

Abstract
Although pressure-area isotherms are commonly measured for lipid monolayers, it is not always appreciated how much they can vary depending on experimental factors. Here, we compare experimental and simulated pressure-area isotherms for dipalmitoylphosphatidylcholine (DPPC) at temperatures ranging between 293.15 K and 323.15 K, and explore possible factors influencing the shape and position of the isotherms. Molecular dynamics simulations of DPPC monolayers using both coarse-grained (CG) and atomistic models yield results that are in rough agreement with some of the experimental isotherms, but with a steeper slope in the liquid-condensed region than seen experimentally and shifted to larger areas. The CG lipid model gives predictions that are very close to those of atomistic simulations, while greatly improving computational efficiency. There is much more variation among experimental isotherms than between isotherms obtained from CG simulations and from the most refined simulation available. Both atomistic and CG simulations yield liquid-condensed and liquid-expanded phase area compressibility moduli that are significantly larger than those typically measured experimentally, but compare well with some experimental values obtained under rapid compression.
INTRODUCTION
Lung surfactant is the surface-active lining of the alveoli, and consists of ∼90% lipids and 5–10% proteins. Of the surfactant lipids, 80–90% are phospholipids, 70–80% of which are phosphatidylcholines, approximately half of which is dipalmitoylphosphatidylcholine (phosphatidylcholine with two palmatic acid tails, also known as dipalmitoyl lecithin) (1). Not only is dipalmitoylphosphatidylcholine (DPPC) the primary component of lung surfactant, but it is also thought to be primarily responsible for the reduction of surface tension in the lungs to near-zero. The surface film is thought to become enriched in DPPC at higher surface pressures due to selective adsorption of DPPC or the squeeze-out of non-DPPC components (2–5). Thus, understanding the response of DPPC to changes in surface area is fundamental to determining the functionality of lung surfactant and how to better design lung surfactant replacements for respiratory distress syndrome, both neonatal and adult.
Despite intensive research, the complex action of natural lung surfactant is poorly understood (6). Measurements of the surface behavior of surfactant films under dynamic compression have been among the most prevalent methods of study of pulmonary surfactant (7). The lipid phase transitions of lung surfactant are believed to be partially responsible for the regulation of surface tension in the lungs (5). A common feature of almost all lung surfactants and model mixtures is the coexistence of a semicrystalline solid phase known as the liquid-condensed (LC) phase and a disordered fluid phase called the liquid-expanded (LE) phase (8). In the LC/LE phase coexistence region, the surface film becomes a mesh of finely divided LC/LE domains, which may impart strength and flexibility (9). Lipid membrane phase transitions are also associated with density fluctuations, which are thought to play a very active role in membrane function (10). DPPC and other phospholipids are known to exhibit very rich phase behavior, which despite much research is not well understood. The current view is that the phase behavior of lipid monolayers displays subtle continuous changes between phases. The richness of phase behavior is indicative of the fact that monolayers are frustrated systems where local and global equilibria compete (11). This frustration is caused in part by the difference in the cross-sectional area of lipid headgroups and lipid tails, which induces a strain on the monolayer.
The defining features of a typical pressure-area isotherm for DPPC, in the proximity of the main phase transition temperature, are shown in Fig. 1 (left). The surface pressure π is calculated as: π = γ0 − γ, where γ0 is the surface tension of pure water and γ is the surface tension of the monolayer-coated air-water interface (12). The monolayer area is typically given in terms of area/lipid. With increasing area and decreasing surface pressure, the phase transitions of the DPPC monolayer proceed in the following order: liquid-condensed (LC); coexistence between the liquid-condensed and liquid-expanded phases (LC-LE); liquid-expanded (LE); and coexistence between the liquid-expanded and gaseous phases (LE-G). The LC-LE phase transition is a first-order transition and is thus ideally represented by a perfectly horizontal plateau; however, experimental coexistence plateaus are only roughly horizontal. Once the monolayer has been compressed into a condensed phase, it becomes relatively incompressible and very low surface tensions (high surface pressures) are achieved with little change in area; thus, the LC portion of the isotherm has a steep slope. When the monolayer is compressed past its limiting area, monolayer collapse occurs. Collapse is signified by a decrease in area at constant surface pressure (a collapse plateau), resulting from the loss of lipids from the monolayer. In general, as the temperature is increased, DPPC isotherms shift to higher surface areas or equivalently higher surface pressures at a fixed area, and the coexistence region becomes less horizontal and is shifted to higher surface pressures (13). As shown in Fig. 1 (right), this behavior is seen in the isotherms of Crane et al. (14), which were obtained at 298.15 K, 303.15 K, and 310.15 K using the captive bubble apparatus. This behavior is attributed to an increase in the thermal motion of the chains at higher temperature, which leads to an increase in surface pressure (15). Phillips and Chapman (16) found the static DPPC pressure-area isotherms obtained at various temperatures differed in the coexistence region, but converged at high (near-zero surface tension) and low (near-zero surface pressure) surface pressures. Similar observations can be seen in the isotherms obtained at various temperatures by Crane et al. (14) using the captive bubble apparatus (Fig. 1, right), and in the film balance experiments of Baldyga and Dluhy (17).
FIGURE 1.

(Left) The defining features of a typical pressure-area isotherm for DPPC near the main transition temperature. The phase regions include the liquid-condensed (LC), liquid-expanded (LE), and the LC-LE and LE-G transition regions. The LC-LE horizontal coexistence region and the horizontal collapse plateau are identified. (Right) Experimental results showing the effect of temperature on the shape of compression and expansion pressure-area isotherms of DPPC. These isotherms are reproduced from those published by Crane et al. (14), at 298.15 K (dotted line), 303.15 K (dashed line), and 310.15 K (solid line). The experimental results presented in this figure (right) and in subsequent figures were obtained using Data Thief III, Ver.1 (191).
Computer simulations of phospholipid systems are of great interest because they can yield molecular-level insight into the structure and dynamics of these systems on a resolution and timescale that may not be feasible experimentally. Coarse-grained simulations have the further advantage of realizing increased simulation times and larger system sizes. Like their experimental counterparts, pressure-area isotherms obtained from simulations of lipid monolayers also vary from study to study. For comparison, simulations of DPPC monolayers using both coarse-grained (CG) and atomistic models are included here, both from the work of other authors and from our own new simulations. To the best of our knowledge, there has not yet been a comprehensive review of the factors that could affect the shape of the pressure-area isotherm, nor a critical comparison of experimental and simulated pressure-area isotherms obtained from varying methods and experimental conditions. Therefore, here, in addition to presenting our new simulation work, we review a broad and diverse sample of the huge number of published isotherms for DPPC monolayers.
The remainder of this article is outlined as follows: First, we provide details of our simulations, then present the simulation results, and finally compare them with experimental results with a brief discussion of factors that might contribute to the observed large variation among experimental results. Although our discussion will focus on DPPC, many of the factors discussed here affect the isotherms of other phospholipids similarly.
SIMULATION METHOD
Our simulations are divided into five categories: coarse-grained (CG) pressure-area isotherm simulations using 1), surface tension coupling; 2), anisotropic pressure coupling; 3), semiisotropic pressure coupling; and 4), the NVT ensemble, as well as 5), atomistic pressure-area isotherm simulations using surface-tension coupling. Simulation parameters are given for each type of simulation below. For all simulations, temperature was maintained by coupling to a Berendsen thermostat with a 1-ps time constant (18). All simulations were run with periodic boundary conditions. All simulations and analysis were performed using GROMACS simulations software (19,20). The GROMACS analysis tool g_energy was used to extract the surface tensions and box dimensions at each time step (21). To obtain surface pressure from our surface tensions, pure water surface tensions of 72.8, 72.5, 72.0, 71.2, 69.6, and 67.9 mN/m were used at temperatures of 293.15 K, 295.15 K, 298.15 K, 303.15 K, 313.15 K, and 323.15 K, which are roughly the surface-tension values given in the CRC Handbook of Chemistry and Physics (22). It should be noted that the simulated surface tensions at the air-water interface actually differ considerably from the experimental values, due to the peculiar nature of water (23,24). Vega and Miguel (25) calculated a surface tension of 54.7 mN/m from their SPC water simulations at 300 K, which underestimates the experimental value by ∼17 mN/m. This could conceivably lead to an overestimation of surface pressures in our isotherms, which are calculated from the experimental surface tension. If this were the case, the low-surface-pressure expansion observed in our simulations at surface pressures near 30 mN/m would actually be occurring at significantly lower surface pressures. However, errors in simulated water/vapor surface tension are thought to have little effect on the measurement of monolayer surface tension, which is dominated by headgroup/water and chain/vapor interactions (26). Thus, it is unlikely that our surface pressures are overestimated significantly. Because sources of error in simulation of water surface tension are likely to be particular to water and not expected to similarly affect the simulation of monolayer surface tensions, we believe that it is more accurate to use the experimental values of water surface tension instead of the simulated ones, in our calculation of monolayer surface pressure.
Experimental results are typically performed under atmospheric pressure, corresponding to a normal pressure of ∼1 bar. An applied normal pressure of 1 bar is commonly used in bilayer studies (27–30). However, the simulation of monolayers requires the use of empty space placed above the monolayer to prevent the monolayer from interacting with the periodic image of the simulation box. Despite the presence of the lipid/vacuum interface, implying a normal pressure of 0 bar, some monolayer studies have used an applied normal pressure of 1 bar (27,31,32). We have simulated several points along the CG isotherm at 298.15 K using both a normal pressure of 0 bar and of 1 bar. Allowing the height of the box to fluctuate with an applied normal pressure of 1 bar leads to shrinkage in the z-dimension, upon lateral expansion, requiring the box size to be manually adjusted by periodic addition of more vacuum space. However, the use of 1 bar vs. 0 bar led to no detectable difference in the isotherm. Therefore, all results presented here will be for simulations performed at 1 bar. It has also been noted that due to large fluctuations in instantaneous pressure on the order of hundreds of atmospheres, in a simulation, 1 bar is essentially equivalent to 0 bar (27,33).
Coarse-grained simulations
For all of our coarse-grained simulations, we utilize the peptide force-field parameters developed by Marrink et al. (34). The area/headgroup for DPPC bilayers using the coarse-grained model of Marrink et al. was found to match the experimental value, and many other properties have been found to match experiment at a quantitative or semiquantitative level (34). The CG model for DPPC has one bead representing the phosphate moiety, one bead representing the choline moiety, two tail beads representing the glycerol linkage, and four beads for each of the tails (each tail bead corresponds to four tail carbons). This model is used in conjunction with the coarse-grained model of Marrink et al. for water, which merges four water molecules into a single coarse-grain bead. The structure files for the CG DPPC monolayers were adapted from the CG structure files given on Marrink's website for DPPC bilayers in the fluid phase (35) and energy-minimized. The resulting fluid phase monolayer files contained two monolayers (composed of 256 lipid each) placed so that their headgroups were initially separated by ∼7 nm of CG water molecules (10,654 CG molecules) and their tail groups were separated by ∼10 nm of empty space. The resulting disordered monolayers were contained in a box of size 12.6847 nm × 12.8295 nm × 23.2 nm. However, in some of our CG simulations, spontaneous box shrinking became an issue, and intermittent addition of vacuum was necessary to prevent the two monolayers from merging into a single bilayer. For all simulations, the following parameters were taken from Marrink's website (35) and have been optimized for the coarse-grained model: short-range electrostatic and van der Waals cutoffs of 1.2 nm, with van der Waals interaction shifting smoothly to Lennard-Jones interaction at 0.9 nm, and with the Lennard-Jones cutoff set to 1.2 nm. The neighbor list was updated every 10 steps using a grid with a 1.2 nm cutoff distance. In all coarse-grained simulations, the energy parameters were saved every 0.4 ps and used for analysis with the GROMACS analysis tool g_energy (21).
Most of our coarse-grained simulations were 20 ns in duration. Marrink and Mark (36) suggested that only a few nanoseconds of simulation time are needed to measure area/lipid for CG simulation. However, our results have shown that ∼10 ns of equilibration time was necessary before areas settled down to steady values. Thus, only the last 10 ns of our 20-ns simulations were used for the calculation of average surface tension and area. The radial distribution functions and angle distributions were also averaged over the last 10 ns of the 20-ns CG simulations. In some cases, near a phase transition, from mostly LE to mostly LC phase and vice versa, simulations appeared to be metastable, and longer simulation times up to 100 ns were necessary. In each case, the last 10 ns of simulation time were used for calculations. At large values of surface tension, the box size diverged and eventually exploded, making movement further down the isotherm to low-surface pressures impossible. The divergence of box size is attributed to the onset of hole formation, followed by expansion and ultimately the rupture of the monolayer. A plot of lateral area versus simulation time is given in the Supplementary Material for a CG simulation displaying uncontrollable box expansion.
Because we are using the original CG model of Marrink et al. (37), all liquid-condensed phases simulated will be untilted. Marrink and co-workers have shown that tilted phases can be simulated using the CG model, if the model is altered to increase the size difference between the head- and tail-group beads. By decreasing the tail-group bead size by 10%, Marrink et al. (37) succeeded in simulating the tilted phase in a DPPC bilayer. It should also be noted that, due to the use of smoother potential functions for CG simulations, the dynamics of CG simulations are significantly faster (of course in computer time, but also even in physical time, as reported by the simulation) than for atomistic simulations. As a result, the effective time, which has been determined from water and lipid lateral diffusion rates, is roughly four times longer than the physical time (34). All times reported in this article will be physical time, as reported by the simulation not the effective times.
Three different pressure-coupling methods were employed: anisotropic, semiisotropic, and surface-tension pressure coupling. Anisotropic pressure coupling allows the box to flex independently in six directions (xx, yy, zz, xy/yx, xz/zx, and yz/zy) in response to a change in the pressure tensor. Semiisotropic pressure coupling only allows the box to change dimension laterally (x/y) and vertically (z). Surface tension coupling is similar to semiisotropic pressure coupling, but it uses normal pressure coupling for the z-direction, whereas the surface tension is coupled to the x/y dimensions of the box. The average surface tension γ(t) is calculated from the difference between the normal and the lateral pressure and the box is allowed to change dimension laterally (x/y) to adjust the surface tension back toward the set value. For more details on each coupling mechanism, the reader is referred to the GROMACS User Manual (21) and relevant simulation articles (27,29,30,33,38).
Surface tension coupling
Simulations with surface tension coupling were run at 293.15 K, 295.15 K, 298.15 K, 303.15 K, and 323.15 K. These simulations were run at several surface tensions varying between −50 and 62.5 mN/m. For all simulations, the z pressure component was set to 1 bar. Berendsen pressure coupling was used with a 1-ps time constant and with all compressibilities set to 5 e–6 bar−1. A timestep of 0.04 ps was used for most simulations. However, simulations undergoing a large change in box size (near the phase transition plateaus) required a smaller time-step of 0.02 ps and longer simulation times. Two types of initial configurations were used:
Independent runs. The simulations hereafter referred to as independent runs involved the independent quenching of each simulation from a state that was initially disordered. These simulations were run with the fluid phase monolayer files described above as the initial configurations. All independent runs lasted 20 ns, except at 295.15 K where runs were 100 ns in length, because 20-ns simulations had not fully converged. In addition, independent runs were also performed at 298.15 K from an initial configuration containing 1024 lipids. This configuration was obtained from the disordered configuration containing 256 lipids/monolayer (described above) by patching four boxes together and performing energy minimization.
Cycling. For each temperature, the fluid phase monolayer was used as a starting configuration for a 200-ns simulation at a surface tension of −50 mN/m. The large negative value of surface tension is physiologically meaningless, but was chosen to ensure that the resulting configuration was well ordered. This resulting configuration was then used as the starting configuration for a 20-ns simulation at zero surface tension, and then the final configuration of this run was used as the starting configuration for the next run at higher surface tension. This process of using the previous run as the starting point for the next run was repeated, stepping down the isotherm to the largest surface tension attainable. When the surface tension reached the largest value possible without a diverging box size, the process was reversed, stepping back up the isotherm until a zero surface tension was reached. This process of cycling enables the simulation of a complete hysteresis loop. At 303.15 K, the cycling simulations were also performed with a simulation time of 100 ns for each run, to test the extent of equilibration of the 20-ns cycling simulations.
