

Abstract
This study hypothesized that decline in sarcoplasmic reticulum (SR) Ca2+ release and maximal SR-releasable Ca2+ contributes to decreased specific force with aging. To test it, we recorded electrically evoked maximal isometric specific force followed by 4-chloro-m-cresol (4-CmC)-evoked maximal contracture force in single intact fibers from the mouse flexor digitorum brevis muscle. Significant differences in tetanic, but not in 4-CmC-evoked, contracture forces were recorded in fibers from aging mice as compared to younger mice. Peak intracellular Ca2+ in response to 4-CmC did not differ significantly. SR Ca2+ release was recorded in whole-cell patch-clamped fibers in the linescan mode of confocal microscopy using a low-affinity Ca2+ indicator (Oregon green bapta-5N) with high-intracellular ethylene glycol-bis(α-aminoethyl ether)-N,N,N′N′-tetraacetic acid (20 mM). Maximal SR Ca2+ release, but not voltage dependence, was significantly changed in fibers from old compared to young mice. Increasing the duration of fiber depolarization did not increase the maximal rate of SR Ca2+ release in fibers from old compared to young mice. Voltage-dependent inactivation of SR Ca2+ release did not differ significantly between fibers from young and old mice. These findings indicate that alterations in excitation-contraction coupling, but not in maximal SR-releasable Ca2+, account for the age-dependent decline in intracellular Ca2+ mobilization and specific force.
INTRODUCTION
A basic problem common to aging mammals is diminished muscular strength. The many factors that account for absolute force in relation to lost muscle mass have been extensively reviewed (1–3) and probably are not directly related to age-related loss in the force-generating capacity of the skeletal muscle per cross-sectional area, or specific force (SF). In vitro studies on contractility showed that when the maximal isometric force for aged mice and rats is normalized to the smaller muscle fiber cross-sectional area, a significant deficit in specific isometric force remains unexplained by the smaller cross-sectional area (4–6). These data suggest that, in addition to reduced cross-sectional area, other factors contribute to muscle weakness in aged mammals; for example, contraction-induced injury (7), posttranslational modifications of contractile proteins (8), and excitation-contraction uncoupling (ECU) (9).
Excitation-contraction coupling (ECC) is a series of ionic and molecular events by which membrane depolarization is converted into skeletal muscle contraction. Two important proteins involved in ECC are the dyhydropyridine receptor (DHPR) and the sarcoplasmic reticulum (SR) ryanodine receptor (RyR). Depolarization of the sarcolemma, associated with an action potential, causes charge movement within the transverse tubules (10) and a conformational change in the DHPR (a voltage-gated Ca2+ channel/voltage sensor) (11). In skeletal muscle, the conformational change in the DHPR results in activation of the RyR at the triadic junction, causing Ca2+ release into the myoplasm and muscle contraction (12).
Peak intracellular Ca2+ transients evoked by sarcolemmal depolarization have been shown to decrease with age (9,13); however, whether alterations in sarcolemmal depolarization-evoked SR Ca2+ release and/or maximal SR-releasable Ca2+ account for this decrease is not known. This study was designed to determine whether the decreased Ca2+ transient in muscle fibers from old mice is due to decreased SR Ca2+ release and associated with diminished maximal SR-releasable Ca2+ in flexor digitorum brevis (FDB) muscle fibers.
In the past, the voltage dependence of the SR Ca2+ release flux in mammalian muscle fibers was inferred from theoretical deconvolutions of the evoked Ca2+ transients (14–18). Here, SR Ca2+ release was measured using a procedure applied to cardiac myocytes and FDB muscle fibers that combines a high intracellular ethylene glycol-bis(α-aminoethyl ether)-N,N,N′N′-tetra-acetic acid (EGTA) concentration (20 mM) and a low-affinity Ca2+ indicator (Oregon green bapta-5N) with laser scanning confocal microscopy recordings (19–21). This approach allows direct measurement of the SR Ca2+ release flux from the recorded fluorescence transients.
We also took advantage of the potency and specificity of the RyR activator 4-chloro-m-cresol (4-CmC) (22–26) to elicit maximal SR Ca2+ release and force in single intact muscle fibers. Unlike caffeine, this compound has been shown to induce Ca2+ release via the RyR without adversely affecting the SR Ca2+ pump or myofibrillar sensitivity (23). It can therefore be used effectively to study the maximal force a fiber can develop in response to maximal SR Ca2+ release, while bypassing the ECC mechanism.
The results of this study indicate that SR Ca2+ release is impaired, and maximal SR-releasable Ca2+ is preserved, which supports the ECU mechanism in aging muscle fibers. Preliminary results were presented at the 50th Annual Meeting of the Biophysical Society (Salt Lake City, UT, 2006).
METHODS
Animals
Flexor digitorum brevis muscles were dissected from young (3- to 6-month) and old (20- to 22-month) FVB (Friend virus B, our colony) or DBA (dilute brown agouti) mice. We have used these strains to study aging muscle (5,6,27), and availability determined the inclusion of the two strains in this work. However, the findings reported are independent of mouse strain (see below). The animals were housed at Wake Forest University School of Medicine and killed by cervical dislocation; animal handling and procedures were approved by the Wake Forest University School of Medicine Animal Care and Use Committee.
Single intact fiber contraction and intracellular Ca2+ recordings
The technique for dissecting single intact fibers followed procedures previously described (6,28). The recording solution consisted of (mM) NaCl 121, KCl 5, CaCl2 1.8, MgCl2 0.5, NaH2PO4 0.4, NaHCO3 24, and glucose 5.5. Solutions were bubbled continuously with a mixture of 5% CO2-95% O2 to achieve a pH of 7.4. The fiber was mounted between a 400A force-transducer (Aurora Scientific, Aurora, Ontario, Canada) (compliance, 1 μm mN−1; resonant frequency, 0.6 kHz) and an adjustable holder, allowing control of fiber position and length, as described in previous studies (6,28). Fibers were stimulated by an electrical field generated between two parallel silver electrodes connected to a Grass S48 stimulator (Astro-Medical, West Warwick, RI). Fiber length was adjusted until maximal force was elicited by a single twitch contraction (LO) under isometric conditions. Suprathreshold square wave pulses of 0.5-ms duration were delivered to elicit twitch contractions. Tetanic contractions were elicited with 0.5-ms square wave pulses delivered in 350-ms trains. Frequency was increased until maximal force was attained. All subsequent tetanic contractions were elicited with the frequency that elicited maximal force, as described (13). All experiments were performed at room temperature (21–23°C). For data acquisition, a personal computer, a D-A and A-D board interface (Molecular Devices, Sunnyvale, CA), and pCLAMP software (Axon Instruments, Union City, CA) were used.
