

Abstract
Amelogenin is the predominant protein found during enamel development and is thought to be the biomineralization protein controlling the unique elongated hydroxyapatite crystals that constitute enamel. The secondary structure of biomineralization proteins is thought to be important in the interaction with hydroxyapatite. Unfortunately, very little data are available on the structure or the orientation of amelogenin, either in solution or bound to hydroxyapatite. The C-terminus contains the majority of the charged residues and is predicted to interact with hydroxyapatite; thus, we used solid-state NMR dipolar recoupling techniques to investigate the structure and orientation of the C-terminus of LRAP, a naturally occurring splice variant of full-length amelogenin. Using 13C{15N} Rotational Echo DOuble Resonance (REDOR), the structure of the C-terminus was found to be largely random coil, both on the surface of hydroxyapatite as well as lyophilized from solution. The orientation of the C-terminal region with respect to hydroxyapatite was investigated for two alanine residues (Ala46 and Ala49) using 13C{31P} REDOR and one lysine residue (Lys52) using 15N{31P} REDOR. The residues examined were found to be 7.0, 5.7, and 5.8 Å from the surface of hydroxyapatite for Ala46, Ala49, and Lys52, respectively. This provides direct evidence that the charged C-terminus is interacting closely with hydroxyapatite, positioning the acidic amino acids to aid in controlling crystal growth. However, solid-state NMR dynamics measurements also revealed significant mobility in the C-terminal region of the protein, in both the side chains and the backbone, suggesting that this region alone is not responsible for binding.
INTRODUCTION
Biominerals such as bone, enamel, and mollusk shells are formed with unusual strength and morphological properties when compared to their mineral counterparts. Although the proteins present during formation have been found to be critical in the resulting biominerals, it is not clear how the proteins interact with the crystal surface and what role protein structure plays in this interaction, though secondary structure is thought to be critical to protein affinity and the ultimate biomineral properties (1–5). Enamel, in particular, is a fascinating noncollagen-based, extracellular biomineral that results in hydroxyapatite (HAP) crystals that are ∼3000 times longer than those found in bone (6). It has been demonstrated that amelogenin, the predominant protein found during enamel formation (>90%), is essential in proper enamel formation (7–12), yet how this control is achieved is not well understood.
Amelogenin is a 180-residue protein, and unlike other biomineralization proteins, it is very hydrophobic, having only eight acidic and five basic amino acids in the entire sequence, located almost entirely in the N- and C-termini (Table 1) (13). Experimental studies indirectly demonstrated the importance of the charged C-terminus in HAP interactions when C-terminal deleted proteins showed reduced HAP interactions (14,15) and growth inhibition (15,16). Leucine-rich amelogenin protein (LRAP) is a naturally occurring 59-residue splice variant of amelogenin that includes only the charged N- and C-termini (Table 1) (17) from the full protein. The sequence conservation of these regions among species in the full protein (18–20) and the combination of the only likely protein-HAP interacting regions into a reduced peptide suggest that this protein may have a significant, though as yet unidentified, function of its own in enamel development. LRAP has not been as well studied as the parent protein, but is seen to be a strong HAP crystal growth inhibitor (20,21), and LRAP and full-length amelogenin were both found to bind the same number of calcium ions from solution (22), providing further support that the regions comprising LRAP are responsible for the interaction of the full-length protein with HAP. In addition to a possible functional role of its own, the truncated LRAP protein serves as an excellent model for the full protein because of its sequence homology with full-length amelogenin and its conservation of residues with the potential to interact with HAP. Additionally, its smaller size allows easy incorporation of selective isotopic labels, which is not possible using the recombinant techniques necessary for larger proteins, enabling experiments which allow the site-specific, molecular-level determination of protein structure and protein-surface interactions for this important class of proteins.
TABLE 1.
Primary structures of amelogenin and LRAP
| LRAP | MPLPPHPGSPGYINLpSYEVLTPLKWYQSMIRQP PLSPILPELPLEAWPATDKTKREEVD |
| Amelogenin | MPLPPHPGSPGYINLpSYEVLTPLKWYQSMIRQP#PLSPILPELPLEAWPATDKTKREEVD |
| #YPSYGYEPMGGWLHHQIIPVLSQQHPPSHTLQPHHHLPVVPAQQPVAPQQPMMPVPGHHSMTPTQHHQPN | |
| IPPSAQQPFQQPFQPQAIPPQSHQPMQPQSPLHPMQPLAPQPPLPPLFSMQ |
Note the conservation of the primary structure of the N- and C-termini in LRAP. The central portion of amelogenin is indicated by #. Charged residues are shown in bold type.
Both LRAP (21,23) and amelogenin (24,25) are seen to form into nanospheres comprised of multiple monomers, which is thought to play a role in the function of the protein and has been studied in some detail. The secondary structure of the protein is likely very important in both the ultimate quaternary structure as well as the resulting function of amelogenin. Unfortunately, very little quantitative secondary structural information has been available in past years due to the lack of available experimental techniques for large, noncrystallizable proteins such as amelogenin (26,27). The importance of the structure of amelogenin to its function is perhaps best highlighted by the fact that in two separate cases, X-linked amelogenesis imperfecta (AIH1) with single amino acid modifications (Tyr21 with Ile or Pro41 with Thr) (12) results in dramatically malformed enamel. Clearly, the protein-crystal interaction has been altered due to these mutations. The observation that the quaternary structure is altered for these mutants (28,29) would suggest that either the secondary or tertiary (or both) structures are also disrupted, but have not yet been investigated. This physiological example demonstrates the critical need for secondary structural information in understanding protein-surface interactions that control biomineralization processes. Infrared (30), circular dichroism (CD) (30,31), solution NMR (32), small-angle neutron scattering (33), and Raman (34) spectroscopic techniques have all been used to look at the secondary structure of full-length amelogenin, but have only provided a very qualitative insight into the overall structure. The results of these studies suggest that the solubilized protein contains some β-sheet and some random-coil regions with little α-helical structure, though determination of the location of these structural motifs within the protein is not possible with most of these techniques. The secondary structure of LRAP was studied by solution-state 1H NMR and CD at low pH and was observed to be random coil under these conditions. The authors were unable to maintain enough solubility to repeat the NMR experiment at more biologically relevant pH, so a quantitative solution structure is unavailable, though the CD spectrum only changed modestly at pH 7.4 (22). Significantly, all of these studies focus on the solubilized protein, not on the protein bound to hydroxyapatite, its functional form in controlling crystal growth processes.
Solid-state NMR has recently been demonstrated as a technique providing quantitative, site-specific structural (35–40) and orientation (41,42) information for biomineralization proteins under biologically relevant conditions both off the surface and bound to HAP and is not limited by the formation of nanospheres. Recently, using solid-state NMR (41) and neutron reflectivity (43), we demonstrated that the C-terminus of LRAP is close enough to hydroxyapatite to control mineral growth, providing some of the first direct evidence of the importance of the charged C-terminus in the protein-crystal interaction. This work extends those studies to investigate not only the orientation of additional residues within the C-terminus but also the secondary structure of the protein in this important interaction region. In this work, we utilized Rotational Echo DOuble Resonance (REDOR) to study the secondary structure and orientation of LRAP under biologically relevant conditions. In addition to structural and orientation studies, protein dynamics ranging from 10−3 to 10−5 s were measured using relaxation measurements, cross-polarization efficiencies, and a Herzfeld Berger (HB) analysis to characterize the mobility of LRAP on HAP, an important component in understanding the protein-mineral interface.
EXPERIMENTAL METHODS
Materials
Labeled amino acids were purchased from Cambridge Isotopes (Andover, MA) and used as received. Solvents were used without further purification. Fmoc-protected labeled amino acids were prepared according to standard procedures (44,45). Briefly, 5.6 mmol amino acid was stirred with 30 mL 10% warm Na2CO3 in a 100-mL round-bottom flask. Fmoc-O-succinimide (6.6 mmol) was added in 20 mL dioxane and stirred. After 24 h, 1.7 mmol Fmoc-O-succinimide was added and the solution was stirred for an additional 24 h. After this stirring, 150 mL of water was added to the solution, which was then washed three times with ether. Then, 150 mL of ethyl acetate was added and the pH was adjusted to 2 with concentrated HCl. The ethyl acetate was washed two times with 1 M HCl and two times with water, after which it was dried over magnesium sulfate and removed under vacuum. The purity of the residual solid was checked with thin-layer chromatography (10:1 toluene/acetic acid) and the amino acid was used without further purification.