Anisotropic and semiisotropic pressure coupling
Anisotropic and semiisotropic pressure simulations were run at 298.15 K and at lateral pressures of 0, −10, −20, −30, and −40 bar. For these simulations, the z-pressure component was set to 1 bar and the off-diagonal pressure components of the anisotropic pressure tensor were all set to 0 bar. Berendsen pressure coupling was used with a 1-ps time constant and with all compressibilities set to 5 e–6 bar−1. For all simulations, a timestep of 0.04 ps was used. These simulations were run independently starting from the disorder configuration, containing 256 lipids/monolayer, described above.
NVT
Two NVT simulations were run at 298.15 K. Both simulations were started form the disordered monolayer configuration, containing 256 lipids/monolayer, described above. The first simulation was run with the initial box size unchanged. The other simulation was run with the box size widened to 14 nm × 14 nm × 23.2 nm and then energy-minimized. For both simulations, a timestep of 0.04 ps was used.
Atomistic simulations
Atomistic simulations were performed using the GROMACS force field (19,20). An atomistic structure file containing a 128-lipid DPPC bilayer was taken from the Tieleman group website (39) and modified to create a system containing two DPPC monolayers composed of 64 lipids each. The monolayers were placed with their headgroups facing each other and initially separated by ∼7 nm of SPC water molecules (9662 molecules) and their tail-groups separated across a periodic boundary by ∼10 nm of empty space. The resulting system was then energy-minimized and used as the starting configuration for each simulation. A 2-fs time step was used and each simulation was run for 10 ns. The bond lengths were constrained using the LINCS algorithm (40). A particle-mesh Ewald summation (41) was used to calculate the electrostatic interactions with a Fourier spacing of 0.12 nm and a fourth-order interpolation. The Coulomb cutoff was set to 0.9 nm and the van der Waals cutoff was set to 1.2 nm. The neighbor list was updated every 10 steps using a grid with a 0.9-nm cutoff distance. Temperature was maintained at 323.15 K with a Berendsen thermostat (18). Surface-tension coupling was used with a Berendsen barostat and a time constant of 1.0 ps with all compressibilities set to 4.5 e−5 bar−1. The z-pressure component was set to 1 bar. The simulations were run at several surface tensions varying between 0 and 60 mN/m. Energies were output every 0.4 ps for the calculation of pressure-area isotherms. Calculations were made over only the last 5 ns of each simulation using the GROMACS analysis tool g_energy (21). The radial distribution functions and angle distributions were also averaged over the last 5 ns of the 10-ns atomistic simulations.
RESULTS
Simulated isotherms
We performed 20-ns cycling coarse-grained simulations of DPPC monolayers, using surface tension coupling, as described in Simulation Method, at 293.15 K, 295.15 K, 298.15 K, 303.15 K, and 323.15 K. The resulting compression and expansion isotherms, for each temperature, are shown in Fig. 2. An increase in temperature results in an upward shift to larger surface pressures, a shortening of the LC-LE coexistence region of both the compression and expansion isotherms, and an increasing slope in the coexistence region of the compression isotherms. With the exception of the isotherm at 323.15 K, which is shifted slightly to the right, all of the isotherms overlap except in the coexistence region. Although some experimental isotherms exhibit large hysteresis loops, the hysteresis seen in our isotherms is much larger than usually seen experimentally (Fig. 1, right), our LC-LE coexistence regions occur at much larger pressures, and our isotherms are also shifted to larger areas/lipid than those seen experimentally. Despite these differences, there are also some similarities. Experimental isotherms show, as seen in the simulations, that as the temperature is increased the coexistence region becomes less horizontal and is shifted to higher surface pressures, although the limiting high-pressure area of the isotherm remains invariant with temperature (Fig. 1, right). At 323.15 K hysteresis can be seen between compression and expansion isotherms at near zero surface tension, suggesting metastability of the LC phase in the expansion isotherm at high surface pressure (Fig. 2).
FIGURE 2.

Our pressure-area isotherms, obtained using cycling of coarse-grained simulations at 293.15 K (squares), 295.15 K (asterisks), 298.15 K (circles), 303.15 K (diamonds), and 323.15 K (triangles). The arrows indicate the direction of cycling. In this and subsequent figures, the error bars (standard error) on our simulated isotherms are roughly the same size as the symbols.
Fig. 3 shows the coarse-grained cycling isotherm at 293.15 K and the corresponding changes in the packing of the C2 tail beads with movement along the isotherm. Hexagonal packing, which is characteristic of the LC phase, is clearly visible at low areas/lipid. Whereas at larger areas/lipid the tail beads display disordered packing typical of the LE phase. As expected, the phase transition region, or plateau region, is accompanied by a visible change in the degree of order of the chain packing.
FIGURE 3.

Coarse-grained pressure-area isotherm obtained by cycling at 293.15 K and corresponding images of the packing of C2 tail beads (from both monolayers) at various points along the isotherm.
We therefore compare our coarse-grained simulations to atomistic simulations, both our own and those obtained by others, as well as to the coarse-grained results of Adhangale et al. (32) all at 323.15 K. In Fig. 4, our coarse-grained results, both from independent quenching and cycling, are compared to our atomistic results from independent quenching, as well as to the atomistic results of Kaznessis et al. (24), Skibinsky et al. (42), and Klauda et al. (26) and to the coarse-grained results of Adhangale et al. (32), and to the experimental results of Crane et al. (14). Kaznessis et al., Skibinsky et al., and Klauda et al. obtained their atomistic pressure-area isotherms using the NVT ensemble in CHARMM. Adhangale et al. used the coarse-grained model developed by Marrink et al. (34), with the NPNγT ensemble in the simulation package NAMD. The experimental pressure-area isotherm of Crane et al. (14) was obtained using a captive bubble apparatus. Our coarse-grained results are very close to those obtained from our atomistic simulations. This indicates that the shift of the pressure-area isotherms to larger areas/lipid (relative to most experimental isotherms) is not an artifact of the coarse-grained model, but occurs for coarse-grained and atomistic simulations alike. Our simulations also resemble the atomistic results of Skibinsky et al. (42) and Klauda et al. (26) and the experimental results of Crane et al. (14), differing slightly in magnitude and slope, whereas the results of Adhangale et al. are shifted to considerably lower area/lipid, and the results of Kaznessis et al. are shifted to considerably lower surface pressures.
FIGURE 4.

Comparison of simulated and experimental pressure-area isotherms at 323.15 K: our independent coarse-grained simulations (□), our cycling coarse-grained simulations (▵), our atomistic simulations (○), the atomistic simulations of Kaznessis et al. (24) (▪), Klauda et al. (26) (▾), Skibinsky et al. (42) (▴), the coarse-grained simulations of Adhangale et al. (32) (•), and the experimental results obtained by Crane et al. (14) using the captive bubble apparatus (+). For simplicity, our simulations are denoted by open symbols and solid lines, experiments are denoted by characters and dashed lines, and solid symbols and dotted lines denote simulations by other groups.
Skibinsky et al. (42) obtained starting configurations for their NVT monolayer simulations at each area, from NPnγT bilayer simulations. This provided a well-equilibrated starting point for the monolayer simulations, which is necessary to obtain an accurate surface pressure in constant volume simulation, which does not allow area to adjust to bring the system to equilibrium. The simulations of Klauda et al. (26) were started from the final coordinates obtained by Skibinsky et al., and run under the same conditions as used by Skibinsky et al. (42) but with the addition of the isotropic-periodic sum method to treat long-range Lennard-Jones interactions. This isotherm agrees very well with the Skibinsky isotherm, only shifted slightly, suggesting that the treatment of long-range LJ interactions has only a small effect on the isotherm. On the other hand, our results were obtained using the NPnγT ensemble with two different starting conditions: independent quenching from a disordered state and cycling (stepping down and back up the isotherm point by point from an initially ordered state). The results of Adhangale were obtained using the same coarse-grained model used in our simulations (the CG model of Marrink et al. (34)), but with long-range electrostatics added in the form of a smooth particle mesh Ewald summation. The large difference between the results of the simulations of Adhangale et al. (32) and our simulations may result from a problem with their periodic boundary conditions, which leads the monolayer to curve substantially at the edges, seemingly suggesting buckling, while maintaining disorder in the acyl chains even at increased surface pressure, where our simulations and experiments show highly ordered tails. The low surface pressures shown by the isotherm of Kaznessis et al. may result from the short simulation time of 1.3 ns, which is not adequate for pressure convergence. Simulation of a DPPC monolayer has also been performed by Mauk et al. (43), using a united-atom model and the CHARMM22.0 force field at 21°C; however, in this very early article, only two points of the isotherm were simulated, and the timescale simulated was only 120 ps, too short to provide reliable results.
Effect of ensemble
For comparison, we ran two NVT simulations at 323.15 K (Fig. 5, diamonds). The first simulation was run without making adjustments to the box size (63.6 Å2/molecule), and the second simulation with the box size increased (76.6 Å2/molecule). When the box size is increased, an unphysical increase in pressure is observed, suggesting that the NVT ensemble does not allow for sufficient pressure relaxation. Other authors have noted the inability of constant-area and constant-volume simulations to equilibrate to appropriate pressures. Simulations of DPPC bilayers performed by Feller et al. (27,38) also show that constant-area simulations tend to predict larger surface pressures at a given surface area than those predicted by constant-surface-tension simulations. Mauk et al. (43) found that the NπT ensemble was more favorable than the NAT ensemble, the latter of which yielded inaccurate equilibrium pressures and chain order. Furthermore, Mauk et al. (43) have suggested that the inaccuracy of NAT simulations of phospholipids monolayers is due to the lack of fluctuations in the periodic cell, which restricts the phospholipids from assuming energetically favorable conformations.
FIGURE 5.

Coarse-grained pressure-area isotherms obtained at 298.15 K using the NVT ensemble (diamonds) and the NPT ensemble with three pressure coupling mechanisms: surface tension (squares), anisotropic (triangles), and semiisotropic (circles).
Enforcing a constant surface area imposes a stronger restriction on the phase space available to the system then does enforcing a constant average pressure (44). Area is an extensive property that does not fluctuate when constrained. On the other hand, pressure is an intensive property, which is constrained as a time-averaged constant with fluctuations allowed. Also a change in pressure can be provoked by small intermolecular displacements, whereas a change in area requires large concerted motions of the lipids. Thus, the system is slow to equilibrate in response to imposed changes in area (44). However, it should be noted that constant-area simulations give reasonable results if the starting conditions are well equilibrated. In their simulations of DPPC bilayers, Feller and Pastor (38) found that order parameters, lateral diffusivities, magnitudes of area fluctuations, area fluctuation decay rates, and bilayer area compressibility moduli did not depend significantly on choice of ensemble (NPNAT versus NPNγT). In more recent studies, DPPC bilayer simulations showed that the pressure-area isotherms obtained using both ensembles were consistent with each other, suggesting the equivalence of the ensembles (42,45).
In addition to surface-tension coupling and NVT simulations, we also performed coarse-grained simulations using anisotropic and semiisotropic pressure coupling methods, to test the accuracy of each method. The isotherms obtained with each coupling method at 298.15 K are shown in Fig. 5. At 298.15 K, each coupling method gives nearly the same isotherm, differing only in LC-LE coexistence region, where they give different slopes. Although the choice of coupling method does not seem to have a big impact, the surface-tension coupling method yields the flattest plateau. Furthermore, in their simulations Feller et al. (27) set surface tension and allowed area to vary, regarding this as the most natural ensemble for simulating lipid/water interfaces. For these reasons surface-tension coupling was chosen as the preferred method and used for the majority of our simulations. Feller and Pastor (38) have suggested that simulation results depend much more on area than on ensemble used, which is consistent with our findings at 298.15 K.
P-N orientation
We calculated the distribution of P-N tilt with respect to the membrane normal from our atomistic simulations at 323.15 K (Fig. 6). For comparison, the P-N tilt in our CG simulations is taken as the tilt of the bond connecting the PO4 and NC3 CG beads with respect to the membrane normal, which is calculated at 298.15 K from simulations on the larger system size (1024 lipids/monolayer), and at 323.15 K for the smaller size of 256 lipids/monolayer. The tilt angle was compared at areas/lipid corresponding to the two endpoints of each isotherm. No change is observed in the coarse-grained P-N tilt angle distribution as the area is changed, at either 298.15 K or 323.15 K. However, the atomistic simulations show a noticeable difference in the P-N tilt distribution as the monolayer is expanded from 56 to 73 Å2/molecule. At 73 Å2/molecule, the distribution is narrower than for the distribution at 56 Å2/molecule and shifted so that although the probability of an angle below 60° is unchanged, the probability of an angle between 60° and 105° is increased, and the probability of an angle between 105° and 160° is decreased. The coarse-grained distributions are similar to the atomistic distribution at 56 Å2/molecule. However, the CG PO4-NC3 tilt distribution does not exhibit the dependence on surface area seen in the atomistic simulations. The coarse-grained distributions show a shift to lower angles as the temperature is increased, and the distribution narrows slightly, excluding angles above 160°. Our atomistic simulations at 323.15 K give a single peak centered at ∼90° at 56 Å2/molecule and at ∼85° at 73 Å2/molecule. Our coarse-grained simulations peak at 90° at 298.15 K and 78° at 323.15 K.
FIGURE 6.

P-N tilt angle distribution for atomistic simulations at 323.15 K with areas 56 Å2/molecule and 73 Å2/molecule, for coarse-grained (CG) simulations with 1028 lipids/monolayer at 298.15 K with areas 48 Å2/molecule and 68 Å2/molecule, and for coarse-grained simulations with 256 lipids/monolayer at 323.15 K with areas 56 Å2/molecule and 71 Å2/molecule. The solid, dark-shaded, and light-shaded lines represent the atomistic simulations, and CG simulations at 298.15 K and 323.15 K, respectively. For each shade, the solid and dotted lines represent the smaller and larger area per lipid, respectively. For clarity, the data shown here has been smoothed using time-averaged values.
Numerous experimental studies, including surface-potential measurements, on phospholipid bilayer systems suggest that the P-N orientation is parallel to the bilayer surface (46,47). A recent sum frequency generation spectroscopy study performed by Ma and Allen (48) suggests that the choline methyl groups are tilted from the surface normal and lie roughly parallel to the air-water interface. The sum frequency generation spectra obtained by Ma and Allen (48) at 12 mN/m (LE phase) and 42 mN/m (LC phase) are similar. These results suggest that the choline headgroup orientation is not significantly different in the LE and LC phases, in accordance with the previously held hypothesis that the overall conformation of the headgroup is not as sensitive to the aggregation state and the nature of the environment as the tails (48,49). The P-N tilt angle distributions obtained from our atomistic and coarse-grained simulations are also centered at or near 90°, in accord with experiments. Our results are also in agreement with previous atomistic simulations of a DPPC monolayer performed by Dominguez et al. (50), which showed that the average angle between the monolayer surface and the P-N vector was 5°. Although the shape of simulated P-N distributions vary, more recent atomistic (31) and coarse-grained (32) simulations have also shown average P-N tilt angles in the proximity of 90° with respect to the membrane normal.
As the DPPC monolayer undergoes a transition from the liquid-expanded to the liquid-condensed phase, the methylene groups of the DPPC tails transform from predominantly gauche conformations to all-trans conformations (48). The lipid tail dihedral distribution was calculated from the four CG tail beads for a system size of 1024 lipids/monolayer. At 298.15 K we found that at 48 Å2/molecule the trans tail configuration (180°) is highly preferred over the gauche configuration (±60°) and at 68 Å2/molecule the trans configuration becomes less favorable and the distribution broadens such that all tail dihedrals are sampled almost equally, as is expected (data not shown).
Radial distribution functions
In Fig. 7, the PO4-PO4, PO4-NC3, NC3-NC3, and C2-C2 radial distribution functions (RDFs) are shown, where PO4 is the phosphate moiety, NC3 is the choline moiety, and C2 is the second CG tail bead from the glycerol linkage (which corresponds to the fifth through eighth carbon atoms from the glycerol linkage); each of these sites is represented by a single coarse-grained bead. Each radial distribution function is normalized so that the integral is equal to the total number of lipids (twice the number of lipids in the case of the C2-C2 distribution because there are two C2 sites/lipid). The atomistic results compared in Fig. 7 were obtained using the following atoms: P, N, and the sixth tail carbon from the glycerol linkage. The two endpoints of each isotherm are selected to observe the effect of surface area on the shape of the radial distributions. Each isotherm used was obtained from independent runs rather then cycling. At 298.15 K (Fig. 7, left) the RDFs are compared at areas of 48 and 68 Å2/molecule for the larger CG system size (1024 lipids/monolayer). At 323.15 K (Fig. 7, center and right) the RDFs are compared at areas of 56 and 71 Å2/molecule for a CG system of size 256 lipids/monolayer and at areas of 56 and 73 Å2/molecule for an atomistic system size of 64 lipids/monolayer. The difference in the areas shown at 298.15 K and 323.15 K reflects the shift in the isotherms to larger areas/lipid as temperature is increased.