Dose-response curves were established for FDB fibers from both young and old mice. To check the viability of the cells and reproducibility of tetanic contraction, a reference trial of 50 tetanic contractions, set at 10-s intervals for 9 min, was performed. The cell was not further tested if a decline in tetanus at any time during this protocol was >10% of the initial amplitude. The test protocol was as follows: 1), three control maximal tetanic contractions set at 2-min intervals; 2), application of 100 μM 4-CmC; 3), washout of the drug; 4), series of tetanic contractions elicited at 2-min intervals until recovery of the maximal contraction force; and 5), application of the next 4-CmC concentration. Steps 2–4 were repeated, substituting progressively greater 4-CmC concentrations—200, 350, 500, 750, and 1000 μM—in Step 2. As preliminary experiments with 2000 μM 4-CmC did not induce any further increase in force, this concentration was not systematically tested.
Intracellular Ca2+ mobilization in response to 1 mM 4-CmC was recorded in enzymatically dissociated FDB fibers. FDB muscles were treated with 2 mg/ml collagenase (Sigma, St. Louis, MO) in a shaking bath at 37°C. After 3 h of enzymatic treatment, they were dissociated into single fibers using Pasteur pipettes of different tip size. Contraction was prevented by incubating the fibers in 50 μM N-benzyl-P-toluene sulfonamide (BTS) for 30 min and then loading with the Ca2+ indicator Fura-FF via the patch pipette in the whole-cell configuration of the patch clamp. Fura-FF, a low-affinity Ca2+ indicator, was used because it is ratiometric and can measure peak intracellular Ca2+ in FDB fibers without saturating (18). Ca2+ fluorescence was recorded using a photomultiplier-based system provided with a random access monochromator (Ram X, PTI). Excitation and emission filters were set at 340/380 nm and 510 nm wavelength (Omega Optical, Brattleboro, VT), respectively. The relation between the fluorescence ratio R (F380/F340) and Ca2+ concentration was calculated, as described (29). Rmax (3.50) and Rmin (0.29) were calculated in FDB fibers equilibrated with 0.02% saponin and 1 mM Ca2+ concentration or in cells incubated for 20–30 min in 10 μM BAPTA AM. As a wide range of Kd values for Ca2+ has been reported in the literature (6.5 μM (18), 19.2 μM (30), 31.5 μM (31), and 35 μM (32)), we decided to calibrate the Fura-FF used for our experiments in the muscle fiber. A Kd value of 51 μM was determined in vitro using microcapillaries, a Ca2+ calibration kit (Molecular Probes-Invitrogen, Carlsbad, CA), and a Fura-FF concentration of 10 μM, as described previously (18). The explanation for this wide range of Kd values is not obvious.
Whole-cell patch-clamp, confocal fluorescence imaging, and SR Ca2+ release calculations
Enzymatically dissociated fibers (see above) were transferred to a small, flow-through Lucite chamber positioned on a microscope stage. Fibers were continuously perfused with the external solution (see below) using a push-pull syringe pump (WPI, Sarasota, FL). Only fibers exhibiting a clean surface and no contracture were used for electrophysiological recordings. Muscle fibers were voltage-clamped using an Axopatch-200B amplifier (Molecular Devices) in the whole-cell configuration of the patch-clamp technique (33). Patch pipettes were pulled from borosilicate glass (Boralex, WPI) using a Flaming Brown micropipette puller (P97, Sutter Instrument, Novato, CA) and then fire-polished to obtain electrode resistances ranging from 450 to 650 kΩ. In the cell-attached configuration, the seal resistance was in the range 1–4.5 GΩ, and in the whole-cell configuration, values ranged between 75 and 120 MΩ (34). The pipette was filled with the following solution (mM): 140 Cs-aspartate, 5 Mg-aspartate2, 20 Cs2EGTA, and 10 HEPES (N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid]), and pH was adjusted to 7.4 with CsOH (34,35). The pipette solution also contained 500 μM Oregon green bapta-5N (OGB-5N) (Invitrogen). The external solution contained (mM) 150 TEA (tetraethylammonium hydroxide)-CH3SO3, 2 MgCl2, 2 CaCl2, 10 Na-HEPES, 0.05 BTS, and 0.001 tetrodotoxin (36,37). Solution pH was adjusted to 7.4 with CsOH. All the experiments were conducted at room temperature (21–22°C). For these experiments we preferred OGB-5N over Fura-FF due to its higher quantum yield and suitability for cell imaging with krypton-argon laser confocal microscopy. The fibers were loaded with OGB-5N via the patch pipette. The dye was allowed to diffuse for 20–30 min before fiber stimulation and after attaining the whole-cell voltage-clamp configuration. Intracellular OGB-5N transients were recorded using a Bio-Rad Radiance 2100 laser scanning confocal microscope (Zeiss, Oberkochen, Germany). Confocal microscopy allowed us to improve the signal/noise ratio under experimental conditions in which myoplasmic Ca2+ concentration was strongly buffered by 20 mM EGTA. The high EGTA concentration in the patch-pipette ensured a resting myoplasmic Ca2+ concentration value that approached the 60 nM existing in the pipette (20,38). This experimental manipulation also ensured a more accurate estimation of the Ca2+ release flux. Fibers were imaged through a C-Apochromat 40× water-immersion objective (NA 1.2, Zeiss) or a 20× Fluar (NA 0.75) using a krypton-argon laser at 488-nm excitation wavelength. The fluorescence emission was measured at 528 ± 25 nm wavelength. For most experiments, the laser was attenuated to 6–12% with a neutral density filter. Fibers were imaged in line-scan (x-t) mode. The fiber was always oriented parallel to the x scan direction. Linescan images were acquired with 256 pixels (0.236 μm/pixel) in the x- and 512 pixels (0.833 ms/pixel) in the t-direction. For image acquisition, we used LaserSharp 2000 software (Bio-Rad, Zeiss), and for the analysis of the image intensity profile, Scion/Image J software (NIH, Bethesda, MD).
To determine the SR Ca2+ release flux, Ca2+ transients were analyzed using the closed-form equilibrium approximation (19) and a single-compartment kinetic model including EGTA, OGB-5N, and Ca2+ as the reactants with rate constants determined in vitro (20,21). The kinetics of the Ca2+ release flux (J(t))was described by the equation
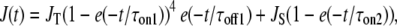 |
(1) |
where JT is the amplitude of a transient component of the total Ca2+ release flux and JS is the amplitude of a steady component of the total Ca2+ release flux.
