

Abstract
The biological and molecular properties of tetrodotoxin (TTX)-sensitive voltage-gated Na+ currents (INa) in murine vas deferens myocytes were investigated using patch-clamp techniques and molecular biological analyses. In whole-cell configuration, a fast, transient inward current was evoked in the presence of Cd2+, and was abolished by TTX (Kd = 11.2 nM), mibefradil (Kd = 3.3 μM), and external replacement of Na+ with monovalent cations (TEA+, Tris+, and NMDG+). The fast transient inward current was enhanced by veratridine, an activator of voltage-gated Na+ channels, suggesting that the fast transient inward current was a TTX-sensitive INa. The values for half-maximal (Vhalf) inactivation and activation of INa were −46.3 mV and −26.0 mV, respectively. RT-PCR analysis revealed the expression of Scn1a, 2a, and 8a transcripts. The Scn8a transcript and the α-subunit protein of NaV1.6 were detected in smooth muscle layers. Using NaV1.6-null mice (NaV1.6−/−) lacking the expression of the Na+ channel gene, Scn8a, INa were not detected in dispersed smooth muscle cells from the vas deferens, while TTX-sensitive INa were recorded in their wild-type (NaV1.6+/+) littermates. This study demonstrates that the molecular identity of the voltage-gated Na+ channels responsible for the TTX-sensitive INa in murine vas deferens myocytes is primarily NaV1.6.
INTRODUCTION
It is well known that activation of voltage-dependent Ca2+ channels and voltage-gated Na+ channels is involved in the generation of action potentials in various types of excitable cells (such as nerve fibers, skeletal muscle fibers, and cardiac myocytes). Although voltage-gated Na+ currents (INa) fail to be recorded in the vast majority of smooth muscle tissues, in some smooth muscles, there are several reports regarding the existence of INa in vascular (rat portal vein, (1); murine portal vein, (2)) and visceral smooth muscle (rat myometrium, (3); guinea-pig ureter, (4); sheep lymphatics, (5)), suggesting that INa are involved in the generation of action potentials. Voltage-gated Na+ channels appear to be selectively expressed in some, but not all smooth muscle, thereby questioning their significance in the physiology of the tissues. Most of INa in these smooth muscles are sensitive to tetrodotoxin (TTX) although the potency of the TTX-sensitivity seems to vary from tissue to tissue. Furthermore, little attention has been given to the molecular properties of TTX-sensitive INa in freshly dispersed smooth muscle myocytes.
Recent studies have revealed that voltage-gated Na+ channels consist of three subunits (expressed as a trimer): namely, an α-subunit (260 kDa) that forms the core protein of the channel (possessing the TTX-binding sites) and two β-subunits (30–40 kDa) that modify the channel function as an auxiliary subunit. To date, 11 isoforms of genes (Scn1a-11a) encoding TTX-sensitive and TTX-insensitive Na+ channels have been identified within a single family of Na+ channels, NaV1.x (6).
It has been reported that in mouse vas deferens, the presence of the fast TTX-sensitive component contributes to the rising phase of action potentials (7). In rat vas deferens smooth muscle cells, TTX-sensitive INa can be detected by use of conventional whole-cell recordings (8). However, the molecular and pharmacological properties of INa in the rodent vas deferens still remain elusive.
In this study, the electrophysiological and pharmacological properties of fast inward currents in murine vas deferens isolated smooth muscle cells were investigated using patch-clamp techniques. The molecular identity of the TTX-sensitive pore-forming subunits was revealed using RT-PCR analysis, in situ hybridization, and immunohistochemistry. Furthermore, INa were compared in NaV1.6-null mice (motor end-plate disease (med) mouse, NaV1.6−/–) lacking the expression of the Na+ channel gene, Scn8a, to INa in their wild-type (NaV1.6+/+) littermates.
MATERIALS AND METHODS
Cell dispersion
All animal experiments were approved by the animal care and use committee of Faculty of Medicine, Kyushu University (Fukuoka, Japan). Male Balb/c mice (8–10 weeks) or C3HeB/FeJ mice (2–3 weeks) were killed by cervical dislocation. Vasa deferentia were removed and immediately placed in physiological salt solution (PSS, see below). Myocytes were freshly isolated by the gentle tapping method (9) and stored at 4°C. Relaxed spindle-shaped cells were normally used for patch-clamp analysis within 3–4 h after isolation.
Recording and data analysis
Patch-clamp experiments (conventional whole-cell configuration) were performed at room temperature (21–23°C), as described previously (10). Glass pipettes of between 3 and 5 MΩ were prepared by a micropipette puller (P-97, Sutter Instruments, Novato, CA). Junction potentials between the bath and pipette solutions were measured with a 3 M KCl reference electrode and were under 1 mV, so that correction for these potentials was not made. The series resistance was compensated at the beginning of each experiment.
The data recording system used was essentially the same as described previously (10). The whole-cell INa data were filtered at 1 kHz by an eight-pole Bessel filter, sampled at 0.1 ms and analyzed on a PowerMac G4 computer (Apple Computer Japan, Tokyo, Japan) using commercial software (Chart v5.0.2, AD Instruments, Castle Hill, Australia).
Steady-state inactivation of INa was assessed using a two-step protocol where the cells were stepped from −70 mV to a range of voltages between −120 and +40 mV for a period of 2 s before a 200 ms step to −10 mV. The peak amplitude of INa evoked by each test pulse was measured and the peak amplitude of INa without application of any conditioning pulse was normalized as one. The lines were drawn by fitting the data to the Boltzmann's equation using the least-squares method,
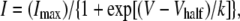 |
where I, Imax, V, Vhalf, and k are the relative amplitude of INa observed at various amplitudes of the conditioning pulse (I) and observed after the application of a conditioning pulse of −70 mV (Imax), amplitude of the conditioning pulse (V), and, where the amplitude of INa was reduced to half (Vhalf), slope factor (k).
Activation curves were derived from the current-voltage relationships. Conductance (G) was calculated using the equation
 |
where INa is the peak current elicited by depolarizing test pulses from −60 to +40 mV from the holding membrane potential of −70 mV and ENa is the equilibrium potential for Na+. Gmax is the maximal Na+ conductance (calculated at potentials above −10 mV). The G/Gmax ratios were plotted against the membrane potential as relative amplitudes.