FIGURE 7.

Radial distribution functions. (Left) Independent coarse-grained (CG) simulations at 298.15 K for the larger system size (1024 lipids/monolayer) at both 48 Å2/molecule (black) and 68 Å2/molecule (red). (A) PO4-PO4 distribution. (D) C2-C2 distribution. (Center and right) Atomistic (atom.) simulations at 323.15 K with 64 lipids/monolayer at both 56 Å2/molecule (black) and 73 Å2/molecule (red) and independent CG simulations at 323.15 K with 256 lipids/monolayer at both 56 Å2/molecule (green) and 71 Å2/molecule (blue). (B) PO4-PO4 distribution; (C) NC3-NC3 distribution; (E) C2-C2 distribution. (F) PO4-NC3 distribution.
At 298.15 K, the CG PO4-PO4 (Fig. 7 A), PO4-NC3 (not shown), and NC3-NC3 (not shown) RDFs show little difference as area is changed from 48 to 68 Å2/molecule; however, the C2-C2 (Fig. 7 D) RDF changes significantly. At 48 Å2/molecule, the C2-C2 RDF reflects the highly ordered tails expected for a system in the LC phase, whereas at 68 Å2/molecule it reflects the disordering of the system. These CG results are in contrast to the atomistic results of Knecht et al. (5) at 293 K, which show that decreasing the area/lipid causes lipids to bind closer together, leading to an increase in the phosphate-phosphate correlation in addition to the increase in tail order observed here. Although our CG radial distribution functions show a clear increase in tail order as area is decreased, unlike the atomistic simulations of Knecht et al, we see only a small increase in the height of the first phosphate-phosphate correlation peak. These results suggest that the coarse-grained model is better at capturing the effect of changing surface area on lipid tails than on lipid headgroups.
At 323.15 K the simulated isotherms are in the expanded phase. The CG C2-C2 (Fig. 7 E) distribution indicates that the tails are slightly more ordered at 56 Å2/molecule than at 71 Å2/molecule. However, both areas/lipid give an RDF that reflects considerably less order than does the LC RDF at 298.15 K and 48 Å2/molecule (Fig. 7 D), and is comparable to the less ordered distribution at 298.15 K and 68 Å2/molecule (Fig. 7 D). At 323.15 K, the CG PO4-PO4 (Fig. 7 B), PO4-NC3 (Fig. 7 F), and NC3-NC3 (Fig. 7 C) RDFs show little difference between the two areas/lipid and are almost identical to those at 298.15 K (Fig. 7 A, PO4-NC3 and NC3-NC3 distributions are not shown), suggesting that temperature has a larger effect on the RDF of lipid tails than that of lipid headgroups.
For the atomistic simulations at 323.15 K (Fig. 7, center and right), a change in surface area from 56 to 73 Å2/molecule does not strongly affect any of the RDFs; however, the distributions do appear to fluctuate more at 73 Å2/molecule. Overall the CG and atomistic radial distribution functions match reasonably well at 323.15 K. Despite some differences, the C2-C2 and C-C (Fig. 7 E), PO4-NC3 and P-N (Fig. 7 F), and PO4-PO4 and P-P (Fig. 7 B) RDFs correlate well. However, the NC3-NC3 and N-N (Fig. 7 C) RDFs differ from each other considerably, whereas the NC3-NC3 (Fig. 7 C) RDF is very similar to the PO4-PO4 RDF (Fig. 7 B), indicating that the coarse-grained model is unable to capture the difference in the N-N and P-P interactions present in the atomistic simulations, which ultimately leads to inaccuracy in the NC3-NC3 RDF. The (inaccurate) similarity between the NC3-NC3 and PO4-PO4 distributions in the CG simulations is a direct result of an oversimplification contained in the CG model. The CG model uses bead types Qd (charged hydrogen-bond donor) and Qa (charged hydrogen-bond acceptor) to represent NC3 and PO4 sites, respectively. Qa-Qa and Qd-Qd Lennard-Jones interactions are both considered intermediate and have the same LJ parameters (25).
The shape and location of the peaks of our atomistic P-N and P-P RDFs correlate well with the atomistic results of Kaznessis et al. (24) for a DPPC monolayer and Sun (31) for a 1,2-dilignoceroylphosphatidylcholine monolayer. Both our P04-NC3 (CG) and P-N (atomistic) RDFs show a strong attraction between choline and phosphate groups, in agreement with the atomistic results of Kaznessis et al. (24). It has been proposed that electrostatic interactions between neighboring choline and phosphate groups are responsible for attraction between neighboring phospholipids (51).
Hole formation
Our simulations show hole formation at areas in the proximity of 100 Å2/lipid, which could represent the onset of the liquid-gas phase transition. For the CG surface tension coupling simulations, at 323.15 K, calculations were made for specified surface tensions between 0 mN/m and 46.6 mN/m, which yielded average surface pressures between 68.8 mN/m and 21.5 mN/m. When the specified surface tension was increased further to 47 mN/m, a jump in area/lipid was observed from ∼71.4 Å2 to ∼129 Å2. As shown in Fig. 8, this jump in area/lipid is accompanied by hole formation, which is not an artifact of the coarse-grained method of simulation, because hole formation was also observed in our atomistic simulations (left). The holes are unstable and expanding, ultimately leading to the rupture of the monolayer. Knecht et al. (5) also saw hole formation in their united-atom simulations of DPPC monolayers. They observed the transient formation of holes at ∼98 Å2/molecule and stable pore formation at ∼105 Å2/molecule. According to Knecht et al. the appearance of holes suggests the onset of the LE-G phase transition. Fluorescence microscopy has revealed that in the LE-G coexistence region the gas phase is present as holes in an interconnected liquid phase (52). Due to limited spatial resolution of fluorescence images, the LE-G coexistence region cannot be directly determined using fluorescence microscopy (5). However, the LE-G phase transition is thought to occur at areas of hundreds of Å2/molecule (53). Knecht et al. propose that the hole formation in their MD simulations corresponds to the sharp transition in the order of lipid chains recently detected by vibrational sum frequency generation spectra at 110 Å2/molecule, which they suggest could be associated with the onset of the gas-liquid coexistence region (5). Knecht et al. also observed LC domain formation away from pore boundaries (5). Whether LC domain formation can be seen in CG simulations at conditions beyond those needed to generate holes has not yet been tested. In contrast to our results and those of Knecht et al. (5), the results of Nielsen et al. (54) using a CG model (which is structurally similar to the model of Marrink et al. (34), but includes long-range electrostatics) showed that at large area/lipid, monolayer lipids become highly disordered and spread on the surface instead of forming holes. In the simulations of Nielsen et al. (54), the entropic benefit of spreading on the surface outweighs the van der Waals interaction energy, which suggests a possible problem with their energy parameterization, which they admit is exploratory and not yet validated. Hole formation has also been observed in atomistic simulations of DPPC bilayers. Leontiadou et al. (28) observed a critical surface tension (∼38 mN/m) above which pores in the bilayer expand becoming unstable and ultimately leading to the rupture of the bilayer. Feller and Pastor (38) have also described large and sudden expansions at a surface tension of 50 mN/m, which may suggest the disruption of the bilayer.
FIGURE 8.

Hole formation in atomistic (left) and coarse-grained (right) simulations at 323.15 K, from the side (top) and corresponding top view (bottom). The lipid tails and glycerol groups are shown in green, the headgroups in red, and the waters in blue. The corresponding surface tensions and simulation times are given below the images.
Effect of bead size
It is generally agreed that the packing of DPPC molecules is determined by the size difference between the head- and tail-groups, with the area required by the headgroup being substantially larger than that required for the tails, leading to packing adjustments such as lipid-chain tilting and headgroup overlap (46,49). The coarse-grained model of Marrink et al. (34) utilizes a Lennard-Jones bead size of σ = 0.47 nm, for all bead types. Thus it does not capture the large difference in limiting area between the phosphatidylcholine headgroup and the acyl chains. To test the effect of the relative size difference between the headgroup and acyl chains on the packing of DPPC monolayers, we ran simulations (results not shown) with the bead size of the tails including the glycerols decreased, while the headgroup bead size remained at 0.47 nm. Our simulations showed that decreasing the tail-bead size by the proper amount allows the monolayer to achieve smaller minimum areas closer to the experimentally determined limiting area, while maintaining the correct packing arrangement. On the other hand, decreasing tail-bead size too much impairs packing and the area is not minimized.
DISCUSSION
Comparing simulated and experimental isotherms
Many studies containing experimentally measured pressure-area isotherms for pure DPPC monolayers have been reported (3,13–17,55–167). However, very few studies compare their isotherms with those of others, and those that do tend to compare with only one or two selected isotherms that resemble their own. A major reason for this lack of comparison is due to the diverse conditions under which isotherms are obtained, making reproducibility problematic. Thus, even though the pressure-area isotherm of a monolayer is a thermodynamic relationship that, like pressure-volume isotherms for bulk substances, ought to be a universal function if measured accurately and under equilibrium conditions, in practice isotherms vary considerably, due to variability in compression rate, type, and geometry of experimental apparatus, and experimental artifacts (leakage, impurities, etc.), as well as pH, ionic strength, and spreading solvent (168). The variation among selected experimental isotherms is illustrated in Fig. 9, at 293.15 K (top left), 295.15 K (top right), 298.15 K (bottom left), and 303.15 K (bottom right) with our simulated isotherms included.
FIGURE 9.

Comparison of simulated CG pressure-area isotherms with various experimental ones at 293.15 K (top left), 295.15 K (top right), 298.15 K (bottom left), and 303.15 K (bottom right).
The complexity of phospholipid phase behavior and the many experimental factors involved can lead to results that are ambiguous and apparently conflicting. The difficulty in finding isotherms obtained under similar conditions has been noted before (16,76). Experimental artifacts can also lead to results that can be easily misinterpreted. Different authors may come to remarkably different, and often contradictory, interpretations of monolayer behavior, involving factors such as collapse mechanism, relaxation times, and the effect of the experimental conditions (spreading method, compression rate, etc.). These differences are not inconsequential; the shape of the isotherm is physiologically relevant, making accurate determination of it very important. For example, the very low surface tension when the film is compressed toward collapse is thought to be a mechanism for preventing alveolar closure at end-expiration (169), and the steep slope of DPPC postcollapse expansion isotherms is thought to be important for alveolar recruitment and stabilization of lung units during inspiration (4). Furthermore, the shape of the isotherm is crucial to obtaining a proper understanding the behavior of the monolayer on the molecular level; for example, the compressibility is determined from the slope of the isotherm (158).
When comparing experimental pressure-area isotherms, there are a few key experimental trends to keep in mind. Varying the dynamic compression rate is not expected to have a large effect (72,158,170), and in many cases the presence of relatively small concentrations of ions leads to little or no change in the isotherm of zwitterionic monolayers (63,140–142,171). At moderate pH, the isotherm shows little sensitivity to pH. However, at low pH, decreased hydrogen-bonding leads to an increase in the maximum surface pressure and can cause a shift to smaller areas due to hindered solvation, and at high pH, solvation is increased and equilibrium is shifted toward the fluid phase (130,143,172). The type of experimental apparatus used is known to have an effect on the shape of pressure-area isotherms, and each type has a unique set of conditions and limitations to take into account (7,12,72,82,151,163,168,173–181). The geometry should be considered because of curvature effects, area available for creep and leakage, and disordering of lipids near walls that all effect the measurement of area/lipid. The potential for leakage is greatest at high temperatures and large dynamic pressures, and is greatest in a conventional Langmuir trough; however, the use of devices such as ribbon barriers help minimize or even eliminate leakage (72,82,174,182). The pulsating bubble surfactometer also suffers from leakage, whereas the captive bubble apparatus is free from the effects of leakage. Leakage leads to a shift in the isotherm to lower surface pressures and a decrease in its slope, which can be mistaken as premature collapse (82). Even in the absence of leakage, creep along the walls can be an issue and problems with contact angle can give erroneously low surface tensions (176,182). Impurities may also arise from many sources including the experimental apparatus itself, and lead to isotherms that do not have a well-defined phase-transition region, are shifted, or do not reach near-zero surface tensions upon end-compression (16,145,148). Careful consideration of the choice of spreading solvent is necessary, because it can have a large effect on the displacement of isotherms along the area/molecule axis and can impair film stability (76,150,179). Polar components are surface-active and may solubilize the lipids, causing a shift in the isotherm to very low areas/lipid due to the loss of lipid from the interface. The effects of compression rate, pH, ionic strength, experimental apparatus, spreading agent, and impurities are discussed in more detail in the Supplementary Material.
As noted by others (43), simulations of phospholipid monolayers are limited to the nanosecond timescale, which cannot account for long time adjustments that the monolayers undergo to reach equilibrium. Thus, the results of computer simulations of phospholipid monolayers must not be interpreted as equilibrium behavior, but rather as dynamic (i.e., metastable or quasiequilibrium). This is important to take into account when comparing simulation results to experimental data. It is important to compare simulation results with dynamic isotherms (isotherms compressed relatively rapidly and thus allowed to reach near-zero surface tensions), rather than static isotherms, which have relaxed to equilibrium and reach substantially lower surface pressures.
In Fig. 9, our simulated pressure-area isotherms are compared to experimental isotherms at 293.15 K (15,55–61) (top left), 295.15 K (62–66) (top right), 298.15 K (14,67–72) (bottom left), and 303.15 K (14,17,73) (bottom right). For each temperature, our simulations were run both independently from an initially disordered state (black triangles) and cycled beginning from an initially ordered state (blue squares). At 303.15 K, the results from cycling simulations are compared for run durations of 20 ns and 100 ns at each point (Fig. 9, bottom right). The experimental conditions for each isotherm are given in Table 1, including compression rate, type of experimental apparatus, subphase composition, pH, and spreading solvent.
TABLE 1.