Protein preparation, purification, and characterization
Proteins were prepared using standard Fmoc chemistry by United Biochemical Research (Seattle, WA). Proteins were purified using prep-scale reverse-phase high-performance liquid chromatography: buffer A, 0.1% trifluoroacetic acid in water; buffer B, 0.1% trifluoroacetic acid in acetonitrile. LRAP eluted at 54% B. Proteins were analyzed for molecular weight and purity using electrospray mass spectroscopy. Previously, the effect of the phosphoserine at position 16 was investigated to confirm that it was not in close proximity to the C-terminal residue being measured, both off and on the surface (41). Since the alteration of the phosphoserine to serine did not affect the orientation of the C-terminus for LRAP-A46, LRAP-A49, and LRAP-K52 were synthesized with a serine replacing pS16, under the assumption that there would again be no effect on the structure.
Sample preparation of the free protein
To prepare a solid-state sample of the free protein, 30 mg of LRAP was dissolved into 1 mL of phosphate buffer, consisting of a solution with 0.15 M NaCl and saturated with respect to hydroxyapatite (PB), diluted to 20 mL with water, and the pH adjusted to 7.4. This was frozen in liquid nitrogen and lyophilized, and the entirety of the resulting powder was packed into the NMR rotor.
Sample preparation of the protein bound to HAP
To prepare the protein sample bound to HAP, 30 mg of LRAP was dissolved into 92 mL of PB (0.33 mg/mL) and the pH was adjusted to 7.4. The protein was added to 100 mg of HAP. The 50-m2/g HAP, prepared (46) and characterized using standard techniques, was washed three times with 10 mL of PB immediately before contacting the protein solution. The protein was allowed to bind for 1 h, after which time the protein-HAP complex was centrifuged, and washed three times with 5 mL of PB. The amount of protein bound was determined by measuring the change in concentration before and after binding and for each wash using ultraviolet absorbance measurements (λ = 275 nm). Typically, 4–6 mg of protein was bound to 100 mg of HAP. The sample was packed into an NMR rotor as a wet paste, by transferring a small quantity into the rotor, spinning to 4 kHz, removing the excess water, and repeating this process until the rotor was full (∼50–70 mg of sample), resulting in a hydrated, surface bound sample.
For the lyophilized, surface-bound sample, the packed hydrated sample was frozen with liquid nitrogen in the rotor and lyophilized. The NMR experiments were always done on the hydrated sample first, and then on the lyophilized sample.
NMR experiments
NMR experiments were performed on a three-channel Chemagnetics Infinity console (Chemagnetics, Varian, Fort Collins, CO) operating at 300 MHz proton frequency. A three-channel, variable-temperature Chemagnetics probe was used, employing a 6-μs 90° pulse for 1H and a 0.5- to 1-ms contact time. Temperatures in the rotor were calibrated using 207Pb(NO3)2 (47). Chemical shifts were referenced to glycine, 177.0 ppm (48).
Dynamics
Spectra and T1ρ measurements were taken at both −80°C (frozen) and at room temperature (RT) (20°C). For HB analysis, a spinning speed of 1.5 kHz was used and 28,800 scans were taken for each sample to allow direct intensity comparisons. The room temperature hydrated, surface-bound samples were also run with additional signal averaging to allow more accurate fitting of the chemical shift anisotropy (CSA) parameters. For T1ρ analysis, 10 lock times were used, from 0.05 to 4.55 ms.
REDOR
XY8 phase cycling was used on both observe and dephasing channels for REDOR experiments (49,50). For all three types of REDOR experiments, 13C{31P}, 15N{31P}, and 13C{15N}, 180° pulses of 13.0–15.0 μs were used for both the observe and dephasing nuclei, and samples were spun at 4 kHz. Two-pulse phase-modulated decoupling (TPPM) (51) with a 65-kHz decoupling field was used throughout. Hydrated samples were run at −80°C, whereas lyophilized samples were run at −45°C. Typically, 4096 scans were taken for shorter dephasing periods and 8192–16,384 scans were taken for longer dephasing times, with a 3-s pulse delay. Data were collected at every 8 or 16 rotor periods, out to 104 rotor periods for 13C{31P} and 13C{15N}. In all cases, the final dephasing curve represents the average of at least three to five repetitions. For the 15N{31P} experiments only, data were collected every 24 rotor periods, out to 104 rotor periods, with a 1-s pulse delay: 42,300 scans were collected for early dephasing times and 84,600 scans for longer dephasing times. For the hydrated 15N{31P} bound to HAP, only 14 ms of dephasing was collected due to a significant loss in signal/noise. REDOR dephasing curves were fit by simulations generated using SIMPSON (52).
RESULTS
Dynamics
Table 2 compares the chemical shift and relaxation parameters determined for several residues in the C-terminus of the protein. For the proteins with labeled methyl group A46(13Cβ) and α-carbon, A49(13Cα), the T1ρ values were >10 ms under lyophilized conditions, consistent with no mobility on the timescale of the T1ρ experiment. For the hydrated samples bound to HAP, A46(13Cβ) had a relaxation time of 4.3 ms, and A49(13Cα) had a faster observed relaxation of 2.5 ms. When frozen, the samples containing backbone carbonyl labels, L42(13C′) and A49(13C′), had relaxation times of >20 ms, as expected for a rigid carbonyl group. Under hydrated conditions, L42(13C′) had a relaxation time of 4.2 ms, and A49(13C′) had a relaxation time of 8.0 ms.
TABLE 2.
Chemical shift and relaxation parameters for the C-terminus of LRAP
| LRAP-L42(C′) | LRAP-A49(C′) | LRAP-A46(Cβ) | LRAP-A49(Cα) | |
|---|---|---|---|---|
| T1ρ (msec) | ||||
| Hydrated | 4.2 ± 1 | 8.0 ± 1 | 4.3 ± 1 | 2.5 ± 1 |
| Frozen | 19.5 ± 1 | 19.5 ± 1 | 10.5 ± 1 | 12.2 ± 1 |
| Ω (ppm) | ||||
| Hydrated | 82 ± 20 | 81 ± 20 | — | — |
| Frozen | 140 ± 20 | 154 ± 20 | — | — |
| η | ||||
| Hydrated | 0.7 ± 0.1 | 0.7 ± 0.1 | — | — |
| Frozen | 0.6 ± 0.1 | 0.6 ± 0.1 | — | — |
| σiso (ppm) | ||||
| Hydrated | 173.7 ± 0.5 | 175.7 ± 0.5 | 17.4 ± 0.5 | 50.5 ± 0.5 |
| Frozen | 173.3 ± 0.5 | 175.2 ± 0.5 | 16.8 ± 0.5 | 50.3 ± 0.5 |
| Lyophilized | 173.9 ± 0.5 | 174.6 ± 0.5 | 17.1 ± 0.5 | 49.8 ± 0.5 |
| Linewidth (ppm) | ||||
| Hydrated | 3.1 ± 0.2 | 4.5 ± 0.2 | 3.3 ± 0.2 | 7.3 ± 0.2 |
| Frozen | 5.5 ± 0.2 | 5.2 ± 0.2 | 5.4 ± 0.2 | 8.9 ± 0.2 |
σiso = (1/3)(σ11 + σ22 + σ22), η = (σ22 − σ11)/(σ33 − σiso), where |σ11 − σiso| ≤ |σ33 − σiso| and Ω = |σ11 − σ33|.