Drugs and solutions
The following solutions were used: PSS containing (mM): Na+ 140, K+ 5, Mg2+ 1.2, Ca2+ 2, glucose 5, Cl− 151.4, HEPES 10, titrated to pH 7.35–7.40 with Tris base. The composition of the pipette solutions was (mM): Cs+ 130, tetraethylammonium (TEA+) 10, Mg2+ 2, Cl− 144, glucose 5, EGTA 5, ATP 5, HEPES 10/Tris (pH 7.35–7.40). Cells were allowed to settle in the small experimental chamber (80 μl in volume). The bath was superfused by gravity throughout the experiments at a rate of 2 ml min−1 from a solution reservoir. Efonidipine (kindly provided by the Nissan Chemical Industries, Funabashi, Japan), mibefradil, nifedipine, and veratridine (from Sigma-Aldrich Japan, Tokyo, Japan) were prepared daily as 100 mM stock solutions in dimethylsulphoxide. The final concentration of dimethylsulphoxide was <0.03% and this concentration of solvent did not affect control membrane currents. Tetrodotoxin (TTX, Sankyo, Tokyo, Japan) and kurtoxin (Peptide Institute, Osaka, Japan) were dissolved in deionized water, aliquoted, and stored frozen (−20°C).
RNA preparation and reverse transcription-polymerase chain reaction analysis
Total RNA was isolated from vas deferens tissue using CONCERT cytoplasmic RNA Purification Reagent (Invitrogen, Carlsbad, CA). First-strand cDNA was synthesized from 4 μg of total RNA using 200 U Superscript II Reverse Transcriptase (Invitrogen). The PCR reaction was performed using 1 μl of cDNA in 45 μl PCR SuperMix High Fidelity (Invitrogen) containing 0.5 μM of each primer. After denaturing at 94°C for 2 min, the cycling conditions were 94°C for 15 s, annealing 65°C for 30 s, and 72°C for 1 min for 35 cycles and then final extension at 72°C for 10 min. An aliquot of the RT-PCR product (10 μl) was analyzed on a 2% agarose gel. Generic subunit-specific primers (Scn2a, 3a, 5a, 10a, and 11a) were designed based on information from murine sequences and the locations of the primers indicated are based on the subunit sequence information obtained from GenBank (Table 1). Scn1a, 4a, 8a, and 9a were adapted from Saleh et al. (2) (for primers see Table 1). Control reactions were carried out in the absence of reverse transcriptase to ensure that the detected products were not the result of possible DNA contamination and by use of corresponding templates as positive controls to ensure that the primers were annealing successfully. All amplicons were of the expected sizes and their identity was confirmed by DNA sequence analysis.
TABLE 1.
Nucleotide sequences for the custom designed primers used to detect the voltage-gated Na+ channel gene isoforms in RT-PCR
| Gene name | REFseq ID | Primer sequence (5′ to 3′) | Gene name | REFseq ID | Primer sequence (5′ to 3′) |
|---|---|---|---|---|---|
| Scn1a | AJ810515 | F-AGCCTCACTGTGACTGTGCC | Scn8a | NM_011323 | F-GGGAGGACGATGAAGACAG |
| R-CTATTCGGAAGCACGTCCTCC | R-TCAGGCAGAACACCGTCAG | ||||
| Scn2a | L42341 | F-GCTTATCTCACTCCGTCATT | Scn9a | XM_196271 | F-CAGCAAAGAGAGACGGAACC |
| R-ACAATCGGGAGGTCCACTAT | R-CCCTCAGTGTCCGTAGAGATT | ||||
| Scn3a | NM_018732 | F-TGTGCTGAAATAACGACTTG | Scn10a | NM_009134 | F-TGTTGTGTGGCAATGGATCT |
| R-TTCACATAATCAATCCCCTT | R-GCACCTCCTGTTCTTTCTGC | ||||
| Scn4a | NM_133199 | F-CATCAAATCCCTGCGCACGC | Scn11a | NM_011887 | F-CAGCTTTGGCTGGTCTTTTC |
| R-ATAAAGATGACGAAGTAAAGG | R-CCTCCTTTTCCTCCCTCAAC | ||||
| Scn51a | NM_021544 | F-ACCACGGTTACACCAGCTTC | |||
| R-CCTCGTGTTCCTTCTTGAGC |
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed as described (11). Briefly, murine Scn8a digoxigenin-labeled “sense” and “antisense” RNA probes were produced from cDNA fragment corresponding to position 241–779 of the Scn8a cDNA sequence according to GenBank accession No. NM_011323. The cDNA fragment isolated from murine cerebellum cDNA library by PCR using 5′-CTG GAG GAC TTT GAC CCG TAC TAT-3′ (Forward) and 5′-AAC ACC GTC AGG ATC ATC ACG TCT-3′ (Reverse) primers, was subcloned into pCR II-TOPO (Invitrogen). The probes were labeled with the DIG-RNA labeling kit (Roche Diagnostics, Indianapolis, IN) according to manufacturer's instruction and used at a 1:1000 dilution in hybridization buffer. Vasa deferentia dissected from adult mice were subjected to overnight hybridization with the riboprobes at 70°C, and subsequently incubated with anti-DIG Fab fragments (Roche Diagnostics) overnight at 4°C. Detection was performed with staining buffer containing nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate (Roche Diagnostics). After staining for 30 min, the samples were sliced by microtome (Leica, Nussloch, Germany) to a 20 μm thickness and the staining was observed by BioZero microscopy (Keyence, Osaka, Japan).
Immunohistochemical studies
Vasa deferentia and cerebrum were embedded in a mixture (2:1) of optimal cutting temperature compound (Tissues-Tek, Sakura, Tokyo, Japan) and 20% sucrose in phosphate-buffered saline (PBS). Tissues in the embedding medium were immediately frozen in liquid nitrogen-cooled hexane. Frozen sections were cut with a cryostat (CM3050S, Leica, Tokyo, Japan) to 6 μm thick sections, and were mounted on silane-precoated glass slides and allowed to dry in air at room temperature for ∼30 min. Sections were fixed in cold acetone for 5 min and washed thoroughly in PBS, permeabilized and blocked with blocking solution (5% fat-free milk, 2% bovine serum albumin (BSA), and 0.1% Triton-X 100 in PBS) for 60 min at room temperature. Primary purified rabbit anti-NaV1.x antibodies (i.e., anti-NaV1.1 antibody, anti-NaV1.2 antibody and anti-NaV1.6 antibody, respectively) were purchased from Chemicon International (Temecula, CA). After washing, sections were incubated with the primary purified rabbit anti-NaV1.x antibody and diluted (1:200) in 1% BSA and 0.1% Triton-X 100 in PBS at 4°C overnight in a humidified chamber. As a negative control, the primary antibody was absorbed with the peptide against which it was made (i.e., blocking peptide). After washing three times (5 min each) in PBS, endogenous peroxidase activity was quenched with 0.3% H2O2 in methanol for 30 min. Sections were washed in PBS and then reacted with Envision system labeled polymer-horseradish peroxidase anti-rabbit (Dako Cytomation, Carpenteria, CA) for 30 min at room temperature, and visualized with 3,3′diaminobenzidine (Dojin Chemicals, Kumamoto, Japan) with 0.01% H2O2. Sections were dehydrated and coverslipped in mounting material (MGK-S, Matsunami Glass, Osaka, Japan).