OGB-5N calibration in vitro and in FDB mouse fibers
OGB-5N Kd for Ca2+ was calculated in vitro using borosilicate micropipettes (1.12 and 1.5 mm internal and external diameter, respectively) and a set of 11 Ca2+ calibration standard solutions (Calbuf-2, WPI), containing 25 μM OGB-5N. Ca2+ concentration in the standard solutions ranged from pCa 4 to 8, and their osmolarity was 300 mOsm. The normalized fluorescence/pCa relationship was plotted, and a Hill equation fitted to data points (n = 6–10 pipettes/Ca2+ concentration). The calculated Kd value was 37 μM for the batch of OGB-5N used. The in vivo calibration was carried out in enzymatically dissociated FDB fibers from young (4–6 months) mice. The fiber preparation followed the protocol described above for fluorescence recordings. Fibers were transferred to the recording chamber and exposed to 50 μM BTS. We found that this concentration of the myosin II ATPase inhibitor completely suppresses contraction in fibers tested at optimal length (data not shown). Fibers were exposed to 0.02% saponin for 1 min and then to the Ca2+ standard solutions containing 25 μM OGB-5N. This procedure allowed the intracellular compartment to equilibrate with the external medium. The maximal fluorescence intensity recorded in each fiber was computed for the dose-response curve. This analysis provided a Kd of 36 μM (n = 15–24 fibers/Ca2+ concentration), which is consistent with the value obtained by in vitro calibration.
STATISTICAL ANALYSIS
Data are presented as mean ± SE, as indicated. Statistical significance among variables was determined using the parametric Student's t-test and Mann Whitney rank sum test run in SigmaPlot 8.0 or SigmaStat 3.0 (SPSS, Chicago, IL). Values of P < 0.05 were considered significant.
RESULTS
4-CmC concentration-force relationship in single intact FDB muscle fiber
The first group of experiments used 4-CmC as the agent to bypass sarcolemma and directly elicit massive SR Ca2+ release. Therefore, we defined potential differences in the drug's ability to evoke contractures at submaximal and maximal concentrations in single intact muscle fibers from young and old mice. Fiber contracture was elicited by various 4-CmC concentrations (100, 200, 350, 500, 750, and 1000 μM) and measured in single intact FDB fibers from young mice (n = 5 fibers from three mice) and old mice (n = 6 fibers from three mice). The single intact fiber preparation has several advantages over a multifiber preparation. First, the contracting fiber can be very rapidly exposed to the perfusion solution. Second, whereas fibers can heterogeneously contribute to whole-muscle force production due to differences in pennation and length, we can be sure the single intact fiber is functioning at optimal length (LO) (6,39). Fig. 1 A shows force of contracture measured in a young, intact, skeletal FDB muscle fiber, activated by increasing concentrations of 4-CmC normalized to maximal force. Tetanic contractions fully recovered between increasing concentrations of 4-CmC. Application of 100 μM 4-CmC did not result in a significant force of contracture. The force of contracture steadily rose from 200 μM to a maximum induced by 750–1000 μM 4-CmC that approximately equaled the amplitude of tetanic contraction (see below). Application of 1 mM 4-CmC elicited force similar to that obtained with 750 μM but more reproducible. Concentrations >1 mM resulted in an incomplete tetanic recovery, indicating that 1 mM is the highest concentration of 4-CmC to accurately elicit maximal force of contracture without inducing damage. Fig. 1 B plots all data points for fibers from young and old mice, and curves are fitted using the equation
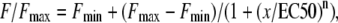 |
(2) |
where F is force; x represents the 4-CmC concentration; EC50 is the 4-CmC concentration that elicits 50% of the maximum response; and n is the Hill slope. The EC50 values for fibers from young and old mice were 295 ± 22 and 274 ± 33 μM, respectively (P > 0.05).
FIGURE 1.

(A) 4-CmC dose-response curve in single intact FDB fibers from young and old mice. Contractures in single mouse FDB fibers in response to 100, 200, 350, 500, 750, and 1000 μM 4-CmC. Marks above indicate exposure time to the drug. (B) Normalized maximal force/4-CmC concentration relationship. EC50 calculated in fibers from young (circles) and old (squares) mice (in μM, mean ± SE): 335 ± 16 (n = 6 fibers from five mice) and 358 ± 7 (n = 6 fibers from five mice), respectively, did not differ significantly (P > 0.05). Data points represent individual fibers. Experimental points were fitted to a Hill equation. Solid and dashed lines are the fitting curves to data points for young and old mice, respectively.
FDB single intact fiber contracture/tetanus
The second set of experiments compared the force developed by a single intact fiber in response to 1 mM 4-CmC-induced contracture or depolarization-mediated maximal tetanus. Both types of responses were expressed as specific force, as described by Gonzalez et al. (6). The 4-CmC contracture recorded in fibers from young (n = 23) and old (n = 8) mice did not differ statistically; however, the tetanic responses induced by field stimulation resulted in significant specific force decrease in old (n = 16) compared to young (n = 12) mice (Fig. 2, A–D) (P < 0.05). Fig. 2 E shows tetanic contraction and contracture specific force for all fibers from young and old mice studied. The age-dependent decline in fiber tetanic specific force confirms previous reports from our lab (6,13). In those publications, we also reported a decreased peak intracellular Ca2+ concentration recorded simultaneously with tetanic contraction. The 4-CmC-evoked Ca2+ release was recorded in enzymatically dissociated and BTS-immobilized FDB fibers from young (n = 8) (Fig. 2 F a) and old (n = 7) (Fig. 2 F b) mice, using Fura-FF as the ratiometric Ca2+ indicator. Fig. 2 G shows no statistically significant difference in the peak intracellular Ca2+ transient recorded in fibers from both age groups. Although these results do not rule out differences in SR Ca2+ content, they suggest that the age-dependent decline in electrically elicited fiber force cannot be explained by significant alterations in maximal SR-releasable Ca2+.
FIGURE 2.

Tetanus and 4-CmC contracture in FDB fibers. (A and C) Tetani recorded in single intact FDB fibers from young (A) and old (C) mice in response to a 350-ms-duration pulse (electrical field stimulation) at a frequency of 100 Hz. (B and D) 4-CmC contractures in fibers from young (B) and old (D) mice. The horizontal line above the curve indicates exposure time to the drug, and the dotted line is the baseline. (E) Specific force for tetanus and contracture in fibers from young and old mice expressed in kPa (mean ± SE). The asterisk indicates a statistically significant difference in tetanus between age groups and between tetanus recorded in old mice compared to 4-CmC contractures in fibers from both young and old mice. (F) Intracellular Ca2+ transients evoked by 1 mM 4-CmC in fibers from young (a) and old (b) mice. The bars above indicate the time the fiber was exposed to the drug. (G) Statistical analysis of the peak intracellular Ca2+ concentration recorded in fibers from young (n = 8 fibers) and old (n = 7 fibers) mice.