The spectra taken for L42(13C′) and A49(13C′) for the HB analysis are shown in Fig. 1. The arrows indicate the isotropic resonance, and the asterisks indicate spinning side bands. The cross-polarization efficiency decreases significantly for both hydrated RT samples (Fig. 1, middle), as compared to the frozen counterparts (Fig. 1, upper), as evidenced by the decreased signal/noise ratio. A significant averaging of the CSA, which is characterized by the changes in the relative height and breadth of the spinning side-band pattern, was also observed, best seen in the comparison of the upper (frozen) and lower (RT) spectra of Fig. 1, as well as in the reduced span (Ω = |δ11 − δ33|) shown in Table 2. The bottom spectrum was signal averaged longer to allow more accurate fitting of the CSA parameters.
FIGURE 1.

Cross-polarization magic angle spinning spectra of carbonyl-labeled proteins hydrated on HAP. (a) LRAP-L42A46. (b) LRAP-A49T53. Upper spectra, frozen (−80°C); middle spectra, RT, lower spectra, RT with more scans for accurate fitting of CSA parameters. Arrows indicate isotropic resonances and asterisks indicate spinning side bands. The carbonyl resonances at both amino acid positions (L42 and A49) show a significant reduction in cross-polarization efficiency (as seen by the loss in signal/noise in the RT spectrum as compared to the frozen spectrum) as well as a significant averaging of the CSA (seen best by comparing the CSA of the upper and lower spectra), indicating significant mobility of the protein backbone in the binding region.
Structure
Structural studies in this work were focused on the C-terminus, where two regions were investigated using the solid-state NMR dipolar recoupling technique REDOR. Table 3 shows the two labeling schemes employed to study the structure by incorporating a backbone 13C=O (C′) at the i residue and a backbone 15N at the i + 4 residue. The best-fit distance, determined with a χ2 analysis, is also shown. Each structure was determined under conditions of 1), the protein bound to the surface, hydrated; 2), the protein bound to the surface, lyophilized; and 3), the protein off the surface.
TABLE 3.
Labeling schemes used for structure and orientation samples
|
Distances (assuming an isolated spin pair)
|
|||||
|---|---|---|---|---|---|
| Name | Labeling scheme | Off | Bound, lyophilized | Bound, hydrated | |
| (Structure, 13C-15N) | |||||
| LRAP-L42A46 | LRAP-PELPLEAWPATDKTKREEVD | 5.3 ± 0.5 | 5.5 ± 0.5 | 5.6 ± 0.5 | Random coil |
| LRAP-A49T53 | LRAP-PELPLEAWPATDKTKREEVD | 5.9 ± 0.5 | 6.1 ± 0.5 | 6.9 ± 1.0 | Random coil |
| (Orientation, 13C-31P or 15N-31P) | |||||
| LRAP-A46* | LRAP-PELPLEAWPATDKTKREEVD | >12 ± 1.0 | 7.0 ± 0.5 | 8.0 ± 0.5 | Next to HAP |
| LRAP-A49 | LRAP-PELPLEAWPATDKTKREEVD | >12 ± 1.0 | 6.0 ± 0.5 | — | Next to HAP |
| LRAP-K52 | LRAP-PELPLEAWPATDKTKREEVD | >10 ± 1.0 | 5.8 ± 0.5 | 4 ± 1.0 | Next to HAP |
Residues in bold indicate isotopic labels. For the structure samples, bold residues represent backbone 13C′, bold underlined residues represent backbone 15N. For the orientation samples, bold italic residues are 13Cβ (A46), 13Cα (A49), or 15Nζ (K52).
Labeling scheme reported previously (41).
The 13C{15N} REDOR dephasing curves are shown in Fig. 2, where the experimental data are represented by symbols and the fits are drawn as lines. Standard deviations, which are based on multiple measurements, are shown only for the lyophilized data for clarity, and represent a maximum error. Fig. 2 a shows the dephasing curves for LRAP-L42A46 which, bound to the surface of HAP, had a measured distance of 5.6 ± 0.5 Å under hydrated conditions (open circles), and an indistinguishable distance of 5.5 ± 0.5 Å under lyophilized conditions (solid diamonds). Off the surface, the distance was 5.3 ± 0.5 Å (solid squares). Random-coil, α-helical, and β-sheet structures are shown for comparison.
FIGURE 2.

REDOR dephasing curves for (a) LRAP-L42A46 and (b) LRAP-A49T53: off the surface (solid squares); on the surface, hydrated (open circles); and on the surface, lyophilized (solid diamonds). Error bars, determined from multiple measurements, are shown only for on the surface, lyophilized data, for clarity, and indicate a maximum error. Dephasing curves for random coil (short-dashed curve), α-helical (dash-dotted curve), and β-sheet (long-dashed curve) structures are also shown for comparison. For both regions studied, the best fits for each set of data are shown with solid lines that are consistent with a largely random-coil structure, with no change in structure when the protein is bound to the surface.
The distances measured for LRAP-A49T53 (Fig. 2 b) were slightly longer than for LRAP-L42A46, with a distance of 6.9 ± 1 Å bound to the surface of HAP under hydrated conditions. After lyophilizing, the distance was slightly shorter at 6.1 ± 0.5 Å. Off the surface, the distance was 5.9 ± 0.5 Å.
Orientation
Table 3 and Fig. 3 show the results orienting three of the residues in the C-terminus of LRAP to the surface of HAP, using either 13C{31P} or 15N{31P} REDOR. Distances shown assume an isolated spin pair, as discussed further (vida infra). For these experiments, a single isotope, either a backbone 13CH (A49) or a sidechain 15NH2 (K52), was introduced and the distance measured from the label to the HAP 31P groups. For both samples off the surface (solid diamonds), there was no measurable dephasing, as expected with no nearby phosphate groups. Shown in Fig. 3, top, the distance from the surface for LRAP-A49(13Cα) under lyophilized conditions (open circles) was 5.7 ± 0.5 Å. The signal/noise ratio for LRAP-A49(13Cα) hydrated and surface-bound was inadequate to obtain a statistically significant dephasing curve. LRAP-K52(15Nζ) under lyophilized conditions (solid squares) was found to be 5.8 ± 0.5 Å from the nearest 31P in the surface of HAP (Fig. 3 b). The first 14 ms of dephasing for LRAP-K52(15Nζ) bound and hydrated (open circles) resulted in a measured distance of 4.0 ± 1.0 Å.
FIGURE 3.

(a) 13C{31P} REDOR dephasing curves for LRAP-A49(13Cα) off the surface (solid diamonds) and on HAP, lyophilized (open circles). LRAP-A49(13Cα) off the surface shows no dephasing, as expected. The initial dephasing of LRAP-A49(13Cα) on HAP would suggest a distance of 5.7 Å from a single 31P in the surface of HAP under lyophilized conditions (solid line). Fitting the data assuming two nearby 31P groups, one 5.7 Å and one 8.0 Å away, also fits the data well (long dashed line). LRAP-A46(13Cβ) (open squares) is shown for comparison (reproduced from works by Shaw and co-workers (41,43)), and shows a longer distance (7.0 Å) under lyophilized conditions (dashed-dotted line). (b) 15N{31P} REDOR dephasing curves for LRAP-K52(15Nζ) off the surface of HAP (solid diamonds), hydrated and bound to HAP crystals (open circles), and lyophilized and bound to HAP crystals (solid squares). The hydrated data are consistent with a 4.0-Å distance from the nearest 31P (solid line). The lyophilized data are most consistent with a single 5.8 Å distance or a combination of 6.0 Å and 7.0 Å (dashed line) 15N–31P distances. A 6.0-Å distance is also shown for comparison (dotted line). LRAP-K52(15NH2) off the surface showed no dephasing, as expected.
DISCUSSION
Structure
The secondary structure of LRAP was determined using REDOR by measuring the distance between isotopically labeled backbone 13C=O (C′) and 15N in the i → i + 4 residues, the distance across a putative hydrogen bond of an α-helix (4.2 Å), as shown (Fig. 2). The dephasing curves are clearly very different for helical structures compared to random-coil and β-sheet structures, and this measurement provides a quantitative determination of global secondary structure. If a β-sheet structure is present, the longer distance (10.6 Å) is outside of the range of the REDOR experiment, allowing for a positive distinction between helical and nonhelical secondary structures. The dephasing curve for a random-coil structure (5.8 Å) is also shown, where the curve was generated by taking a linear combination of every distance at 0.1-Å intervals from 4.2 Å to 10.6 Å, and is distinguishable from both β-sheet and α-helical structures.
