

Abstract
We studied the low-frequency terahertz spectroscopy of two photoactive protein systems, rhodopsin and bacteriorhodopsin, as a means to characterize collective low-frequency motions in helical transmembrane proteins. From this work, we found that the nature of the vibrational motions activated by terahertz radiation is surprisingly similar between these two structurally similar proteins. Specifically, at the lowest frequencies probed, the cytoplasmic loop regions of the proteins are highly active; and at the higher terahertz frequencies studied, the extracellular loop regions of the protein systems become vibrationally activated. In the case of bacteriorhodopsin, the calculated terahertz spectra are compared with the experimental terahertz signature. This work illustrates the importance of terahertz spectroscopy to identify vibrational degrees of freedom which correlate to known conformational changes in these proteins.
INTRODUCTION
The relationship between the collective, low-frequency vibrational motions of proteins and their subsequent biological processes is a subject of long-standing interest in spectroscopy and dynamics. Collective motions can involve subdomains of hundreds of atoms with corresponding frequencies at the lowest end of the far infrared spectrum from 0 to 100 cm−1, which is the terahertz regime (THz). When considering protein dynamics and the pathways to conformational change, these frequencies are thought to be pivotal (1–5). For example, a comparison of low-frequency normal mode motions and the directions of large-amplitude fluctuations from molecular dynamics simulations indicates clear similarities (6,7). In particular, the calculated root mean-square (RMS) amplitudes of the atomic displacement from a normal mode analysis of bovine pancreatic trypsin inhibitor contrasted with motions obtained from a molecular dynamics simulation based on the same potential energy function suggested that a normal mode description can provide a reliable description of the internal motions of proteins (8,9).
Experimental methods that directly probe these low-frequency motions include neutron scattering (10–15), ultrafast optical heterodyne-detected Raman-induced Kerr-effect spectroscopy (16), Fourier transform infrared (FTIR) and two-dimensional infrared spectroscopy (17–21), THz spectroscopy (22–24), and normal mode calculations (2,4,6,8,25–28). In all of these studies lies the same fundamental question: what is the activation pathway whereby vibrational energy flows, in a concerted manner, to produce a conformational change in the protein?
In a recent study, Keskin and co-workers found that in general members of the same fold family share common dynamical motions, and in particular their lowest frequency modal shapes are nearly superimposable (29). The aim of this work is to compare the lowest frequency vibrational motions of two functionally different but geometrically similar proteins, rhodopsin and bacteriorhodopsin. We note that this work cannot explicitly point to light activation pathways in these systems, simply because the initial step to protein activation is coupled to electronic and structural changes in the retinal chromophore. However, our work does illustrate possible low-frequency motions that are accessible during the subsequent steps in the activation process and further illustrates similar structural and dynamical elements of the two systems.
Physiologically bacteriorhodopsin and rhodopsin are structurally similar but functionally distinctive proteins. Both are seven-member helical transmembrane proteins with a covalently attached light-activated chromophore, retinal. However, the biological function of each protein is unique. Light activation of bacteriorhodopsin serves as a proton pump from the cytosol to the extracellular space of the bacterial membrane. The resulting proton gradient is utilized to drive the synthesis of adenine triphosphate (ATP). During the photoactivation process, isomerization of the all-trans retinal to 13-cis-retinal produces a large displacement of the C13 methyl group of the retinal; this subsequently produces major conformational changes in bacteriorhodopsin, particularly in the E and F helices and the cytoplasmic loops (30–32). Based on structural studies of the dark state and photointermediates, it has been suggested that the dark state of bacteriorhodopsin has an open channel on the extracellular surface. Upon light activation, the protein undergoes a conformational change resulting in the opening of a cytoplasmic channel. This will then allow a proton to enter into the interior of the protein; therefore, the opening and closing of a channel within the protein provides a vectoral mechanism for proton transport (31,33).
Rhodopsin, on the other hand, is a low acuity photopigment. In this case, light activation creates photoisomerization of 11-cis to all-trans retinal and consequently the salt bridge between the protonated shift base and Glu-113 on helix III is neutralized. As a result of this charge neutralization, the interactions between helix III and helices VII, I, and II are released; thus the proteins undergo a series of conformational changes resulting in the metarhodopsin II intermediate, which exhibits enhanced binding with the G-protein, transducin. (17,18,30,32,34–36). This binding initiates the photosignal transduction pathway, which is thought to be similar to other G-protein signaling pathways.
Previous studies of bacteriorhodopsin and rhodopsin by vibrational spectroscopy have illustrated that the low-frequency vibrational motions are anharmonic and that there is a fair degree of vibrational coupling both within the protein itself and external to the environment (17–19,23,37–43). In the case of bacteriorhodopsin, the spectroscopic absorption features depend on the degree of hydration, suggesting a coupling to the water degrees of freedom (23,37,43). Moreover, Mathies and co-workers have found that the excited state of retinal will couple to lower frequency modes, by an intramolecular vibrational redistribution process, and this is influenced by the binding pocket of the protein (39,42). In terms of rhodopsin, the vibrational spectrum of the Lumi intermediate suggests that there is also strong coupling between the retinal all-trans chromophore and the binding pocket of the protein (40). Finally, FTIR difference spectroscopy has revealed that there is a coupling between the lipid vibrational degrees of freedom and that of the intermediate metarhodopsin I degrees of freedom and the intramolecular interactions between helices 3 and 5 of the protein (17).
The computational consideration of both rhodopsin and bacteriorhodopsin remains an active area as well. Previously, Whitmire and colleagues performed a limited normal mode analysis of the terahertz spectra for the full-length (dark state) bacteriorhodopsin and found that the absorbance increased rapidly with increasing frequency, without strong spectroscopic features (23,43). A similar result was also obtained for the D96N mutant of bacteriorhodopsin. This study concluded that the lowest frequency vibrational modes resulted in large-scale motions, but a correlation of these motions to subsequent biological function did not appear. In terms of a reduced representation of protein structure using a Gaussian network model approach, Rader and co-workers were able to illustrate that specific residues in rhodopsin stabilize the dark state (44).