In immunofluorescence studies, sections were incubated with the primary purified rabbit anti-NaV1.x antibody (i.e., anti-NaV1.1 antibody, anti-NaV1.2 antibody, and anti-NaV1.6 antibody, respectively) as described, and the primary antibody was absorbed with the peptide against which it was made (i.e., blocking solution; 2% goat IgG, 1% BSA, and 0.1% Triton-X 100 in PBS). After washing in PBS (three washes for 5 min each), sections were incubated with phycoerythrin-conjugated goat anti-rabbit IgG (sc-3752; Santa Cruz Biotechnology, Santa Cruz, CA) diluted to 1:30 in PBS with 2% normal goat serum, 1% BSA, and 0.1% Triton-X 100 in PBS for 1 h at room temperature under dark conditions. Sections were then washed in PBS a further three times (5 min duration each). Coverslips were mounted onto slides by use of fluorescence mounting medium and slides were viewed by fluorescent microscopy (Biozero BZ-8000, Keyence). Mouse cerebrum was used as a positive control for anti-NaV1.1 and anti-NaV1.2 antibodies.
NaV1.6-null mice
Heterozygous Scn8a-motor end-plate disease (med) mice maintained on a C3HeB/FeJ background were obtained from The Jackson Laboratories (Bar Harbor, ME). The med mutation produces complete loss of NaV1.6 expression. To compare animals homozygous for med (NaV1.6−/–) with wild-type (NaV1.6+/+) littermates, genotyping was performed with individual pups from the intercrosses of heterozygote (NaV1.6+/–) mice, and then NaV1.6−/– or NaV1.6+/+ mice were selected for further examination. Genotyping of NaV1.6 was performed by PCR as described previously (12). PCR amplification was achieved with 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s using the following specific primers: for the wild-type allele, 5′-GGA GCA AGG TTC TAG GCA GCT TTA AGT GTG-3′ and 5′-GTC AAA GCC CCG GAC GTG CAC ACT CAT TCC-3′ and for the mutant allele, 5′-TCC AAT GCT ATA CCA AAA GTC CC-3′ and 5′-GGA CGT GCA CAC TCA TTC CC-3′. PCR products were separated on a 1.5% agarose gel, allowing the resolution of a 230-basepair product for the wild-type allele and a 194-basepair product for the mutant allele.
Statistics
Statistical analyses were performed using analysis of variance tests (two-factor with replication). Changes were considered significant at P < 0.05 (*, P < 0.05; **, P < 0.01). Data are expressed as mean with the standard deviation (mean ± SD).
RESULTS
Depolarization elicits three types of inward currents in murine vas deferens myocytes
After establishing a conventional whole-cell configuration in smooth muscle cells freshly isolated from murine vas deferens, application of a depolarizing step to −10 mV from a holding potential of −70 mV produced an inward current, which reached a peak and then gradually decayed (Fig. 1 A). Application of nifedipine (10 μM) dramatically reduced the peak amplitude of the inward current. In the presence of nifedipine, a transient inward current superimposed upon a slowly inactivating component was obtained. Further application of Cd2+ (100 μM) abolished the slow components of the current, leaving a fast transient component (Fig. 1 A).
FIGURE 1.

Three components of voltage-dependent inward currents in murine vas deferens myocytes. (A) Representative current traces in the absence and presence of several drugs (nifedipine (10 μM), Cd2+ (100 μM), and TTX (1 μM), respectively) measured using a conventional whole-cell configuration. The bath solution was PSS and the recording pipette was filled with a Cs+-TEA+ solution containing EGTA (5 mM). Each current was evoked by 200 ms depolarization steps from a holding potential of −70 to −10 mV. (B) Three different components of voltage-dependent inward currents, namely, a nifedipine-sensitive component, a nifedipine-insensitive but Cd2+-sensitive component, and a TTX-sensitive component. The nifedipine-sensitive component was obtained by subtracting the current recorded in the presence of nifedipine (10 μM) from the control current recorded in the absence of drugs. The nifedipine-insensitive but Cd2+-sensitive component was obtained by subtracting the current recorded in the presence of nifedipine (10 μM) and Cd2+ (100 μM) from the current in the presence of nifedipine (10 μM) alone. The TTX-sensitive component was obtained by subtracting the current in the presence of nifedipine (10 μM), Cd2+ (100 μM), and TTX (1 μM) from the current in the presence of only nifedipine and Cd2+. (C) Current-voltage relationships of the three components obtained in panel B from the same cell. The peak amplitude of the membrane current was measured in each condition.
To classify these components, a method for pharmacological subtraction of the leak and the capacitive currents was performed (10). As shown in Fig. 1 B, subtracting the current in the presence of nifedipine (10 μM) from the control current yielded a nifedipine-sensitive inward current, which was evoked at potentials more positive than −40 mV. The maximum peak amplitude was obtained at ∼−10 mV and was reduced at more positive potentials, showing a bell-shaped current-voltage relationship (Fig. 1 C). Subtracting the current in the presence of nifedipine (10 μM) and Cd2+ (100 μM) from that in the presence of nifedipine alone yields a nifedipine-insensitive but Cd2+-sensitive inward current. This current appeared at potentials more positive than −40 mV and peaked at 0 mV, demonstrating a bell-shaped current-voltage relationship (Fig. 1 C). By subtracting the current in the presence of TTX (1 μM), nifedipine (10 μM) and Cd2+ (100 μM) from that in the presence of nifedipine and Cd2+, a TTX-sensitive component was obtained.
To examine the possibility that TTX-resistant Na+ currents were included in the nifedipine-insensitive and Cd2+-sensitive component, the electrophysiological properties of the Cd2+-sensitive component were investigated in the presence of TTX (1 μM) and nifedipine (10 μM). Fig. 2 A shows the time course when the extracellular Na+ concentration in the bath solution was changed to an isoosmotic solution of TEA+. When extracellular Na+ was replaced with TEA+, the peak amplitude of the nifedipine-insensitive and TTX-resistant inward current was not affected (n = seven, three different animals). Similar results were obtained when the same amount of Na+ was replaced with Tris+ (n = six cells, three different animals, Fig. 2 B) or N-methyl-D-glucamine (NMDG+) (n = six cells, three different animals, Fig. 2 B). The nifedipine-insensitive and TTX-resistant inward currents were abolished by subsequent application of Cd2+. These results suggest that the nifedipine-insensitive, TTX-resistant and Cd2+-sensitive component seems to possess no Na+ permeability. Since TTX-resistant Na+ currents are reported to be highly Cd2+-sensitive (13,14), RT-PCR was performed to determine the presence of TTX-resistant Na+ channel genes (Scn5a, Scn10a, and Scn11a). Specific primers for the amplification were designed to produce cDNA fragments to each isoform gene for α-subunits (see Table 1). Positive amplicons for Scn5a (heart cells), 10a (dorsal root ganglion) and 11a (dorsal root ganglion) were detected, whereas in vas deferens, none of amplicons was detected (Fig. 2 C). All amplicons were sequenced to confirm their identity.
