

Abstract
A new class of 2-oxo-tetrahydro-1,8-naphthyridine-based protein farnesyltransferase inhibitors were synthesized and found to inhibit protein farnesyl transferase from the malaria parasite with potencies in the low nanomolar range. The compounds were much less potent on mammalian protein prenyltransferases. Two of the compounds block the growth of malaria growth in culture with potencies in the sub-micromolar range. Some of the compounds also were found to be much more metabolically stable than previously described tetrahydroquinoline-based protein farnesyltransferase inhibitors.
Keywords: malaria, P. falciparum, anti-malarials, protein farnesyltransferase, drug discovery
Malaria remains one of the key parasitic diseases present today with more than 400 million acute illnesses and at least 2–3 million deaths annually throughout the tropical and subtropical regions of the world. The malaria parasite is able to become resistant to chemotherapeutic agents. For example, in many places there is a major problem of resistance to chloroquine, a 4-aminoquinoline that has been the mainstay of treatment for malaria for many years. Hence, there is urgent need for the development of new drugs against new molecular targets 1.
Protein prenylation has been vigorously studied over the past ~15 years because it is found on several signaling proteins (including heterotrimeric G proteins) that connect cell surface receptors to intracellular effectors, and also on Ras proteins, one of the most common oncoproteins found in human tumors. Protein farnesyltransferase (PFT) transfers the farnesyl group from farnesyl diphosphate to the SH of the cysteine near the C-terminus of proteins such as Ras. PFT inhibitors (PFTIs) have been extensively developed as anti-cancer agents because of their ability to block tumor growth in experimental animals. We have been pursuing PFTIs as anti-malarial and anti-trypanosome agents because these compounds are much more toxic to these parasites than to mammalian cells, and there is a large number of lead compounds from which to launch a anti-parasite drug discovery program (piggy-back medicinal chemistry) 2–4. We have developed tetrahydroquinoline-based PFTIs as anti-malarials with low nanomolar potency on parasite PFT and on parasites cultured in vitro 3–7. One of our lead compounds, 1 (Figure 1), kills Plasmodium falciparum growth in vitro with an ED50 of 16 nM, shows high oral bioavailability and is able to cure rats infected with rodent malaria 6.
Figure 1.

Compound 1 is a tetrahydroquinoline-based PFTI that is metabolized by cytochrome P450 to give compound 2. Compound 3 shows the general structure of the 2-oxo-tetrahydro-1,8-naphthyridine-based PFTIs prepared in the current study.
Unfortunately, high doses of 1, 50 mg/kg, were required for cures in rodents because of rapid compound clearance. In vitro microsome metabolism studies suggest that the major culprit is cytochrome P450-catalyzed loss of the imidazole-containing side chain (which binds to the active site Zn2+ of PFT) leading to 2 (Figure 1). This reaction starts either with P450-catalyzed hydrogen atom abstraction from the CH2 group attached to N1 of the tetrahydroquinoline ring to give a C-centered radical or enzyme-catalyzed oxidation of N1 to give the N-centered radical cation. Regardless of the mechanism, we envisioned that placement of an oxo group at the 2-position of the tetrahydroquinoline ring and a N in place of C-8 would reduce P450-catalyzed radical formation due to a rise in the oxidation potential of the N1 lone pair electrons (due to involvement of the lone pair in resonance with the carbonyl and the pyridine N). Thus, we set out to prepare 2-oxo-tetrahydro-1,8-naphthyridine-based PFTIs exemplified by 3 (Figure 1). Consideration of the x-ray structure of tetrahydroquinoline PFTIs bound to mammalian PFT and a homology model of the active site of malarial PFT 4, 6, it appears that addition of the 2-oxo and 8-aza groups to the tetrahydroquinoline scaffold would be tolerated.
Compounds were prepared following the synthetic sequence illustrated in Scheme 1. Ethyl chloronicotinate 5 was prepared from 2-chloro nicotinic acid 4, and installation of the imidazole was accomplished by nucleophilic substitution conditions to give 7. This was followed by reduction of the ester group and subsequent oxidation to yield 8. The Wittig olefination of compound 8 with Boc protected phosphonoacetate 9 gave 10 followed by catalytic hydrogenation over palladium in methanol to give 11. Subsequent bromination with Br2 in acetic acid afforded 6-bromo analogue 12, which was converted to the corresponding 6-cyano derivative 13 by treatment with zinc cyanide and tetrakis(triphenylphosphine)palladium in dimethylformamide. Removal of Boc group with trifluoroacetic acid in dichloromethane afforded the key intermediate 3-amino-6-cyano-2-oxo-tetrahydro-1,8-naphthyridine 14. Completion of target molecules was accomplished following a 2-step sequence of reductive amination and sulfonamide formation. If sulfonation was carried out first followed by alkylation of the sulfonamide N with R2Br, the observed product was the enamine with a double bond in the 3,4-position of the lactam ring (due to elimination of the sulfinate). Full synthetic details are available as Supplementary Data.