Recent work on modeling the activated states of G-protein-coupled receptors by Niv and co-workers has found that in addition to helix VI movement, helices III and V appear to play a prominent role in the activation process (45). A recent 40 ns simulation of rhodopsin in an explicit membrane bilayer found that after 10 ns of simulation time helices I, II, V, and VII changed their tilt angles and the cytoplasmic loops (in particular CIII) showed significant motion. Moreover, this simulation found a wide range of coupling between the helices and the lipid matrix, suggesting that the low-frequency motions are significantly affected by the protein environment (46). This observation was confirmed by a later 118 ns simulation (47).
Taken together, these studies point to the importance of understanding the low-frequency vibrational motions of transmembrane proteins, even at a harmonic level, to determine probable pathways for vibrationally mediated conformational changes. Of course, it would be ideal to include anharmonic effects in any calculation of the vibrational degrees of freedom for these two protein systems, and we are currently working on this issue. However, due to the complexity of the anharmonic calculation on larger systems, this is beyond the scope of this work.
MATERIALS AND METHODS
Sample preparation
Bacteriorhodopsin is isolated in the form of purple membrane patches (48). These patches consist of 25% lipid and 75% protein. We found the uniformity of the films to be critical for the THz measurements. Solutions of bacteriorhodopsin were spun down to pellet form (35 min at 32 K rpm), dissolved in doubly deionized water, and sonicated for 1 h to fully dissolve the aggregates. A 2% glycerol solution of the sonicated media was pipetted onto the substrate and rapidly dried in a desiccant box that was constantly purged with dry nitrogen gas. One-half of the substrate was left clean to be used as a reference. The wild-type protein film had thicknesses of ∼200 μm and an optical density of A568 = 2. The sample was held in a hydration-controlled cell kept at 80% relative humidity at 21°C with hydration maintained by maintaining contact with a saturated salt solution.
Terahertz time domain spectroscopy
The experimental absorption coefficient measurements were made from bacteriorhodopsin films using the terahertz time domain spectroscopy (THzTDS) of Whitmire and co-workers (23). The THzTDS measurements used an unamplified titanium-sapphire mode-locked laser width of 790 nm, pulse width of 65 fs, and average power of 300 mW for generation and detection of the THz pulses. Coherent detection of the output field transient is accomplished with an optically gated antenna receiver or by electrooptic detection (49,50). The THzTDS systems used were enclosed in a dry nitrogen purged box to diminish THz absorption due to ambient humidity. All data shown here are of the absorbance as defined by Abs = −2log |Esample/Ereference|, where Esample (Ereference) is the field transmitted through the sample (reference). The frequency dependence of the absorption coefficient was verified for samples with a thickness ranging from 40 to 240 microns without any significant variation in the slope.
Calculation of the terahertz spectroscopy
The calculation of the terahertz vibrational spectrum for both proteins was accomplished using the CHARMM22 empirical force field (51). For rhodopsin, we began with the x-ray crystal structure of Teller et al. (Protein Data Bank (PDB) ID: 1HZX.pdb) (52), whereas for bacteriorhodopsin we utilized the x-ray crystal structure, 1C3W.pdb. In both proteins, we stripped away the carbohydrates and bound water molecules; however, we included the covalently attached retinal ligand in our calculations of the spectroscopy. All missing residues were added with PDBviewer, and both structures were geometry optimized until the gradient of the energy was within 0.00001 kcal/(mol Å).
The rhodopsin retinal parameters used in the calculation of the energy minimization and in the calculation of the harmonic vibrational frequencies are given by Saam and co-workers (53), and the bacteriorhodopsin retinal force field parameters are given by Tajkhorshid and co-workers (54). For both structures, the mass-weighted Hessian was diagonalized to obtain the harmonic normal mode frequencies. The resulting eigenvalues from this diagonalization were all greater than or nearly equal to zero, indicating the structures were sufficiently energy minimized to produce a stable local/global minimum.
In the case of rhodopsin it was particularly difficult to minimize the structure to a local/global configuration, and typically we would find one imaginary frequency at −6.61 cm−1, indicating a conformational transition state-like intermediate. To reach a global minimum, we further minimized the entire structure using an adopted basis Newton Raphson algorithm for an additional 16,580 steps. At each 1000 steps, the Hessian was recalculated to monitor the lowest frequency until we were finally able to arrive at a local/global minimum where the lowest frequency was 0.000029 cm−1. This is an important point: although the normal mode frequencies and spectroscopy changed relatively little between the two structures (intermediate and local/global minimum), the normal mode motions and the corresponding molecular conclusions were significantly altered.
We calculated the intensity of the vibrational fundamental, υ′ = υ″ + 1, of each normal mode, Qi, as proportional to the square of the transition dipole moment derivative, δμi/δQi, and numerically evaluated according to (28,55)
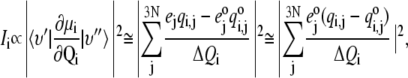 |
(1) |
where the sum is over all 3N Cartesian coordinate displacements, 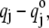 derived from the eigenvectors of the Hessian by dividing by square root of the atomic masses and ΔQi represents the RMS displacement of atoms in mode Qi, arbitrarily set to 1 in the final approximation of Eq. 1. Since CHARMM assigns a static partial charge,
derived from the eigenvectors of the Hessian by dividing by square root of the atomic masses and ΔQi represents the RMS displacement of atoms in mode Qi, arbitrarily set to 1 in the final approximation of Eq. 1. Since CHARMM assigns a static partial charge,  according to atom type regardless of bond length, the electrical anharmonicity is by definition zero, making the latter approximation necessary. Eq. 1 yields the integrated intensity and therefore, for comparisons with experiment, we used a Lorentzian function to describe each line shape, as given by the following formula:
according to atom type regardless of bond length, the electrical anharmonicity is by definition zero, making the latter approximation necessary. Eq. 1 yields the integrated intensity and therefore, for comparisons with experiment, we used a Lorentzian function to describe each line shape, as given by the following formula:
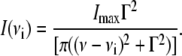 |
(2) |
In this case, the lines are uniformly broadened to a temperature of 4 K, and Γ is defined as the full width at half-maximum. With the inclusion of vibrational anharmonic coupling between modes, we would be able to account for anharmonic line broadening. However, our frequency calculations are strictly at the harmonic level. Improvements that include mechanical anharmonicity are currently underway but are tedious for systems of this size. Therefore, all lines are uniformly broadened and are at the harmonic level of theory.
