

Abstract
We show that fluorescence lifetime imaging microscopy (FLIM) of green fluorescent protein (GFP) molecules in cells can be used to report on the local refractive index of intracellular GFP. We expressed GFP fusion constructs of Rac2 and gp91phox, which are both subunits of the phagocyte NADPH oxidase enzyme, in human myeloid PLB-985 cells and showed by high-resolution confocal fluorescence microscopy that GFP-Rac2 and GFP-gp91phox are targeted to the cytosol and to membranes, respectively. Frequency-domain FLIM experiments on these PLB-985 cells resulted in average fluorescence lifetimes of 2.70 ns for cytosolic GFP-Rac2 and 2.31 ns for membrane-bound GFP-gp91phox. By comparing these lifetimes with a calibration curve obtained by measuring GFP lifetimes in PBS/glycerol mixtures of known refractive index, we found that the local refractive indices of cytosolic GFP-Rac2 and membrane-targeted GFP-gp91phox are ∼1.38 and ∼1.46, respectively, which is in good correspondence with reported values for the cytosol and plasma membrane measured by other techniques. The ability to measure the local refractive index of proteins in living cells by FLIM may be important in revealing intracellular spatial heterogeneities within organelles such as the plasma and phagosomal membrane.
Fluorescence lifetime imaging microscopy (FLIM) has become a robust technique in biochemistry and cell biology for the Förster resonance energy transfer detection of molecular interactions between protein molecules labeled with donor and acceptor members of the fluorescent protein (FP) family (1,2). Besides a sensitivity to molecular interactions (e.g., via energy transfer or collisions), fluorescence lifetimes of fluorophores are generally also dependent on other microenvironment parameters such as pH, viscosity, refractive index, and the presence of ions. Whereas viscosity has been shown not to affect the fluorescence lifetime of green fluorescent protein (GFP) (3), Suhling et al. and Borst et al. have reported that the refractive index of the microenvironment does influence the fluorescence lifetimes of GFP (4) and cyan and yellow fluorescent proteins (FPs) (5). FP lifetimes were further shown by these groups to satisfy the Strickler-Berg relationship (Eq. 1) between refractive index and fluorescence lifetime (6),
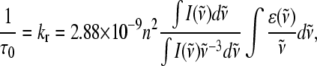 |
(1) |
in which τ0 is the natural radiative lifetime (related to the fluorescence lifetime τ via the fluorescence quantum yield φ = τ/τ0), kr is the radiative rate constant, n is the refractive index, I is the fluorescence emission,  is the wavenumber, and ɛ is the extinction coefficient.
is the wavenumber, and ɛ is the extinction coefficient.
Although FLIM on FP chimeras is now one of the most suitable optical microscopy techniques to investigate molecular interactions between proteins in living cells, little is known about the effect of different intracellular refractive indices on FP fluorescence lifetimes. To investigate whether the Strickler-Berg relationship is satisfied by GFP in living cells, which would imply that FLIM can be used to sense local intracellular refractive indices, we stably expressed enhanced GFP fusion constructs of Rac2 and gp91phox in human myeloid PLB-985 cells by retroviral transduction (see Supplementary Material). These cells also expressed a monomeric red FP (7) chimera of p67phox which might serve as a Förster resonance energy transfer acceptor in future experiments aimed at studying the molecular interactions between gp91phox or Rac2 and p67phox in living cells. Gp91phox, p67phox, and Rac2 are all subunits of the multimeric phagocyte NADPH oxidase enzyme that plays a critical role in the innate immune response against invading microorganisms (8). Upon NADPH oxidase activation, which occurs when leukocytes ingest microorganisms by phagocytosis, oxygen is reduced by gp91phox to superoxide (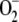 ). Superoxide is subsequently converted to other reactive oxygen species such as peroxide and hypochlorite, which contribute to the killing of the phagocytosed microbes. We have previously investigated (9,10) the dynamic behavior of GFP-Rac2 in resting and phagocytosing PLB-985 cells by fluorescence correlation spectroscopy and fluorescence recovery after photobleaching (FRAP) experiments, respectively. Whereas GFP-Rac2 displays a random translational diffusion in the cytosol in resting cells (10), we found that it is continuously being translocated to the phagosomal membrane in cells ingesting zymosan particles (9). At the membrane of the phagosome, Rac2 has been shown to bind to membrane-embedded gp91phox, which is a necessary interaction for NADPH oxidase activation to occur (11). In this study, we investigated GFP-Rac2 and GFP-gp91phox by FLIM because their intracellular locations in resting cells (Rac2 is cytosolic and gp91phox is membrane-bound) might give rise to different fluorescence lifetimes caused by different local refractive indices.