The measured distances found for LRAP-L42A46 are consistent with a largely random-coil structure (Fig. 2), with essentially no change under all conditions investigated (off the surface, on the surface hydrated, and on the surface lyophilized) indicating that no structural change occurs in this region during binding. Based on a distance slightly shorter than that expected for a random-coil structure, there is possibly a slight helical content in this region of the protein (36,37). Here, random coil is defined as a large distribution of structures (from 4.2 to 10.6 Å) rather than a single unique structure. However, dephasing curves that include the best-fit average with a distribution of 1 or 2 Å (i.e., 5.6 ± 1 Å in the case of LRAP-L42A46, bound to HAP, hydrated) also fit the data well, suggesting that the random-coil state may not experience all torsion angles equally but may merely consist of a distribution about an average that does not fall into either an α-helix or a β-sheet secondary structure. A distribution of structures may also be supported by the large observed carbonyl linewidths of >3 ppm (Table 2). A single unique structure would typically have linewidths of <2 ppm, though it should be noted that other broadening mechanisms could be responsible (53).
LRAP-A49T53 off the surface and bound to HAP, lyophilized, have slightly longer measured distances than LRAP-L42A46 whose fits are indistinguishable from the dephasing curve for a random coil. The experimental data for hydrated LRAP-A49T53 bound to the surface have enough experimental uncertainty to make the distinction between random coil and β-sheet less clear. However, based on the random coil observed in the residues immediately to the N-terminus of these residues (L42A46), coupled with the fact that LRAP-A49T53 both off the surface and bound to the surface, lyophilized, are clearly best fit with a random coil and are not in a β-sheet conformation, and that distinct dephasing is observed that would not be observed for the β-sheet structure, we hypothesize that the hydrated sample also has a random-coil structure.
To provide additional support to this interpretation, the chemical shifts of both A49C′ and A49Cα were compared under hydrated and lyophilized conditions. Although absolute chemical shifts are difficult to interpret on a highly charged surface such as HAP, relative chemical shifts can be compared for qualitative insights. The C′ and Cα chemical shifts change very predictably, from 2 to 4 ppm or even more (54,55), in the presence of a structural change. For alanine, the chemical shift for both of these carbons would be expected to change by ∼2–2.5 ppm in the event of a conformational change from random coil to β-sheet (54). However, the chemical shifts of both the C′ and Cα were found to be unchanged in going from the hydrated to the lyophilized state (Table 2). The absence of any significant change in chemical shift for the lyophilized and hydrated surface-immobilized samples provides strong support for our interpretation that the hydrated, surface-immobilized protein at the LRAP-A49T53 position is best described by a random-coil structure.
The nearly unchanged chemical shift for the surface-immobilized hydrated and hydrated/frozen samples (Table 2) for both proteins is an additional (qualitative) indication of an unchanged secondary structure. The observation of a random-coil structure is consistent with previous computational models (17) and CD solution studies of LRAP, which indicated a largely random-coil structure (22). The data presented in this work represent some of the first quantitative, site-specific structural data of an amelogenin protein, bound to hydroxyapatite under biologically relevant conditions.
Orientation
The orientation of the C-terminus of LRAP was determined by incorporating single isotopic labels into the backbone or side chain of LRAP (Table 3) and measuring the distance from that labeled residue (either 13C or 15N) to any nearby 31P groups in the surface of HAP. Under lyophilized conditions, LRAP-A49(13Cα) was determined to be 5.7 ± 0.5 Å from the nearest 31P groups in the HAP surface. LRAP-K52(15Nζ), a residue more likely to interact directly with the 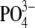 groups in the HAP surface, was also found to be near the surface, 5.8 Å from the nearest
groups in the HAP surface, was also found to be near the surface, 5.8 Å from the nearest  group. Previously, the 13Cβ group of A46 was determined to be close to the surface of HAP based on the measurement of 8.0 ± 0.5 Å to the surface under hydrated conditions and 7.0 ± 0.5 Å to the surface under lyophilized conditions (41), supported by modeling studies that showed the same alanine to be 7.3 Å from the nearest surface phosphate group (43). Neutron reflectivity experiments were also in agreement, showing that the region of LRAP-42→49 was 5–10 Å from the surface (43). The proximity of multiple groups in the C-terminus to the surface of HAP demonstrated in this work confirms the importance of the charged C-terminus in binding LRAP to the surface of HAP.
group. Previously, the 13Cβ group of A46 was determined to be close to the surface of HAP based on the measurement of 8.0 ± 0.5 Å to the surface under hydrated conditions and 7.0 ± 0.5 Å to the surface under lyophilized conditions (41), supported by modeling studies that showed the same alanine to be 7.3 Å from the nearest surface phosphate group (43). Neutron reflectivity experiments were also in agreement, showing that the region of LRAP-42→49 was 5–10 Å from the surface (43). The proximity of multiple groups in the C-terminus to the surface of HAP demonstrated in this work confirms the importance of the charged C-terminus in binding LRAP to the surface of HAP.
The distances that were fit to the dephasing curves above assume a simple isolated spin pair (i.e., 1-13C → 1-31P). However, we know this is not the case based on the lattice structure of HAP, where each label could be within 10 Å (the limit of detection of REDOR) of as many as six  groups. Unfortunately, one 13C in close proximity to more than one 31P group cannot be treated as an average of the two distances, but has to be modeled explicitly. NMR models incorporating multiple distances were investigated and show that at the distances under consideration, within the error of the measurement, at least one 31P is within the indicated distance. For instance, a single distance of 5.7 Å or combined distances of 5.7 Å and 7.0 Å, 5.7 Å and 8.0 Å, or 5.7 Å and 9.0 Å all fit the dephasing curves of the A49(13Cα)-31P within experimental error. Adding a third 31P group maintains the result that one of the 31P groups is 5.7 Å from the A49(13Cα). Recent work has also suggested that 31P-31P homonuclear couplings may contribute somewhat to the dephasing curve and the resulting distance models (42,56). This was also investigated using the 600 Hz homonuclear coupling suggested for HAP and was found to have minimal impact for the longer distances found in our system. Interactions of K52(15Nζ) with multiple 31P groups were also investigated. Combined distances of 6.0 Å and 7.0 Å, as well as 6.0 Å and 8.0 Å or 6.0 Å and 9.0 Å fit the experimental data equally well. Our models also suggested that the 31P-31P coupling did not contribute significantly to the 15N{31P} dephasing curve. Thus, for our system, with distances of ∼6.0 Å or longer from the surface, assuming a single, isolated spin pair does result in an accurate measure to the nearest 31P. This allows the nearest point to the surface of HAP to be determined independent of a complex multispin system. In trying to describe the more complex spin system, the distances that are proposed represent only some of the possible orientation models. However, it should be emphasized that many different physical models could accurately reflect our data, and in the absence of further constraints, it would be impossible to determine precisely which model most accurately describes this system. Due to the structural heterogeneity, as well as the multiple faces of HAP to which LRAP is likely to bind, it is very likely that the best model would be a distribution of many distances, rather than only one or two distances. However, the data unequivocally demonstrate that multiple sites in the C-terminus, including a basic residue, are oriented next to the surface of HAP, establishing its importance in binding LRAP to HAP. Computational methods are currently being developed that utilize the structural constraints determined here to produce an energy-minimized three-dimensional model of the interaction of the C-terminus with the surface of HAP and should provide insight into the possible range of conformations in the side-chain orientations.