Scheme 1.

Reagents, conditions and yields: (a) SOCl2, EtOH, 80%; (b) Et3N, DMF, 60%; (c) LiAlH4, THF, 70%; (d) MnO2, CH2Cl2, 75% ; (e) Tetramethylguanidine, CH2Cl2, 65%; (f) H2/Pd-C, CH3OH, 50%; (g) Br2, CH3COOH, 65%; (h) Zn(CN)2, Pd(PPh3)4, 35%; (i) 20% CF3COOH, CH2Cl2, 100%; (j) R2-CHO, NaCNBH3, CH3OH, 55–60%; (k) R1-SO2Cl, DIPEA, CH2Cl2, 15–20%.
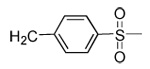
Our previous structure-activity data on THQ-based inhibitors of malarial PFT led to the discovery of compounds with R1 = N-methyl-4-imidazolyl or 2-pyridyl (i.e. 1) as being potent inhibitors of malarial PFT 7. In table 1, we report anti-malarial results obtained with 2-oxo-tetrahydro-1,8-naphthyridine-based PFTIs with R1 = N-methyl-4-imidazolyl or 2-pyridyl and with variation of the R2 group. Compounds with R1 = N-methyl-4-imidazolyl conferred the best in vitro activity against Plasmodium falciparum PFT (18 and 20 showed 98% and 95% inhibition at 50 nM, respectively) compared to compounds with R1 = 2-pyridyl (21 and 19 showed 88% and 48% inhibition at 50 nM, respectively). We also tested the compounds for their ability to block the growth of Plasmodium falciparum in human red blood cell cultures. Values of ED50, the concentration of compound that reduces parasite growth by 50%, are listed in Table 1. Two malarial strains were studied, 3D7, which is chloroquine resistant and K1, which is chloroquine sensitive. Compounds 18, 20 and 21 showed good potency, with values of ED50 in the 175–420 nM range (Table 1). These compounds are also the most potent in the series studied on inhibiting Plasmodium falciparum PFT in vitro. Testing on malarial PFT and on parasites was carried out as described 6.
Table 1.
Structure, anti-malarial activity and microsomal metabolism of 2-oxo-tetrahydro-1,8-naphthyridine-based PFTIs
| Compd. | X | R1 | R2 | Malaria PFT enzyme % inhibition at the indicated inhibitor concentration (nM) | ED50 for inhibition of parasite growth in vitro (nM)1 | Microsome metabolism half time (min)2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 500 | 50 | 5 | 0.5 | 3D7 | K1 | Naph | THQ | ||||
| 16 | Br | 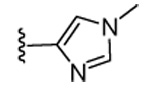 |
H | 74 | 28 | 0 | 0 | >5000 | >5000 | ND | ND |
| 17 | CN | 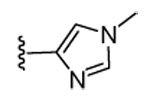 |
H | 94 | 66 | 32 | 15 | >5000 | >5000 | ND | ND |
| 18 | CN | 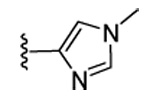 |
 |
99 | 98 | 80 | 27 | 420 | 300 | 52 | 15.6 |
| 19 | CN | 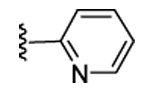 |
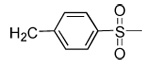 |
91 | 48 | 3 | 0 | 3100 | >5000 | 15 | ND |
| 20 | CN | 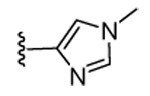 |
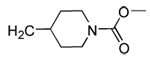 |
98 | 95 | 71 | 16 | 350 | 175 | >120 | 5.4 |
| 21 | CN | 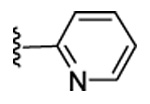 |
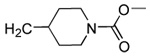 |
97 | 88 | 33 | 0 | 320 | 310 | ND | 3.8 |
ED50 is the concentration of compound that 50% inhibits the growth of parasites (chloroquine sensitive strain 3D7 or chloroquine resistant strain K1) in red blood cell cultures (measured according to ref. 6).