Vector field representation of normal mode motion
To better visualize the collective low-frequency normal mode motions of rhodopsin and bacteriorhodopsin, we calculated a vector field representation for selected vibrational frequencies. Our method is similar to that of Thomas et al. (56) in that a vector field is calculated over a partition of the protein. However in our case, we partitioned the protein at each amino acid instead of on a cubic lattice, as in the work of Thomas et al. In our work, the center of each amino acid partition was defined to be the Cα carbon, and all atomic displacement normal mode vectors within a 4 Å radius were translated to this position. We then calculated an overall vector field for each Cα carbon through vector addition of each composite atomic motion. We note that our atomic displacement vectors describing the vibrational motions are mass weighted. The program to perform this work is written in Perl and is designed to interface with the visual molecular dynamics program (57). Request for this, or other programs, to reproduce these calculations can be made to greguric@umbc.edu.
RESULTS AND DISCUSSION
Fig. 1 illustrates the energy-minimized structure of rhodopsin and bacteriorhodopsin from which the harmonic vibrational frequencies were calculated. The Cα RMS of each structure, compared with the corresponding x-ray crystallographic structure, is 2.12 Å and 1.01 Å for rhodopsin and bacteriorhodopsin, respectively. Although the minimization procedure maintained the orientation and structure of the seven transmembrane helices, most of the loops are shifted. In particular, for rhodopsin the cytoplasmic loops CII and CIII, which are thought to be important for binding with transducin (58), differ from their crystallographic structures. It has been reported by Li and co-workers that these loops have a relatively high B-factor (59), and we likewise find larger amplitude vibrational motions within these loops.
FIGURE 1.

(left) The energy-minimized structure of bacteriorhodopsin and (right) the energy-minimized structure of rhodopsin. The coloring in each figure is as follows, bacteriorhodopsin: Helices A (red), B (yellow), C (light green), D–E (dark green), and F (blue); rhodopsin: Helix I (orange), II (yellow), III (light green), IV–V (dark green), VI (light blue), and VII–VIII (dark blue). The retinal was included in all calculations but is not illustrated in this figure.
Fig. 2 a illustrates an overlay of the energy-minimized structure of each protein with the corresponding crystallographic structure, and Fig. 2 b compares the transition state of rhodopsin (one negative frequency at −6.61 cm−1) with the fully minimized structure of rhodopsin. Although the Cα RMS is 1.8 Å between these two structures, one can see from Fig. 2 b that further minimization of the rhodopsin significantly altered the cytoplasmic loops, and this had a secondary effect on the hydrogen-bonding network of the protein and on the electrostatic surface of the loops. Although the main chain to main chain hydrogen bonding decreased by 3%, the main chain to side chain and the side chain to side chain hydrogen bonding increased by 6% and 16%, respectively, in the fully minimized structure. Moreover because the majority of the charged residues reside on these loops, the electrostatic surfaces of the cytoplasmic loops were also affected.
FIGURE 2.

(a) (Left panel) Overlay of the crystal structure in blue and the energy-minimized structure of bacteriorhodopsin in red and (right panel) overlay of the crystal structure (blue) and energy-minimized structure of rhodopsin (red). The retinal is not illustrated in the figures but is maintained in the calculations. (b) Overlay of the transition state of rhodopsin (red) with the fully minimized structure of rhodopsin (blue).
From the minimized structures of bacteriorhodopsin (wild-type and a D96N mutant) and rhodopsin, we calculated the corresponding terahertz spectra. In the bacteriorhodopsin systems, we calculate 291 normal mode frequencies, whereas for rhodopsin we calculate 476 normal mode frequencies between 0 to 50 cm−1. The lowest frequency obtained from diagonalization of the Hessian for the bacteriorhodopsin systems was −0.02 cm−1 (wild-type) and −0.11 cm−1 (D96N mutant), whereas for rhodopsin the lowest frequency we obtained was 0.000029 cm−1. Figs. 3 (wild-type) and 4 (D96N mutant) represent our calculated terahertz spectra for the bacteriorhodopsin systems, and Fig. 5 shows the calculated spectrum for the rhodopsin protein.
FIGURE 3.

Theoretical terahertz spectra of wild-type bacteriorhodopsin. A Lorentzian representation of the spectra is given as a solid black line. Underneath this is a stick representation of each normal mode calculated, with those highlighted in bold graphically illustrated in Fig. 8.
FIGURE 5.

Theoretical terahertz spectra of rhodopsin. A Lorentzian representation of the spectra is given as a solid black line. Underneath this is a stick representation of each normal mode calculated, with those highlighted in bold graphically illustrated in Fig. 8.
In each figure a Lorentzian representation (Eq. 2) for each normal mode frequency is illustrated as a solid line, and each individual mode frequency is given below in stick representation. The bold lines in each figure represent modes with unusually high dipole derivative values; therefore, we selected these modes for further investigation (Table 1). To determine differences and similarities between the three proteins studied, we plot the rhodopsin and bacteriorhodopsin spectra together in Fig. 6. The calculated intensities in Fig. 6 are weighted by the molecular weight of each protein, and in Figs. 3–5 the intensities have been normalized to the same scale. Fig. 7 illustrates the experimental terahertz spectra of bacteriorhodopsin.