FIGURE 2.

No Na+ permeability could be detected in the nifedipine-insensitive and TTX-resistant inward current which was blocked by Cd2+. (A) The time course of the inward currents in the presence of nifedipine (10 μM) and TTX (1 μM) when the extracellular Na+ was replaced with TEA+ (the inward currents were elicited by repetitive depolarizing pulses from −70 mV to −10 mV). The peak amplitude of the inward current was measured under each experimental condition. (B) Columns indicate the relative mean values of the peak amplitude of the inward current elicited by a step pulse from −70 mV to −10 mV. The peak amplitude of the inward current was normalized as one just before extracellular Na+ was replaced with several isoosmotic solutions (TEA+, Tris+, and NMDG+). Data are expressed as mean ± SD (n = 6–7 cells, three different animals). (C) RT-PCR was performed as described in the Materials and Methods and a ladder was used to indicate the size of the amplified fragments. Specific primers for Scn gene isoforms (Scn5a, 10a, and 11a) were used and mRNA was extracted from freshly dissected murine heart (H), dorsal root ganglion (D), and vas deferens (V).
TTX-sensitivity of the transient inward currents
To characterize further the transient TTX-sensitive inward current, in a series of experiments, Cd2+ (100 μM) was added to the bath solution. The peak amplitude of the transient TTX-sensitive inward current was maintained for at least 30 min when test depolarization pulses were applied at 20 s intervals (the peak amplitude of the transient inward current at 20 min being 98% (n = 37 cells, 12 different animals) of the value determined after reaching steady state). Therefore, all experiments were performed within 20 min after the peak amplitude of the transient TTX-sensitive inward current had become stable at −70 mV. In the presence of 100 μM Cd2+, TTX caused a concentration-dependent inhibition of the transient inward current (Kd = 11.2 nM; n = 4–10 cells, Fig. 3, B and C). On removal of TTX, this inhibitory action was reversed to the control level.
FIGURE 3.

The effects of TTX on the transient inward currents. (A) Representative traces of the transient inward currents eliminated by preapplication of nifedipine (10 μM) and Cd2+ (100 μM) with increasing concentrations of TTX. (B) Concentration-response relationship for TTX on the transient inward currents. The peak amplitude of the transient inward current elicited by a step pulse to −10 mV from a holding potential of −70 mV just before application of TTX was normalized as one (i.e., control). The curve was drawn by fitting the following equation using the least-squares method: Relative amplitude of the transient inward current = 1/{1 + (D/Kd) nH}, where Kd, D, and nH are the inhibitory dissociation constant, the concentration of TTX (nM), and Hill's coefficient, respectively. The following values were used for the fitted curve: Kd = 11.2 nM, nH = 1.0. Each symbol indicates the mean of 4–10 observations with the mean ± SD shown by the vertical lines. Some of the SD bars are smaller than the symbol. (C) The current-voltage relationships in the absence and presence of TTX (10 nM) when Cd2+ (100 μM) was present. The current amplitude was measured as the peak amplitude of the transient inward current at each membrane potential.
Voltage-dependent properties of the transient inward current
The transient TTX-sensitive inward current shows rapid activation and inactivation. The decay of the transient inward current was fitted to a single exponential in the whole range of potentials studied (Fig. 4 A). The time constant of the current decay (τ) decreased monotonically from 4.0 ± 0.8 ms at −20 mV to 0.7 ± 0.1 ms at +10 mV (Fig. 4 B). The steady-state inactivation and activation curves for the transient TTX-sensitive inward currents were obtained (conditioning pulse duration, 2 s; holding membrane potential, −70 mV). Inactivation of the transient TTX-sensitive inward currents occurred with conditioning pulses positive to −60 mV and the 50% inactivation potential (Vhalf), evaluated by fitting to the Boltzmann's equation, was −46.3 ± 0.6 mV (n = five cells, three different animals). The activation curves were obtained from the current-voltage relationships fitted to the Boltzmann's equation, and the 50% activation potential (Vhalf) was −26.0 ± 0.7 mV (n = seven cells, five different animals). The average activation and inactivation values are summarized in Fig. 4 C.
FIGURE 4.

Voltage-dependent activation and inactivation of the transient inward currents. (A) The declining phase of the transient inward current elicited by pulses from the holding potential of −70 mV to each indicated membrane potential (−20, −10, 0, and +10 mV) was fitted to a single exponential (mean inactivation time constant (τ), 4.3, 1.3, 0.9, and 0.6 ms, respectively). (B) Each column shows the mean value of the time constant (τ) of the transient inward current measured at each membrane potential with mean ± SD (n = five cells). (C) The activation and the steady-state inactivation curves of the transient inward currents. The steady-state inactivation curve was obtained using a double pulse protocol, fitting to the Boltzmann's equation (see Materials and Methods). Each symbol indicates the mean of five observations with mean ± SD. The activation curve was obtained from the current-voltage relationship of the transient inward current, fitting to the Boltzmann's equation (see Materials and Methods). Each symbol indicates the mean of seven observations with mean ± SD. Some of the SD bars are smaller than the symbol.
Na+ serves as a charge carrier for the transient inward currents
To estimate the ion selectivity of the TTX-sensitive transient inward currents, extracellular Na+ concentration was changed to an isoosmotic solution of TEA+. When extracellular Na+ was replaced with TEA+, the peak amplitude of the TTX-sensitive transient inward currents markedly decreased (Fig. 5, A and B) and on reintroduction and removal of TEA+, the peak amplitude of the TTX-sensitive transient inward current readily recovered to the control level (Fig. 5, A and B). Similar results were obtained when the same amount of Na+ was replaced with Tris+ or NMDG+ (data not shown). These results suggest that the TTX-sensitive transient inward current is carried mainly by Na+ (Fig. 5 C, n = five cells, three different animals).
FIGURE 5.