Given is the half-time for loss of parent compound when incubated with mouse liver microsomes according to the procedure given in ref. 6. Compounds tested are: 1) Column labeled Naph, the 2-oxo-tetrahydro-1,8-naphthyridines shown in the table; 2) Column labeled THQ, the corresponding tetrahydroquinoline-based PFT (analogs of 1) with the same R1 and R2 groups as the indicated 2-oxo-tetrahydro-1,8-naphthyridine. ND is not determined.

Next, we tested the stability of the 2-oxo-tetrahydro-1,8-naphthyridine-based inhibitors toward metabolism by mouse liver microsomes in vitro. For comparison, in Table 1 we also include previous data for in vitro metabolism of the corresponding tetrahydroquinolines. Compound 19 was found to be about 3-fold more stable than the corresponding tetrahydroquinoline. For compound 20, the increase in stability is dramatic, > 20-fold. In vitro microsome metabolism studies were carried out as described 6. All compounds in Table 1 were found to be stable after incubation in aqueous buffer at pH 2 for 24 hr.
We tested 4 of the compounds for inhibition of mammalian protein prenyltransferases, and results are shown in Table 2.
Table 2.
Inhibition of rat PFT and protein geranylgeranyltranferase-I (PGGT-I) by 2-oxo-tetrahydro-1,8-naphthyridine-based PFTIs
It can be seen that the compounds are reasonably selective for the malarial versus rat PFT. For example, compounds 17, 18 and 20 at 5 nM inhibits malarial PFT by 32, 80 and 71%, but inhibit rat PFT at this concentration only by 0, 0 and 7%, respectively. All 4 compounds are very poor inhibitors of rat PGGT-I.
In conclusion, we have developed a new class of PFT inhibitors based on the 2-oxo-tetrahydro-1,8-naphthyridine scaffold that are more potent on malaria PFT than on the mammalian enzyme. These compounds are metabolically more stable than the tetrahydroquinoline lead compounds. The route of synthesis of these novel compounds was also developed. The most potent compounds in the series inhibit the malarial PFT with potencies in the low nanomolar range and kill cultured parasites in the hundreds of nanomolar range. Future studies will focus on the oral bioavailability and anti-malaria efficacy of these compounds in rodents.
Supplementary Material
Acknowledgements
This work was supported by funds fro the National Institutes of Health (AI054384) and the Medicines for Malaria Venture (Geneva).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nwaka S, Hudson A. Nat. Rev. Drug Discov. 2006;5:941. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- 2.Gelb MH, Brunsveld L, Hrycyna CA, Michaelis S, Tamanoi F, Van Voorhis WC, Waldmann H. Nat. Chem. Biol. 2006;2:518. doi: 10.1038/nchembio818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckner FS, Eastman RT, Yokoyama K, Gelb MH, Van Voorhis WC. Curr. Opin. Investig. Drugs. 2005;6:791. [PubMed] [Google Scholar]
- 4.Eastman RT, Buckner FS, Yokoyama K, Gelb MH, Van Voorhis WC. J. Lipid Res. 2006;47:233. doi: 10.1194/jlr.R500016-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Nallan L, Bauer KD, Bendale P, Rivas K, Yokoyama K, Horney CP, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, Hamilton A, Sebti S, Windsor WT, Weber PC, Buckner FS, Chakrabarti D, Gelb MH, Van Voorhis WC. J. Med. Chem. 2005;48:3704. doi: 10.1021/jm0491039. [DOI] [PubMed] [Google Scholar]
- 6.Van Voorhis WC, Rivas KL, Bendale P, Nallan L, Horney C, Barrett LK, Bauer KD, Smart BP, Ankala S, Hucke O, Verlinde CLMJ, Chakrabarti D, Strickland C, Yokoyama K, Buckner FS, Hamilton AD, Williams DK, Lombard LJ, Floyd D, Gelb MH. Antimicrob. Agents Chemother. 2007;51:3659. doi: 10.1128/AAC.00246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bendale P, Olepu S, Suryadevara PK, Bulbule V, Rivas KL, Nallan L, Smart BP, Yokoyama K, Ankala S, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, Buckner FS, Chakrabarti D, Verlinde CLMJ, Van Voorhis WC, Gelb MH. J. Med. Chem. 2007;50:4585. doi: 10.1021/jm0703340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokoyama K, Zimmerman K, Scholten J, Gelb MH. J. Biol. Chem. 1997;272:3944. doi: 10.1074/jbc.272.7.3944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.