Na+ channel likely occur beyond the lidocaine binding site. These additional contacts will require further studies using site-directed mutagenesis.
Ranolazine Targetted the Open Na+ Channel Preferentially, whereas Lidocaine Targetted Both the Open and the Inactivated Na+ Channel
With respect to the IC50 values for the open rNav1.4 Na+ channel, ranolazine (2.43 μM) is 8.6-fold more potent than lidocaine (20.9 μM) (Wang et al., 2004), indicating that the open Na+ channel reacts more strongly with ranolazine than with lidocaine. Block of sustained currents by ranolazine in Nav1.4, Nav1.5, and Nav1.7 Na+ channel isoforms has a Hill coefficient close to unity (ranging from 1.12 to 1.28; Table 1). Recovery of the open-channel block by ranolazine is much slower than that of lidocaine at the holding potential (Fig. 8B; τ = 558 ms versus 292 ms) (Wang et al., 2004), demonstrating that ranolazine dissociates more slowly from its receptor in the inactivation-deficient mutant channel upon repolarization than lidocaine does.
For the inactivated rNav1.4 Na+ channel block, however, ranolazine is weaker (by a factor of 3.9-fold) than lidocaine, with IC50 values of 75 and 19 μM, respectively. The ratio of the IC50 value for the inactivated block over the IC50 value for the open-channel block by ranolazine and lidocaine is 31-and 0.9-fold for rNav1.4 Na+ channels, respectively. This large potency ratio of the inactivated- over the open-channel block by ranolazine is unique and is very different from that of LAs (Hille, 1977). We do not know why the inactivated Na+ channel is resistant to ranolazine block. One possibility is that ranolazine has no direct access to its receptor when the channel is in closed, inactivated configuration. This is possible, because ranolazine is larger than traditional LAs, which may gain access to their receptor within the inner cavity via the hydrophobic pathway even when the channel is in the closed state (Hille, 2001). Such hydrophobic pathway may be relatively narrow and therefore greatly limit the access of ranolazine.
Ranolazine Block of Na+ Channels Was Strongly Use-Dependent during Repetitive Pulses
All wild-type Na+ channel isoforms studied in this report exhibited significant use-dependent block by 100 μM ranolazine during repetitive pulses. Because ranolazine targetted the open state of Na+ channels selectively, the use-dependent phenotype was therefore probably due to the block of the open Na+ channel, produced cumulatively during repetitive pulses. The on-rate (8.2 μM−1 s−1) and the off-rate (22 s−1) for the open-channel block of inactivation-deficient rNav1.4 mutant channels yields the KD value of 2.68 μM, which was close to the IC50 value for ranolazine block of persistent late Na+ currents (Fig. 6). We used a high concentration of ranolazine to study its use-dependent phenotype because of its slow on-rate. At lower ranolazine concentrations, the level of this use-dependent block was much lower (Fredj et al., 2006). The involvement of inactivated channels in this use-dependent phenotype should be minimal, judging from the potency ratio of 31 between the open and inactivated block by ranolazine. Another finding strongly supporting this contention is that the pulse duration required to elicit significant use-dependent block was relatively short (Fig. 4); a 2-ms duration is sufficient to generate maximal use-dependent block of Na+ currents. We therefore conclude that the cumulative use-dependent block found in wild-type Na+ channel isoforms (Fig. 3, 5) by ranolazine is due primarily to the accumulation of the open-channel block during repetitive pulses.
Potential Therapeutics versus Adverse Effects of Drugs That Target Na+ Channel Isoforms Nonselectively: Kinetic Considerations
Because ranolazine preferentially blocks sustained cardiac Na+ currents, Fredj et al. (2006) suggested that ranolazine may provide a distinct advantage in prevention and treatment of cardiac arrhythmias. Small persistent late Na+ currents have also been found in various excitable tissues, either due to tissue injury (e.g., reperfusion, stroke, or nerve injury) or Na+ channel genetic defects (LQT-3 syndromes, hyperkalemic periodic paralysis, severe myoclonic epilepsy of infancy, or inherited erythromelalgia). Ranolazine block of the open hNav1.7 and rNav1.4 Na+ channel isoforms has IC50 values of 1.7 and 2.4 μM, respectively, which are smaller than the 6.2 μM found for hNav1.5 (Table 1) and are at the lower end of the therapeutic plasma concentration range of 2 to 6 μM. This finding suggests that the open states of many other Na+ channel isoforms could also be the target of ranolazine. As a result, ranolazine may have clinical applications beyond its current use for chronic angina.
Most LAs and class 1 antiarrhythmic agents are known to target both the open and inactivated Na+ channels. However, few Na+ channel blockers target the open state of Na+ channels selectively. For example, flecainide seems to block the open Na+ channel exclusively, with minimal interactions with its resting and inactivated states (Wang et al., 2003a; Ramos and O’leary, 2004). The on- and off-rates of flecainide for the open rNav1.4 Na+ channel are 14.9 μM−1 s−1 and 12.2 s−1, respectively, with a calculated KD value of 0.82 μM. For comparison, we found that the ranolazine on-rate value (8.2 μM−1 s−1) for the open rNav1.4 Na+ channel is 1.8-fold slower than that for flecainide under identical ionic conditions. Because ranolazine and flecainide have a similar molecular weight (427.5 versus 414.3), the slower on-rate for ranolazine could be due to its unique three-dimensional structure. Alternatively, the open Na+ channel pore could have a “cut-off” dimension for drugs that target the LA receptor within the inner cavity. In other words, drugs slightly larger than flecainide may have a significantly slower on-rate because of the narrow open Na+ channel pore.