TABLE 1.
Description of normal mode motion of the three proteins
| Frequency | Rhodopsin | Bacteriorhodopsin | D96N bacteriorhodopsin mutant |
|---|---|---|---|
| RHO: 10.67 cm−1 | Cytoplasmic loops and cytoplasmic side of helix V (From Ile-217 to loops) outward movement toward membrane and helix VIII. | Cytoplasmic loops and all helices move in an accordion (up-down) fashion. | Cytoplasmic loops and all helices move outward toward membrane. |
| Brho: 10.37 cm−1 | |||
| Brho_mutant: 8.28 cm−1 | |||
| RHO: 22.08 cm−1 | Cytoplasmic loops and helices I, IV, VI, VII, and VIII all move outward. | All helices move outward toward membrane. | Helices B, C, F, and G all move in an accordion (up-down) fashion. |
| Brho: 21.95 cm−1 | |||
| Brho_mutant: 19.84 cm−1 | All loops move outward. | ||
| RHO: 39.7 cm−1 | Helix II (downward) Helix III (upward) Helix IV (downward) Helix V (upward) Helix VI (outward) Cytoplasmic loops (upward) and extracellular loops (downward) | All helices move in-out toward membrane. | All helices move in-out toward membrane. |
| Brho: 22.93 cm−1 | |||
| Brho_mutant: 33.83 cm−1 | All loops move in-out toward membrane. | All loops move in-out toward membrane. | |
| RHO: 45.24 cm−1 | Extracellular loops and helical region near extracellular surface move outward. Helices III–VII move outward. | Extracellular loops Helix A (in-out) Helix B (in-out) Helix C (up-down) Helix D (in-out) Helix F (out) | Entire protein moves in-out of membrane. |
| Brho: 37.7 cm−1 | |||
| Brho_mutant: 34.2 cm−1 | |||
| RHO: 48.52 cm−1 | Extracellular surface moves in an accordion (up-down) fashion. Helices V–VII move in an accordion (up-down) fashion. | Extracellular loops (large excursions) Helix D (in and out) Helix E (in and out) Helix F (in and out, Val-183 in particular) | Extracellular loops (large excursions) Helices A, B, C Helix E (in particular V132), Helix F Helix G (in particular L202). |
| Brho: 40.68 cm−1 | |||
| Brho_mutant: 48.6 cm−1 | All helical movement is in an accordion (up-down) fashion. |
Brho, bacteriorhodopsin.
FIGURE 6.

Overlay of the three theoretical terahertz spectra of rhodopsin (solid), bacteriorhodopsin (long dash), and the D96D mutant of bacteriorhodopsin (short dash).
FIGURE 4.

Theoretical terahertz spectra of the D96N mutation of bacteriorhodopsin. A Lorentzian representation of the spectra is given as a solid black line. Underneath this is a stick representation of each normal mode calculated, with those highlighted in bold graphically illustrated in Fig. 8.
FIGURE 7.

Overlap of the bacteriorhodopsin experimental spectra (open diamonds) with the predicted terahertz spectra from Fig. 3.
A comparison between the calculated rhodopsin and bacteriorhodopsin terahertz vibrational spectra illustrates that there are significant similarities, which is confirmed by an analysis of selected normal mode motions (Table 1 and Fig. 8). For example, in the lowest terahertz region analyzed (8–13 cm−1) the vibrational motion is mainly confined to the cytoplasmic loop region of both proteins with some additional movements of the extracellular loops. In the midterahertz region studied (20–40 cm−1), the movement goes downward from the cytoplasmic loops to include new helical motions. In this case both proteins illustrate movement of the helices outward toward the membrane bilayer. However, as the frequency increases the movement begins to change from a breathing against the membrane to a stretching and compression of the helices within the membrane. At the highest frequencies probed (40–50 cm−1), the movement is now confined mainly to the extracellular loops of the protein systems.
FIGURE 8.

Vector representation of selected normal mode motions for the three proteins. The color scheme for the B-factors is as follows: blue illustrates areas of low mobility and red designates areas of high mobility.
It is important to point out that in the case of the rhodopsin intermediate-like structure (with a lowest frequency of −6.6 cm−1) the normal modes correspond to motions that are opposite to those in the fully minimized structure. For the rhodopsin intermediate structure, we found that the lowest frequency motion (8–13 cm−1) was confined to the extracellular loop region whereas the higher frequencies (40–50 cm−1) corresponded to motion in the cytoplasmic loops. Previously we found that the low-frequency terahertz spectrum is highly dependent on the hydrogen-bonding network of the system (55). Because the cytoplasmic loops of rhodopsin are flexible and changed during minimization, this had a secondary effect on the hydrogen bonding of the system.
In general, the fully minimized system had a slightly higher hydrogen-bonding network, in particular in the side chains of the cytoplasmic loops. Because the cytoplasmic loops are also highly charged, this had an additional effect on the electrostatic nature of this region of the protein. We believe that this is the cause of the difference in the normal mode motions between the intermediate and fully minimized structures of rhodopsin and points to the conclusion that changes in the cytoplasmic loop region may assist to control the dynamics of the protein. This is in support of experimental data that suggest a correlation between the cytoplasmic loop mobility and the equilibrium between active metarhodopsin II and inactive metarhodopsin I (30).
However, normal mode movements in both the fully minimized rhodopsin and bacteriorhodopsin systems are surprisingly similar, with the higher frequencies corresponding to motions in the extracellular loops and extending downward to the cytoplasmic surface as the frequencies decrease. Subramaniam and Henderson have suggested that the unilluminated state of bacteriorhodopsin exists in an equilibrium mixture between two conformations (31). In one conformation, the cytoplasmic half of the protein is opened and creates a proton channel. In the other conformation the extracellular side of the protein is open and creates another proton channel. In unilluminated wild-type bacteriorhodopsin, the equilibrium is shifted toward the conformation which has accessibility to the extracellular side of the protein, and this conformation dominates in the dark state (31). However in the MN intermediates of bacteriorhodopsin, the cytoplasmic side of the protein opens and this then creates the necessary channel to allow the capture, translocation, and later release of a proton (33). It is also well known that the cytoplasmic surface of the protein, and in particular the EF loop, undergoes significant conformational change upon photoactivation (30,31,37,60).