Direct measurement of SR Ca2+ release in voltage-clamped FDB muscle fibers
Fig. 3 A illustrates OGB-5N transients in FDB fibers from young and old mice detected by confocal microscopy in linescan mode. Fibers were voltage-clamped at −90 mV and depolarized by a command pulse to 60 mV for 80 ms. The pixel intensity profile superimposed on the images exhibits a peak that rapidly decays, followed by a “steady” phase until the end of fiber depolarization. The OGB-5N transient shape is similar to the SR Ca2+ release waveform obtained using a deductive mathematical algorithm (40,41). The amplitude of the peak and the steady phases of the OGB-5N transients are smaller in the old fiber than the young. Although the pulse duration in our regular protocol was 40 ms (see below), in this case, we prolonged the depolarizing pulse to better display differences in the “steady” phase between these recordings. Records in Fig. 3, A and B, were fitted and superimposed on the model equations, previously described (21), in fibers from young and old mice (Fig. 3, C and D, respectively, dashed lines). The time course of the predicted Ca2+ concentration closely matches that of the measured ΔF/F fluorescence transients. The corresponding free Ca2+ concentrations were calculated and represented in Fig. 3 E for the records depicted in Fig. 3, A–D. The calculated free Ca2+ concentration for the young fiber is similar to that previously reported for FDB fibers from young normal mice under similar experimental conditions (21), but obviously lower than in the presence of 0.2 mM EGTA in the pipette solution (9). Additionally, both free Ca2+ concentration and SR Ca2+ release flux are significantly lower in fibers from old compared to young mice. Experimental and theoretical Ca2+ transients exhibit a slow return to baseline (Fig. 3, C–E) during repolarization, which indicates that the rate of cytosolic Ca2+ removal by the SR and sarcolemmal Ca2+ transport mechanisms was negligible under these recording conditions (19). The predicted SR flux traces in Fig. 3 F, measured from the baseline, reach peak (JT) and steady (JS) values of 180 and 112 μM ms−1, respectively, for young mice, and 82 and 36 μM ms−1 for old mice; the mean ± SE of 12 fibers per age group were 182 ± 25 and 121 ± 16 μM ms−1 for JT and JS in fibers from young and 98 ± 33 and 43 ± 3.8 μM ms−1 for the same parameters in fibers from old mice. The 46% decrease in the peak Ca2+ flux (JT) closely matches the 48% reduction in the experimental ΔF/F OGB-5N transients.
FIGURE 3.

OGB-5N transients in FDB fibers from young and old mice. (A and B) OGB-5N fluorescence transients recorded in confocal linescan mode in whole-cell, patch-clamped FDB fibers in response to an 80-ms command pulse to 60 mV (holding potential, −90 mV). The internal solution contained high EGTA (20 mM) and a low-affinity Ca2+ indicator (OGB-5N). (C and D) OGB-5N transients (solid lines) fitted to, and superimposed on, the predictions of the model equations (dashed lines) in fibers from young (C) and old (D) mice, respectively. (E and F) Free Ca2+ concentration and SR Ca2+ release flux corresponding to the fibers represented in A–D. The values for the kinetic parameters τon1 (ms), τon2 (ms), and τoff1 (ms) were, respectively, 0.23, 0.025, and 2.4 for young mice, and 0.21, 0.029, and 2.7 for old mice.
Fig. 4 shows the analysis of a complete set of recordings in fibers from young and old mice, voltage-clamped at −90 mV (Vh), and depolarized by command pulses from −60 to 80 mV. Fig. 4 A compares OGB-5N transients at selected voltages from −30 to 50 mV every 20 mV, the interval corresponding to the steepest part of the fluorescence-voltage curve (Fig. 4 B). The amplitudes of both peak and steady phases are lower in fibers from old compared to young mice. Fig. 4 B plots the peak OGB-5N transient recorded with a 10-mV interval in fibers from young and old mice. To analyze the voltage dependence of the OGB-5N signal, data points were fitted to a Boltzmann equation of the form
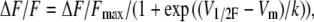 |
(3) |
where ΔF/Fmax is the maximal normalized fluorescence; Vm is the membrane potential; V1/2F is the half-activation potential; and k is the steepness of the curve. ΔF/Fmax was 0.61 ± 0.07 and 0.33 ± 0.05; V1/2F was −14 ± 1.2 and −17 ± 1.9 mV; and k was 13 ± 1.2 and 14 ± 1.5, for young (n = 35 fibers) and old (n = 37 fibers) mice, respectively. Differences between fibers from young and old mice are statistically significant (P < 0.05) for ΔF/Fmax, but not for V1/2F and k.
FIGURE 4.

Voltage dependence of OGB-5N fluorescent signal in FDB fibers from young and old mice. (A) OGB-5N transients elicited by 40-ms command pulses at variable voltages. Traces illustrate the signal recorded at −30, −10, 10, 30, and 50 mV. Dotted lines indicate the baseline. (B) Peak OGB-5N transient/Vm relationship in fibers from young (circles) and old (squares) mice. Data points were fitted to a Boltzmann equation of the form F = Fmax/(1 + exp(V1/2 − Vm)/k), where Fmax is the maximal fluorescence; V1/2 is the fluorescent half-activation potential; Vm is the membrane potential; and k is the steepness of the curve. Fmax, half-activation potential, and steepness of curves were, respectively, 0.79, −1.09, and 3.37 for young mice and 0.39, 0.87, and 2.25 for old mice.
To determine whether differences in the peak and steady phases of SR Ca2+ release reported above depend on pulse duration, we recorded OGB-5N transients at 80 mV within a wide range of pulse durations (1.0, 2.5, 5.0, 10, 20, 40, 80, 100, and 200 ms) in fibers from young and old mice (Fig. 5, A and B). Differences in maximal OGB-5N transients were statistically significant within the whole range of pulse durations, which indicates that the age-dependent decline in SR Ca2+ release is not increased by prolonging RyR activation.
FIGURE 5.

OGB-5N peak transient dependence on pulse duration and peak/steady ratio dependence on membrane voltage. Various pulse durations tested in fibers from young (Aa) and old (Ab) mice, voltage-clamped at −90 mV (holding potential), and pulsed to 80 mV. (B) Maximum OGB-5N transient-pulse duration relationship in fibers from young (circles) and old (squares) mice. (C) Peak/steady ratio measured in OGB-5N transients in response to 40-ms pulses.
The peak and steady phases of the SR Ca2+ release waveform have been attributed to calcium- and voltage-mediated SR Ca2+ release, respectively (18,41–43). Fig. 5 C represents the peak/steady ratio of traces recorded at 50 mV (40-ms duration) in fibers from young and old mice. Fibers from young mice (n = 25) exhibit a shallow voltage dependence, as reported for rat fibers (44), and this relationship increases almost twofold in fibers from old mice (n = 27). As both peak and steady phases of SR Ca2+ release are decreased in fibers from old compared to young mice (Fig. 4), a more pronounced decrease in the steady phase determines a significant increase in the peak/steady flux ratio. These results are consistent with the reported decrease in DHPRα1 subunit expression in aging mammalian skeletal muscle (5,45,46).