Experimental conditions used to obtain pressure-area isotherms
| Temp (°C) | Rate | EA | Subphase | Spreading solvent | |
|---|---|---|---|---|---|
| Ahuja and Möbius (55) | 20 | Discontinuous | FRT | Pure water | Chloroform with 2% ethanol |
| Bordi et al. (56) | 20 | 0.1 cm/min | LBW | Water +0.145 M NaCl, pH 7.2 | Chloroform/methanol (1:1) |
| Borissevitch et al. (57) | 20 | 2 mN/m × min | LW | Pure water, pH 5.9 | Chloroform |
| Dubreil et al. (58) | 20 | 3 cm/min | LW | Phosphate buffer, pH 7.2 | Chloroform |
| Miñones et al. (59) | 20 | 8.2 Å2/molecule × min | LBW | Water, pH6 (adjusted with HCl) | Chloroform/ethanol (4:1) |
| Sández et al. (60) | 20 | 27 cm2/min | LT | Citrate, phosphate, and sodium borate buffer, pH 7 | Chloroform/ethanol (4:1) |
| Williams et al. (61) | 20 | 0.5 cm2/min | LBW | Water + 0.15 M NaCl, pH 5.6 | Chloroform/methanol (4:1) |
| Yun et al. (15) | 20 | 7.5 cm2/min | LBW | Pure water | Chloroform |
| Dynarowicz-Łątka et al. (62) | 22 | 30 cm2/min | LT | Pure water | Chloroform/methanol (9:1) |
| Hunt et al. (63) | 22 | 5.1 cm2/min | LW | Pure water | n-Hexane/ethanol (9:1) |
| Rana et al. (64) | 22 | 0.5 cm2/min | LBW | Water + 0.15 M NaCl, pH 5.6 | Chloroform/methanol (4:1) |
| Slotte and Mattjus (65) | 22 | <6 Å2/molecule × min | TMT | Pure water | Hexane/2-propanol (3:2) |
| Taneva et al. (66) | 22 | 40 cm2/min | LWRB | Water + 0.15 M NaCl, pH 7 | 1-Propanol/0.5 M sodium acetate (1:1) |
| Crane et al. (14) | 25,30,50 | 2.5–5 Å2/molecule × min | CB | 10 mM HEPES, 1.5 mM CaCl2, 0.15 M NaCl, pH 7 | Chloroform/methanol (1:2) |
| Gladston and Shah (67) | 25 | Discontinuous | MWB | Water + 0.9% NaCl, pH 5.6 | Chloroform/methanol/water (80:35:5) |
| Kanintronkul et al. (68) | 25 | 1 cm/min | LW | Carbonate buffer, pH 9 | Chloroform |
| Lee et al. (69) | 25 | 4.6 Å2/molecule × min | LBW | Pure water | Chloroform/methanol (9:1) |
| Nakahara et al. (70) | 25 | 10.3 Å2/molecule × min | LW | Water + 0.15 M NaCl, pH 2 | n-Hexane/ethanol (9:1) |
| Shen et al. (71) | 25 | 1.5 cm/min | LB | Pure water, pH 6.5 | Chloroform |
| Tabak et al. (72) | 25 | ≤96 Å2/molecule × min | LWRB | Pure water | Hexane/ethanol (9:1) |
| Tabak et al. (72) | 25 | N/A | Spread | Pure water | Hexane/ethanol (9:1) |
| Baldyga and Dluhy (17) | 30 | Not specified | JLFB | Water + 0.15 M NaCl, pH 5.6 | Chloroform |
| Maskarinec et al. (73) | 30 | Not specified | LW | Pure water | Chloroform |
Rate of compression, type of apparatus, subphase composition/pH, and spreading solvent used to obtain the isotherms reproduced in Fig. 9. (Abbreviations used: EA, experimental apparatus; LT, Langmuir trough; MWB, modified Wilhelmy balance; LW, Langmuir-Wilhelmy balance; LWRB, Langmuir-Wilhelmy balance with a ribbon barrier; LB, Langmuir-Blodgett balance; LBW, Langmuir-Blodgett with a Wilhelmy plate; FRT, Fromherz-type round trough; TMT, Teflon-milled trough; JLFB, Joyce-Loebl film balance; CB, captive bubble method; Spread, equilibrium spreading in a beaker.)
These experimental isotherms in Fig. 9 vary greatly from one to the next in shape and magnitude. All of the isotherms presented here were obtained at moderate pH, except those of Kanintronkul et al. (68) (pH 9) and Nakahara et al. (70) (pH 2), both at 298.15 K. pH is not expected to be a major factor affecting the isotherms that were obtained at moderate pH values, for which the monolayer is thought to be insensitive to pH. The isotherm of Kanintronkul et al. (68) is shifted to a larger area/lipid relative to the other isotherms; it also displays elevated surface pressures at large areas/lipid, and does not display well-defined phase transitions. This can likely be attributed to increased solvation and a shift in equilibrium toward the fluid phase, resulting from the high pH. In contrast, the isotherm of Nakahara et al. (70) is shifted to lower areas/lipid reflecting hindered solvation attributed to the acidic medium.
No defining trends associated with the type of apparatus used are evident from the isotherms shown in Fig. 9. All of the isotherms obtained at 293.15 K and 295.15 K were obtained in a trough (see Table 1), yet much variation among them remains. At 298.15 K and 303.15 K, all pressure-area isotherms were obtained with a trough, except for the isotherms reported by Crane et al. (14), which utilized the captive bubble apparatus. Despite this, the isotherms presented by Crane et al. (14) do not have any defining features that distinguish them from the other isotherms presented here. Leakage could be an issue in any of the experiments except those of Crane et al. (because of the use of the captive bubble apparatus), the equilibrium isotherm of Tabak et al. (72) (because spreading inside a beaker was used), and the dynamic isotherms of Tabak et al. (72) and Taneva et al. (66) (because of the use of a ribbon barrier). Furthermore, experiments performed without the use of a Wilhelmy plate or with discontinuous compression may be especially susceptible to leakage. Thus, leakage is a likely factor attributing to the large variation between the experimental isotherms shown here.
Dynamic compression rate appears to play a role in the slope of the isotherms at high surface pressures (low areas/lipid). The slope tends to become steeper as compression rate is increased. Isotherms compressed the quickest, such as those obtained by Bordi et al. (56), Williams et al. (62), Rana et al. (64), Slotte and Mattjus (65), and Crane et al. (14) have the steepest slopes. This is made more evident by the magnitude of the area compressibility moduli calculated for these isotherms (discussed in detail in the next section). Note that although slower compression leads to better equilibration, it does not necessarily produce more accurate isotherms. Additionally, isotherms that compressed quickly better mimic the physiological conditions.
The spreading solvent is typically not thought to have a large effect when used in a trough, which is open to air circulation and takes up a relatively large surface area. Nevertheless, in comparing these isotherms, spreading solvent does appear to have played a major role. At 293.15 K, the isotherms obtained by Borissevitch et al. (57), Dubreil et al. (58), Ahuja and Möbius (55), and Yun et al. (15) all reach relatively low surface pressures at end compressions of roughly 42, 48, 50, and 55 mN/m, respectively. Collapse does not appear to have been reached before measurement was halted for the isotherms of Dubreil et al. (58) and Ahuja and Möbius (55), and it remains uncertain what the actual collapse pressure would have been. For all of these isotherms the spreading solvent was pure or almost pure chloroform (98% in the case of (55)). At 295.15 K, the isotherm obtained by Dynarowicz-Łątka et al. (62) used the highest concentration of chloroform in the spreading solvent (90% by volume), and also has the lowest maximum surface pressure (highest minimum surface tension). At 298.15 K, slightly low dynamic maximum surface pressures are obtained by Nakahara et al. (70) (∼64 mN/m), Kanintronkul et al. (68) (∼65 mN/m), Shen et al. (71) (∼65 mN/m), and Gladston and Shah (67) (∼67 mN/m). Of these, the isotherms obtained by Kanintronkul et al. (68) and Shen et al. (71) used pure chloroform as a spreading agent, and that of Gladston and Shah (67) utilized 80% chloroform. Gladston and Shah (67) claim that the onset of film collapse actually occurred at ∼44 mN/m as indicated by an inflection point far before the plateau at ∼67 mN/m (68). This change in slope could be attributed to the squeeze-out of chloroform causing some of the DPPC molecules to be removed from the monolayer, leading to an underestimation of collapse pressure. At 303.15 K, the isotherm obtained by Baldgya and Dluhy (17) displays the lowest maximum pressure and uses pure chloroform as a spreading agent.
At 293.15 K the isotherms of Bordi et al. (56), Borissevitch et al. (57), and Yun et al. (15) are all shifted to lower areas/lipid relative to the other isotherms shown, with the isotherm of Bordi et al. (56) and Borissevitch et al. (57) reaching areas/lipid even smaller than the limiting area of 39 Å2/molecule. The isotherms of Borissevitch et al. (57) and Yun et al. (15) were obtained with a spreading solvent that was pure chloroform, and that of Bordi et al. (56) was obtained with a 1:1 chloroform/methanol solution. At 298.15 K, the isotherms obtained by Gladston and Shah (67), and of Lee et al. (69) are shifted to low areas/lipids, with the former reaching areas/lipid that are smaller than the limiting area. These isotherms were obtained using chloroform-methanol spreading solutions containing 66.7% and 90% chloroform by volume, respectively. At 303.15 K, the isotherms obtained by Baldgya and Dluhy (17) and Maskarinec et al. (73) are shifted to areas/lipid that are smaller than the limiting areas. Both of these isotherms were obtained using pure chloroform as a spreading agent. The isotherm of Maskarinec et al. (73) displays a collapse plateau at a very low area/lipid (∼30 Å2/molecule), suggesting that DPPC has been lost from the monolayer before the collapse plateau is reached. The shift in these isotherms could result from the use of pure or almost pure chloroform as the spreading solvent. It should be noted that chloroform is known to be surface active due to its polarity (150).
Thus, spreading solvent effects may contribute significantly to the observed variation between isotherms (76). The use of chloroform as a spreading solvent appears to shift the isotherms to lower areas/lipid and also decrease the surface pressure at collapse. This suggests the possible loss of lipid from the monolayer, perhaps through the removal of some DPPC molecules from the monolayer with the squeeze-out of chloroform, leading to a decrease of area and the appearance of premature collapse. Whatever the mechanism, the use of chloroform clearly impedes the ability of the monolayer to reach near-zero surface tensions. Others have noted that by increasing the amount of chloroform in the subphase, the amount of DPPC lost from the film increases dramatically, indicating that the presence of chloroform can impair film stability (180). According to Wüstneck et al. (150), the interaction between DPPC molecules is strongly depressed by the presence of chloroform, which is in incorporated into the monolayer and causes an apparent increase in the molecular area. At low pressures, this causes a shift of the isotherm to larger areas and an increase in minimum surface pressure (150). However, as surface pressure is increased, the chloroform is squeezed out, taking DPPC molecules along and shifting the isotherm to low areas/lipid (150). Wüstneck et al. (150) found that when chloroform is present at the surface in large enough concentrations, a plateau corresponding to the squeeze-out of chloroform can be visualized at ∼50 mN/m. The increased molecular area at low pressures (due to the presence of chloroform in the monolayer) combined with the decreased molecular areas at high pressure (due to the loss of DPPC with chloroform from the monolayer) results in an isotherm that is broader, changing gradually with surface pressure. Other experimental spreading solvents may also cause a shift in the isotherm and the ratio of polar and nonpolar components in the spreading solvent is of critical importance (76). At 295.15 K, the isotherm of Taneva et al. (66) is shifted to small areas/lipid, and reaches an area smaller than the limiting area. Taneva et al. (66) note that the use of the propanol/sodium acetate solvent appears to have contributed to a shift in the isotherm of pure DPPC to low areas/lipid, perhaps due to partial dissolution of DPPC into the subphase or incomplete dissociation of aggregates formed in the spreading solution (66).
Although spreading solvent appears to play a role, with so many experimental factors involved, it is impossible to be sure what leads to a shift in area/lipid or shape of a given isotherm. At 295.15 K, the isotherm obtained by Dynarowicz-Łątka et al. (62) was obtained without the use of a Wilhelmy plate, and is thus at a higher risk of film leakage, which could also explain the low surface tension at collapse. At 298.15 K, the compression isotherm of Gladston and Shah (67) displays an inflection point at ∼44 mN/m, which may be a result from the choice of spreading agent, but could also be attributed to leakage, which may be magnified by the use of discontinuous compression or by the experimental apparatus. Also at 298.15 K, the isotherm obtained by Nakahara et al. (70) is shifted to an area/lipid that is smaller than the limiting area. The isotherm of Nakahara et al. (70) appears to exhibit material leakage from the monolayer, as indicated by a change in slope before the collapse plateau is reached. This isotherm is also distinct from the other isotherms shown in Fig. 9, because it is obtained at low pH. At 303.15 K, the isotherm of Baldgya and Dluhy (17) appears to exhibit an inflection point (at ∼40 Å2/molecule), which could be caused by the onset of collapse at low collapse pressure (∼60 mN/m), or by film leakage.
It is difficult to say which of the isotherms shown in Fig. 9 are reliable, especially with so much variation among them and in the methods used to obtain them. It is more feasible to identify those that are most likely to exhibit experimental artifacts. Isotherms obtained at high and low pH ((68) (pH 9) and (70) (pH 2) at 298.15 K), dynamic isotherms apparently exhibiting early collapse (Miñones et al., (59) Sández et al. (60), and Yun et al. (15) at 293.15 K, Dynarowicz-Łątka et al. (62) at 295.15 K, Gladston and Shah (67) and Kanintronkul et al. (68) at 298.15 K), isotherms that are suspected of exhibiting spreading-solvent artifacts (Taneva et al. (66) 295.15 K), and isotherms that are shifted to areas/lipid that are smaller than the limiting area of 39 Å2/molecule (Borissevitch et al. (57) and Bordi et al. (56) at 293.15 K, Taneva et al. (66) at 295.15 K, Nakahara et al. (70) and Gladston and Shah (67) at 298.15 K, Baldgya and Dluhy (17) and Maskarinec et al. (73) at 303.15 K) are most likely to be misleading. This leaves the isotherms obtained by Ahuja and Möbius (55), Dubreil et al. (58), and Williams et al. (61) 293.15 K; Hunt et al. (63), Rana et al. (64), and Slotte and Mattjus (65) at 295.15 K; Lee et al. (69), Shen et al. (71), and Tabak et al. (72) at 298.15 K; and Crane et al. (14) at 298.15 K, 303.15 K, and 323.15 K as possibly the most trustworthy isotherms. These isotherms all exhibit the same general shape but vary in placement along the area/lipid axis and slope, with the latter likely being affected by compression rate.
From Fig. 9, it is clear that the simulated isotherms give areas that are too large, with limiting areas near 49 Å2/molecule—a phase-transition plateau that is shifted upward to much higher surface pressures than those seen experimentally, and an overly large hysteresis loop. The steep slope upon expansion is typical of experimental expansion isotherms, which exhibit a sudden drop in pressure. The LE portion of the compression isotherm is steeper than the posttransition slopes seen in the experimental isotherms. Furthermore, the length of the coexistence region is much smaller for simulated compression isotherms than in experimental ones. Our LC-LE phase-coexistence plateaus occur at higher surface pressures than do those predicted experimentally and the surface pressures of our plateaus increase with increasing temperature, suggesting that the transition temperature for our simulated monolayers is likely too low. These factors indicate that our simulations do not accurately reproduce the behavior seen in experimental isotherms.
It should also be noted that some differences between simulations and experiments could be due to the absence of chain tilting in the simulations, because this absence leads to changes in area with increased pressure that are too small (174). Also, experimental factors, particularly the choice of spreading solvent, not included in our simulations, may explain why our predicted isotherms exhibit abrupt changes and steep slopes that are uncharacteristic of experimental isotherms. Other atomistic (23,27) and coarse-grained models (54) also give pressure-area isotherms that are shifted to higher area/lipid relative to experimental values. Feller et al. (27) attribute the differences between their results and experiments to difficulties in the evaluation of surface pressure, which depends on the accurate determination of long-range forces between atoms and has large instantaneous fluctuations (27). They also suggest that the accuracy of simulated isotherm could be improved by better potential energy parameterization or by incorporation of long-range forces. Moreover, because the coarse-grained model lumps roughly four acyl-tail carbon atoms into each tail bead, it is not able to capture the sensitivity of the phase transition temperature to chain length. Phillips and Chapman (16) showed that subtracting two methylene groups from each chain shifts the isotherm by an amount that is equivalent to raising the temperature by 20 K. However, coarse-grained lipids differing by only one or two methylene groups are represented by the same CG structure and thus cannot predict such effects. Also, as noted by Feller et al. (27), surface tension depends on the accurate determination of long-range forces, which are not considered by the coarse-grained model. However, it is also important to consider inherent limitations associated with simulated isotherms due to system size and timescale limitations.
Area compressibility modulus
The compressibility (Cs) of the DPPC monolayer can be calculated from the slope of the pressure-area isotherm according to
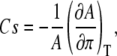 |
(1) |
where A and π are the area and surface pressure, respectively (165). The area compressibility modulus (Cs−1) is the reciprocal of the compressibility. Typical experimental values of the area compressibility modulus for DPPC monolayers are 10–50 mN/m for LE films, 100–250 mN/m for LC films, and >250 mN/m for solid films (165,166). Here, the condensed and expanded phase moduli are approximated from the slopes of the experimental isotherms shown in Fig. 9, using linear regression. Any moduli falling outside of the typical range are reported in Table 2. To avoid mislabeling a solid-phase modulus as a high value for the condensed phase modulus, in Table 2 we only report moduli for isotherms that did not have a kink, because a kink might indicate a transition to solid phase. Comparing the isotherms given in Table 2 with the experimental conditions listed in Table 1, we notice that the isotherms giving LC moduli larger than typical condensed phase values were all compressed rapidly at rates of 0.5 cm2/min or at rates <6 Å2/molecule × min, excluding the isotherm of Ahuja and Möbius (55), which was compressed discontinuously. The rapid compression of these isotherms appears to be associated with their steep slopes and corresponding high compressibility moduli. Many studies have suggested that there is little or no variation in the shape of dynamic isotherms as compression rate is varied (72,158,170). However, at high surface pressures, as the slope of the isotherm becomes nearly vertical, changes in the slope of the isotherm that may appear small can significantly increase the area compressibility modulus. Furthermore, faster compression rates are known to lead to the formation of smaller LC and LE domains, due to diffusion-limited growth (170). It is conceivable that this change in domain size could alter the compressibility of the monolayer.