groups. Unfortunately, one 13C in close proximity to more than one 31P group cannot be treated as an average of the two distances, but has to be modeled explicitly. NMR models incorporating multiple distances were investigated and show that at the distances under consideration, within the error of the measurement, at least one 31P is within the indicated distance. For instance, a single distance of 5.7 Å or combined distances of 5.7 Å and 7.0 Å, 5.7 Å and 8.0 Å, or 5.7 Å and 9.0 Å all fit the dephasing curves of the A49(13Cα)-31P within experimental error. Adding a third 31P group maintains the result that one of the 31P groups is 5.7 Å from the A49(13Cα). Recent work has also suggested that 31P-31P homonuclear couplings may contribute somewhat to the dephasing curve and the resulting distance models (42,56). This was also investigated using the 600 Hz homonuclear coupling suggested for HAP and was found to have minimal impact for the longer distances found in our system. Interactions of K52(15Nζ) with multiple 31P groups were also investigated. Combined distances of 6.0 Å and 7.0 Å, as well as 6.0 Å and 8.0 Å or 6.0 Å and 9.0 Å fit the experimental data equally well. Our models also suggested that the 31P-31P coupling did not contribute significantly to the 15N{31P} dephasing curve. Thus, for our system, with distances of ∼6.0 Å or longer from the surface, assuming a single, isolated spin pair does result in an accurate measure to the nearest 31P. This allows the nearest point to the surface of HAP to be determined independent of a complex multispin system. In trying to describe the more complex spin system, the distances that are proposed represent only some of the possible orientation models. However, it should be emphasized that many different physical models could accurately reflect our data, and in the absence of further constraints, it would be impossible to determine precisely which model most accurately describes this system. Due to the structural heterogeneity, as well as the multiple faces of HAP to which LRAP is likely to bind, it is very likely that the best model would be a distribution of many distances, rather than only one or two distances. However, the data unequivocally demonstrate that multiple sites in the C-terminus, including a basic residue, are oriented next to the surface of HAP, establishing its importance in binding LRAP to HAP. Computational methods are currently being developed that utilize the structural constraints determined here to produce an energy-minimized three-dimensional model of the interaction of the C-terminus with the surface of HAP and should provide insight into the possible range of conformations in the side-chain orientations.
Although hydrated conditions are desirable to most accurately simulate biological conditions, for both A49(13Cα) and K52(15Nζ), residual mobility of the protein at reduced temperatures (−80°C) limited the measurement of only the first 14 ms of dephasing for K52(15Nζ) and none of the dephasing curve for A49(13Cα), due to the severe reduction in signal intensity. Despite this, qualitative comparisons of the data on the hydrated surface-bound protein can still be made. For instance, it has been observed that lyophilization of the LRAP-A46(13Cβ) sample caused a significant decrease (1 Å) in the distance from the surface (41). LRAP-K52(15Nζ) showed a significant increase in the distance upon lyophilization (5.8 Å, up from 4 Å) based on the first 14 ms of dephasing. Although the significant reduction in signal intensity indicates that under hydrated conditions, part of the sample is not being observed, the observation of a significant change in the distance from the surface in the presence of water indicates a significant role for water. If one assumes that these distances are representative of the entire sample, a significant change in distance to the surface was observed. If it represents just one (less mobile) orientation that is a small fraction of the entire protein population, it still suggests that water plays a significant role, as a large part of the unobserved signal has a different orientation based on the lyophilized data. It could also be a combination of these two effects, and although investigating this phenomenon is beyond the scope of this study, in either case, the orientations of the A46(13Cβ) and K52(15Nζ) side chains were altered with respect to the surface of HAP with the removal of water, highlighting the importance of water in the interaction of LRAP with the HAP surface. The lack of structural change upon lyophilization suggests that water has more influence on the side-chain interaction than on the protein structure.
Dynamics
To further understand the role of water, mobility was examined more critically using additional dynamic studies, including both a HB analysis, cross-polarization efficiencies, and T1ρ relaxation measurements. The HB analysis determines protein dynamics by measuring motionally averaged chemical shift tensors. The CSA is defined by three principle elements, σ11, σ22, and σ33, where the isotropic chemical shift is defined as σiso = (1/3)(σ11 + σ22 + σ33). and can be determined directly from the NMR spectrum. A very useful parameter obtained from lineshape studies is the span, Ω = |σ11–σ33|. If motions are present on timescales much larger than the CSA, ≫20 kHz for carbonyl carbons at 7.06 T, the CSA will become motionally averaged (57), reducing the magnitude of the span. Cross-polarization efficiencies and relaxation measurements are sensitive to motions on the timescale of 10−3–10−5 s (58), extending the timescale of motions to which NMR is sensitive.
Relaxation measurements revealed the motion of A49(13Cα) and A46(13Cβ) to be significantly faster than the corresponding lyophilized samples and, likewise, the backbones of L42(13C′) and A49(13C′) were also shown to relax much more quickly under hydrated conditions (Table 2), indicating motion on the kilohertz timescale in the presence of water. Additionally, the HB analysis showed not only a large decrease in the cross-polarization efficiency, but also a significantly motionally averaged CSA for both L42(13C′) and A49(13C′), as seen in Fig. 1 and Table 2, with the Ω reduced by ∼40%, and the spinning side-band pattern converging to the isotropic line. These observations suggest a significant mobility in this region of the protein, bracketing the timescale of the motion to 10−3–10−5 s. In a previous solid-state NMR study, it was found that 70% of the protein in the fetal enamel matrix exhibited very fast motion (59), possibly consistent with the observations made here. To compare to another well studied biomineralization protein, statherin had similar T1ρ values and saw significant reductions in cross-polarization intensity, similar to LRAP and suggesting a similar frequency of motion. However, the CSA of hydrated, surface-immobilized statherin was not motionally averaged, unlike the observations for LRAP. This suggests that LRAP has a larger amplitude of motion than statherin. Statherin phosphoserine groups, which were proposed to be responsible for binding, were observed to be very rigid under hydrated conditions. Mobility increased with distance from this region, but was still found to be restricted based on the dynamics of backbone carbonyl carbons (36,53). The motionally averaged CSA observed for the C-terminus of LRAP indicates a different (less rigid) binding mode from that observed for statherin, and may also indicate a significant role for dynamics in the function of surface-immobilized amelogenins. The dynamic, structural, and orientation data are summarized in Fig. 4.
FIGURE 4.

Structural and orientation data for LRAP determined in this study indicate that residues L42–T53 are in a random-coil configuration in the presence of water when bound to HAP under biologically relevant conditions. Three residues within that region, A46, A49, and K52, are oriented next to the surface of HAP, providing direct evidence that the charged residues of the C-terminus (indicated in approximate locations by − and + signs) are close enough to the HAP surface to exert influence on crystal growth. The mobility (arrows) in this region also suggests that another part of the protein, possibly in the N-terminus, as indicated, but as yet unidentified, is important in stabilizing the strong protein-crystal interaction.
Interaction mechanism
It has been suggested in the literature that a well defined secondary structure would enhance, or is possibly necessary for, crystal regulation (2,3,5,6,60–63), though this has been difficult to establish due to the lack of structural data for bound proteins. In support of this hypothesis, in one of the only quantitative structural studies for a naturally occurring biomineralization protein, salivary statherin was found to have a very tight helix in the protein-surface interaction region when it was bound to the surface of HAP (36,37). The random-coil structure found for the C-terminal binding region of LRAP is not consistent with this theory, and a comparison of the physical properties of LRAP and statherin proved insightful. The pIs for the 15 terminal residues of the respective proposed binding regions are similar for the two proteins (statherin: DpSpSEEKFLRRIGRFG, pI 4.68; and LRAP: EAWPATDKTKREEVD, pI 4.65). However, statherin has five consecutive acidic residues followed by less localized basic residues, whereas LRAP has acidic and basic residues interspersed throughout the C-terminal 15 residues. The charge distribution of the acidic and basic residues may be significant for the observed structure of LRAP on HAP. Interestingly, though, a mutation study of the basic residues of statherin showed no change in structure when the mutant proteins were bound to the surface (64), suggesting that these residues are not critical in aiding structural recognition motifs for statherin. Mutation studies, as well as studies investigating the effects of binding pH and temperature, are needed to further understand the role of electrostatics, charge distribution, and structure for the interaction of LRAP with HAP.