Ion-selectivity and pharmacological properties of the transient inward currents. (A) The time course of the transient inward currents when the extracellular Na+ was replaced with TEA+ (the transient inward currents were elicited by repetitive depolarizing pulses from −70 mV to −10 mV). The peak amplitude of the transient inward current was measured under each experimental condition. (B) Original current traces before (control, (a)), after application of Na+-free solution, (b) and washout (c) as indicated in A. (C) Columns indicate the relative mean values of the peak amplitude of the transient inward current was elicited by a step pulse from −70 mV to −10 mV. The peak amplitude of the transient inward current was normalized as one just before extracellular Na+ was replaced with isoosmotic solution of TEA+. Asterisks indicate a statistically significant difference, demonstrated using a paired t-test (P < 0.01). Data are expressed as mean ± SD (n = five cells). (D) Histogram summarizing the relative effects of various blockers and activators on the peak amplitude of the TTX-sensitive Na+ current in the presence of 100 μM Cd2+. The peak amplitude of the TTX-sensitive Na+ current was normalized as one just before the application of divalent cations and drugs (i.e., control). Ni2+ (100–300 μM; n = four cells), Cd2+ (up to 500 μM; n = six cells), efonidipine (1–10 μM; n = four cells), kurtoxin (100 nM; n = six cells), and veratridine (1–10 μM; n = six cells). Each column shows the relative amplitude of the inward current (mean value ± SD). Asterisks indicate a statistically significant difference, using a paired t-test (P < 0.01). (E) Relationships between relative inhibition of the peak amplitude of the transient inward current and the concentration of mibefradil. The peak amplitude of INa was elicited by a step pulse to −10 mV from −70 mV. The curve was fitted using following equation and the least-squares method: Relative amplitude of the transient inward current = 1/{1 + (D/Kd) nH}, where Kd, D, and nH are the dissociation constant, concentration of mibefradil (μM), and Hill's coefficient, respectively. The following values were used for the fitted curve: Kd = 3.3 μM, nH = 1.0, and n = 4–6 cells.
Pharmacological properties of the transient TTX-sensitive inward INa
As summarized in Fig. 5 D, the effects of several channel blockers (Ni2+, Cd2+, efonidipine and kurtoxin) on the peak amplitude of the TTX-sensitive transient inward INa were investigated. None of the channel blockers had a significant effect on the amplitude of the inward current; however, veratridine, an activator of voltage-gated Na+ channels, enhanced the peak amplitude of the TTX-sensitive transient inward INa in a concentration-dependent manner (1–10 μM). In contrast, mibefradil inhibited the peak amplitude of the transient inward current in a concentration-dependent manner (Kd = 3.3 μM; n = 4–6 cells, Fig. 5 E).
Molecular expression of Na+ channels
To determine the molecular nature of voltage-gated Na+ channels, samples of RNA were obtained from murine heart and vas deferens. It is well established that six isoforms of genes for voltage-gated Na+ channels (Scn1a, 2a, 3a, 4a, 8a, and 9a) are highly TTX-sensitive (6). Since INa in murine vas deferens were abolished by TTX, these results strongly suggest that TTX-sensitive voltage-gated Na+ channels are functionally expressed in that tissue. To investigate the precise subtype of channel present, specific primers for the amplification of TTX-sensitive voltage-gated Na+ channels were designed to produce cDNA fragments to each isoform gene for α-subunits (Scn1a, 2a, 3a, 4a, 8a, and 9a, see Table 1). Positive amplicons for Scn1a, 2a, 3a, 4a, and 8a were detected in cardiac myocytes, while in vas deferens, only Scn1a, 2a, and 8a were detected (Fig. 6 A). All amplicons were sequenced to confirm their identity.
FIGURE 6.

Molecular identification of voltage-gated Na+ channel subunits expressed in mouse vas deferens. (A) RT-PCR was performed as described in the Materials and Methods and a ladder was used to indicate the size of the amplified fragments. Specific primers for Scn gene isoforms (Scn1a, 2a, 3a, 4a, 8a, and 9a) were used and mRNA was extracted from freshly dissected murine heart (H) and vas deferens (V). (B) Whole-mount in situ hybridization with a DIG-labeled Scn8a antisense probe revealed Scn8a expression in the smooth muscle layers. Arrows indicate the positive signals in smooth muscle myocytes. Black bar represents 100 μm. (C) The expanded figure in panel B. Solid bar represents 30 μm. (D) No staining is obtained with the Scn8a sense riboprobe used as a control. Solid bar represents 100 μm. (E) The expanded figure in panel D. Solid bar represents 30 μm.
Expression of Scn8a mRNA
To study the distribution of Scn8a mRNA throughout the mouse vas deferens organ, whole-mount in situ hybridization was performed in fixed tissue using Scn8a-specific DIG-labeled sense and antisense riboprobes. The antisense riboprobe displayed distinct intense markings in smooth muscle layer tissues compared with sense probes, suggesting that smooth muscle myocytes are candidates for Scn8a expression (Fig. 6, B and C).
Immunohistochemical localization of NaV1.6
To localize a molecular marker for NaV1.6, immunohistochemical studies were performed. NaV1.6 immunoreactivity is clearly visible in the smooth muscle layers of the vas deferens (Fig. 7 A), while no specific immunoreactive signal was seen when the primary antibody was preabsorbed with the immunizing NaV1.6 antigen (Fig. 7 B). Immunohistochemistry using nonimmune rabbit IgG instead of primary antibody also gave a negative result (data not shown). Similarly, as shown in Fig. 7 C, NaV1.6 immunoreactivity is clearly visible in the smooth muscle bundles. In contrast, no specific immunoreactive signal was seen when the primary antibody was preabsorbed with the immunizing NaV1.6 antigen (Fig. 7 D). Although Scn1a and Scn2a amplicons were detected in the vas deferens, note that immunoactivity of NaV1.1 (Fig. 8 A) and NaV1.2 (Fig. 8 C) were not detected in the smooth muscle layers of murine vas deferens, but immunoactivity of NaV1.1 (Fig. 8 B) and NaV1.2 (Fig. 8 D) were clearly visible in mouse cerebrum as a positive control.
FIGURE 7.

Fluorescent images of immunoreactivity for NaV1.6 in the smooth muscle layers of mouse isolated vas deferens. (A) NaV1.6 immunoactivity; clear membranous staining was observed in the tissues of the smooth muscle layer. (B) Negative-control: use of NaV1.6 antibody preabsorbed with the immunizing antigen (BP) never yields immunostaining. Bar (solid line) represents 100 μm. (C) NaV1.6 immunoactivity; clear membranous staining was observed within the smooth muscle layers of murine vas deferens. (D) Negative-control: use of NaV1.6 antibody preabsorbed with the immunizing antigen (BP) never yielded immunostaining. Open bar represents 100 μm.