Ranolazine was shown to restore normal cardiac action potentials after the removal of the persistent late Na+ currents at its therapeutic concentration range (Belardinelli et al., 2006). Noble and Noble (2006) suggested that an ideal therapeutic cardiac Na+ channel blocker should block only the late component of sodium currents, because block of peak cardiac Na+ currents would itself promote arrhythmia by slowing conduction and facilitating re-entry. In principle, the possibility of adverse effects increases significantly if a drug has rapid access to the normal open channel during the initial phase of action potentials. Therefore, the slow action of ranolazine may be preferable to the action of other fast-open Na+ channel blockers, because it may leave the initial action potential intact but will attenuate the following high-frequency discharges found under pathological conditions. Although these features may grant ranolazine a greater safety margin, we stress that extrapolation of kinetic results to clinical efficacy must be made cautiously, as ranolazine may also have a striking selectivity for the block of atrial cardiac sodium channels (Burashnikov et al., 2007).
Acknowledgments
We thank Drs. Roland Kallen, Norbert Klugbauer, and James Trimmer for kindly providing hNav1.5, hNav1.7, and rNav1.4 Na+ channel clones, respectively.
This work was supported by National Institutes of Health grant GM48090 (to G.K.W. and S.-Y.W.).
ABBREVIATIONS
- LA
local anesthetic
- rNav1.4
the rat skeletal muscle Na+ channel
- hNav1.5
the human cardiac Na+ channel
- hNav1.7
the human neuronal type 1.7 Na+ channels
- HEK
human embryonic kidney
- rNav1.4-WCW
L435W/L437C/A438W mutant
Footnotes
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
References
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Belardinelli L, Wu L, Fraser H, Zygmunt AC, Burashnikov A, Diego JM, Fish JM, Cordeiro JM, Goodrow RJ, Jr, et al. Electrophysiologic properties and antiarrhythmic actions of a novel antianginal agent. J Cardiovasc Pharmacol Ther. 2004;9(Suppl 1):S65–S83. doi: 10.1177/107424840400900106. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. Ion Channels and Disease. Academic Press; London: 2000. Voltage-gated Na+ channels; pp. 67–96. [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl 4):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC. Ion-channel defects and aberrant excitability in myotonia and periodic paralysis. Trends in Neurosci. 1996;19:3–10. doi: 10.1016/0166-2236(96)81859-5. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Mackie K. Local anesthetics. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. Macmillan Publishing Company; New York: 2001. pp. 367–384. [Google Scholar]
- Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation. 2006;113:2462–2472. doi: 10.1161/CIRCULATIONAHA.105.597500. [DOI] [PubMed] [Google Scholar]
- Cota G, Armstrong CM. Sodium channel gating in clonal pituitary cells: the inactivation step is not voltage dependent. J Gen Physiol. 1989;94:213–232. doi: 10.1085/jgp.94.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edrich T, Wang SY, Wang GK. State-dependent block of human cardiac HNav1.5 sodium channels by propafenone. J Membr Biol. 2005;207:35–43. doi: 10.1007/s00232-005-0801-4. [DOI] [PubMed] [Google Scholar]
- Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty E, Neher ME, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sinauer Associates Inc; Sunderland, MA: 2001. Classical mechanisms of block; pp. 503–536. [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty MM, Edgerton GB, Shah RD, Hanck DA, Fozzard HA, Lipkind GM. Charge at the lidocaine binding site residue phe-1759 affects permeation in human cardiac voltage-gated sodium channels. J Physiol. 2007;581:741–755. doi: 10.1113/jphysiol.2007.130161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101:12706–12711. doi: 10.1073/pnas.0404915101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92(Suppl 4):iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepine CJ, Wolff AA. A controlled trial with a novel anti-ischemic agent, ranolazine, in chronic stable angina pectoris that is responsive to conventional antianginal agents. Ranolazine Study Group. Am J Cardiol. 1999;84:46–50. doi: 10.1016/s0002-9149(99)00190-3. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Ramos E, O’leary ME. State-dependent trapping of flecainide in the cardiac sodium channel. J Physiol. 2004;560:37–49. doi: 10.1113/jphysiol.2004.065003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schram G, Zhang L, Derakhchan K, Ehrlich JR, Belardinelli L, Nattel S. Ranolazine: ion-channel-blocking actions and in vivo electrophysiological effects. Br J Pharmacol. 2004;142:1300–1308. doi: 10.1038/sj.bjp.0705879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- Wang GK, Russell C, Wang SY. State-dependent block of wild-type and inactivation-deficient Na+ channels by flecainide. J Gen Physiol. 2003a;122:365–374. doi: 10.1085/jgp.200308857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S-Y, Bonner K, Russell C, Wang GK. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated Sodium Channels. Biophys J. 2003b;85:911–920. doi: 10.1016/S0006-3495(03)74530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S-Y, Mitchell J, Moczydlowski E, Wang GK. Block of inactivation-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway. J Gen Physiol. 2004;124:691–701. doi: 10.1085/jgp.200409128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JN, Baker M. Voltage-gated sodium channels. Curr Opin Pharmacol. 2001;1:17–21. doi: 10.1016/s1471-4892(01)00007-8. [DOI] [PubMed] [Google Scholar]
- Wood JN, Boorman JP, Okuse K, Baker MD. Voltage-gated sodium channels and pain pathways. J Neurobiol. 2004;61:55–71. doi: 10.1002/neu.20094. [DOI] [PubMed] [Google Scholar]
- Wright SN, Wang S-Y, Kallen RG, Wang GK. Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophys J. 1997;73:779–788. doi: 10.1016/S0006-3495(97)78110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]