). Superoxide is subsequently converted to other reactive oxygen species such as peroxide and hypochlorite, which contribute to the killing of the phagocytosed microbes. We have previously investigated (9,10) the dynamic behavior of GFP-Rac2 in resting and phagocytosing PLB-985 cells by fluorescence correlation spectroscopy and fluorescence recovery after photobleaching (FRAP) experiments, respectively. Whereas GFP-Rac2 displays a random translational diffusion in the cytosol in resting cells (10), we found that it is continuously being translocated to the phagosomal membrane in cells ingesting zymosan particles (9). At the membrane of the phagosome, Rac2 has been shown to bind to membrane-embedded gp91phox, which is a necessary interaction for NADPH oxidase activation to occur (11). In this study, we investigated GFP-Rac2 and GFP-gp91phox by FLIM because their intracellular locations in resting cells (Rac2 is cytosolic and gp91phox is membrane-bound) might give rise to different fluorescence lifetimes caused by different local refractive indices.
We first verified the subcellular localization of GFP-Rac2 in GFP-Rac2/p67phox-mRFP PLB-985 cells and GFP-gp91phox in PLB-985 cells by confocal fluorescence microscopy. As shown in Fig. 1 A, GFP-Rac2 is indeed cytosolic in resting cells. This was further confirmed by fluorescence loss in photobleaching experiments, which showed that repetitive photobleaching of a small cytosolic region causes all of the fluorescence in these cells to disappear (see Supplementary Material). As expected, confocal microscopy showed that p67phox-mRFP is also cytosolic in resting GFP-Rac2/p67phox-mRFP PLB-985 cells (results not shown).
FIGURE 1.

Confocal fluorescence images (A and C) of GFP-Rac2/p67phox-mRFP (A) and GFP-gp91phox (C) PLB-985 cells. Corresponding bright-field images are shown in B and D.
As shown in Fig. 1 C, GFP-gp91phox is mainly localized to the plasma membrane but also to intracellular vesicles of ∼0.5 μm in diameter. This is consistent with reported subcellular fractionation assays on PLB-985 cells (12) and with previous microscopy studies of immunofluorescently-labeled gp91phox in fixed PLB-985 cells (13) and also shows that N-terminal tagging of gp91phox with GFP does not prevent the targeting of gp91phox to its functional sites, i.e., the plasma membrane and vesicular membranes. In COS-7 and Chinese hamster ovary cells (which, in contrast to PLB-985 cells differentiated into neutrophil-like cells, are nonphagocytic cells), GFP-gp91phox has also been reported to localize in the plasma membrane and intracellular membranes (14).
We next performed frequency-domain FLIM experiments on resting GFP-gp91phox, GFP-gp91phox/p67phox-mRFP, and GFP-Rac2/p67phox-mRFP PLB-985 cells using a wide-field fluorescence microscope equipped with a Lambert Instruments Fluorescence Attachment for lifetime imaging (see Supplementary Material). A blue light-emitting diode (λmax = 468 nm) modulated at 40 MHz was used to excite GFP. Fluorescence detection was performed by a combination of a modulated (40 MHz) image intensifier and a charge-coupled device camera, providing a spatial resolution of ∼0.35 μm/pixel in FLIM images. A narrow emission bandpass filter (520/35 nm) was used to allow detection of GFP only and suppress any fluorescence emission from mRFP attached to p67phox. FLIM measurements were calibrated by a 10-μM solution of rhodamine 6G, the lifetime of which was set to 4.11 ns (15). Fig. 2, B and D, show representative FLIM images of resting GFP-gp91phox and GFP-Rac2/p67phox-mRFP cells, respectively.