In the case of rhodopsin it is known from extensive work using site-directed spin labeling and electron paramagnetic resonance spectroscopy (36,61,62), FTIR (17,18), and fluorescence (30) spectroscopy that the cytoplasmic ends of helices III, VIII, and VI move outward and in the case of helix VI, they also rotate clockwise upon light activation. Further mutagenesis and antibody work suggest that helix VII moves outward. Taken all together, these results indicate that upon light activation, the cytoplasmic region of the protein opens up for interaction with transducin (63,64). We found that between 20 and 40 cm−1 there is modest to extensive movement of all helices that could be connected to the lowest frequency motion in the cytoplasmic side of the protein. However, it remains to be seen if the movements of these helices are coupled to each other or to the movement of the cytoplasmic and extracellular loops. Perhaps our vibrational analysis can assist in a further study of the vibrational energy coupling between the low-frequency terahertz motions in the dark state of bacteriorhodopsin and, due to the similarities in the normal mode motions, by analogy for rhodopsin as well.
In general it was more difficult to analyze the bacteriorhodopsin systems, as the normal mode frequencies corresponded to overall motions of the helices in both an accordion fashion perpendicular to the membrane plane and in-and-out parallel to the membrane plane. Surprisingly, whereas much of the normal mode motion was similar between the wild-type and the D96N mutant of bacteriorhodopsin, the frequencies corresponding to these normal modes were distinctive. One difference between the wild-type and mutant systems was found at 19.8 cm−1. This frequency corresponded to an overall accordion movement of the helices in the mutant protein, whereas the wild-type system had a similar mode at 20.19 cm−1 but with a much lower intensity. Two close peaks at 21 and 23 cm−1 had much higher intensity and corresponded to movement of the helices outward, parallel to the membrane plane.
Another distinction we found was the doublet at 37 and 40 cm−1 in the wild-type system, which corresponded to outward and accordion movement of the helices. Although the mutant system exhibits similar normal mode motions that correspond to this doublet, this region of the spectrum is missing the higher intensities. This is consistent with a previous terahertz study of bacteriorhodopsin and the D96N mutant in which it was found that the D96N mutant system is less intense than the corresponding wild-type protein (23). It is known that the lifetime of the intermediate M state of the D96N mutant increases relative to the wild-type protein, and it was speculated that conformational flexibility within the wild-type protein could be involved with the shorter lifetime. Our calculated spectra are strictly for the dark state proteins, and we find that their relative normal mode motions are similar in both cases.
We also compared the calculated terahertz spectrum of wild-type bacteriorhodopsin with that of the corresponding experimental spectrum and found that the overall trend is similar (Fig. 7). We point out that the calculations are on a single molecule in the gas phase whereas the experimental spectrum is from a thin film sample. From Fig. 7 we find that in both cases the average dependency of the intensity increases linearly with the frequency. Moreover, our calculated spectrum of bacteriorhodopsin seems to be in better agreement with the experimental data than that of the previous calculations of Whitmire et al. (23).
CONCLUSIONS
Low-frequency terahertz spectroscopy offers an advantage to probe motions that are involved in conformational activation pathways of biomolecules. However, even modest size proteins, such as bacteriorhodopsin, often have a multitude of normal mode frequencies, and hence densities of states, in this region of the spectrum. It is a difficult problem to determine which of these many normal mode motions accessed in this region are key motions along bioactivated pathways. Here we present the calculated and measured terahertz spectroscopy for two geometrically similar proteins, rhodopsin and bacteriorhodopsin. Each protein consists of seven transmembrane helices connected by extracellular and cytoplasmic loops. Regardless of their geometric similarities, the biological function and photoactivation pathways of both proteins are distinctive. However in terms of low-frequency terahertz spectroscopy, both proteins are surprisingly similar.
We find that the calculated lowest frequencies correspond to movement in the cytoplasmic region of the proteins and as the frequency increases the normal mode movements flow down the helices toward the extracellular region of the protein systems. Moreover, even the overall movement of the helices is similar in both protein systems. The calculations are compared against experimentally resolved terahertz spectroscopy to illustrate that the resolution of terahertz signatures of biomolecules is greatly assisted by even a simplistic computational analysis. These findings can be readily tested by performing THz spectroscopy on rhodopsin/bacteriorhodopsin samples where the extracellular motion is constrained, such as for samples prepared as layer films. The constrained extracellular environment would dampen the low-frequency terahertz signatures but leave the higher frequencies unchanged.
The important question remains as to whether the identified modes that illustrate possible vibrational energy transfer pathways from cytoplasmic to extracellular surface are physiological relevant. Out of computational necessity, the calculations were preformed on isolated proteins; therefore, important contributions due to hydration and membrane effects were neglected. It is known that the level of hydration can affect the bacteriorhodopsin photocycle (23,37,65), and there is spectroscopic evidence that the nature of the lipid membrane will also affect the photocycle of both proteins (18,66–68). Moreover there are compelling atomic force microscopy studies to suggest that rhodopsin is a functional dimer in the rod outer segment membrane (69–71). Questions such as the effects of the internal and the external environment on the terahertz spectroscopy of these proteins cannot be addressed at this time, but remain an ongoing area of active research. However, recent work on the terahertz spectroscopy of condensed phase biological molecules suggests that environmental effects can alter the terahertz signatures and corresponding molecular motions in this region of the spectrum (5,55,72).