TABLE 2.
Experimental and simulated area compressibility moduli
| Temperature | Cs−1 | Area | Phase | |
|---|---|---|---|---|
| Typical experimental values* | Varies | 100–250 mN/m | Varies | LC |
| Independent runs | 298.15 K | ∼363 mN/m | 47.5 Å2 | LC |
| Larger system size | 298.15 K | ∼316 mN/m | 47.8 Å2 | LC |
| Ahuja and Möbius (55) (experimental) | 293.15 K | ∼326 mN/m | 44 Å2 | LC |
| Williams et al. (61) (experimental) | 293.15 K | ∼290 mN/m | 47.9 Å2 | LC |
| Rana et al. (64) compression (experimental) | 295.15 K | ∼252 mN/m | 54.1 Å2 | LC |
| Rana et al. (64) expansion (experimental) | 295.15 K | ∼279 mN/m | 46.7 Å2 | LC |
| Slotte and Mattjus (65) (experimental) | 295.15 K | ∼279 mN/m | 45.4 Å2 | LC |
| Crane et al. (14) (experimental) | 298.15 K | ∼293 mN/m | 44.6 Å2 | LC |
| Crane et al. (14) compression (experimental) | 303.15 K | ∼313 mN/m | 45.9 Å2 | LC |
| Crane et al. (14) expansion (experimental) | 303.15 K | ∼265 mN/m | 47.1 Å2 | LC |
| Typical experimental values† | Varies | 10–50 mN/m | Varies | LE |
| Independent runs | 298.15 K | ∼169 mN/m | 63.9 Å2 | LE |
| Independent runs | 323.15 K | ∼115–360 mN/m | 56.2–71.2 Å2 | LE |
| Larger system size | 298.15 K | ∼41.5 mN/m | 67.7 Å2 | LE |
| Atomistic | 323.15 K | ∼120–268 mN/m | 56.0–72.5 Å2 | LE |
| Adhangale and Gaver (32) (coarse-grained) | 323.15 K | ∼92–227 mN/m | 45.0–56.2 Å2 | LE |
| Skibinsky et al. (42) (atomistic) | 323.15 K | ∼64.5–128 mN/m | 54–80 Å2 | LE |
| Crane et al. (14) compression (experimental) | 303.15 K | ∼58 mN/m | 68.9 Å2 | LE |
| Crane et al. (14) expansion (experimental) | 303.15 K | ∼60 mN/m | 69 Å2 | LE |
| Crane et al. (14) (experimental) | 323.15 K | ∼67.3–168 mN/m | 57.2–80.7 Å2 | LE |
Moduli approximated from our atomistic and coarse-grained (CG) simulations, as well as from our CG simulations with the larger system size of 1024 lipids/monolayer, and approximated from the experimental and simulated isotherms of others.
The steep slopes of our simulated isotherms yield moduli that are larger than typical experimental values, and these values at 298.15 K and 323.15 K are given in Table 2 along with moduli approximated from the slopes from other simulated and experimental isotherms at 323.15 K, from Fig. 4. The corresponding areas at which the moduli were calculated are also given. For our simulation isotherms at 323.15 K, a range of areas is given, because the moduli were evaluated at multiple points. The moduli were calculated by assembling results from independent runs at each pressure (as described in Simulation Method) and not from the cycling isotherms, because the slope of the expansion portion of the cycling isotherms is clearly too steep to give results that are comparable to experimental values. At 298.15 K, the two endpoints of each isotherm, corresponding to smallest and largest area/lipid simulated, were selected to represent LC and LE phase moduli. At a temperature of 323.15 K, the entire isotherm is in the expanded phase, and for this case, the modulus was evaluated at each point along the isotherms. For comparison with our results, area compressibility moduli were approximated from the slopes of other atomistic (42), coarse-grained (32), and experimental (14) isotherms at 323.15 K. When evaluating the isotherm of Adhangale et al. (32), the modulus was not approximated at the lowest area because there is a large jump in area between this and subsequent points.
As seen in Table 2, the compressibility moduli obtained for both coarse-grained and atomistic simulations do not correlate well with those typically obtained from experiments. At 298.15 K, the LE modulus obtained from our independent runs for monolayers composed of 256 lipids fell into a range expected for LC films, while our LC modulus was also too high, falling into the range of values expected for a solid film. At 323.15 K, although the entire isotherm is considered to be expanded, the compressibility moduli obtained from our coarse-grained (256 lipids/monolayer) and atomistic (64 lipids/monolayer) isotherms are again too large—once more falling into the range expected for LC and even solid films. The values obtained from our atomistic simulations differ little from those obtained from our coarse-grained simulations, although they give a narrower range of moduli which are slightly improved at low areas. The coarse-grained and atomistic isotherms of Adhangale et al. (32) and Skibinsky et al. (42) also give moduli that are higher than those typically expected from experiments. However, it should be noted that the values obtained by Skibinsky et al. (42) correlate very well with those obtained from the experimental isotherm of Crane et al. (14), which also yield values of compressibility modulus that are larger than those typical of expanded films.
For our larger system size (1024 lipids/monolayer), we obtained an LC modulus that is lower by ∼15% than for the 256 lipids/monolayer isotherm, but still larger than the typical experimental values. However, the LE modulus is greatly improved in the larger system size, falling within the experimental range expected for LE isotherms. These results show that increasing system size decreases the area compressibility modulus, or conversely increases compressibility. This is to be expected because for a larger system size, the surface can wrinkle, adding to its ability to fluctuate in area, and thus increasing compressibility. Atomistic simulations performed on small bilayer patches also yield moduli that are significantly larger than the experimental estimates (183). Marrink et al. (34) found moduli for a coarse-grained DPPC bilayer at 323 K, of 260 ± 40 mN/m for a bilayer composed of 6400 lipids and 400 ± 30 mN/m for a bilayer composed of 256 lipids. The difference in moduli for the two system sizes was attributed to the contribution of undulatory modes in the large system. Imposing a small box size is known to lead to artificial rigidity and suppressed undulations (34,36,37,44,184). Monolayers and bilayers have different bending constants, and thus their undulations differ in magnitude, which should lead to different area compressibility moduli for monolayers and bilayers. However, they can be expected to react similarly to system size constraints. Applying a surface tension will decrease undulations, and thereby reduce the undulatory contribution to the compressibility (184). Thus, finite size effects will decrease with increasing surface tension.
Effects of system size, timescale, and hysteresis
There have been many studies of finite-size effects in lipid bilayers. De Vries et al. (44) found that for constant volume simulations of DPPC bilayers, the surface tension, electron density profile across the bilayer, and the carbon-deuterium order parameters, all converged to system-size-independent and time-independent values for a system size as small as 36 lipids/leaflet and a simulation time as short as 4 ns. De Vries et al. (44) suggest that some finite size effects may be seen for systems larger than 36 lipids/leaflet. However, these are primarily due to the appearance of long-wavelength undulations. Klauda et al. (185) also found that a system size of 72 lipids (36/leaflet) was large enough to calculate accurately the structural properties (such as electron density profiles and deuterium order parameters) for a DPPC bilayer. In an earlier study, Lindahl and Edholm (184) found a slight system-size dependence in the area/lipid when a cutoff method was used for evaluation of long-range electrostatics, with a system of 64 lipids differing by 1.5 Å2 from a system 16 times as large. However, when a particle-mesh Ewald summation was used this difference was cut to <1 Å2 (186). In a recent study of DOPC bilayers Castro-Román et al. (187) found that finite-size effects contributed very little to membrane structure, with virtually no differences observed between different system sizes in their neutron and x-ray scattering factors and scattering-length density profiles. Instead they suggest that force field inaccuracies account for large structural discrepancies between simulation and experiment.
Although finite size has little effect on the properties of leaflets composed of 36 or more lipids in the single-phase region, there are more serious finite-size effects when two phases coexist. Experimentally, each coexisting region of liquid-condensed or liquid-expanded phase extends over distances of thousands of Ångstroms. Simulating these biphasic systems in a box consisting of only hundreds of lipids lends concern over the magnitude of the line tension between such small domains and the correspondence of simulation results to experiment. The small size of simulated LC and LE domains raises uncertainty over whether such domains are stable. Experimentally, above the phase transition temperature, small nuclei can form known as hetero-phase fluctuations (188). Due to the small size of these nuclei, there exists a large line tension, which opposes the thermodynamic driving force for the phase transition (37). In their simulations of CG DPPC bilayers, Marrink et al. (37) observed fast fluctuations due to formation and disappearance of small clusters of the condensed gel phase. Marrink et al. (37) also observed long-lived fluid domains that remained trapped and metastable over a microsecond timescale with small defects persisting on even longer timescales. From their bilayer simulations, Marrink et al. (37) calculated a line tension between liquid crystalline and gel domains of 3 ± 2 pN, which matches within uncertainty the experimental value of 4 pN estimated by the kinetic model of Kharakoz and Shlyapnikova (188) for small gel clusters appearing in DPPC vesicles. For CG DPPC bilayers, Marrink et al. (37) found that regions smaller than a critical nucleus size of 10–40 lipids/monolayer, depending on the temperature, were unstable. The system size of our simulations is large enough to contain domains larger than the critical nucleus size reported by Marrink et al., but the nanosecond timescale is shorter than that observed for metastable domains, suggesting that for our simulations the structures that form in the two-phase region are metastable. The metastable nature of two-phase structures in our simulations is also evident from the large hysteresis seen between our compression and expansion isotherms. Although the time- and length-scales are much different, it is important to keep in mind that dynamic experimental isotherms are also metastable. Experiments (189) have shown that the kinetics of the order-disorder transition are strongly dependent on heating and cooling rate, and under nonequilibrium conditions intermediate structures may form that differ from the equilibrium structure.
To study the effect of system size we ran simulations of monolayers composed of 256 lipids and 1024 lipids at 298.15 K. Isotherms obtained from independent runs containing 256 lipids (black triangles) and 1024 lipids (purple circles) are compared in the bottom-left-hand side of Fig. 9. Both system sizes gave the same isotherm, except at surface pressures <30 mN/m, where the larger system began to expand. The larger system also exhibited the onset of hole formation sooner (at a higher surface pressure) than the smaller system. These results correlate well with the finding of Knecht et al. (5) for an atomistic DPPC monolayer, which showed that in the LC–LE coexistence region increasing system size had little effect on the overall lipid order; however, the rupture of the monolayer occurred at a smaller molecular area.
The effects of timescale were also studied by comparing 20-ns and 100-ns cycling simulations at 303.15 K. The isotherms obtained from these simulations are shown in the bottom-right-hand corner of Fig. 9. Although the increased simulation time yields little difference in the shape and position of the compression and expansion isotherms, there is a notable difference in the hysteresis. The 100-ns cycling simulations undergo the LC-LE phase transition sooner (i.e., at higher pressure) than do the 20-ns simulations, decreasing the size of the observed hysteresis loop. If the timescale of these simulations were increased arbitrarily, the hysteresis loops would be expected to narrow and eventually reach a true equilibrium value. However, even for the slower cycling, there is a marked difference between the hysteresis loops seen in our simulations and those of typical experiments. Although some experimental isotherms yield large hysteresis loops ((67); Fig. 9 bottom left), most experimental hysteresis loops are much smaller ((64,14); Fig. 9, top and bottom right) than those seen in our simulations. Given the huge difference in time and length scales of our simulations compared to experiments, it would be computationally infeasible to carry out simulations that come significantly closer to attainment of the equilibrium isotherm (37).
CONCLUSIONS
Although many experimental pressure-area isotherms for DPPC monolayers have been reported, there is a large variation among them to which many factors might contribute, making comparison difficult and misinterpretation easy. We can make educated guesses about what causes a given isotherm to display a shift or characteristic shape; however, with so many complex factors involved, the cause of the variations among experimental isotherms remains somewhat ambiguous. A high concentration of chloroform in the spreading solvent appears to be associated with a shift in the isotherm to low areas/lipid and a decreased ability of the monolayer to reach near-zero surface tensions. This could be due to the removal of DPPC molecules from the monolayer with chloroform upon compression, leading to a decrease in area and the appearance of premature collapse. High pH appears to shift the isotherm to a larger area/lipid due to increased solvation, while low pH shifts the isotherm to lower areas/lipid reflecting hindered solvation. No defining trends associated with the type of apparatus used are evident from the isotherms studied here. Isotherms obtained in a trough show much variation and those obtained with the captive bubble apparatus exhibit no clear defining features that distinguish them from those obtained in a trough. Dynamic compression rate appears to play a role in the slope of the isotherms at high surface pressures (low areas/lipid), with a steeper slope and correspondingly larger area compressibility modulus as compression rate is increased. However, the large variability in experimental isotherms remains largely unexplained. Thus, it is clear that there is a need for some standardization to make experimental isotherms more interpretable and to make comparisons, both to simulated isotherms and among experimental ones, feasible.
Values of area compressibility modulus obtained for both coarse-grained and atomistic simulations (ours and those of others) overestimate those typically obtained from experiments, although the disagreement diminishes somewhat as simulation box size increases. Thus, it is conceivable that a simulation of a macroscopic system size could produce moduli within the range of typical experimental values. Furthermore, experimental isotherms tend to show higher moduli when obtained by more rapid compression, which might also help explain the relatively high moduli obtained from simulations, which of course are obtained under very rapid compressions compared to typical experiments.
PO4-PO4, PO4-NC3, and NC3-NC3 radial distribution functions (RDFs) show little difference between the LC and LE phases, while C2-C2 distributions show a significant decrease in tail order as the monolayer is expanded, indicating that the structure of the DPPC headgroups is affected much less by the phase transition than is the structure of the DPPC tails. Furthermore, P-N tilt angle distributions obtained from our atomistic and coarse-grained simulations give an average P-N orientation that is parallel to the interface and is not significantly affected by the LC-LE phase transition. In accord with experimental observations, this provides further evidence that the DPPC headgroup region is not strongly affected by the transition from LC to LE phase.
The coarse-grained NC3-NC3 and atomistic N-N RDFs differ considerably from each other and the coarse-grained NC3-NC3 RDF matches closely that of PO4-PO4, although the corresponding atomistic N-N and P-P RDFs differ considerably. Thus, the coarse-grained model is unable to capture the difference between N-N and P-P interactions present in the atomistic simulations, which ultimately leads to inaccuracy in the coarse-grained NC3-NC3 RDF. Furthermore, the first correlation peak of the atomistic P-P distribution broadens as the monolayer undergoes a transition from the LC to LE (5). This distinction is not seen in our coarse-grained PO4-PO4 radial distribution function at 298.15 K. These results suggest that the coarse-grained model is better at capturing the effect of changing surface pressure on lipid tails than on lipid headgroups.
Despite some limitations, molecular simulation could be a key to obtaining a more detailed understanding of the complex mechanisms involved in the phase transitions of DPPC, of other physiologically relevant lipids, and of mixtures of lipids and proteins. Simple coarse-grained models, such as that developed by Marrink et al., are powerful tools for studying such systems, on length- and timescales that are difficult or impossible to obtain using atomistic simulation. Using the coarse-grained model of Marrink et al., we were able to quickly obtain compression and expansion isotherms for DPPC at five different temperatures and visualize the changes in packing from hexagonal to disordered as the DPPC monolayer underwent a phase change from the liquid-condensed (LC) to the liquid-expanded (LE) state. These fast and simple simulations provide a tool for comparison to experiment and clarification of the possible mechanisms involved in the rich phase behavior of DPPC. However, there are limitations, shown by the fact that even the atomistic simulated isotherms tend to be shifted to higher areas/lipid than experimental ones and do not exhibit the correct shape. To obtain more accurate simulated isotherms, more work is needed on either potential energy parameterization or the evaluation of long-range forces, for both coarse-grained and atomistic models.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We thank Senthil Kandasamy for his assistance and for insightful discussions.
This work was made possible by financial support from the University of Michigan.
Editor: Kathleen B. Hall.
References
- 1.Akino, T. 1992. Lipid components of the surfactant system. In Pulmonary Surfactant: From Molecular Biology to Clinical Practice. B. Robertson, L. M. G. van Golde, and J. J. Batenburg, editors. Elsevier Science Publishers, Amsterdam, The Netherlands.