FIGURE 2.

Fluorescence intensity (A and C) and FLIM (B and D) images of GFP-gp91phox (A and B) and GFP-Rac2/p67phox-mRFP (C and D) PLB-985 cells. The lifetime scale bar ranges from 1 to 4 ns.
It is clear from these images that the average lifetime of membrane-bound GFP-gp91phox is significantly reduced compared to cytosolic GFP-Rac2 in PLB-985 cells. By averaging FLIM data of many cells, the fluorescence lifetime histograms shown in Fig. 3 were obtained. The highly overlapping blue and green histograms indicate that the presence of p67phox-mRFP does not influence the lifetime of GFP-gp91phox in GFP-gp91phox/p67phox-mRFP cells. We therefore also assume that the red curve in Fig. 3 is representative for GFP-Rac2 only, despite the presence of p67phox-mRFP in these cells. Gaussian fitting of the histograms in Fig. 3 resulted in average lifetimes of 2.31 ± 0.25 ns and 2.70 ± 0.20 ns for GFP-gp91phox and GFP-Rac2, respectively. To relate the observed fluorescence lifetimes of the different GFP chimeras in PLB-985 cells to different refractive indices, we constructed a calibration curve by measuring the fluorescence lifetime of GFP in PBS/glycerol mixtures of varying refractive index (see Supplementary Material), in analogy with a previous study (4). Using this calibration curve, the lifetimes for GFP-Rac2 in the cytosol and GFP-gp91phox in membranes correspond to local refractive indices of 1.38 ± 0.04 and 1.46 ± 0.06, respectively. These values closely resemble recently reported refractive indices for the cytosol, e.g., n = 1.36 (16) and n = 1.36−1.39 (17), and previous estimates for the plasma membrane, e.g., n = 1.46−1.60 (18) using phase microscopy techniques. Interestingly, in FLIM experiments using GFP physisorbed to polystyrene microspheres (n = 1.59), we found that the average fluorescence lifetime of GFP close to the PS surface is ∼1.84 ns (results not shown), which according to our GFP calibration curve corresponds to a local refractive index of 1.60. These experiments therefore validate our FLIM results on GFP chimeras in PLB-985 cells.
FIGURE 3.

Fluorescence lifetime histograms of GFP-gp91phox (blue), GFP-gp91phox/p67phox-mRFP (green), and GFP-Rac2/p67phox-mRFP (red) PLB-985 cells. Curves represent FLIM data recorded from 32 (blue), 28 (green), and 65 (red) cells.
In conclusion, we have demonstrated that FLIM enables the local refractive index of GFP chimeras in living cells to be measured. FLIM may therefore be valuable in studies aimed at investigating local heterogeneities in cellular structures such as membranes (19).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
Financial support from the Landsteiner Foundation for Blood Transfusion Research (Amsterdam, The Netherlands) is gratefully acknowledged. We thank Dr. Lydia Henderson (University of Bristol, UK) for the GFP-gp91phox vector. H.-J.v.M. thanks Dr. Thomas Jovin and Dr. Donna Arndt-Jovin for their hospitality and for stimulating FLIM discussions during a three-months visit to their laboratory at the Max Planck Institute for Biophysical Chemistry (Göttingen, Germany), which was financially supported (short-term fellowship ASTF No. 259-2005 to H.-J.v.M.) by the European Molecular Biology Organization (Heidelberg, Germany).
Editor: Egward H. Egelman.