Finally, the experimental terahertz dielectric response for large proteins was found to be broad and nearly linear with considerable similarity to the density of modes but has little correlation with the previously calculated intensities. This earlier finding has caused some concern that the dominant contribution to the response was not due to the collective structural modes but rather was a glass-like response of the mobile side chains. The results presented in this work demonstrate that indeed the frequency-dependent response for a single molecule is surprisingly smooth and the features may be further washed out by an ensemble averaging over the thermally accessible conformations in a macrosample. These computational results do suggest that dielectric experiments should not rely solely on side-chain relaxational response but should also include collective mode contributions in the final analysis.
Editor: Brian R. Dyer.
References
- 1.Tama, F., and Y.-H. Sanejouand. 2001. Conformational change of proteins arising from normal mode calculations. Protein Eng. 14:1–6. [DOI] [PubMed] [Google Scholar]
- 2.Jaaskelainen, S., C. S. Verma, R. Hubbard, P. Linko, and L. S. D. Caves. 1998. Conformational change in the activation of lipase: an analysis in terms of low-frequency normal modes. Protein Sci. 7:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markelz, A. G., A. Roitberg, and E. J. Heilweil. 2000. Pulsed terahertz spectroscopy of DNA, bovine serum albumin and collagen between 0.1 and 2.0 THz. Chem. Phys. Lett. 320:42–48. [Google Scholar]
- 4.Tama, F., F. X. Gadea, O. Marques, and Y.-H. Sanejouand. 2000. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 41:1–7. [DOI] [PubMed] [Google Scholar]
- 5.Knab, J., J.-Y. Chen, and A. G. Markelz. 2006. Hydration dependence of conformational dielectric relaxation of lysozyme. Biophys. J. 90:2576–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandrov, V., U. Lehnert, N. Echols, D. Milburn, D. Engelman, and M. Gerstein. 2005. Normal modes for predicting protein motions: a comprehensive database assessment and associated web tool. Protein Sci. 14:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amadei, A., A. B. M. Linnsen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 8.Brooks, B. R., and M. Karplus. 1983. Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA. 80:6571–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ichiye, T., and M. Karplus. 1991. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 11:205–217. [DOI] [PubMed] [Google Scholar]
- 10.Bizzarri, A. R., A. Paciaroni, C. Arcangeli, and S. Cannistraro. 2001. Low-frequency vibrational modes in proteins: a neutron scattering investigation. Eur. Biophys. J. 30:443–449. [DOI] [PubMed] [Google Scholar]
- 11.Gabel, F., D. Bicout, U. Lehnert, M. Tehei, M. Weik, and G. Zaccai. 2002. Protein dynamics studied by neutron scattering. Q. Rev. Biophys. 35:327–367. [DOI] [PubMed] [Google Scholar]
- 12.Bu, Z., D. A. Neumann, S.-H. Lee, C. M. Brown, D. Engelman, and C. C. Han. 2000. A view of dynamics changes in the molten globule-native folding step by quasielastic neutron scattering. J. Mol. Biol. 301:525–536. [DOI] [PubMed] [Google Scholar]
- 13.Joti, Y., A. Kitaob, and N. Goa. 2004. Molecular simulation study to examine the possibility of detecting collective motion in protein by inelastic neutron scattering. Physica B (Amsterdam). 350:E627–E630. [Google Scholar]
- 14.Koutsopoulos, S., J. van der Oost, and W. Norde. 2005. Temperature-dependent structural and functional features of a hyperthermostable enzyme using elastic neutron scattering. Proteins. 61:377–384. [DOI] [PubMed] [Google Scholar]
- 15.Gabel, F., M. Weik, P. Masson, F. Renault, D. Fournier, L. Brochier, B. P. Doctor, A. Saxena, I. Silman, and G. Zaccai. 2005. Effects of Soman inhibition and of structural differences on cholinesterase molecular dynamics: a neutron scattering study. Biochem. J. 89:3303–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giraud, G., J. Karolin, and K. Wynne. 2003. Low-frequency modes of peptides and globular proteins in solution observed by ultrafast OHD-RIKES spectroscopy. Biophys. J. 85:1903–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beck, M., T. P. Sakmar, and F. Siebert. 1998. Spectroscopic evidence for interaction between transmembrane helices 3 and 5 in rhodopsin. Biochemistry. 37:7630–7639. [DOI] [PubMed] [Google Scholar]
- 18.Vogel, R., and F. Siebert. 2003. Fourier transform IR spectroscopy study for new insights into molecular properties and activation mechanisms of visual pigment rhodopsin. Biopolymers. 72:133–148. [DOI] [PubMed] [Google Scholar]
- 19.Vogel, R., J. J. Ruprecht, C. Villa, T. Mielke, G. F. Schertle, and F. Siebert. 2004. Rhodopsin photoproducts in 2D crystals. J. Mol. Biol. 338:597–609. [DOI] [PubMed] [Google Scholar]
- 20.Chung, H. S., M. Khalil, A. W. Smith, Z. Ganim, and A. Tokmakoff. 2005. Conformational changes during the nanosecond-to-millisecond unfolding of ubiquitin. Proc. Natl. Acad. Sci. USA. 102:612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, J., H.-W. He, Q. Wang, and Y.-B. Yan. 2006. Sequential events in ribonuclease a thermal unfolding characterized by two-dimensional infrared correlation spectroscopy. Protein Pept. Lett. 13:33–40. [DOI] [PubMed] [Google Scholar]
- 22.Siegel, P. H. 2004. Terahertz technology in biology and medicine. IEEE Trans Microw. Theory Tech. 52:2438–2447. [Google Scholar]
- 23.Whitmire, S. E., D. Wolpert, A. G. Markelz, J. R. Hilebrecht, J. Galan, and R. R. Brige. 2003. Protein flexibility and conformational state: a comparison of collective vibrational modes of wild-type and D96N bacteriorhodopsin. Biophys. J. 85:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmuttenmaer, C. A. 2004. Exploring dynamics in the far-infrared with terahertz spectroscopy. Chem. Rev. 104:1759–1780. [DOI] [PubMed] [Google Scholar]