- 2.Keough, K. M. W. 1992. Physical chemistry of pulmonary surfactant in the terminal air spaces. In Pulmonary Surfactant: From Molecular Biology to Clinical Practice. B. Robertson, L. M. G. van Golde, and J. J. Batenburg, editors. Elsevier Science Publishers, Amsterdam, The Netherlands.
- 3.Notter, R. H., S. A. Tabak, S. Holcomb, and R. D. Mavis. 1980. Postcollapse dynamic surface pressure relaxation of binary surface films containing dipalmitoyl phosphatidylcholine. J. Colloid Interface Sci. 74:370–377. [Google Scholar]
- 4.Notter, R. H., R. Taubold, and R. D. Mavis. 1982. Hysteresis in saturated phospholipid films and its potential relevance for lung surfactant function in vivo. Exp. Lung Res. 3:109–127. [DOI] [PubMed] [Google Scholar]
- 5.Knecht, V., M. Müller, M. Bonn, S. J. Marrink, and A. E. Mark. 2005. Simulation studies of pore and domain formation in a phospholipid monolayer. J. Chem. Phys. 122:24704–24712. [DOI] [PubMed] [Google Scholar]
- 6.Barnes, G. T., and I. R. Gentle. 2005. Interfacial Science: An Introduction. Oxford University Press, New York.
- 7.Notter, R. H. 1984. Surface chemistry of pulmonary surfactant: the role of individual components. In Pulmonary Surfactant. B. Robertson, L. M. G. van Golde, and J. J. Batenburg, editors. Elsevier Science Publishers, Amsterdam, The Netherlands.
- 8.Alonso, C., T. Alig, J. Yoon, F. Bringezu, H. Warriner, and J. A. Zasadzinski. 2004. More than a monolayer: relating lung surfactant structure and mechanics to composition. Biophys. J. 87:4188–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nag, K., J. Perez-Gil, M. L. F. Ruano, L. A. D. Worthman, J. Stewart, C. Casals, and K. M. W. Keough. 1998. Phase transitions in films of lung surfactant at the air-water interface. Biophys. J. 74:2983–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouritsen, O. G. 1991. Theoretical-models of phospholipid phase-transitions. Chem. Phys. Lipids. 57:179–194. [DOI] [PubMed] [Google Scholar]
- 11.Luty, T., C. J. Eckhardt, and J. Lefebvre. 1999. Langmuir monolayers as disordered solids: disorder and elastic fluctuations in mesophases. J. Chem. Phys. 111:10321–10329. [Google Scholar]
- 12.Kwok, D. Y., D. Vollhardt, R. Miller, D. Li, and A. W. Neumann. 1994. Axisymmetric drop shape analysis as a film balance. Colloids Surf. A Physicochem. Eng. Aspect. 88:51–58. [Google Scholar]
- 13.Vilallonga, F. 1968. Surface chemistry of L-α-dipalmitoyl lecithin at the air-water interface. Biochim. Biophys. Acta. 163:290–300. [DOI] [PubMed] [Google Scholar]
- 14.Crane, J. M., G. Putz, and S. B. Hall. 1999. Persistence of phase coexistence in disaturated phosphatidylcholine monolayers at high surface pressure. Biophys. J. 77:3134–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yun, H., Y. W. Choi, N. J. Kim, and D. Sohn. 2003. Physicochemical properties of phosphatidylcholine (PC) monolayers with different alkyl chains, at the air/water interface. Bull. Korean Chem. Soc. 24:377–383. [Google Scholar]
- 16.Phillips, M. C., and D. Chapman. 1968. Monolayer characteristics of saturated 1,2-diacyl phosphatidylcholines (lecithins) and phosphatidylethanolamines at the air-water interface. Biochim. Biophys. Acta. 163:301–313. [DOI] [PubMed] [Google Scholar]
- 17.Baldyga, D. D., and R. A. Dluhy. 1998. On the use of deuterated phospholipids for infrared spectroscopic studies of monomolecular films: a thermodynamic analysis of single and binary component phospholipid monolayers. Chem. Phys. Lipids. 96:81–97. [DOI] [PubMed] [Google Scholar]
- 18.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. Dinola, and J. R. Hawk. 1984. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 19.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 20.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- 21.van der Spoel, D., A. R. van Buuren, E. Apol, P. J. Meulenhoff, D. P. Tieleman, A. L. T. M. Sijbers, B. Hess, K. A. Feenstra, E. Lindahl, R. van Drunen, and H. J. C. Berendsen. 2001. Gromacs User Manual version 3.0. Nijenborgh, Groningen, The Netherlands. http://www.gromacs.org/.
- 22.Weast, R. C. 1981. Handbook of Chemistry and Physics. A Ready-Reference Book of Chemical and Physical Data, 61st Ed. Robert C. Weast, editor. CRC Press, Boca Raton, FL.
- 23.Zakharov, V. V., E. N. Brodskaya, and A. Laakonen. 1998. Surface properties of water clusters: a molecular dynamics study. Mol. Phys. 95:203–209. [Google Scholar]
- 24.Kaznessis, Y. N., S. Kim, and R. G. Larson. 2002. Simulations of zwitterionic and anionic phospholipid monolayers. Biophys. J. 82:1731–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vega, C., and E. de Miguel. 2007. Surface tension of the most popular models of water by using the test-area simulation method. J. Chem. Phys. 126:154707–154716. [DOI] [PubMed] [Google Scholar]
- 26.Klauda, J. B., X. Wu, R. W. Pastor, and B. R. Brooks. 2007. Long-range Lennard-Jones and electrostatic interactions in interfaces: application of the isotropic periodic sum method. J. Phys. Chem. B. 111:4393–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feller, S. E., Y. Zhang, and R. W. Pastor. 1995. Computer simulation of liquid/liquid interfaces. II. Surface tension-area dependence of a bilayer and monolayer. J. Chem. Phys. 103:10267–10276. [Google Scholar]
- 28.Leontiadou, H., A. E. Mark, and S. J. Marrink. 2004. Molecular dynamics simulations of hydrophilic pores in lipid bilayers. Biophys. J. 86:2156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiu, S. W., M. Clark, V. Balaji, S. Subramaniam, H. L. Scott, and E. Jakobsson. 1995. Incorporation of surface tension into molecular dynamics simulation of an interface: a fluid phase lipid bilayer membrane. Biophys. J. 69:1230–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.López Cascales, J. J., T. F. Otero, A. J. Fernández Romero, and L. Camacho. 2006. Phase transition of a DPPC bilayer induced by an external surface pressure: from bilayer to monolayer behavior. A molecular dynamics study. Langmuir. 22:5818–5824. [DOI] [PubMed] [Google Scholar]
- 31.Sun, F. 2002. Constant normal pressure, constant surface tension, and constant temperature molecular dynamics simulation of hydrated 1,2-dilignoceroylphosphatidylcholine monolayer. Biophys. J. 82:2511–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adhangale, P. S., and D. P. Gaver III. 2006. Equation of state for a coarse-grained DPPC monolayer at the air/water interface. Mol. Phys. 104:3011–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang, Y., S. E. Feller, B. R. Brooks, and R. W. Pastor. 1995. Computer simulation of liquid/liquid interfaces. I. Theory and application to octane water. J. Chem. Phys. 103:10252–10266. [Google Scholar]
- 34.Marrink, S. J., A. H. de Vries, and A. E. Mark. 2004. Coarse-grained model for semiquantitative lipid simulations. J. Phys. Chem. B. 108:750–760. [Google Scholar]
- 35.Marrink, S. J. 2004. University of Groningen. Department of Biophysical Chemistry. http://rugmd4.chem.rug.nl/∼marrink/coarsegrain.html.
- 36.Marrink, S. J., and A. E. Mark. 2001. Effect of undulations on surface tension in simulated bilayers. J. Phys. Chem. B. 105:6122–6127. [Google Scholar]
- 37.Marrink, S. J., J. Risselada, and A. E. Mark. 2005. Simulation of gel phase formation and melting in lipid bilayers using a coarse-grained model. Chem. Phys. Lipids. 135:223–244. [DOI] [PubMed] [Google Scholar]
- 38.Feller, S. E., and R. W. Pastor. 1999. Constant surface tension simulations of lipid bilayers; the sensitivity of surface areas and compressibilities. J. Chem. Phys. 111:1281–1287. [Google Scholar]
- 39.Tieleman, D. 2002. University of Calgary. Department of Biological Sciences. http://moose.bio.ucalgary.ca/download.html.
- 40.Hess, B., H. Bekker, H. J. C. Berendsen, and G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 41.Essman, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 42.Skibinsky, A., R. M. Venable, and R. W. Pastor. 2005. A molecular dynamics study of the response of lipid bilayers and monolayers to trehalose. Biophys. J. 89:4111–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mauk, A. W., E. L. Chaikof, and P. J. Ludovice. 1998. Structural characterization of self-assembled lipid monolayers by N pi T simulation. Langmuir. 14:5255–5266. [Google Scholar]
- 44.De Vries, A. H., I. Chandrasekhar, W. F. van Gunsteren, and P. H. Hünenberger. 2005. Molecular dynamics simulations of phospholipid bilayers: influence of artificial periodicity, system, size and simulation time. J. Phys. Chem. B. 109:11643–11652. [DOI] [PubMed] [Google Scholar]
- 45.Venable, R. M., A. Skibinsky, and R. W. Pastor. 2006. Constant surface tension molecular dynamics simulations of lipid bilayers with trehalose. Mol. Simul. 32:849–855. [Google Scholar]
- 46.Gennis, R. B. 1989. Biomembranes: Molecular Structure and Function. Springer-Verlag, New York.
- 47.Möhwald, H. 1990. Phospholipid and phospholipid-protein monolayers at the air/water interface. Annu. Rev. Phys. Chem. 41:441–476. [DOI] [PubMed] [Google Scholar]
- 48.Ma, G., and H. C. Allen. 2006. DPPC Langmuir monolayer at the air-water interface: probing the tail and headgroups by vibrational sum frequency generation spectroscopy. Langmuir. 22:5341–5349. [DOI] [PubMed] [Google Scholar]
- 49.Hauser, H., I. Pascher, R. H. Pearson, and S. Sundell. 1981. Preferred conformation and molecular packing of phosphatidylethanolamine and phosphatidylcholine. Biochim. Biophys. Acta. 650:21–51. [DOI] [PubMed] [Google Scholar]
- 50.Dominguez, H., M. Smondyrev, and M. L. Berkowitz. 1999. Computer simulations of phosphatidylcholine monolayers at air/water and CCl4/water interfaces. J. Phys. Chem. B. 103:9582–9588. [Google Scholar]
- 51.Pasenkiewicz-Gierula, M., Y. Takaoka, H. Miyagawa, K. Kitamura, and A. Kusumi. 1999. Charge pairing of headgroups in phosphatidylcholine membranes: a molecular dynamics simulation study. Biophys. J. 76:1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lösche, M., E. Sackmann, and H. Möhwald. 1983. A fluorescence microscopic study concerning the phase-diagram of phospholipids. Ber. Bunsenges. Phys. Chem. 87:848–852. [Google Scholar]
- 53.Kaganer, V. M., H. Möhwald, and P. Dutta. 1999. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 71:779–819. [Google Scholar]
- 54.Nielsen, S. O., C. F. Lopez, P. B. Moore, J. C. Shelley, and M. L. Klein. 2003. Molecular dynamic investigations of lipid Langmuir monolayers using a coarse-grained model. J. Phys. Chem. B. 107:13911–13917. [Google Scholar]
- 55.Ahuja, R. C., and D. Möbius. 1992. Photophysical properties of a pyrene-labeled phospholipid in matrix monolayers at the gas/water interface. Langmuir. 8:1136–1144. [Google Scholar]
- 56.Bordi, F., F. D. Luca, C. Cametti, A. Naglieri, R. Misasi, and M. Sorice. 1999. Interactions of mono- and di-sialogangliosides with phospholipids in mixed monolayers at the air-water interface. Colloids Surf. B Biointerfaces. 13:135–142. [Google Scholar]
- 57.Borissevitch, G. P., M. Tabak, and O. N. Oliveira. 1996. Interaction of dipyridamole with lipids in mixed Langmuir monolayers. Biochim. Biophys. Acta. 1278:12–18. [DOI] [PubMed] [Google Scholar]
- 58.Dubreil, L., V. Vié, S. Beaufils, D. Marion, and A. Renault. 2003. Aggregation of puroindoline in phospholipid monolayers spread at the air-liquid interface. Biophys. J. 85:2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miñones Jr, J., J. M. Rodríguez Patino, O. Conde, C. Carrera, and R. Seoane. 2002. The effect of polar groups on structural characteristics of phospholipids monolayers spread at the air-water interface. Colloids Surf. A Physicochem. Eng. Aspect. 203:273–286. [Google Scholar]
- 60.Sández, M. I., A. Suárez, and A. Gil. 2002. Surface pressure-area isotherms and fluorescent behavior of phospholipids containing labeled pyrene. J. Colloid Interface Sci. 250:128–133. [DOI] [PubMed] [Google Scholar]
- 61.Williams, A. D., J. M. Wilkin, and R. A. Dluhy. 1995. An investigation of miscibility in monolayer films of phosphocholine-phosphoglycerol binary mixtures. Colloids Surf. A Physicochem. Eng. Aspect. 102:231–245. [Google Scholar]
- 62.Dynarowicz-Łątka, P., V. Rosilio, P. Boullanger, P. Fontaine, M. Goldmann, and A. Baszkin. 2005. Influence of a neoglycolipid and its PEO-lipid moiety on the organization of phospholipids monolayers. Langmuir. 21:11941–11948. [DOI] [PubMed] [Google Scholar]
- 63.Hunt, R. D., M. L. Mitchell, and R. A. Dluhy. 1989. The interfacial structure of phospholipids monolayer films: an infrared reflectance study. J. Mol. Struct. 214:93–109. [Google Scholar]
- 64.Rana, F. R., A. J. Mautone, and R. A. Dluhy. 1993. Surface chemistry of binary mixtures of phospholipids in monolayers. Infrared studies of surface composition at varying surface pressures in a pulmonary surfactant model system. Biochemistry. 32:3169–3177. [DOI] [PubMed] [Google Scholar]
- 65.Slotte, J. P., and P. Mattjus. 1995. Visualization of lateral phases in cholesterol and phosphatidylcholine monolayers at the air-water interface—a comparative study with two different reporter molecules. Biochim. Biophys. Acta. 1254:22–29. [DOI] [PubMed] [Google Scholar]
- 66.Taneva, S., T. McEachren, J. Stewart, and K. M. W. Keough. 1995. Pulmonary surfactant protein SP-A with phospholipids in spread monolayers at the air-water interface. Biochemistry. 34:10279–10289. [DOI] [PubMed] [Google Scholar]
- 67.Gladston, M., and D. O. Shah. 1967. Surface properties and hysteresis of dipalmitoyl lecithin in relation to the alveolar lining layer. Biochim. Biophys. Acta. 137:255–263. [DOI] [PubMed] [Google Scholar]
- 68.Kanintronkul, Y., T. Srikhirin, C. Angsuthanasombat, and T. Kerdcharoen. 2005. Insertion behavior of the Bacillus thuringiensis Cry4Ba insecticidal protein into lipid monolayers. Arch. Biochem. Biophys. 442:180–186. [DOI] [PubMed] [Google Scholar]
- 69.Lee, Y. L., J. Y. Lin, and C. H. Chang. 2006. Thermodynamic characteristics and Langmuir-Blodgett deposition behavior of mixed DPPA/DPPC monolayers at air/liquid interfaces. J. Colloid Interface Sci. 296:647–654. [DOI] [PubMed] [Google Scholar]
- 70.Nakahara, H., S. Nakamura, H. Kawasaki, and O. Shibata. 2005. Properties of two-component Langmuir monolayer of single chain perfluorinated carboxylic acids with dipalmitoylphosphatidylcholine (DPPC). Colloids Surf. B Biointerfaces. 41:285–298. [DOI] [PubMed] [Google Scholar]
- 71.Shen, Y., Y. Tang, A. Xie, J. Zhu, S. Li, and Y. Zhang. 2006. Studies on the behaviors of dipalmitoylphosphatidylcholine and bilirubin in mixed monolayer at the air/water interface. Appl. Surf. Sci. 252:5861–5867. [Google Scholar]
- 72.Tabak, S. A., R. H. Notter, J. S. Ultman, and S. M. Dinh. 1977. Relaxation effects in the surface pressure behavior of dipalmitoyl lecithin. J. Colloid Interface Sci. 60:117–125. [Google Scholar]
- 73.Maskarinec, S. A., J. Hannig, R. C. Lee, and K. Y. C. Lee. 2002. Direct observation of poloxamer 188 insertion into lipid monolayers. Biophys. J. 82:1453–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dumaual, A. C., L. J. Jenski, and W. Stillwell. 2000. Liquid crystalline/gel state phase separation in docosahexaenoic acid-containing bilayers and monolayers. Biochim. Biophys. Acta. 1463:395–406. [DOI] [PubMed] [Google Scholar]
- 75.Hoda, K., Y. Ikeda, H. Kawasaki, K. Yamada, R. Higuchi, and O. Shibata. 2006. Mode of interaction of ganglioside Langmuir monolayer originated from echinoderms: three binary systems of ganglioside/DPPC, ganglioside/DMPE, and ganglioside/cholesterol. Colloids Surf. B Biointerfaces. 52:57–75. [DOI] [PubMed] [Google Scholar]
- 76.Munden, J. W., and J. Swarbrick. 1973. Effect of spreading solvent on monolayer characteristics of dipalmitoyl lecithin. J. Colloid Interface Sci. 42:657. [Google Scholar]
- 77.Miñones Jr, J., J. Miñones, O. Conde, J. M. Rodriguez Patino, and P. Dynarowicz-Łątka. 2002. Mixed monolayers of amphotericin B-dipalmitoyl phosphatidyl choline: study of complex formation. Langmuir. 18:2817–2827. [Google Scholar]