The relative binding strengths of LRAP and statherin yielded additional insight. Both proteins are found to bind strongly to HAP. Adsorption isotherm studies have shown that the affinity to HAP for amelogenin (19.7 × 105 L/mol) (65) is slightly higher than for statherin (11.1 × 105 L/mol) (66,67). Both LRAP (20,21) and full-length amelogenin (15,20,68,69) are also found to be inhibitors of HAP growth. At a coverage of Q = 3.3 × 10−8 mol/m2, full-length amelogenin has similar or slightly higher growth inhibition (∼30% (15)) than statherin ((∼20%) 70), and LRAP is an even better inhibitor than full-length amelogenin (20). The comparison of the relative affinity and inhibition for the two proteins indicates that a specific structural motif does not necessarily result in a stronger interaction and the overall electrostatic interaction may play a more significant role in some cases, as has also been suggested previously (71).
It is also interesting that the acidic decapeptide (TDKTKREEVD) showed a significant reduction in binding affinity (6.2 × 103 L/mol), providing further evidence that this region alone is not responsible for the affinity of amelogenin to HAP (16). The mobility in this region of the immobilized LRAP protein observed here supports this interpretation. One explanation consistent with our work would be that of a cooperative stabilization, such as contact points in other parts of the protein aiding in binding, which might lessen a requirement for a specific structure. Though loss of structure could also explain a reduced binding affinity for the decapeptide, and the structure was never determined, the decapeptide would be expected to have a random-coil structure, identical to that observed for the full LRAP protein and removing that as a variable. Taking the combination of both of these studies provides compelling evidence that the C-terminus is not solely responsible for LRAP's high affinity to HAP.
Based on the random-coil structure observed for LRAP, the current results are most consistent with an electrostatic interaction mechanism, as has been suggested previously for amelogenin (72), rather than a lattice-matching mechanism. Previous computational models are also consistent with multiple protein conformations and multiple side-chain orientations, which still (or as a consequence) result in acidic carbonyl groups being aligned next to Ca2+ ions in the HAP lattice (43,73). The postulation that a structural motif is necessary for a strong interaction, although appealing, does not appear to be the case for LRAP. With only two data points containing quantitative structural data for biomineralization proteins (statherin and LRAP), it is difficult to make any sweeping conclusions. A larger structural database of proteins bound to surfaces will be necessary to shed further light onto protein-binding mechanisms. Further investigation of amelogenin structure and specific regions interacting with the HAP surface is currently underway and will continue to shed light on the mechanisms of enamel formation, protein-crystal interactions, and crystal regulation.
Extrapolating these results directly to the full protein is tempting, but must be approached with caution. Although the sequence conservation between LRAP and amelogenin in the N- and C-termini is significant, the absence of the large hydrophobic segment could affect secondary structural characteristics in the protein extremities. Particularly, the large number of glutamine residues present in the full protein could potentially interact with HAP, providing additional stabilization or an alternate binding mechanism. However, studies to date have demonstrated that this region is largely internalized in the full protein under biologically relevant conditions, suggesting that the binding regions are restricted to the N- and C-termini. Additionally, the large number of proline residues in the full protein will also affect secondary structure. It remains to be seen whether the centralized proline residues would affect the structure at the N- and C-termini. There has been some evidence in the literature that segments of the full protein maintain their structural conformations as protein fragments (31), providing support in extending the results found in this work to the full protein. Ultimately, although LRAP is a very good model for the full protein, it is only a model. Structural and orientation studies of the full protein are currently being investigated, guided by the insight gained with the more readily studied model protein.
CONCLUSIONS
Solid-state NMR dipolar recoupling experiments were used to determine that the C-terminus of the amelogenin, LRAP, has a largely random-coil structure when bound to the surface of HAP. This structure orients three of the residues in this region, including two alanines and one of the basic lysine residues, next to the surface of HAP, demonstrating the importance of the C-terminus in binding LRAP to the surface of HAP, and suggesting that an electrostatic mechanism is most consistent with the interaction of the C-terminus with HAP. Solid-state NMR dynamics studies revealed significant mobility throughout the region investigated, suggesting that this portion of the protein is not the only region required for the strong affinity of LRAP to HAP. Further studies are underway to understand the role of structure in the interaction of the C-terminus of LRAP with HAP, as well as to determine other protein-HAP interaction regions.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Dental and Craniofacial Research grant DE-015347 and was performed at Pacific Northwest National Laboratory, operated by Battelle for the United States Department of Energy.
Editor: Arthur G. Palmer 3rd.
References
- 1.Addadi, L., J. Moradian-Oldak, H. Furedi-Milhofer, S. Weiner, and A. Veis. 1992. Stereochemical aspects of crystal regulation in calcium phosphate-associated mineralized tissues. In Chemistry and Biology of Mineralized Tissues. H. Slavkin and P. Price, editors. Elsevier Science, Amsterdam.153–162.
- 2.Addadi, L., and S. Weiner. 1985. Interactions between acidic proteins and crystals: stereochemical requirements in biomineralization. Proc. Natl. Acad. Sci. USA. 82:4110–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeOliveira, D. B., and R. A. Laursen. 1997. Control of calcite crystal morphology by a peptide designed to bind to a specific surface. J. Am. Chem. Soc. 119:10627–10631. [Google Scholar]
- 4.Fincham, A. G., E. C. Lau, J. P. Simmer, and M. Zeichner-David. 1992. Amelogenin biochemistry: form and function. In Chemistry and Biology of Mineralized Tissues. H. C. Slavkin and P. Price, editors. Elsevier Science, Amsterdam. 187–201 p.
- 5.Lowenstam, H. A., and S. Weiner. 1989. On Biomineralization. Oxford University Press, New York.
- 6.Hunter, G. 1996. Interfacial aspects of biomineralization. Curr. Opin. Solid State Mater. Sci. 1:430–435. [Google Scholar]
- 7.Diekwisch, T., S. David, P. B., Jr., V. Santos, and H. C. Slavkin. 1993. Antisense inhibition of AMEL translation demonstrates supramolecular controls for enamel HAP crystal growth during embryonic mouse molar development. Development. 117:471–482. [DOI] [PubMed] [Google Scholar]
- 8.Gibson, C. W., Z. A. Yuan, B. Hall, G. Longenecker, E. H. Chen, T. Thyagarajan, T. Sreenath, J. T. Wright, S. Decker, R. Piddington, G. Harrison, and A. B. Kulkarni. 2001. Amelogenin-deficient mice display an amelognesis imperfecta phenotype. J. Biol. Chem. 276:31871–31875. [DOI] [PubMed] [Google Scholar]
- 9.Paine, M., D. Zhu, W. Luo, P. Bringas, M. Goldberg, S. White, Y. Lei, M. Sarikaya, H. Fong, and M. Snead. 2000. Enamel biomineralization defects result from alterations to amelogenin self-assembly. J. Struct. Biol. 132:191–200. [DOI] [PubMed] [Google Scholar]