Ca2+-dependent inactivation of SR Ca2+ release in fibers from young and old mice
To determine whether the decreased SR Ca2+ flux results from an increase in Ca2+-dependent inactivation of SR Ca2+ release with aging, we applied a double pulse protocol (Fig. 6 A, bottom) (47–49). The peak or inactivating component of the test pulse was largest when very negative prepulses were applied. If the prepulse evoked measurable Ca2+ release, it suppressed the inactivating component of the subsequent test pulse without affecting the steady phase. Fig. 6 A compares OGB-5N transients elicited by prepulses from −80 to 80 mV with 20-mV intervals in FDB fibers from young and old mice. The threshold for OGB-5N transients and suppression of the peak during the second pulse was −40 mV and between −20 and 0 mV, respectively, for fibers from young (n = 15 fibers) and old (n = 17 fibers) mice. Fig. 6 B represents inactivation of the peak SR Ca2+ flux as a function of prepulse voltage in fibers from young and old mice. The peak component was normalized to the amplitude of the signal recorded in response to the prepulse to −100 mV (49) in fibers from young and old mice and fitted to a Boltzman equation of the form
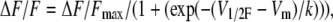 |
(4) |
where ΔF/Fmax took a value of 1; Vm is the potential during the prepulse; V1/2F is the half-inactivation potential; and k is the steepness of the curve. V1/2F values were −28 ± 3.2 mV and −25 ± 2.9 mV, and k values were 9.6 ± 1.2 and 10.7 ± 1.4 in fibers from young and old mice, respectively. No statistically significant differences were found for any of the inactivation parameters fitted to the experimental points (P > 0.05). These results indicate that the age-dependent decline in SR Ca2+ release reported above is not accounted for by alterations in Ca2+-dependent inactivation of SR Ca2+ flux.
FIGURE 6.

Ca2+-dependent inactivation of OGB-5N transients in fibers from young and old mice. A double-pulse protocol was used to explore Ca2+-dependent inactivation of the OGB-5N transients (bottom). (A) OGB-5N transients elicited by prepulses from −80 to 80 mV compared with 20-mV intervals in FDB fibers from young and old mice. (B) Inactivation of the peak SR Ca2+ flux represented as a function of prepulse voltage in fibers from young and old mice. The peak component was normalized to the amplitude of the signal recorded in response to the prepulse to −100 mV and fitted to a Boltzman equation (Eq. 4).
DISCUSSION
This work reports that 1), electrically evoked, but not 4-CmC-evoked, contraction produces significant differences in specific force recorded in fibers from old and young mice; 2), peak SR Ca2+ flux is reduced in fibers from old compared to young mice; 3), the steady component of SR Ca2+ flux decreases more markedly with aging than the peak SR Ca2+ release, and, therefore, the Ca2+ release flux peak/steady ratio is larger in old compared to young fibers; and 4), Ca2+-dependent inactivation of SR Ca2+ release does not differ between fibers from young and old mice. These results indicate that voltage-mediated SR Ca2+ release is impaired in fibers from old mice, and SR Ca2+ depletion does not seem to account for this effect, as the maximal SR-releasable Ca2+ does not differ in fibers from young and old mice.
Voltage- and 4-CmC-evoked contraction in single intact fibers from young and old mice
This study took advantage of 4-CmC as a potent ryanodine receptor agonist (23,25,50) to investigate maximal releasable Ca2+ by measuring single-fiber specific force and peak intracellular Ca2+. We tested a wide range of concentrations to examine age-dependent changes in fiber sensitivity to 4-CmC. Additionally, analysis of normalized force-[4-CmC] relationships suggests that the amount of Ca2+ released in response to maximal and submaximal 4-CmC concentrations is similar in fibers from young and old mice. It may be argued that fibers from aging mice exhibit lower SR Ca2+ release in response to 4-CmC, and the myofilaments' increased sensitivity for Ca2+ would offset the difference between young and old fibers. The literature does not support this speculation, because the pCa50 measured in skinned fibers from young adult and old mice was reported as 5.9 and 5.8, respectively (51). Peak intracellular Ca2+ recording, though not a direct measurement of SR Ca2+ release, indicates that the release flux is not altered when the ECC mechanism is bypassed. These results, together with the decreased tetanic force in response to electrical stimulation, indicate that voltage-mediated SR Ca2+ release, but not maximal SR-releasable Ca2+, is impaired in aging muscle fibers. A recent study suggests that fragmented and uncoupled SR stores a Ca2+ pool sensitive to caffeine but not to sarcolemmal depolarization in aging muscle (52). However, segregated Ca2+ release is unlikely to explain our results, due to the fact that ultramicroscopic studies of mouse (53) or human muscle fibers do not report the presence of feet (RyR) beyond the SR junctional face (54).
Direct measurement of SR Ca2+ release in FDB fibers
The method used here to measure directly SR Ca2+ release has some advantages compared to those previously used (see below). High myoplasmic EGTA concentration prevents fiber movement, which is highly convenient for intracellular Ca2+ transient and sarcolemmal current recording with the patch-clamp technique. High EGTA also restricts the increase in myoplasmic free Ca2+ concentration to within a few hundred nanometers of the SR Ca2+ release sites (55). High concentration of this exogenous Ca2+ buffer dominates the endogenous buffer capacity of the fiber, and the SR Ca2+ release recordings become independent of intrinsic Ca2+ binding sites (19,20,55). Troponin C, parvalbumin, and SERCA's Kd for Ca2+ are in the low micromolar–high nanomolar range, whereas EGTA's Kd for Ca2+ is in the low nanomolar range (17,55–57). The significant difference in Ca2+ affinity and the high myoplasmic concentration allow EGTA to overwhelm the endogenous buffer capacity (20,55).