- 78.Taneva, S. G., and K. M. W. Keough. 1995. Calcium ions and interactions of pulmonary surfactant proteins SP-B and SP-C with phospholipids in spread monolayers at the air/water interface. Biochim. Biophys. Acta. 1236:185–195. [DOI] [PubMed] [Google Scholar]
- 79.Hoda, K., H. Nakahara, S. Nakamura, S. Nagadome, G. Sugihara, N. Yoshino, and O. Shibata. 2006. Langmuir monolayer properties of the fluorinated-hydrogenated hybrid amphiphiles with dipalmitoylphosphatidylcholine (DPPC). Colloids Surf. B Biointerfaces. 47:165–175. [DOI] [PubMed] [Google Scholar]
- 80.Nakahara, H., S. Nakamura, S. Lee, G. Sugihara, and O. Shibata. 2005. Influence of a new amphiphilic peptide with phospholipids monolayers at the air-water interface. Colloids Surf. A Physicochem. Eng. Aspect. 270–271:52–60. [Google Scholar]
- 81.Nakahara, H., S. Nakamura, K. Nakamura, M. Inagaki, M. Aso, R. Higuchi, and O. Shibata. 2005. Cerebroside Langmuir monolayers originated from the echinoderms. I. Binary systems of cerebrosides and phospholipids. Colloids Surf. B Biointerfaces. 42:157–174. [DOI] [PubMed] [Google Scholar]
- 82.Tabak, S. A., and R. H. Notter. 1977. Modified technique for dynamic surface pressure and relaxation measurements at the air-water interface. Rev. Sci. Instrum. 48:1196–1201. [Google Scholar]
- 83.Matsumoto, M., Y. Tsujii, K. I. Nakamura, and T. Yoshimoto. 1996. A trough with radial compression for studies of monolayers and fabrication of Langmuir-Blodgett films. Thin Solid Films. 280:238–243. [Google Scholar]
- 84.Lehmler, H. J., M. Jay, and P. M. Bummer. 2000. Mixing of partially fluorinated carboxylic acids and the hydrocarbon analogs with dipalmitoylphosphatidylcholine at the air-water interface. Langmuir. 16:10161–10166. [DOI] [PubMed] [Google Scholar]
- 85.Shapovalov, V. L., E. A. Kotova, T. I. Rokitskaya, and Y. N. Antonenko. 1999. Effect of gramicidin A on the dipole potential on phospholipid membranes. Biophys. J. 77:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wiedmann, T. S., and K. R. Jordan. 1991. Interaction of cyclosporine A with dipalmitoylphosphatidylcholine at the air-water interface. Langmuir. 7:318–322. [Google Scholar]
- 87.Hass, H., W. Caetano, G. P. Borissevitch, M. Tabak, M. I. Mosquera Sanchez, O. N. Oliveira Jr, E. Scalas, and M. Goldmann. 2001. Interaction of dipyridamole with phospholipids monolayers at the air-water interface: surface pressure and grazing incidence x-ray diffraction studies. Chem. Phys. Lett. 335:510–516. [Google Scholar]
- 88.Wüstneck, N., R. Wüstneck, V. B. Fainerman, R. Miller, and U. Pison. 2001. Interfacial behavior and mechanical properties of spread lung surfactant protein/lipid layers. Colloids Surf. B Biointerfaces. 21:191–205. [DOI] [PubMed] [Google Scholar]
- 89.Flach, C. R., A. Gericke, K. M. W. Keough, and R. Mendelsohn. 1999. Palmitoylation of lung surfactant protein SP-C alters surface thermodynamics, but not protein secondary structure or orientation in 1,2-dipalmitoylphosphatidylcholine Langmuir films. Biochim. Biophys. Acta. 1416:11–20. [DOI] [PubMed] [Google Scholar]
- 90.Piknova, B., W. R. Schief, V. Vogel, B. M. Discher, and S. B. Hall. 2001. Discrepancy between phase behavior of lung surfactant phospholipids and the classical model of surfactant function. Biophys. J. 81:2172–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lance, M. R., C. Washington, and S. S. Davis. 1996. Evidence for the formation of amphotericin B-phospholipid complexes in Langmuir monolayers. Pharmacol. Res. 13:1008–1014. [DOI] [PubMed] [Google Scholar]
- 92.Ronzon, F., B. Desbat, J. P. Chauvet, and B. Roux. 2002. Behavior of a GPI-anchored protein in phospholipid monolayers at the air-water interface. Biochim. Biophys. Acta. 1560:1–13. [DOI] [PubMed] [Google Scholar]
- 93.Yu, H., and S. W. Hui. 1992. Merocyanine 540 as a probe to monitor the molecular packing of phosphatidylcholine: a monolayer epifluorescence microscopy and spectroscopy study. Biochim. Biophys. Acta. 1107:245–254. [DOI] [PubMed] [Google Scholar]
- 94.Cavalli, A., G. Borissevitch, M. Tabak, and O. N. Oliveira, Jr. 1996. Interaction of dibucaine with lipids in Langmuir monolayers. Thin Solid Films. 284–285:731–734. [Google Scholar]
- 95.Györvary, E., W. M. Albers, and J. Peltonen. 1999. Miscibility in binary monolayers of phospholipids and linker lipid. Langmuir. 15:2516–2524. [Google Scholar]
- 96.Chen, K. B., C. H. Chang, Y. M. Yang, and J. R. Maa. 2000. On the interaction of dipalmitoyl phosphatidylcholine with normal long-chain alcohols in a mixed monolayer: a thermodynamic study. Colloids Surf. A Physicochem. Eng. Aspect. 170:199–208. [Google Scholar]
- 97.Chou, T. H., and C. H. Chang. 2000. Thermodynamic behavior and relaxation processes of mixed DPPC/cholesterol monolayers at the air/water interface. Colloids Surf. B Biointerfaces. 17:71–79. [Google Scholar]
- 98.Zhao, L., and S. S. Feng. 2006. Effects of cholesterol component on molecular interactions between paclitaxel and phospholipids within the lipid monolayer at the air-water interface. J. Colloid Interface Sci. 300:314–326. [DOI] [PubMed] [Google Scholar]
- 99.Lehmler, H. J., and P. M. Bummer. 2005. Interaction of partially fluorinated long-chain nicotinate with dipalmitoylphosphatidylcholine. J. Lipid Res. 46:2415–2422. [DOI] [PubMed] [Google Scholar]
- 100.Nakahara, H., S. Nakamura, T. Hiranita, H. Kawasaki, S. Lee, G. Sugihara, and O. Shibata. 2006. Mode of interaction of amphiphilic α-helical peptide with phosphatidylcholines at the air-water interface. Langmuir. 22:1182–1192. [DOI] [PubMed] [Google Scholar]
- 101.Li, J. B., J. Krägel, A. V. Makievski, V. B. Fainermann, R. Miller, and H. Möhwald. 1998. A study of mixed phospholipid/β-casein monolayers at the water/air surface. Colloids Surf. A Physicochem. Eng. Aspect. 142:355–360. [Google Scholar]
- 102.Ma, J., S. Koppenol, H. Yu, and G. Zografi. 1998. Effects of cationic and hydrophobic peptide, KL4, on model lung surfactant lipid monolayers. Biophys. J. 74:1899–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vollhardt, D., V. B. Fainerman, and S. Siegel. 2000. Thermodynamic and textural characterization of DPPG phospholipids monolayers. J. Phys. Chem. B. 104:4115–4121. [Google Scholar]
- 104.Roche, Y., P. Peretti, and S. Bernard. 2006. Influence of the chain length of ubiquinones on their interaction with DPPC in mixed monolayers. Biochim. Biophys. Acta. 1758:468–478. [DOI] [PubMed] [Google Scholar]
- 105.Neville, F., M. Cahuzac, O. Konovalov, Y. Ishitsuka, K. Y. C. Lee, I. Kuzmenko, G. M. Kale, and D. Gidalevitz. 2006. Lipid headgroup discrimination by antimicrobial peptide LL-37: insight into mechanism of action. Biophys. J. 90:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee, K. Y. C., A. Gopal, A. von Nahmen, J. A. Zasadzinski, J. Majewski, G. S. Smith, P. B. Howes, and K. Kjaer. 2002. Influence of palmitic acid and hexadecanol on the phase transition temperature and molecular packing of dipalmitoylphosphatidyl-choline monolayers at the air-water interface. J. Chem. Phys. 116:774–783. [Google Scholar]
- 107.Nakahara, H., S. Lee, G. Sugihara, and O. Shibata. 2006. Mode of interaction of hydrophobic amphiphilic α-helical peptide/dipalmitoylphosphatidylcholine with phosphatidylglycerol or palmitic acid at the air-water interface. Langmuir. 22:5792–5803. [DOI] [PubMed] [Google Scholar]
- 108.Hiranita, T., S. Nakamura, M. Kawachi, H. M. Courrier, T. F. Vandamme, M. P. Krafft, and O. Shibata. 2003. Miscibility behavior of dipalmitoylphosphatidylcholine with a single partially fluorinated amphiphile in Langmuir monolayers. J. Colloid Interface Sci. 265:83–92. [DOI] [PubMed] [Google Scholar]
- 109.Maruta, T., K. Hoda, M. Inagaki, R. Higuchi, and O. Shibata. 2005. Langmuir monolayers of cerebroside originated from Linckia laevigata: Binary systems of cerebrosides and phospholipids. Colloids Surf. B Biointerfaces. 44:123–142. [DOI] [PubMed] [Google Scholar]
- 110.Fang, N., and V. Chan. 2003. Chitosan-induced restructuration of a mica-supported phospholipids bilayer: an atomic force microscopy study. Biomacromolecules. 4:1596–1604. [DOI] [PubMed] [Google Scholar]
- 111.Ito, H., T. H. Morton, and V. Vodyanoy. 1989. Small odorant molecules affect steady state properties of monolayers. Thin Solid Films. 180:1–13. [Google Scholar]
- 112.Pathirana, S., W. C. Neely, and V. Vodyanoy. 1998. Condensing and expanding effects of the odorants (+)- and (−)-cavone on phospholipids monolayers. Langmuir. 14:679–682. [Google Scholar]
- 113.Miñones, J., Jr., P. Dynarowicz-Łątka, O. Conde, J. Miñones, E. Iribarnegaray, and M. Casas. 2003. Interactions of amphotericin B with saturated and unsaturated phosphatidylcholines at the air/water interface. Colloids Surf. B Biointerfaces. 29:205–215. [Google Scholar]
- 114.Miñones Conde, J., J. Miñones Trillo, and J. M. Rodríguez Patino. 2006. Structural characteristics of dipalmitoylphosphatidylcholine and hydrolysates from sunflower protein isolate mixed monolayers spread at the air-water interface. J. Phys. Chem. B. 110:11582–11591. [DOI] [PubMed] [Google Scholar]
- 115.Eeman, M., A. Berquand, Y. F. Dufrêne, M. Paquot, S. Dufour, and M. Deleu. 2006. Penetration of surfactin into phospholipid monolayers: nanoscale interfacial organization. Langmuir. 22:11337–11345. [DOI] [PubMed] [Google Scholar]
- 116.Andrade, C. A. S., A. Baszkin, N. S. Santos-Magalhães, L. C. B. B. Coelho, and C. P. de Melo. 2005. Mixed monolayers of Bauhinia monandra and concanavalin A lectins with phospholipids, part II. J. Colloid Interface Sci. 289:379–385. [DOI] [PubMed] [Google Scholar]
- 117.Maloney, K. M., and D. W. Grainger. 1993. Phase separated anionic domains in tertiary mixed lipid monolayers at the air-water interface. Chem. Phys. Lipids. 65:31–42. [DOI] [PubMed] [Google Scholar]
- 118.Davies, R. J., and M. N. Jones. 1992. The thermal behavior of phosphatidylcholine-glycophorin monolayers in relation to monolayer and bilayer internal pressure. Biochim. Biophys. Acta. 1103:8–12. [DOI] [PubMed] [Google Scholar]
- 119.Gonçalves da Silva, A. M., and R. I. S. Romão. 2005. Mixed monolayers involving DPPC, DODAB and oleic acid and their interaction with nicotinic acid at the air-water interface. Chem. Phys. Lipids. 137:62–76. [DOI] [PubMed] [Google Scholar]
- 120.Ayudhya, C. I. N., V. Prachayasittikul, and H. J. Galla. 2004. Binding of chimeric metal-binding green fluorescent protein to lipid monolayer. Eur. Biophys. J. 33:522–534. [DOI] [PubMed] [Google Scholar]
- 121.Kubo, I., S. Adachi, H. Maeda, and A. Seki. 2001. Phosphatidylcholine monolayers observed with Brewster angle microscopy and π-A isotherms. Thin Solid Films. 393:80–85. [Google Scholar]
- 122.Li, J., R. Miller, and H. Möhwald. 1996. Characterization of phospholipids layers at liquid interfaces. 2. Comparison of isotherms of insoluble and soluble films of phospholipids at different fluid/water interfaces. Colloids Surf. A Physicochem. Eng. Aspect. 114:123–130. [Google Scholar]
- 123.Kozarac, Z., B. Cosovic, D. Möbius, and M. Dobric. 2000. Interaction of polysaccharides with lipid monolayers. J. Colloid Interface Sci. 226:210–217. [Google Scholar]
- 124.Krasteva, N., D. Vollhardt, G. Brezesinski, and H. Möhwald. 2001. Effect of sugars and dimethyl sulfoxide on the structure and phase behavior of DPPC monolayers. Langmuir. 17:1209–1214. [Google Scholar]
- 125.Dahmen-Levison, U., G. Brezesinski, and H. Möhwald. 1998. Specific adsorption of PLA2 at monolayers. Thin Solid Films. 327:616–620. [Google Scholar]
- 126.Wege, H. A., J. A. Holgado-Terriza, M. J. Gálvez-Ruiz, and M. A. Cabrerizo-Vílchez. 1999. Development of a new Langmuir-type pendant-drop film balance. Colloids Surf. B Biointerfaces. 12:339–349. [Google Scholar]
- 127.Morgan, H., D. M. Taylor, and O. N. Oliveira, Jr. 1991. Proton transport at the monolayer-water interface. Biochim. Biophys. Acta. 1062:149–156. [DOI] [PubMed] [Google Scholar]
- 128.Ohe, C., Y. Ida, S. Matsumoto, T. Sasaki, Y. Goto, M. Noi, T. Tsurumaru, and K. Itoh. 2004. Investigations of polymyxin B-phospholipid interactions by vibrational sum frequency generation spectroscopy. J. Phys. Chem. B. 108:18081–18087. [Google Scholar]
- 129.Hildago, A. A., W. Caetano, M. Tabak, and O. N. Olivera, Jr. 2004. Interaction of two phenothiazine derivatives with phospholipid monolayers. Biophys. Chem. 109:85–104. [DOI] [PubMed] [Google Scholar]
- 130.Gong, K., S. S. Feng, M. L. Go, and P. H. Soew. 2002. Effects of pH on the stability and compressibility of DPPC/cholesterol monolayers at the air-water interface. Colloids Surf. A Physicochem. Eng. Aspect. 207:113–125. [Google Scholar]
- 131.Feng, S. S., K. Gong, and J. Chew. 2002. Molecular interactions between a lipid and an antineoplastic drug paclitaxel (taxol) within the lipid monolayer at the air-water interface. Langmuir. 18:4061–4070. [Google Scholar]
- 132.Koppenol, S., H. Yu, and G. Zografi. 1997. Mixing of saturated and unsaturated phosphatidylcholines and phosphatidylglycerols in monolayers at the air/water interface. J. Colloid Interface Sci. 189:158–166. [Google Scholar]
- 133.Mansour, H., D. S. Wang, C. S. Chen, and G. Zografi. 2001. Comparison of bilayer and monolayer properties of phospholipids systems containing dipalmitoylphosphatidylglycerol and dipalmitoylphosphatidylinositol. Langmuir. 17:6622–6632. [Google Scholar]
- 134.Lewis, D., and J. Hadgraft. 1990. Mixed monolayers of dipalmitoylphosphatidylcholine with Azone or oleic acid at the air-water interface. Int. J. Pharm. 65:211–218. [Google Scholar]
- 135.Rolland, J. P., C. Santaella, and P. Vierling. 1996. Molecular packing of highly fluorinated phosphatidylcholines in monolayers. Chem. Phys. Lipids. 79:71–77. [DOI] [PubMed] [Google Scholar]
- 136.Wójtowicz, K., W. I. Gruszecki, M. Walicka, and J. Barwicz. 1998. Effect of amphotericin B on dipalmitoylphosphatidylcholine membranes: calorimetry, ultrasound absorption and monolayer technique studies. Biochim. Biophys. Acta. 1373:220–226. [DOI] [PubMed] [Google Scholar]
- 137.Taneva, S. G., and K. M. W. Keough. 1994. Dynamic surface properties of pulmonary surfactant proteins SP-B and SP-C and their mixtures with dipalmitoylphosphatidylcholine. Biochemistry. 33:14660–14670. [DOI] [PubMed] [Google Scholar]
- 138.Hildebran, J. N., J. Goerke, and J. A. Clements. 1979. Pulmonary surface film stability and composition. J. Appl. Physiol. Respir. Environ. Exercise Physiol. 47:604–611. [DOI] [PubMed] [Google Scholar]
- 139.Aston, M. S., T. M. Herrington, and T. F. Tadros. 1995. Interfacial properties of mixtures of lecithin with a block copolymer surfactant at the water/air and water/oil interface. Colloid Polym. Sci. 273:444–452. [Google Scholar]
- 140.Shapovalov, V. L. 1998. Interaction of DPPC monolayer at air-water interface with hydrophobic ions. Thin Solid Films. 327:599–602. [Google Scholar]
- 141.Zaitsev, S. Y., V. P. Vereschetin, V. P. Zubov, W. Zeiss, and D. Möbius. 1996. Ionic selectivity of valinomycin in the dipalmitoylphosphatidylcholine monolayers. Thin Solid Films. 285:667–670. [Google Scholar]
- 142.Wiegart, L., B. Struth, M. Tolan, and P. Terech. 2005. Thermodynamic and structural properties of phospholipid Langmuir monolayers on hydrosol surfaces. Langmuir. 21:7349–7357. [DOI] [PubMed] [Google Scholar]
- 143.Liu, H., J. G. Turcotte, and R. H. Notter. 1995. Thermotropic behavior of structurally-related phospholipids and phosphonolipid analogs of lung surfactant glycerophospholipids. Langmuir. 11:101–107. [Google Scholar]
- 144.Ross, M., C. Steinem, H. J. Galla, and A. Janshoff. 2001. Visualization of chemical and physical properties of calcium-induced domains in DPPC/DPPS Langmuir-Blodgett layers. Langmuir. 17:2437–2445. [Google Scholar]
- 145.Watkins, J. C. 1968. The surface properties of pure phospholipids in relation to those of lung extracts. Biochim. Biophys. Acta. 152:293–306. [DOI] [PubMed] [Google Scholar]
- 146.Clements, J. A., and D. F. Tierney. 1964. Handbook of Physiology: Respiration, Vol. 2, Sec. 3. American Physiological Society, Washington, DC.