FIGURE 8.

Comparative fluorescent images of immunoreactivity for NaV1.1 and NaV1.2 in both the smooth muscle layers of mouse isolated vas deferens and mouse cerebrum. (A) No NaV1.1 immunoactivity within the smooth muscle layers of murine vas deferens. (B) Positive control: use of NaV1.1 antibody in murine cerebrum. (C) No NaV1.2 immunoactivity within the smooth muscle layers of murine vas deferens. (D) Positive control: use of NaV1.2 antibody in murine cerebrum. Open bar represents 100 μm.
NaV1.6-null mice (med mouse) lacking expression of Na+ channel gene Scn8a
To investigate further the molecular and functional contributions of INa in smooth muscle cells from murine vas deferens, homozygous mice with a null allele of NaV1.6 (med mice, NaV1.6−/–; (15)) were used. As shown in Fig. 9 A, in wild-type mice (NaV1.6+/+), TTX-sensitive INa were evoked by application of depolarizing rectangular pulses from a holding potential of −70 mV (n = five, three different animals), showing similar electrophysiological and pharmacological properties (voltage-dependency in Fig. 9 B, TTX-sensitivity (Kd = 10.1 nM; n = 5–6 cells) etc.) with those recorded in Balb/c mice. Conversely, in NaV1.6−/– mice, INa were not detected (n = seven, three different animals; Fig. 9 A). These results imply that the molecular properties of INa observed in vas deferens smooth muscle cells are closely related to NaV1.6.
FIGURE 9.

Comparison of electrophysiological properties of the transient inward currents in NaV1.6−/− and NaV1.6+/+ murine vas deferens myocytes. (A) Representative traces of the transient inward currents eliminated in the presence of 10 μM nifedipine and 100 μM Cd2+ at the indicated membrane potentials. (B) The current-voltage relationships in NaV1.6−/− and NaV1.6+/+ mice when 10 μM nifedipine and 100 μM Cd2+ were present. The current amplitude was measured as the peak amplitude of the transient inward current at each membrane potential.
DISCUSSION
In this study, it has been demonstrated that voltage-dependent inward currents in freshly dispersed smooth muscle cells from mouse vas deferens consist of three different components, namely, a fast activating current followed by two types of slow activating currents. This work establishes that the fast component of inward current flows through TTX-sensitive voltage-gated Na+ channels in murine vas deferens smooth muscle cells, and that the molecular identity of these channels is likely to be NaV1.6.
Characterization of voltage-gated Na+ channels
These observations regarding the electrophysiological and pharmacological properties of the fast component of the inward current can be summarized as follows:
The fast component of the inward current was abolished by TTX (Kd = 11.2 nM) and substitution of Na+ with TEA+, Tris+, and NMDG+.
The peak amplitude of the fast component was enhanced by veratridine, a selective voltage-gated Na+ channel activator, in a concentration-dependent manner (1–10 μM).
The peak amplitude of the fast component was not sensitive to Ni2+ (100–300 μM), Cd2+ (500 μM), efonidipine (1–10 μM), kurtoxin (100 nM), and nifedipine (10 μM).
The fast component of the inward current shows fast kinetics in both its activation and inactivation in biophysical properties.
The decay of the inward current was voltage-dependent, with rapid inactivation between −20 mV and +10 mV.
Taken together, the fast component of the inward current in murine vas deferens appears to be carried through the TTX-sensitive voltage-gated Na+ channels. The two slow activating components of voltage-dependent inward currents were shown to be voltage-dependent Ca2+ currents since both were abolished by Cd2+ (100 μM) and substitution of Ca2+ with Co2+ (2 mM). One of the slow activating components was a nifedipine-sensitive Ca2+ current (i.e., L-type Ca2+ current). In contrast, another slow activating component of the voltage-dependent inward currents was a nifedipine-insensitive inward current.
It has been reported that TTX-resistant voltage-gated Na+ channels, such as NaV1.8 and NaV1.9, were blocked greatly by Cd2+ (13,14). Thus, in these experiments, the electrophysiological and molecular properties of the nifedipine-insensitive and TTX-resistant inward current, which was abolished by Cd2+, were investigated. The peak amplitude of the inward current was not affected by isometric substitution of Na+ with TEA+, Tris+ and NMDG+, suggesting a lack of Na+-permeable component. In RT-PCR analysis, the three TTX-resistant voltage-gated Na+ channel amplicons (Scn5a, 10a, and 11a) were not detected in mRNA signal level. These results suggest that TTX-resistant voltage-gated Na+ channels were not included in the nifedipine-insensitive and TTX-resistant inward current in mouse vas deferens myocytes.
In the presence of TTX and nifedipine, Holman et al. (7) demonstrated that small but significant amplitudes of action potentials were detectable in murine vas deferens. If the nifedipine-insensitive Ca2+ currents measured in this study are contributing to action potentials in murine vas deferens, this might account for Holman's observations. Interestingly, in rat vas deferens, the nifedipine-insensitive Ca2+ current was not observed; only the presence of the nifedipine-sensitive Ca2+ currents (8). Further studies are needed to clarify the nature and properties of the nifedipine-insensitive and TTX-resistant inward currents in the vas deferens.
Similarities and differences between the functional properties of voltage-gated Na+ channels in vascular and visceral smooth muscles
Recently, electrophysiological and molecular properties of INa in smooth muscle cells freshly isolated from murine portal vein have been characterized and TTX inhibited the peak amplitude of INa with a similar potency to that in the vas deferens ((2); murine portal vein, Kd = 12 nM; murine vas deferens, Kd = 11 nM). Apart from the TTX-sensitivity, INa in vas deferens have many different properties compared to those measured in portal vein. For instance, mibefradil (100 μM) caused no effect on INa in portal vein (2) while INa in the vas deferens were abolished by mibefradil in a concentration-dependent manner (Kd = 3.3 μM). Interestingly, in recombinant voltage-gated Na+ channel studies, it has been shown that mibefradil suppresses TTX-sensitive (NaV1.2, NaV1.4, NaV1.7; (16)) and TTX-insensitive Na+ channels (NaV1.5; (16,17)), showing Kd values at submicromolar concentrations. In murine portal vein, Vhalf values of inactivation and activation for INa were shown to be +5 mV and −52 mV, respectively. Conversely, in murine vas deferens, Vhalf values of inactivation and activation of INa were −46 mV and −26 mV, respectively, showing the different biophysical properties of voltage-gated Na+ channels. Although the decay of INa was voltage-dependent in both tissues, the values of the inactivation time constant (τ) at each membrane potential (from −10 mV to +20 mV) in portal vein were much larger than those in vas deferens. It is not clear, however, whether this difference is related to the different space-clamp speed due to the different recording modes of patch-clamp techniques (amphotericin B-perforated patch versus conventional whole-cell mode). In murine portal vein, the TTX-sensitive INa, which were resistant to nicardipine (1 μM) or Cd2+ (1 mM), were observed in 16% of cells (30 from 192 cells). However, most of the smooth muscle cells from the vas deferens show TTX-sensitive INa (∼95%, 98 from 103 cells), suggesting a much higher channel density. In the murine portal vein, a significant membrane hyperpolarization (10 mV) was evoked by the application of TTX. In contrast, TTX consistently caused depolarization (9 mV) in murine vas deferens using microelectrode techniques (7), although it is not certain whether the discrepancy is related to the difference of the tissues. Taking into account these results, we suggest that the properties of INa in the vas deferens are different from those in the portal vein with a similar TTX-sensitivity. Furthermore, in rat vas deferens, biophysical properties of TTX-sensitive INa (the values of Vhalf for inactivation and activation were −63 mV and −19 mV, (8)) were also different from those in mouse vas deferens, suggesting that the properties of INa may vary between species.