SR Ca2+ release was measured in rat muscle fibers about a decade ago (14,15,44) and more recently in the mouse (17,18,49,58). These studies calculate Ca2+ release flux by applying an inductive (17) or deductive (41,59) mathematical approach to global intracellular Ca2+ transients. We used a method described in detail previously for cardiac myocytes and adult FDB muscle fibers (19,21) to investigate SR Ca2+ flux in aging muscle fibers. It takes advantage of the buffer capacity of high intracellular EGTA concentrations and the low affinity of the indicator OGB-5N for Ca2+, and confocal microscopy can be used in the linescan mode. The Ca2+ input flux waveform we calculated resembles that reported using a Ca2+ removal model fit method in mouse (59), rat (44), and frog (12,41) fibers. A large, fast component, inactivated by a depolarizing prepulse, is followed by a smaller, sustained phase until the end of fiber depolarization (42). We did not detect a decline in the amplitude of this second phase of Ca2+ release flux, interpreted as SR Ca2+ depletion, in contrast to reports in the literature (60). More recently, an ∼80% decrease in luminal Ca2+ content was recorded within 100 ms of membrane depolarization to 50 mV in mouse interosseus muscle (18). In this work, we did not observe a decline in the plateau phase of the Ca2+ release flux in FDB fibers subjected to prolonged depolarization (200 ms) (Fig. 5, A and B). It has been proposed that a strong Ca2+ flux through the DHPR in response to the increased driving force for Ca2+ upon fiber repolarization, which occurs at the Ca2+ tail current, transiently increases intracellular Ca2+ flux (19). We recorded intracellular Ca2+ flux in fibers in which SR calcium release was completely blocked by 30-min incubation in 5 μM ryanodine, or Ca2+ flow through the DHPR was fully blocked with a combination of La3+ and Cd2+ added to the bath solution (34). These experiments recorded no contribution of the Ca2+ flux through the DHPR to intracellular Ca2+ (data not shown). Why we did not record the depletion reported previously is not obvious. We speculate that the Vaseline-gap voltage clamp applied to mammalian muscle fibers does not allow full recovery of SR Ca2+ content between pulses, probably due to the large dialysis of intracellular components through both ends of the fiber (34). However, this argument does not explain the decline in Ca2+ release flux recorded in mouse interosseus muscle fibers voltage-clamped with the two-microelectrode technique (18). Another speculation is that increasing cytosolic Ca2+ accumulation in response to prolonged depolarization in our high EGTA condition offsets the decline in the “steady” phase of the Ca2+ release flux. A function representing the increasing accumulation of myoplasmic Ca2+ concentration would be necessary to counterbalance SR Ca2+ depletion during a prolonged depolarization. However, the pioneer work by Pape and co-workers in cut frog muscle fibers equilibrated with 20 mM EGTA showed a fast increase in free Ca2+ concentration near the SR Ca2+ release sites followed by a slow and sustained decay (55).
Values of Ca2+ release flux recorded here are comparable to those reported in the literature: ∼200 μM/ms using furaptra as the Ca2+ indicator in extensor digitorum longus (EDL) muscle fiber (17), ∼200 μM/ms using Fura-FF in interosseus muscle fiber (18), and ∼300 μM/ms using OGB-5N in FDB fibers (21). These values are almost fourfold higher than those reported in rat fibers (see Discussion in (18)).
Differences in SR Ca2+ release between fibers from young and old mice
Previous work from our laboratory showed a decrease in EDL, soleus (6), and FDB (13) muscle fiber SF with aging, and proposed that ECU explains it (61,62). Other theories have been proposed to account for the loss in SF; for example, contraction-induced injury (7) and posttranslational modifications of contractile proteins (8). Although contraction-induced injury has been reported in humans and reproduced in animal models after lengthening contractions, SF deficits have been recorded in the absence of this stress, which suggests that other mechanisms must be operating as well. The lack of SF alterations in manually skinned mouse EDL fibers (51,63) argues against the proposal that posttranslational modifications in aging muscle switch contractile proteins from strongly to weakly bound actomyosin (8). The ECU hypothesis is based on the age-dependent decrease in DHPRα1 subunit expression, charge movement, and intracellular Ca2+ transients (9,45); however, until now, Ca2+ release flux has not been directly measured in fibers from aging mice.
This study reports a decrease in the peak and steady components of SR Ca2+ flux in old compared to young fibers. The peak/steady-state ratio is larger in old than in young fibers and reflects a greater reduction in the steady than in the peak component. Similar differences have been reported for frog compared to rat muscle fibers (44). Whether changes in single RyR conductance among young and old fibers are significant and play a role in the decay in the SR Ca2+ flux is not known. Single-channel conductance has been measured only in RyR1 from young mammalian species (64) but not in aging muscle. The lower steady component of SR Ca2+ flux in old compared to young mice could result from lower values of DHPRα1 subunit density in the transverse tubule membrane, RyR1 density in the SR membrane, open probability for DHPR and/or RyR1, or Ca2+ driving force. In addition, channel mistargeting/triad disorganization in aging muscle (see below) could lead to impaired DHPR/RyR1 interaction. Decreased DHPRα1 subunit expression with aging has been reported in pooled muscles from rat (45), mouse (5), and rabbit (46). Persistent expression of this subunit in aged human skeletal muscle biopsy (65) raises concern about how representative a small muscle biopsy is of the whole musculature. No alterations in RyR1 density have been reported in muscles from aging rat (45) or mouse (5). Open channel probability has not been recorded in either DHPRα1 subunit or RyR1 from aging mammals; therefore, we do not know whether it contributes to the decline in SR Ca2+ flux with aging. Potential alterations in driving force are difficult to assess, since SR Ca2+ content in muscle fibers from young and old mammals has not been measured, and, consequently, the Ca2+ gradient across the SR membrane is not known. However, the lack of significant difference between 4-CmC-evoked contracture amplitudes in single intact muscle fibers from young and old mice reported here suggests that the SR does not undergo significant Ca2+ depletion with aging.
The effect of depolarizing prepulses of various amplitudes on SR Ca2+ release flux was analyzed. Putative Ca2+-dependent inactivation of SR Ca2+ release does not differ significantly in fibers from young and old mice; therefore, alterations in Ca2+-dependent SR Ca2+ release inactivation cannot account for the smaller SR Ca2+ flux recorded in young versus old fibers.
A recent ultramicroscopic study supports the tenet that progressive disorganization of the ECC apparatus in aging human skeletal muscle may account for the decline in performance (54). This work reports disarrangement of the sarcotubular-SR membrane network, characterized by longitudinally oriented tubules, triads not correctly targeted at the I-A band junction, and a high frequency of dyads, together with a decreased number of triads per muscle fiber surface, which would lead to fewer Ca2+ release units (54). Disorganized and/or missing calcium release units might contribute to the decreased SR Ca2+ flux reported here.
In summary, only indirect information on SR Ca2+ release in skeletal muscle from senescent mammals existed until now. This study contributes direct quantification of 4-CmC- and voltage-evoked SR Ca2+ release in FDB fibers from young adult and senescent mice. These results indicate that voltage-mediated SR Ca2+ release is impaired in fibers from old mice, and that SR Ca2+ depletion does not account for this finding, as an extra pool of SR-releasable Ca2+ was mobilized in response to direct RyR1 activation by 4-CmC.
Acknowledgments
We are grateful to Dr. Julio Vergara (Department of Physiology, University of California at Los Angeles) for providing us with the single-compartment computer model and input on data analysis.
This study was supported by grants from the National Institutes of Health, National Institute on Aging (AG13934 and AG15820) and the Muscular Dystrophy Association to Osvaldo Delbono, and by the Wake Forest University Claude D. Pepper Older Americans Independence Center (P30-AG21332).
Ramón Jiménez-Moreno and Zhong-Min Wang contributed equally to this work.
Editor: David D. Thomas.