- 10.Lyngstadaas, S. P., S. Risnes, B. S. Sproat, P. S. Thrane, and H. P. Prydz. 1995. A synthetic, chemically modified ribozyme eliminates amelogenin, the major translation product in developing mouse enamel in vivo. EMBO J. 14:5224–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu, D., M. L. Paine, W. Luo, J. Pablo Bringas, and M. L. Snead. 2006. Altering biomineralization by protein design. J. Biol. Chem. 281:21173–21182. [DOI] [PubMed] [Google Scholar]
- 12.Ravassipour, D. B., P. S. Hart, T. C. Hart, A. V. Ritter, M. Yamauchi, C. Gibson, and J. T. Wright. 2000. Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J. Dent. Res. 79:1476–1481. [DOI] [PubMed] [Google Scholar]
- 13.Simmer, J. P., E. C. Lau, C. C. Hu, T. Aoba, M. Lacey, D. Nelson, M. Zeichner-David, M. L. Snead, H. C. Slavkin, and A. G. Fincham. 1994. Isolation and characterization of a mouse amelogenin expressed in Escherichia coli. Calcif. Tissue Int. 54:312–319. [DOI] [PubMed] [Google Scholar]
- 14.Moradian-Oldak, J., N. Bouropoulos, L. Wang, and N. Gharakhanian. 2002. Analysis of self-assembly and apatite binding properties of amelogenin protein lacking the hydrophilic C-terminal. Matrix Biol. 21:197–205. [DOI] [PubMed] [Google Scholar]
- 15.Aoba, T., M. Fukae, T. Tanabe, M. Shimizu, and E. C. Moreno. 1987. Selective adsorption of porcine-amelogenins onto hydroxyapatite and their inhibitory activity on hydroxyapatite growth in supersaturated solutions. Calcif. Tissue Int. 41:281–289. [DOI] [PubMed] [Google Scholar]
- 16.Aoba, T., E. C. Moreno, M. Kresak, and T. Tanabe. 1989. Possible roles of partial sequences at N- and C-termini of amelogenin in protein-enamel mineral interaction. J. Dent. Res. 68:1331–1336. [DOI] [PubMed] [Google Scholar]
- 17.Gibson, C. W., E. Golub, W. Ding, H. Shimokawa, M. Young, J. Termine, and J. Rosenbloom. 1991. Identification of the leucine-rich amelogenin peptide (LRAP) as the translation product of an alternatively spliced transcript. Biochem. Biophys. Res. Commun. 174:1306–1312. [DOI] [PubMed] [Google Scholar]
- 18.Fincham, A. G., A. B. Belcourt, J. D. Termine, W. T. Butler, and W. C. Cothran. 1983. Amelogenins: sequence homologies in enamel-matrix proteins from three mammalian species. Biochem. J. 211:149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fincham, A. G., J. Moradian-Oldak, and J. P. Simmer. 1999. The structural biology of the developing dental enamel matrix. J. Struct. Biol. 136:270–299. [DOI] [PubMed] [Google Scholar]
- 20.Moradian-Oldak, J., J. Tan, and A. G. Fincham. 1998. Interaction of amelogenin with hydroxyapatite crystals: an adherence effect through amelogenin molecular self-association. Biopolymers. 46:225–238. [DOI] [PubMed] [Google Scholar]
- 21.Habelitz, S., P. K. DenBesten, S. J. Marshall, G. W. Marshall, and W. Li. 2006. Self-assembly and effect on crystal growth of the leucine-rich amelogenin peptide. Eur. J. Oral Sci. 114(Suppl. 1):315–319. [DOI] [PubMed] [Google Scholar]
- 22.Le, T. Q., M. Gochin, J. D. B. Featherstone, W. Li, and P. K. DenBesten. 2006. Comparative calcium binding of leucine-rich amelogenin peptide and full-length amelogenin. Eur. J. Oral Sci. 114(Suppl. 1):320–326. [DOI] [PubMed] [Google Scholar]
- 23.Moradian-Oldak, J. 2001. Amelogenins: assembly, processing and control of crystal morphology. Matrix Biol. 20:293–305. [DOI] [PubMed] [Google Scholar]
- 24.Fincham, A. G., J. Moradian-Oldak, T. G. H. Diekwisch, D. M. Lyaruu, J. T. Wright, P. B. Jr, and H. C. Slavkin. 1995. Evidence for amelogenein “nanospheres” as functional components of secretory-stage enamel matrix. J. Struct. Biol. 115:50–59. [DOI] [PubMed] [Google Scholar]
- 25.Fincham, A. G., J. Moradian-Oldak, J. P. Simmer, P. Sarte, E. C. Lau, T. Diekwisch, and H. C. Slavkin. 1994. Self-assembly of a recombinant amelogenin protein generates supramolecular structures. J. Struct. Biol. 112:103–109. [DOI] [PubMed] [Google Scholar]
- 26.Moradian-Oldak, J., C. Du, and G. Falini. 2006. On the formation of amelogenin microribbons. Eur. J. Oral Sci. 114(Suppl. 1):289–296. [DOI] [PubMed] [Google Scholar]
- 27.Moradian-Oldak, J., and M. Goldberg. 2005. Amelogenin supra-molecular assembly in vitro compared with the architecture of the forming enamel matrix. Cells Tissues Organs. 181:202–218. [DOI] [PubMed] [Google Scholar]
- 28.Moradian-Oldak, J., M. L. Paine, Y. P. Lei, A. G. Fincham, and M. L. Snead. 2000. Self-assembly properties of recombinant engineered amelogenin proteins analyzed by dynamic light scattering and atomic force microscopy. J. Struct. Biol. 131:27–37. [DOI] [PubMed] [Google Scholar]
- 29.Paine, M. L., Y. Lei, K. Dickerson, and M. L. Snead. 2002. Altered amelogenin self-assembly based on mutations observed in human X-linker amelogenesis imperfecta (AIH1). J. Biol. Chem. 277:17112–17116. [DOI] [PubMed] [Google Scholar]
- 30.Renugopalakrishnan, V., E. S. Strawich, P. M. Horowitz, and M. J. Glimcher. 1986. Studies of the secondary structure of amelogenin from bovine tooth enamel. Biochemistry. 25:4879–4887. [DOI] [PubMed] [Google Scholar]
- 31.Goto, Y., E. Kogure, T. Takagi, S. Aimoto, and T. Aoba. 1993. Molecular conformation of porcine amelogenin in solution: three folding units at the N-terminal, central, and C-terminal regions. J. Biochem. (Tokyo). 113:55–60. [DOI] [PubMed] [Google Scholar]
- 32.Aoba, T., K. Kawano, and E. C. Moreno. 1990. Molecular conformation of porcine amelogenins and its significance in protein-mineral interaction: 1H NMR photo-CIDNP study. J. Biol. Buccale. 18:189–194. [PubMed] [Google Scholar]
- 33.Matsushima, N., Y. Izumi, and T. Aoba. 1998. Small-angle X-ray scattering and computer-aided molecular modeling studies of 20 kDa fragment of porcine amelogenin: does amelogenin adopt an elongated bundle structure. J. Biochem. (Tokyo). 123:150–156. [DOI] [PubMed] [Google Scholar]
- 34.Zheng, S., A. T. Tu, V. Renugopalakrishnan, E. Strawich, and M. J. Glimcher. 1987. A mixed β-turn and β-sheet structure for bovine tooth enamel amelogenein: Raman spectroscopic evidence. Biopolymers. 26:1809–1813. [DOI] [PubMed] [Google Scholar]
- 35.Long, J. R., J. L. Dindot, H. Zebroski, S. Kiihne, R. H. Clark, A. A. Campbell, P. S. Stayton, and G. P. Drobny. 1998. A peptide that inhibits hydroxyapatite growth is in an extended conformation on the crystal surface. Proc. Natl. Acad. Sci. USA. 95:12083–12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long, J. R., W. J. Shaw, P. S. Stayton, and G. P. Drobny. 2002. Structure and dynamics of hydrated statherin on hydroxyapatite as determined by solid-state NMR. Biochemistry. 40:15451–15455. [DOI] [PubMed] [Google Scholar]
- 37.Shaw, W., J. Long, J. Dindot, A. Campbell, P. Stayton, and G. Drobny. 2000. Determination of statherin N-terminal peptide conformation on hydroxyapatite crystals. J. Am. Chem. Soc. 122:1709–1716. [Google Scholar]
- 38.Goobes, G., R. Goobes, O. Schueler-Furman, D. Baker, P. S. Stayton, and G. P. Drobny. 2006. Folding of the C-terminal bacterial binding domain in statherin upon adsorption onto hydroxyapatite crystals. Proc. Natl. Acad. Sci. USA. 103:16083–16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long, J. R., N. Oyler, G. P. Drobny, and P. S. Stayton. 2002. Assembly of α-helical peptide coatings on hydrophobic surfaces. J. Am. Chem. Soc. 124:6297–6303. [DOI] [PubMed] [Google Scholar]
- 40.Goobes, R., G. Goobes, W. J. Shaw, G. P. Drobny, C. T. Campbell, and P. S. Stayton. 2006. Binding and structural characteristics of statherin mutants adsorbed onto hydroxyapatite: role of the basic amino acids. Biochemistry. In press.