- 147.Bi, X., C. R. Flach, J. Pérez-Gil, I. Plasencia, D. Andreu, E. Oliveira, and R. Mendelsohn. 2002. Secondary structure and lipid interactions of the N-terminal segment of pulmonary surfactant SP-C in Langmuir films: IR reflection-adsorption spectroscopy and surface pressure studies. Biochemistry. 41:8385–8395. [DOI] [PubMed] [Google Scholar]
- 148.Klopfer, K. J., and T. K. Vanderlick. 1996. Isotherms of dipalmitoylphosphatidylcholine (DPPC) monolayers: features revealed and features obscured. J. Colloid Interface Sci. 182:220–229. [Google Scholar]
- 149.Aroti, A., E. Leontidis, E. Maltseva, and G. Brezesinski. 2004. Effects of Hofmeister anions on DPPC Langmuir monolayers at the air-water interface. J. Phys. Chem. B. 108:15238–15245. [Google Scholar]
- 150.Wüstneck, N., R. Wüstneck, V. B. Fainerman, U. Pison, and R. Miller. 2000. Influence of over-compressed spread L-dipalmitoyl phosphatidylcholine films and the influence of solvent vapor in the gas phase on Π/A isotherms measured by using the captive bubble technique. Colloids Surf. A Physicochem. Eng. Aspect. 164:267–278. [Google Scholar]
- 151.Schürch, S., H. Bachofen, J. Goerke, and F. Possmayer. 1989. Surface properties of rat pulmonary surfactant studied with the captive bubble method: adsorption, hysteresis and stability. J. Appl. Physiol. 67:2389–2396.2606846 [Google Scholar]
- 152.Cruz, A., L. A. Worthman, A. G. Serrano, C. Casals, K. M. W. Keough, and J. Pérez-Gil. 2000. Microstructure and dynamic surface properties of surfactant protein SP-B/dipalmitoylphosphatidylcholine interfacial films spread from lipid-protein bilayers. Eur. Biophys. J. 29:204–213. [DOI] [PubMed] [Google Scholar]
- 153.Notter, R. H., S. A. Tabak, S. Holcomb, and R. D. Mavis. 1979. Postcollapse dynamic surface pressure relaxation in binary surface films containing dipalmitoyl phosphatidylcholine. J. Colloid Interface Sci. 74:370–377. [Google Scholar]
- 154.Bredlow, A., H. J. Galla, and L. D. Bergelson. 1992. Influence of fluorescent lipid probes on the packing of their environment. A monolayer study. Chem. Phys. Lipids. 62:293–301. [Google Scholar]
- 155.Crane, J. M., and S. B. Hall. 2001. Rapid compression transforms interfacial monolayers of pulmonary surfactant. Biophys. J. 80:1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Broniec, A., A. U. Gjerde, A. B. Ølmheim, and H. Holmsen. 2007. Trifluperazine causes a disturbance in glycerophospholipid monolayers containing phosphatidylserine (PS): effects of pH, acyl unsaturation, and proportion of PS. Langmuir. 23:694–699. [DOI] [PubMed] [Google Scholar]
- 157.Su, Y., L. Qingzhong, L. Chen, and Z. Yu. 2007. Condensation effect of cholesterol, stigmasterol, and sitosterol on dipalmitoylphosphatidylcholine in molecular monolayers. Colloids Surf. A Physicochem. Eng. Aspect. 293:123–129. [Google Scholar]
- 158.Jyoti, A., R. M. Prokop, J. Li, D. Vollhardt, D. Y. Kwok, R. Miller, H. Möhwald, and A. W. Neumann. 1996. An investigation of the compression rate dependence on the surface pressure-surface area isotherm for a dipalmitoyl phosphatidylcholine monolayer at the air/water interface. Colloids Surf. A Physicochem. Eng. Aspect. 116:173–180. [Google Scholar]
- 159.Notter, R. H., S. A. Tabak, and R. D. Mavis. 1980. Surface properties of binary mixtures of some pulmonary surfactant components. J. Lipid Res. 21:10–22. [PubMed] [Google Scholar]
- 160.Wüstneck, R., N. Wüstneck, D. O. Grigoriev, U. Pison, and R. Miller. 1999. Stress relaxation behavior of dipalmitoyl phosphatidylcholine monolayers spread on the surface of a pendant. Colloids Surf. B Biointerfaces. 15:275–288. [Google Scholar]
- 161.Wüstneck, R., N. Wüstneck, and U. Pison. 2003. Surface dilatational behavior of pulmonary surfactant components spread on the surface of a captive bubble. 3. Dipalmitoyl phosphatidylcholine, surfactant protein C, and surfactant protein B. Langmuir. 19:7521–7527. [Google Scholar]
- 162.Jyoti, A., R. M. Prokop, and A. W. Neumann. 1997. Manifestation of the liquid-expanded/liquid-condensed phase transition of a dipalmitoylphosphatidylcholine monolayer at the air-water interface. Colloids Surf. B Biointerfaces. 8:115–124. [Google Scholar]
- 163.Li, J. B., R. Miller, D. Vollhardt, G. Weidemann, and H. Möhwald. 1996. Isotherms of phospholipid monolayers measured by the pendant drop technique. Colloid Polym. Sci. 274:995–999. [Google Scholar]
- 164.Munden, J. W., and J. Swarbrick. 1973. Time dependent surface behavior of dipalmitoyl lecithin and lung alveolar surfactant monolayers. Biochim. Biophys. Acta. 291:344–350. [DOI] [PubMed] [Google Scholar]
- 165.Kodama, M., O. Shibata, S. Nakamura, S. Lee, and G. Sugihara. 2004. A monolayer study on three binary mixed systems of dipalmitoyl phosphatidylcholine with cholesterol, cholestanol, and stigmasterol. Colloids Surf. B Biointerfaces. 33:211–226. [Google Scholar]
- 166.Vitovic, P., D. P. Nikolelis, and T. Hianik. 2006. Study of calix[4]resorcinarene-dopamine complexation in mixed phospholipid monolayers formed at the air-water interface. Biochim. Biophys. Acta. 1758:1852–1861. [DOI] [PubMed] [Google Scholar]
- 167.Hoda, K., H. Kawasaki, N. Yoshino, C. H. Chang, Y. Morikawa, G. Sugihara, and O. Shibata. 2006. Mode of interaction of two fluorinated-hydrogenated hybrid amphiphiles with dipalmitoylphosphatidylcholine (DPPC) at the air-water interface. Colloids Surf. B Biointerfaces. 53:37–50. [DOI] [PubMed] [Google Scholar]
- 168.Longo, M. L., A. M. Bisagno, J. A. N. Zasadzinski, R. Bruni, and A. J. Waring. 1993. A function of lung surfactant protein SP-B. Science. 261:453–456. [DOI] [PubMed] [Google Scholar]
- 169.Robertson, B., and L. M. G. van Golde. 1984. Postscript: Surfactant research; current concepts and perspectives for the future. In Pulmonary Surfactant. B. Robertson, L. M. G. van Golde, and J. J. Batenburg, editors. Elsevier Science Publishers, Amsterdam, The Netherlands.
- 170.Nag, K., C. Boland, N. Rich, and K. M. L. Keough. 1991. Epifluorescence microscopic observation of monolayers of dipalmitoylphosphatidylcholine: dependence of domain size on compression rates. Biochim. Biophys. Acta. 1068:157–160. [DOI] [PubMed] [Google Scholar]
- 171.Shah, D. O., and J. H. Schulman 1965. Binding of metal ions to monolayers of lecithins plasmalogen cardiolipin and diacetyl phosphate. J. Lipid. Res. 6:341–349. [PubMed] [Google Scholar]
- 172.Liu, H., R. Z. Lu, J. G. Turcotte, and R. H. Notter. 1994. Dynamic interfacial properties of surface-excess films of phospholipids and phosphonolipid analogs. 1. Effects of pH. J. Colloid Interface Sci. 167:378–390. [Google Scholar]
- 173.Nagle, J. F. 1976. Theory of lipid monolayer and bilayer phase transitions: effect of headgroup interactions. J. Membr. Biol. 27:233–250. [DOI] [PubMed] [Google Scholar]
- 174.Notter, R. H. 2000. Lung Surfactants: Basic Science and Clinical Applications. Marcel Dekker, New York.
- 175.van Golde, L. M. G., J. J. Batenburg, and B. Robertson. 1994. The pulmonary surfactant system. News Physiol. Sci. 9:13–20. [DOI] [PubMed] [Google Scholar]
- 176.Barrow, R. E., and B. A. Hills. 1979. Critical assessment of the Wilhelmy method in studying lung surfactants. J. Physiol. 295:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Enhorning, G. 1977. Pulsating bubble technique for evaluating pulmonary surfactant. J. Appl. Physiol. Respir. Environ. Exercise Physiol. 43:198–203. [DOI] [PubMed] [Google Scholar]
- 178.Schürch, S., H. Bachofen, J. Goerke, and F. Green. 1992. Surface properties of rat pulmonary surfactant studied with the captive bubble method: adsorption, hysteresis and stability. Biochim. Biophys. Acta. 1103:127–136. [DOI] [PubMed] [Google Scholar]
- 179.Goerke, J., and J. Gonzales. 1981. Temperature dependence of dipalmitoyl phosphatidylcholine monolayer stability. Appl. Physiol. Respir. Environ. Exercise Physiol. 51:1108–1114. [DOI] [PubMed] [Google Scholar]
- 180.Putz, G., J. Goerke, S. Schürch, and J. A. Clements. 1994. Evaluation of pressure-driven captive bubble surfactometer. J. Appl. Physiol. 76:1417–1424. [DOI] [PubMed] [Google Scholar]
- 181.Putz, G., J. Goerke, H. W. Taeusch, and J. A. Clements. 1994. Comparison of captive and pulsating bubble surfactometers with use of lung surfactants. J. Appl. Physiol. 76:1425–1431. [DOI] [PubMed] [Google Scholar]
- 182.Notter, R. H., and P. E. Morrow. 1975. Pulmonary surfactant: a surface chemistry viewpoint. Ann. Biomed. Eng. 3:119–159. [DOI] [PubMed] [Google Scholar]
- 183.Anézo, C., A. H. de Vries, H. D. Höltje, D. P. Tieleman, and S. J. Marrink. 2003. Methodological issues in lipid bilayer simulations. J. Phys. Chem. B. 107:9424–9433. [Google Scholar]
- 184.Lindahl, E., and O. Edholm. 2000. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 79:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Klauda, J. B., B. R. Brooks, and R. W. Pastor. 2006. Dynamical motions of lipids and a finite size effect in simulations of bilayers. J. Chem. Phys. 125:144710–144717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wohlert, J., and O. Edholm. 2004. The range and shielding of dipole-dipole interactions in phospholipid bilayers. Biophys. J. 87:2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Castro-Román, F., R. W. Benz, S. H. White, and D. J. Tobias. 2006. Investigation of finite system-size effects in molecular dynamics simulations of lipid bilayers. J. Phys. Chem. B. 110:24157–24164. [DOI] [PubMed] [Google Scholar]
- 188.Kharakoz, D. P., and E. A. Shlyapnikova. 2000. Thermodynamics and kinetics of the early steps of solid-state nucleation in the fluid lipid bilayer. J. Phys. Chem. B. 104:10368–10378. [Google Scholar]
- 189.Erbes, J., A. Gabke, G. Rapp, and R. Winter. 2000. Kinetics of phase transformations between lyotropic mesophases of different topology: a time-resolved synchrotron x-ray diffraction study using the pressure-jump relaxation technique. Phys. Chem. Chem. Phys. 2:151–162. [Google Scholar]
- 190.Schürch, S., F. Possmayer, S. Cheng, and A. M. Cockshutt. 1992. Pulmonary SP-A enhances adsorption and appears to induce surface sorting of lipid extract surfactant. Am. J. Physiol. 263:L210–L218. [DOI] [PubMed] [Google Scholar]
- 191.Tummers, B. 2005. Data Thief III, Ver.1. Http://www.datathief.org/.