Molecular properties of voltage-gated Na+ channels in smooth muscles
In cultured human smooth muscle cells, Jo et al. (18) reported that the expression of Scn9a (coding for NaV1.7) is responsible for the majority of TTX-sensitive INa. Saleh et al. (2) reported that, in murine portal vein, RT-PCR analysis revealed three voltage-gated Na+ channel amplicons (Scn7a, 8a and 9a) in mRNA signal level, in addition to the proteins encoded by Scn8a (NaV1.6) and Scn9a (NaV1.7). In this study, Vhalf for inactivation and activation of INa were −46 mV and −26 mV, respectively, and were close to those observed in recombinant NaV1.6, coexpressed with β-subunits (β1 and β2 subunits, Vhalf for inactivation and activation were −51 mV and −17 mV, respectively (19); β1 subunit, Vhalf for activation was −26 mV (20)). INa in murine vas deferens also possess TTX- and mibefradil-sensitivities similar to those in recombinant NaV1.6. Furthermore, we were able to detect amplicons of appropriate size and sequence for Scn1a (NaV1.1), Scn2a (NaV1.2), and Scn8a (NaV1.6), although the transcript for Scn9a (NaV1.7) was not detected. When a specific antibody for each NaV1.x antigen (anti-NaV1.1 antibody and anti-NaV1.2 antibody) was used, both NaV1.1 and NaV1.2 immunoreactivities are clearly visible in mouse cerebrum. In contrast, no immunoactivity of NaV1.1 and NaV1.2 was detected in the smooth muscle layer of murine vas deferens, suggesting that the channel proteins for NaV1.1 and NaV1.2 are unlikely to be expressed. It is not certain whether other tissues (such as peripheral nerves) may contaminate the mRNA sample of vas deferens smooth muscle, and thereby showing transcripts with mRNA levels which would not normally be found in the smooth muscles themselves.
In these experiments, we have detected mRNA for Scn8a and NaV1.6 proteins in smooth muscle layers of mouse vas deferens, indicating the possible presence of NaV1.6 proteins in these myocytes. Furthermore, NaV1.6-null mice (med mouse, NaV1.6−/–), lacking the expression of the Na+ channel gene, Scn8a, were used to investigate the molecular properties of INa in murine vas deferens, and compared to currents recorded from NaV1.6+/+ wild-type and Balb/c mice. In the presence of nifedipine (10 μM) and Cd2+ (100 μM), INa were not detected in NaV1.6−/– mouse vasa deferentia, while INa were recorded in NaV1.6+/+ wild-type littermates. These results strongly suggest that INa in mouse vas deferens are primarily due to activation of NaV1.6 channels.
Physiological roles of voltage-gated Na+ channels in smooth muscles
Since voltage-gated Na+ channels in human smooth muscle cells were only expressed in the culture conditions but not in freshly dispersed bronchial smooth muscle cells, it is thought that voltage-gated Na+ channels may be involved in de-differentiation and proliferation (18). Thus, under various pathophysiological conditions, such as vascular injury, atherosclerosis, and asthma, the possible expression of voltage-gated Na+ channels is worth considering. Surprisingly though, functional expression of INa has also been reported in a wide variety of freshly isolated smooth muscle cells (see Introduction).
Holman et al. (7) reported that a small membrane depolarization caused an enhancement of voltage-gated Na+ channel activity, thereby inducing an increase of Na+ influx and intracellular Na+ concentration ([Na+]i). The increase in [Na+]i alters the driving force for Na+, the rate of movement of Na+ by the Na+/K+ pump and then the Na+/Ca2+ exchange, which may subsequently raise intracellular Ca2+ concentration ([Ca2+]i) in mouse vas deferens (7). Similarly, it is reported that voltage-gated Na+ channels in murine portal vein play an important role in modulating contractility through a mechanism involving reverse mode Na+/Ca2+ exchange (2).
In the mouse vas deferens, it is reported that individual smooth muscle cells are probably coupled electrically, but that there are few, if any, low resistance pathways in the longitudinal direction (21). In the mouse vas deferens, the excitatory junction potential (EJP) amplitude was more variable in surface cells, suggesting that fewer sites contribute to the EJP and that there is poor or no electrical coupling of small EJPs (22). Furthermore, recently, it has been reported that surface smooth muscle cells of the mouse isolated vas deferens were poorly electrically coupled, suggesting that surface cells are somewhere toward the electrically uncoupled extreme (23). It is possible that the activation of INa may be required to generate sufficient inward current density in a short period of time to facilitate action potential propagation within the smooth muscle, given the presumed scarcity of electrical coupling between cells in the vas deferens. Further studies are necessary to clarify the precise physiological roles and significance of voltage-gated Na+ channels in smooth muscles that express them.
In conclusion, we found that voltage-gated inward currents in smooth muscle cells freshly dispersed from mouse vas deferens are composed of three different membrane currents; namely, nifedipine-sensitive currents (L-type Ca2+ channels), nifedipine-insensitive Cd2+-sensitive currents, and TTX-sensitive INa (voltage-gated Na+ channels; Nav1.6). This study also provides novel evidence that the Nav1.6 channel is the major TTX-sensitive Na+ channel expressed in murine vas deferens smooth muscle cells.
Acknowledgments
We thank Dr. Hidemasa Furue, Kyushu University, Graduate School of Medical Sciences, Department of Physiology, Fukuoka, Japan, for the helpful technical advice. We also thank Mr. Hiroshi Fujii, Kyushu University, Faculty of Medicine, Department of Pathology, Fukuoka, Japan, for his invaluable help with histological experiments.