- 41.Shaw, W., A. Campbell, M. Paine, and M. Snead. 2004. The COOH terminus of the amelogenin, LRAP, is oriented next to the hydroxyapatite surface. J. Biol. Chem. 279:40263–40266. [DOI] [PubMed] [Google Scholar]
- 42.Gibson, J. M., V. Raghunathan, J. M. Popham, P. S. Stayton, and G. P. Drobny. 2005. A REDOR NMR study of a phosphorylated statherin fragment bound to hydroxyapatite crystals. J. Am. Chem. Soc. 127:9350–9351. [DOI] [PubMed] [Google Scholar]
- 43.Shaw, W. J., K. F. Ferris, S. Krueger, U. Perez-Salas, V. Silin, D. J. McGillivray, A. A. Campbell, M. L. Paine, and M. L. Snead. 2004. The orientation of an amelogenin on hydroxyapatite determined using neutron scattering, solid state NMR and computational methods. Proc. Int. Conf. Chem. Biol. Mineralized Tissues, 8th, Banff, Alberta, Canada. 150–153.
- 44.Carpino, L. A., and G. Y. Han. 1972. The 9-fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 37:3404–3409. [Google Scholar]
- 45.Wiejak, S., E. Masiukiewicz, and B. Rzeszotarska. 1999. A large scale synthesis of mono- and di-urethane derivatives of lysine. Chem. Pharm. Bull. (Tokyo). 47:1489–1490. [Google Scholar]
- 46.Ebrahimpour, A., M. Johnsson, C. F. Richardson, and G. H. Nancollas. 1993. The characterization of hydroxyapatite preparations. J. Colloid Interface Sci. 159:158–163. [Google Scholar]
- 47.Bielecki, A., and D. P. Burum. 1995. Temperature dependence of 207Pb MAS for VT MAS spectra of solid lead nitrate. An accurate, sensitive thermometer for VT MAS. J. Magn. Reson. A. 116:215–220. [Google Scholar]
- 48.Duncan, T. M. 1997. Chemical Shift Tensors. Farragut Press, Ithaca, NY.
- 49.Gullion, T., and J. Schaefer. 1989. Rotational-echo double-resonance NMR. J. Magn. Reson. 81:196–200. [DOI] [PubMed] [Google Scholar]
- 50.Gullion, T., and J. Schaefer. 1991. Elimination of resonance offset effects in rotational-echo, double-resonance NMR. J. Magn. Reson. 92:439–442. [Google Scholar]
- 51.Bennett, A., C. Rienstra, M. Auger, K. V. Lakshmi, and R. G. Griffen. 1995. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 103:6951–6958. [Google Scholar]
- 52.Bak, M., J. T. Rasmussen, and N. C. Nielsen. 2000. SIMPSON: a general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 147:296–330. [DOI] [PubMed] [Google Scholar]
- 53.Shaw, W. J., J. R. Long, A. A. Campbell, P. S. Stayton, and G. P. Drobny. 2000. A solid state NMR study of dynamics in a hydrated salivary peptide adsorbed to hydroxyapatite. J. Am. Chem. Soc. 122:7118–7119. [Google Scholar]
- 54.Szilagyi, L. 1995. Chemical shifts in proteins come of age. Prog. Nucl. Magn. Reson. Spectrosc. 27:325–443. [Google Scholar]
- 55.Wishart, D. S., B. D. Sykes, and F. M. Richards. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222:311–333. [DOI] [PubMed] [Google Scholar]
- 56.Raghunathan, V., J. M. Gibson, G. Goobes, J. M. Popham, E. A. Louie, P. S. Stayton, and G. P. Drobny. 2006. Homonuclear and heteronuclear NMR studies of a statherin fragment bound to hydroxyapatite crystals. J. Phys. Chem. B. 110:9324–9332. [DOI] [PubMed] [Google Scholar]
- 57.Herzfeld, J., and A. E. Berger. 1980. Sideband intesities in NMR spectra of samples spinning at the magic angle. J. Phys. Chem. 73:6021–6030. [Google Scholar]
- 58.Schaefer, J., E. O. Stejskal, and R. Buchdahl. 1977. Magic-angle 13C NMR analysis of motion in solid glassy polymers. Macromolecules. 10:384–405. [Google Scholar]
- 59.Termine, J. D., and D. A. Torchia. 1980. 13C-1H magnetic double resonance study of fetal enamel matrix proteins. Biopolymers. 19:741–750. [DOI] [PubMed] [Google Scholar]
- 60.Sicheri, F., and D. S. C. Yang. 1995. Ice-binding structure and mechanism of an antifreeze protein from winter flounder. Nature. 375:427–431. [DOI] [PubMed] [Google Scholar]
- 61.Wierzbicki, A., C. S. Sikes, J. D. Madura, and B. Drake. 1994. Atomic force microscopy and molecular modeling of protein and peptide binding to calcite. Calcif. Tissue Int. 54:133–141. [DOI] [PubMed] [Google Scholar]
- 62.Hauschka, P. V., and S. A. Carr. 1982. Calcium-dependent α-helical structure in osteocalcin. Biochemistry. 21:2538–2547. [DOI] [PubMed] [Google Scholar]
- 63.Gururaja, T. L., and M. J. Levine. 1996. Solid-phase synthesis and characterization of human salivary statherin: a tyrosine-rich phosphoprotein inhibitor of calcium phosphate precipitation. Pept. Res. 9:283–289. [PubMed] [Google Scholar]
- 64.Goobes, R., G. Goobes, W. J. Shaw, G. P. Drobny, C. T. Campbell, and P. S. Stayton. 2007. Thermodynamic roles of basic amino acids in statherin recognition of hydroxyapatite. Biochemistry. 46:4725–4733. [DOI] [PubMed] [Google Scholar]
- 65.Bouropoulos, N., and J. Moradian-Oldak. 2003. Analysis of hydroxyapatite surface coverage by amelogenin nanospheres following the Langmuir model for protein adsorption. Calcif. Tissue Int. 72:599–603. [DOI] [PubMed] [Google Scholar]
- 66.Johnsson, M., C. F. Richardson, E. J. Bergey, M. J. Levine, and G. H. Nancollas. 1991. The effects of human salivary cystatins and statherin on hydroxyapatite crystallization. Arch. Oral Biol. 36:631–636. [DOI] [PubMed] [Google Scholar]
- 67.Raj, P. A., M. Johnsson, M. J. Levine, and G. H. Nancollas. 1992. Salivary statherin. Dependence on sequence, charge, hydrogen bonding potency, and helical conformation for adsorption to hydroxyapatite and inhibition of mineralization. J. Biol. Chem. 267:5968–5976. [PubMed] [Google Scholar]
- 68.Doi, Y., E. D. Eanes, H. Shimokawa, and J. D. Termine. 1984. Inhibition of seeded growth of enamel apatite crystals by amelogenin and enamelin proteins in vitro. J. Dent. Res. 63:98–105. [DOI] [PubMed] [Google Scholar]
- 69.Beniash, E., J. P. Simmer, and H. C. Margolis. 2005. The effect of recombinant mouse amelogenins on the formation and organization of hydroxyapatite crystals in vitro. J. Struct. Biol. 149:182–190. [DOI] [PubMed] [Google Scholar]
- 70.Reference deleted in proof.
- 71.Gerbaud, V., D. Pignol, E. Loret, J. A. Bertrand, Y. Berland, J.-C. Fontecilla-Camps, J.-P. Canselier, N. Gabas, and J.-M. Verdier. 2000. Mechanism of calcite crystal growth inhibition by the N-terminal undecapeptide of lithostathine. J. Biol. Chem. 275:1057–1064. [DOI] [PubMed] [Google Scholar]
- 72.Iijima, M., Y. Moriwaki, T. Takagi, and J. Moradian-Oldak. 2001. Effects of bovine amelogenins on the crystal morphology of octacalcium phosphate in a model system of tooth enamel formation. J. Cryst. Growth. 222:615–626. [Google Scholar]
- 73.Chen, X., Q. Wang, J. Shen, H. Pan, and T. Wu. 2007. Adsorption of leucine-rich amelogenin protein on hydroxyapatite (001) surface through -COO− claws. J. Phys. Chem. C. 111:1284–1290. [Google Scholar]