References
- 1.Dirks, A. J., and C. Leeuwenburgh. 2005. The role of apoptosis in age-related skeletal muscle atrophy. Sports Med. 35:473–483. [DOI] [PubMed] [Google Scholar]
- 2.Karakelides, H., and K. Sreekumaran Nair. 2005. Sarcopenia of aging and its metabolic impact. Curr. Top. Dev. Biol. 68:123–148. [DOI] [PubMed] [Google Scholar]
- 3.Kandarian, S. C., and R. W. Jackman. 2006. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve. 33:155–165. [DOI] [PubMed] [Google Scholar]
- 4.Brooks, S. V., and J. A. Faulkner. 1988. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol. 404:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renganathan, M., M. L. Messi, and O. Delbono. 1998. Overexpression of IGF-1 exclusively in skeletal muscle prevents age-related decline in the number of dihydropyridine receptors. J. Biol. Chem. 273:28845–28851. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez, E., M. L. Messi, and O. Delbono. 2000. The specific force of single intact extensor digitorum longus and soleus mouse muscle fibers declines with aging. J. Membr. Biol. 178:175–183. [DOI] [PubMed] [Google Scholar]
- 7.Brooks, S. V., and J. A. Faulkner. 1990. Contraction-induced injury: recovery of skeletal muscles in young and old mice. Am. J. Physiol. 258:C436–C442. [DOI] [PubMed] [Google Scholar]
- 8.Lowe, D. A., J. T. Surek, D. D. Thomas, and L. V. Thompson. 2001. Electron paramagnetic resonance reveals age-related myosin structural changes in rat skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 280:C540–C547. [DOI] [PubMed] [Google Scholar]
- 9.Wang, Z.-M., M. L. Messi, and O. Delbono. 2000. L-type Ca2+ channel charge movement and intracellular Ca2+ in skeletal muscle fibers from aging mice. Biophys. J. 78:1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider, M. F., and W. K. Chandler. 1973. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature. 242:244–246. [DOI] [PubMed] [Google Scholar]
- 11.Rios, E., and G. Brum. 1987. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 325:717–720. [DOI] [PubMed] [Google Scholar]
- 12.Melzer, W., A. Herrmann-Frank, and H. C. Luttgau. 1995. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta. 1241:59–116. [DOI] [PubMed] [Google Scholar]
- 13.González, E., M. L. Messi, Z. Zheng, and O. Delbono. 2003. Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J. Physiol. 552:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delbono, O., and E. Stefani. 1993. Calcium transients in single mammalian skeletal muscle fibres. J. Physiol. 463:689–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia, J., and M. F. Schneider. 1993. Calcium transients and calcium release in rat fast-twitch skeletal muscle fibres. J. Physiol. 463:709–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szentesi, P., V. Jacquemond, L. Kovacs, and L. Csernoch. 1997. Intramembrane charge movement and sarcoplasmic calcium release in enzymatically isolated mammalian skeletal muscle fibres. J. Physiol. 505:371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baylor, S. M., and S. Hollingworth. 2003. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 551:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ursu, D., R. P. Schuhmeier, and W. Melzer. 2005. Voltage-controlled Ca2+ release and entry flux in isolated adult muscle fibres of the mouse. J. Physiol. 562:347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song, L. S., J. S. Sham, M. D. Stern, E. G. Lakatta, and H. Cheng. 1998. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J. Physiol. 512:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woods, C. E., D. Novo, M. DiFranco, and J. L. Vergara. 2004. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J. Physiol. 557:59–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woods, C. E., D. Novo, M. DiFranco, J. Capote, and J. L. Vergara. 2005. Propagation in the transverse tubular system and voltage dependence of calcium release in normal and mdx mouse muscle fibres. J. Physiol. 568:867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zorzato, F., E. Scutari, V. Tegazzin, E. Clementi, and S. Treves. 1993. Chlorocresol: an activator of ryanodine receptor-mediated Ca2+ release. Mol. Pharmacol. 44:1192–1201. [PubMed] [Google Scholar]
- 23.Westerblad, H., F. H. Andrade, and M. S. Islam. 1998. Effects of ryanodine receptor agonist 4-chloro-m-cresol on myoplasmic free Ca2+ concentration and force of contraction in mouse skeletal muscle. Cell Calcium. 24:105–115. [DOI] [PubMed] [Google Scholar]
- 24.Choisy, S., C. Huchet-Cadiou, and C. Leoty. 1999. Sarcoplasmic reticulum Ca2+ release by 4-chloro-m-cresol (4-CmC) in intact and chemically skinned ferret cardiac ventricular fibers. J. Pharmacol. Exp. Ther. 290:578–586. [PubMed] [Google Scholar]
- 25.Herrmann-Frank, A., M. Richter, and F. Lehmann-Horn. 1996. 4-Chloro-m-cresol: a specific tool to distinguish between malignant hyperthermia-susceptible and normal muscle. Biochem. Pharmacol. 52:149–155. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann-Frank, A., M. Richter, S. Sarkozi, U. Mohr, and F. Lehmann-Horn. 1996. 4-Chloro-m-cresol, a potent and specific activator of the skeletal muscle ryanodine receptor. Biochim. Biophys. Acta. 1289:31–40. [DOI] [PubMed] [Google Scholar]
- 27.Payne, A. M., Z. Zheng, E. Gonzalez, Z. M. Wang, M. L. Messi, and O. Delbono. 2004. External Ca2+-dependent excitation-contraction coupling in a population of ageing mouse skeletal muscle fibres. J. Physiol. 560:137–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lannergren, J., and H. Westerblad. 1987. The temperature dependence of isometric contractions of single, intact fibres dissected from a mouse foot muscle. J. Physiol. 390:285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grynkiewicz, G., M. Poenie, and R. W. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Role of specific intracellular signaling pathways. J. Clin. Invest. 96:1473–1483. [PubMed] [Google Scholar]
- 30.Wokosin, D. L., C. M. Loughrey, and G. L. Smith. 2004. Characterization of a range of fura dyes with two-photon excitation. Biophys. J. 86:1726–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomax, R. B., C. Camello, F. Van Coppenolle, O. H. Petersen, and A. V. Tepikin. 2002. Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J. Biol. Chem. 277:26479–26485. [DOI] [PubMed] [Google Scholar]
- 32.Barg, S., X. Ma, L. Eliasson, J. Galvanovskis, S. O. Gopel, S. Obermuller, J. Platzer, E. Renstrom, M. Trus, D. Atlas, J. Striessnig, and P. Rorsman. 2001. Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Biophys. J. 81:3308–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamill, O. P., A. Marty, E. Neher, B. Sakmann, and F. J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- 34.Wang, Z. M., M. L. Messi, and O. Delbono. 1999. Patch-clamp recording of charge movement, Ca2+ current and Ca2+ transients in adult skeletal muscle fibers. Biophys. J. 77:2709–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams, B. A., T. Tanabe, A. Mikami, S. Numa, and K. G. Beam. 1990. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 346:569–572. [DOI] [PubMed] [Google Scholar]
- 36.Delbono, O. 1992. Calcium current activation and charge movement in denervated mammalian skeletal muscle fibres. J. Physiol. 451:187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delbono, O., M. Renganathan, and M. L. Messi. 1997. Regulation of mouse skeletal muscle L-type Ca2+ channel by activation of the insulin-like growth factor-1 receptor. J. Neurosci. 17:6918–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pusch, M., and E. Neher. 1988. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflugers Arch. 411:204–211. [DOI] [PubMed] [Google Scholar]
- 39.Sugi, H. 1998. Current Methods in Muscle Physiology. Advantages, Problems and Limitations. Oxford University Press, New York.