- 25.Tama, F., W. Wriggers, and C. L. Brooks III. 2002. Exploring global distortions of biological macromolecules and assemblies from low-resolution structural information and elastic network theory. J. Mol. Biol. 321:297–305. [DOI] [PubMed] [Google Scholar]
- 26.Brooks, B. R., D. Janezic, and M. Karplus. 1995. Harmonic analysis of large systems. J. Comput. Chem. 16:1522–1542. [Google Scholar]
- 27.Balog, E., T. Becker, M. Oettl, R. Lechner, R. Daniel, J. Finney, and J. C. Smith. 2004. Direct determination of vibrational density of states change on ligand binding to a protein. Phys. Rev. Lett. 93:028103. [DOI] [PubMed] [Google Scholar]
- 28.Korter, T. M., R. Balu, M. B. Campbell, M. C. Beard, S. K. Gregurick, and E. J. Heilweil. 2006. Terahertz spectroscopy of solid serine and cysteine. Chem. Phys. Lett. 418:65–70. [Google Scholar]
- 29.Keskin, O., R. L. Jernigan, and I. Bahar. 2000. Proteins with similar architecture exhibit similar large-scale dynamic behavior. Biophys. J. 78:2093–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexiev, U., I. Rimke, and T. Pohlmann. 2003. Elucidation of the nature of the conformational changes in the EF-interhelical loop in bacteriorhodopsin and of the helix VIII on the cytoplasmic surface of bovine rhodopsin: a time-resolved fluorescence depolarization study. J. Mol. Biol. 328:705–719. [DOI] [PubMed] [Google Scholar]
- 31.Subramanian, S., and R. Henderson. 2000. Crystallographic analysis of protein conformational changes in the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 1460:157–165. [DOI] [PubMed] [Google Scholar]
- 32.Okada, T., and K. Palczewski. 2001. Crystal structure of rhodopsin: implications for vision and beyond. Curr. Opin. Struct. Biol. 11:420–426. [DOI] [PubMed] [Google Scholar]
- 33.Kataoka, M., and H. Kamikubo. 2000. Structures of photointermediates and their implications for the proton pump mechanism. Biochim. Biophys. Acta. 1460:166–176. [DOI] [PubMed] [Google Scholar]
- 34.Ruprecht, J. J., T. Mielke, R. Vogel, C. Villa, and G. F. Schertler. 2004. Electron crystallography reveals the structure of metarhodopsin I. EBMO J. 23:3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Getmanova, E., A. B. Patel, J. Klein-Seetharaman, M. C. Loewen, P. J. Reeves, N. Friedman, M. Sheves, S. O. Smith, and H. G. Khorana. 2004. NMR spectroscopy of phosphorylated wild-type rhodopsin: mobility of the phosphorylated C-terminus of rhodopsin in the dark and upon light activation. Biochemistry. 43:1126–1133. [DOI] [PubMed] [Google Scholar]
- 36.Farrens, D. L., C. Altenback, K. Yang, W. L. Hubbell, and H. G. Khorana. 1996. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 274:768–770. [DOI] [PubMed] [Google Scholar]
- 37.Kamikubo, H., T. Oka, Y. Imamoto, F. Tokunaga, J. K. Lanyi, and M. Kataoka. 1997. The last phase of the reprotonation switch in bacteriorhodopsin: the transition between the M-type and the N-type protein conformation depends on hydration. Biochemistry. 36:12282–12287. [DOI] [PubMed] [Google Scholar]
- 38.Yan, E. C. Y., Z. Ganim, M. A. Kazmi, B. W. W. Chang, T. P. Sakmar, and R. A. Mathies. 2004. Resonance Raman analysis of the mechanism of energy storage and chromophore distortion in the primary visual photoproduct. Biochemistry. 43:10867–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCamant, D. W., P. Kukura, and R. A. Mathies. 2005. Femtosecond stimulated Raman study of excited state evolution in bacteriorhodopsin. J. Phys. Chem. 109:10449–10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ujj, L., F. Jager, and G. H. Atkinson. 1998. Vibrational spectrum of the Lumi intermediate in the room temperature rhodopsin photo-reaction. Biophys. J. 74:1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim, J. E., D. Pan, and R. A. Mathies. 2003. Picosecond dynamics of G-protein coupled receptor activation in rhodopsin from time-resolved UV resonance Raman spectroscopy. Biochemistry. 42:5169–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim, J. E., and R. A. Mathies. 2002. Anti-Stokes Raman study of vibrational cooling dynamics in the primary photochemistry of rhodopsin. J. Phys. Chem. A. 106:8508–8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markelz, A. G., S. E. Whitmire, J. Hillebrecht, and R. Birge. 2002. THz time domain spectroscopy of biomolecular conformational modes. Phys. Med. Biol. 47:3797–3805. [DOI] [PubMed] [Google Scholar]
- 44.Rader, A. J., G. Anderson, B. Isin, H. G. Khorana, I. Bahar, and J. Klein-Seetharaman. 2004. Identification of core amino acids stabilizing rhodopsin. Proc. Natl. Acad. Sci. USA. 101:7246–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niv, M. Y., L. Skrabanek, M. Filizola, and H. Weinstein. 2006. Modeling activated states of GPCRs: the rhodopsin template. J. Comput. Aided Mol. Des. 20:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crozier, P. S., M. J. Stevens, L. R. Forrest, and T. B. Woolf. 2003. Molecular dynamics simulation of dark-adapted rhodopsin in an explicit membrane bilayer: coupling between local retinal and larger scale conformational change. J. Mol. Biol. 333:493–514. [DOI] [PubMed] [Google Scholar]
- 47.Pitman, M. C., A. Grosfield, F. Suits, and S. E. Feller. 2005. Role of cholesterol and polyunsaturated chains in lipid-protein interactions: molecular dynamics simulation of rhodopsin in a realistic membrane environment. J. Am. Chem. Soc. 127:4576–4577. [DOI] [PubMed] [Google Scholar]
- 48.Oesterhelt, D., and W. Stoeckenius. 1974. Isolation of the cell membrane of halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 31:667–678. [DOI] [PubMed] [Google Scholar]
- 49.Grischkowsky, D., and N. Katzenellenbogen. 1991. Femtosecond Pulses of Terahertz Radiation: Physics and Applications. OSA, Washington, DC.