References
- 1.Festy, F., S. M. Ameer-Beg, T. Ng, and K. Suhling. 2007. Imaging proteins in vivo using fluorescence lifetime microscopy. Mol. Biosyst. 3:381–391. [DOI] [PubMed] [Google Scholar]
- 2.Van Munster, E. B., and T. W. J. Gadella. 2005. Fluorescence lifetime imaging microscopy. Adv. Biochem. Eng. Biotechnol. 95:143–175. [DOI] [PubMed] [Google Scholar]
- 3.Suhling, K., D. M. Davis, and D. Phillips. 2002. The influence of solvent viscosity on the fluorescence decay and time-resolved anisotropy of green fluorescent protein. J. Fluoresc. 12:91–95. [Google Scholar]
- 4.Suhling, K., J. Siegel, D. Phillips, P. M. W. French, S. Lévêque-Fort, S. E. D. Webb, and D. M. Davis. 2002. Imaging the environment of green fluorescent protein. Biophys. J. 83:3589–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borst, J. W., M. A. Hink, A. van Hoek, and A. J. W. G. Visser. 2005. Effects of refractive index and viscosity on fluorescence and anisotropy decays of enhanced cyan and yellow fluorescent proteins. J. Fluoresc. 15:153–160. [DOI] [PubMed] [Google Scholar]
- 6.Strickler, S. J., and R. A. Berg. 1962. Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 37:814–882. [Google Scholar]
- 7.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross, A. R., and A. W. Segal. 2004. The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta. 1657:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Bruggen, R., E. Anthony, M. Fernandez-Borja, and D. Roos. 2004. Continuous translocation of Rac2 and the NADPH oxidase component p67phox during phagocytosis. J. Biol. Chem. 279:9097–9102. [DOI] [PubMed] [Google Scholar]
- 10.Van Manen, H.-J., R. Van Bruggen, D. Roos, and C. Otto. 2006. Single-cell optical imaging of the phagocyte NADPH oxidase. Antioxid. Redox Signal. 8:1509–1522. [DOI] [PubMed] [Google Scholar]
- 11.Bokoch, G. M., and T. Zhao. 2006. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid. Redox Signal. 8:1533–1548. [DOI] [PubMed] [Google Scholar]
- 12.Yu, L., F. R. DeLeo, K. J. Biberstine-Kinkade, J. Renee, W. M. Nauseef, and M. C. Dinauer. 1999. Biosynthesis of flavocytochrome b558. Gp91phox is synthesized as a 65-kDa precursor (p65) in the endoplasmic reticulum. J. Biol. Chem. 274:4364–4369. [DOI] [PubMed] [Google Scholar]
- 13.Zhen, L., L. Yu, and M. C. Dinauer. 1998. Probing the role of the carboxyl terminus of the gp91phox subunit of neutrophil flavocytochrome b558 using site-directed mutagenesis. J. Biol. Chem. 273:6575–6581. [DOI] [PubMed] [Google Scholar]
- 14.Murillo, I., and L. M. Henderson. 2005. Expression of gp91phox/Nox2 in COS-7 cells: cellular localization of the protein and the detection of outward proton currents. Biochem. J. 385:649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanley, Q. S., V. Subramaniam, D. J. Arndt-Jovin, and T. M. Jovin. 2001. Fluorescence lifetime imaging: multi-point calibration, minimum resolvable differences, and artifact suppression. Cytometry. 43:248–260. [DOI] [PubMed] [Google Scholar]
- 16.Curl, C. L., C. J. Bellair, T. Harris, B. E. Allman, P. J. Harris, A. G. Stewart, A. Roberts, K. A. Nugent, and L. M. D. Delbridge. 2005. Refractive index measurements in viable cells using quantitative phase-amplitude microscopy and confocal microscopy. Cytometry A. 65:88–92. [DOI] [PubMed] [Google Scholar]
- 17.Choi, W., C. Fang-Yen, K. Badizadegan, S. Oh, N. Lue, R. R. Dasari, and M. S. Feld. 2007. Tomographic phase microscopy. Nat. Methods. 4:717–719. [DOI] [PubMed] [Google Scholar]
- 18.Beuthan, J., O. Minet, J. Helfmann, M. Herrig, and G. Müller. 1996. The spatial variation of the refractive index in biological cells. Phys. Med. Biol. 41:369–382. [DOI] [PubMed] [Google Scholar]
- 19.Treanor, B., P. M. P. Lanigan, S. Kumar, C. Dunsby, I. Munro, E. Auksorius, F. J. Culley, M. A. Purbhoo, D. Phillips, M. A. A. Neil, D. N. Burshtyn, P. M. W. French, and D. M. Davis. 2006. Microclusters of inhibitory killer immunoglobulin-like receptor signaling at natural killer cell immunological synapses. J. Cell Biol. 174:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]