- 40.Melzer, W., E. Rios, and M. F. Schneider. 1986. The removal of myoplasmic free calcium following calcium release in frog skeletal muscle. J. Physiol. 372:261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melzer, W., E. Rios, and M. F. Schneider. 1987. A general procedure for determining the rate of calcium release from the sarcoplasmic reticulum in skeletal muscle fibers. Biophys. J. 51:849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider, M. F., and B. J. Simon. 1988. Inactivation of calcium release from the sarcoplasmic reticulum in frog skeletal muscle. J. Physiol. 405:727–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Csernoch, L., V. Jacquemond, and M. F. Schneider. 1993. Microinjection of strong calcium buffers suppresses the peak of calcium release during depolarization in frog skeletal muscle fibers. J. Gen. Physiol. 101:297–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirokova, N., J. Garcia, G. Pizarro, and E. Rios. 1996. Ca2+ release from the sarcoplasmic reticulum compared in amphibian and mammalian skeletal muscle. J. Gen. Physiol. 107:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renganathan, M., M. L. Messi, and O. Delbono. 1997. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J. Membr. Biol. 157:247–253. [DOI] [PubMed] [Google Scholar]
- 46.Ryan, M., B. M. Carlson, and K. Ohlendieck. 2000. Oligomeric status of the dihydropyridine receptor in aged skeletal muscle. Mol. Cell Biol. Res. Commun. 4:224–229. [DOI] [PubMed] [Google Scholar]
- 47.Delbono, O. 1995. Ca2+ modulation of sarcoplasmic reticulum Ca2+ release in rat skeletal muscle fibers. J. Membr. Biol. 146:91–99. [PubMed] [Google Scholar]
- 48.Garcia, J., and M. F. Schneider. 1995. Suppression of calcium release by calcium or procaine in voltage clamped rat skeletal muscle fibres. J. Physiol. 485:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szentesi, P., L. Kovacs, and L. Csernoch. 2000. Deterministic inactivation of calcium release channels in mammalian skeletal muscle. J. Physiol. 528:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choisy, S., C. Huchet-Cadiou, and C. Leoty. 2000. Differential effects of 4-chloro-m-cresol and caffeine on skinned fibers from rat fast and slow skeletal muscles. J. Pharmacol. Exp. Ther. 294:884–893. [PubMed] [Google Scholar]
- 51.Brooks, S. V., and J. A. Faulkner. 1994. Isometric, shortening, and lengthening contractions of muscle fiber segments from adult and old mice. Am. J. Physiol. 267:C507–C513. [DOI] [PubMed] [Google Scholar]
- 52.Weisleder, N., M. Brotto, S. Komazaki, Z. Pan, X. Zhao, T. Nosek, J. Parness, H. Takeshima, and J. Ma. 2006. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 174:639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franzini-Armstrong, C. 1991. Simultaneous maturation of transverse tubules and sarcoplasmic reticulum during muscle differentiation in the mouse. Dev. Biol. 146:353–363. [DOI] [PubMed] [Google Scholar]
- 54.Boncompagni, S., L. d'Amelio, S. Fulle, G. Fano, and F. Protasi. 2006. Progressive disorganization of the excitation-contraction coupling apparatus in aging human skeletal muscle as revealed by electron microscopy: a possible role in the decline of muscle performance. J. Gerontol. A Biol. Sci. 61:995–1008. [DOI] [PubMed] [Google Scholar]
- 55.Pape, P. C., D. S. Jong, and W. K. Chandler. 1995. Calcium release and its voltage dependence in frog cut muscle fibers equilibrated with 20 mM EGTA. J. Gen. Physiol. 106:259–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zot, H. G., and J. D. Potter. 1982. A structural role for the Ca2+-Mg2+ sites on troponin C in the regulation of muscle contraction. Preparation and properties of troponin C depleted myofibrils. J. Biol. Chem. 257:7678–7683. [PubMed] [Google Scholar]
- 57.Dode, L., B. Vilsen, K. Van Baelen, F. Wuytack, J. D. Clausen, and J. P. Andersen. 2002. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 3 isoforms by steady-state and transient kinetic analyses. J. Biol. Chem. 277:45579–45591. [DOI] [PubMed] [Google Scholar]
- 58.Szentesi, P., C. Collet, S. Sarkozi, C. Szegedi, I. Jona, V. Jacquemond, L. Kovacs, and L. Csernoch. 2001. Effects of dantrolene on steps of excitation-contraction coupling in mammalian skeletal muscle fibers. J. Gen. Physiol. 118:355–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schuhmeier, R. P., and W. Melzer. 2004. Voltage-dependent Ca2+ fluxes in skeletal myotubes determined using a removal model analysis. J. Gen. Physiol. 123:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider, M. F., B. J. Simon, and G. Szucs. 1987. Depletion of calcium from the sarcoplasmic reticulum during calcium release in frog skeletal muscle. J. Physiol. 392:167–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delbono, O., K. S. O'Rourke, and W. H. Ettinger. 1995. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 148:211–222. [DOI] [PubMed] [Google Scholar]
- 62.Delbono, O. 2002. Molecular mechanisms and therapeutics of the deficit in specific force in ageing skeletal muscle. Biogerontology. 3:265–270. [DOI] [PubMed] [Google Scholar]
- 63.Plant, D. R., and G. S. Lynch. 2002. Excitation-contraction coupling and sarcoplasmic reticulum function in mechanically skinned fibres from fast skeletal muscles of aged mice. J. Physiol. 543:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith, J. S., T. Imagawa, J. Ma, M. Fill, K. P. Campbell, and R. Coronado. 1988. Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J. Gen. Physiol. 92:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryan, M., G. Butler-Browne, I. Erzen, V. Mouly, L. E. Thornell, A. Wernig, and K. Ohlendieck. 2003. Persistent expression of the α1S-dihydropyridine receptor in aged human skeletal muscle: implications for the excitation-contraction uncoupling hypothesis of sarcopenia. Int. J. Mol. Med. 11:425–434. [PubMed] [Google Scholar]