This work was supported by the Japan Science and Technology Agency (grant No. 1139-2007) and the Japanese Society for the Promotion of Science (Exploratory Research grant No. 19650733), both to N.T., who was also supported by the Japanese Society for Scientist Exchange Program between the Japan Society for the Promotion of Science and The Royal Society (grant No. 2006-1-36-RS). H.-L.Z. was supported by the Japan Society for the Promotion of Science (grant No. FY2007) and the JSPS Postdoctoral Fellowship for Foreign Researcher (under N.T., by grant No. P-07196). T.C.C. is supported by the Wellcome Trust and R.D.W. is an A. J. Clark Studentship holder of British Pharmacological Society.
Editor: Richard W. Aldrich.
References
- 1.Mironneau, J., C. Martin, S. Arnaudeau, K. Jmari, L. Rakotoarisoa, I. Sayet, and C. Mironneau. 1990. High-affinity binding sites for [3H] saxitoxin are associated with voltage-dependent sodium channels in portal vein smooth muscle. Eur. J. Pharmacol. 184:315–319. [DOI] [PubMed] [Google Scholar]
- 2.Saleh, S., S. Y. Yeung, S. Prestwich, V. Pucovský, and I. Greenwood. 2005. Electrophysiological and molecular identification of voltage-gated sodium channels in murine vascular myocytes. J. Physiol. 568:155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amédée, T., J. F. Renaud, K. Jmari, A. Lombet, J. Mironneau, and M. Lazdunski. 1986. The presence of Na+ channels in myometrial smooth muscle cells is revealed by specific neurotoxins. Biochem. Biophys. Res. Commun. 137:675–681. [DOI] [PubMed] [Google Scholar]
- 4.Muraki, K., Y. Imaizumi, and M. Watanabe. 1991. Sodium currents in smooth muscle cells freshly isolated from stomach fundus of the rat and ureter of the guinea pig. J. Physiol. 442:351–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollywood, M. A., K. D. Cotton, K. D. Thornbury, and N. G. McHale. 1997. Tetrodotoxin-sensitive sodium current in sheep lymphatic smooth muscle. J. Physiol. 15:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldin, A. L. 2001. Resurgence of sodium channel research. Annu. Rev. Physiol. 63:871–894. [DOI] [PubMed] [Google Scholar]
- 7.Holman, M. E., M. A. Tonta, H. C. Parkington, and H. A. Coleman. 1995. Tetrodotoxin-sensitive action potentials in smooth muscle of mouse vas deferens. J. Auton. Nerv. Syst. 52:237–240. [DOI] [PubMed] [Google Scholar]
- 8.Belevych, A. E., A. V. Zima, I. A. Vladimirova, H. Hirata, A. Jurkiewicz, N. H. Jurkiewicz, and M. F. Shuba. 1999. TTX-sensitive Na+ and nifedipine-sensitive Ca2+ channels in rat vas deferens smooth muscle cells. Biochim. Biophys. Acta. 1419:343–352. [DOI] [PubMed] [Google Scholar]
- 9.Teramoto, N., and A. F. Brading. 1996. Activation by levcromakalim and metabolic inhibition of glibenclamide-sensitive K channels in smooth muscle cells of pig proximal urethra. Br. J. Pharmacol. 118:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu, H. L., G. D. Hirst, Y. Ito, and N. Teramoto. 2005. Modulation of voltage-dependent Ba2+ currents in the guinea-pig gastric antrum by cyclic nucleotide-dependent pathways. Br. J. Pharmacol. 146:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nomura, M., and E. Li. 1998. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 393:786–790. [DOI] [PubMed] [Google Scholar]
- 12.Kohrman, D. C., J. B. Harris, and M. H. Meisler. 1996. Mutation detection in the med and medJ alleles of the sodium channel Scn8a. Unusual splicing due to a minor class AT-AC intron. J. Biol. Chem. 271:17576–17581. [DOI] [PubMed] [Google Scholar]
- 13.Roy, M. L., and T. Narahashi. 1992. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J. Neurosci. 12:2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leffler, A., R. I. Herzog, S. D. Dib-Hajj, S. G. Waxman, and T. R. Cummins. 2005. Pharmacological properties of neuronal TTX-resistant sodium channels and the role of a critical serine pore residue. Pflugers Arch. 451:454–463. [DOI] [PubMed] [Google Scholar]
- 15.Burgess, D. L., D. C. Kohrman, J. Galt, N. W. Plummer, J. M. Jones, B. Spear, and M. H. Meisler. 1995. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease”. Nat. Genet. 10:461–465. [DOI] [PubMed] [Google Scholar]
- 16.McNulty, M. M., and D. A. Hanck. 2004. State-dependent mibefradil block of Na+ channels. Mol. Pharmacol. 66:1652–1661. [DOI] [PubMed] [Google Scholar]
- 17.Strege, P. R., C. E. Bernard, Y. Ou, S. J. Gibbons, and G. Farrugia. 2005. Effect of mibefradil on sodium and calcium currents. Am. J. Physiol. 289:G249–G253. [DOI] [PubMed] [Google Scholar]
- 18.Jo, T., T. Nagata, H. Iida, H. Imuta, K. Iwasawa, J. Ma, K. Hara, M. Omata, R. Nagai, H. Takizawa, T. Nagase, and T. Nakajima. 2004. Voltage-gated sodium channel expressed in cultured human smooth muscle cells: involvement of SCN9A. FEBS Lett. 567:339–343. [DOI] [PubMed] [Google Scholar]
- 19.Smith, M. R., R. D. Smith, N. W. Plummer, M. H. Meisler, and A. L. Goldin. 1998. Functional analysis of the mouse Scn8a sodium channel. J. Neurosci. 18:6093–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou, W., and A. L. Goldin. 2004. Use-dependent potentiation of the NaV1.6 sodium channel. Biophys. J. 87:3862–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holman, M. E., G. S. Taylor, and T. Tomita. 1977. Some properties of the smooth muscle of mouse vas deferens. J. Physiol. 266:751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blakeley, A. G., P. H. Dunn, and S. A. Petersen. 1989. Properties of excitatory junction potentials and currents in smooth muscle cells of the mouse vas deferens. J. Auton. Nerv. Syst. 27:47–56. [DOI] [PubMed] [Google Scholar]
- 23.Young, J. S., K. L. Brain, and T. C. Cunnane. 2007. The origin of the skewed amplitude distribution of spontaneous excitatory junction potentials in poorly coupled smooth muscle cells. Neuroscience. 145:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]