- 50.Chen, Q., M. Tani, Z. P. Jiang, and X.-C. Zhang. 2001. Electro-optic transceivers for terahertz-wave applications. J. Opt. Soc. Am. B. 18:823–831. [Google Scholar]
- 51.MacKerell, A. D., N. Banavali, and N. Foloppe. 2000. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 56:257–265. [DOI] [PubMed] [Google Scholar]
- 52.Teller, D. C., T. Okada, C. A. Behnke, K. Palczewski, and R. E. Stenkamp. 2001. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry. 40:7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saam, J., E. Tajkhorshid, S. Hayashi, and K. Schulten. 2002. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys. J. 83:3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tajkhorshid, E., J. Baudry, K. Schulten, and S. Suhai. 2000. Molecular dynamics study of the nature and origin of retinal's twisted structure in bacteriorhodopsin. Biophys. J. 78:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siegrist, K., C. R. Buchner, I. Mandelbaum, A. R. Hight-Walker, R. Balu, S. K. Gregurick, and D. F. Plusquellic. 2006. High-resolution terahertz spectroscopy of crystalline trialanine: extreme sensitivity to beta-sheet structure and co-crystallized water. J. Am. Chem. Soc. 128:5764–5775. [DOI] [PubMed] [Google Scholar]
- 56.Thomas, A., K. Hinsen, M. J. Field, and D. Perahia. 1999. Tertiary and quaternary conformational changes in aspartate transcarbamylase: a normal mode study. Proteins. 34:96–112. [DOI] [PubMed] [Google Scholar]
- 57.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 58.Konig, B., A. Arendt, J. H. McDowell, M. Kahlert, P. A. Hargrave, and K. P. Hofmann. 1998. Three cytoplasmic loops of rhodopsin interact with transducin. Proc. Natl. Acad. Sci. USA. 86:6878–6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li, J., P. C. Edwards, M. Burghammer, C. Villa, and G. F. Schertle. 2004. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 343:1409–1438. [DOI] [PubMed] [Google Scholar]
- 60.Sass, H. J., G. Buldt, R. Gessenich, D. Henh, D. Neff, R. Schlesinger, J. Berendzen, and P. Ormos. 2000. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 406:649–653. [DOI] [PubMed] [Google Scholar]
- 61.Langen, R., K. Cai, C. Altenback, H. G. Khorana, and W. L. Hubbell. 1999. Structural features of the C-terminal domain of bovine rhodopsin: a site-directed spin-labeling study. Biochemistry. 38:7918–7924. [DOI] [PubMed] [Google Scholar]
- 62.Altenback, C., K. Cai, J. Klein-Seetharaman, H. G. Khorana, and W. L. Hubbell. 2001. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 316 in helix 8 and residues in the sequence 60–75, covering the cytoplasmic end of helices TM1 and TM2 and their connection loop CL1. Biochemistry. 40:15493–15500. [DOI] [PubMed] [Google Scholar]
- 63.Abdulaev, N. G., and K. D. Ridge. 1999. Light-induced exposure of the cytoplasmic end of transmembrane helix seven in rhodopsin. Proc. Natl. Acad. Sci. USA. 95:12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ridge, K. D., N. G. Abdulaev, M. Sousa, and K. Palczewski. 2003. Phototransduction: crystal clear. Trends Biochem. Sci. 28:479–487. [DOI] [PubMed] [Google Scholar]
- 65.Groma, G. I., L. Kelemen, A. Kulcsar, M. Lakatos, and G. Varo. 2001. Photocycle of dried acid purple form of bacteriorhodopsin. Biophys. J. 81:3432–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hendler, R. W., S. M. Barnett, S. Dracheva, S. Bose, and I. W. Levin. 2003. Purple membrane lipid control of bacteriorhodopsin conformational flexibility and photocycle activity: an infrared spectroscopic study. Eur. J. Biochem. 270:1920–1925. [DOI] [PubMed] [Google Scholar]
- 67.Hu, K., Y. Sun, D. Chen, and Y. Zhang. 2000. The effect of lipid environment in purple membrane on bacteriorhodopsin. J. Photochem. Photobiol. B. 58:163–169. [DOI] [PubMed] [Google Scholar]
- 68.Lee, A. G. 2004. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 1666:62–87. [DOI] [PubMed] [Google Scholar]
- 69.Liang, Y., D. Fotiadis, S. Filipek, D. Saperstein, K. Palczewski, and A. Engel. 2003. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 278:21655–21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fotiadis, D., B. Jastrzebska, A. Philippsen, D. Muller, K. Palczewski, and A. Engel. 2006. Structure of the rhodopsin dimer: a working model for G-protein-coupled receptors. Curr. Opin. Struct. Biol. 16:252–259. [DOI] [PubMed] [Google Scholar]
- 71.Filipek, S., K. Krzysko, D. Fotiadis, Y. Liang, D. Saperstein, A. Engel, and K. Palczewski. 2004. A concept of G protein activation by G protein-coupled receptor dimers: the transducin/rhodopsin interface. Photochem. Photobiol. Sci. 3:1–12. [DOI] [PubMed] [Google Scholar]
- 72.Chen, J.-Y., J. Knab, J. Cerne, and A. G. Markelz. 2005. Large oxidation dependence observed in terahertz dielectric response for cytochrome c. Phys. Rev. E. 72:040901. [DOI] [PubMed] [Google Scholar]